Biocytin Recovery and 3D Reconstructions of Filled Hippocampal CA2 Interneurons
* These authors contributed equally
In This Article
Summary
The protocol outlined here describes the immunofluorescence analysis, biocytin recovery and high-quality reconstructions of hippocampal CA2 interneurons following the intracellular electrophysiological recordings in vitro, allowing neuronal characterization and ultimately fine neuronal anatomy to be studied.
Abstract
How cortical network activity processes information is of importance to a large number of basic and clinical scientific questions. The protocol described here identifies the basic building blocks of this circuitry. The in-depth studies of cortical regions will ultimately provide other scientists with the circuit components needed for an understanding of how the brain acquires, processes and stores information and what goes wrong in disease, while the electrophysiological and morphological data are widely used by computational neuroscientists in the construction of model networks that explore information processing. The protocol outlined here describes how biocytin-filled cells recorded in the CA2 region of the hippocampus are recovered and then reconstructed in 3D. Additionally, the protocol describes the demonstration of calcium binding protein or peptide content in recorded interneurons.
Introduction
The cortex and hippocampus are structures of such complexity that the classification of neuronal subtypes1,2,3,4, maps of the connections between them5,6,7,8,9,10,11 and how this circuitry supports cognitive functions12,13,14,15 are still under intense study and the subject of continuing debate. For example, to understand the details and complexities of the circuitry and to coordinate data obtained from many different studies, it is extremely helpful to be able to define and describe the components, but it remains a matter for debate how many different classes of neurons exist, or even whether it is possible to define all neurons as belonging to a specific class. Computational tools that can build and test circuits with varying degrees of complexity are being developed16,17,18, but central to these endeavors is the need for detailed studies of the cell types and of the properties of the connections between them. A large amount of information about the local circuitry of the neocortex of adult rats has already been collected using the protocol described here10,19,20,21,22. Although the "wiring diagram" is far from complete, some clear patterns or rules have emerged. Moreover, although some details vary, these rules are common to two mammalian species (rat and cat) and across several neocortical regions, allowing the development of basic building blocks that are likely to be equally applicable to human neocortex. The technique described here is used to extend our understanding of the functional map of cortical circuitry by identifying the presynaptic and postsynaptic neurons involved in connections in the regions that have not been studied in detail before, using a protocol that allows excellent tissue preservation and remarkable staining recovery in adult brain tissue. Data on the local circuitry in CA2 and neuronal properties in this subfield have been collected with this method by combining intracellular electrophysiological recordings (paired recordings with sharp microelectrodes) with biocytin filling, immunofluorescence, histological procedures and highly-detailed neuronal reconstructions, allowing direct comparison with the neighboring CA regions23,24,25.
The technique described in this article has been developed over the years to obtain detailed neuronal anatomy allowing the correct classification of the cells and high quality and accurate reconstructions of both their dendritic and axonal arbors, data that can be correlated with electrophysiological data collected from paired recordings using sharp electrodes. The histological protocol has been optimized to preserve the ultrastructure of the neurons and obtain excellent recovery of both dendritic (including spines) and axonal arbors. For example, the principle of the double fixation technique by firstly immersing in a fixative solution and secondly post-fixing in osmium tetroxide gives a good contrast for light microscopy26. A small amount of glutaraldehyde and picric acid solution are added to the fixative solution to enhance antibody penetration and to preserve the cells' ultrastructure as suggested in a previous study27. The permeabilization of the brain slices using the freeze-thaw method combined with cryo-protection with sucrose, rather than a traditional detergent, also provides optimal preservation of the tissues for detailed morphological analysis of the recorded cells. In addition, the visualization particularly of very fine structures is improved by reducing background staining, with the incubations with hydrogen peroxide (H2O2) and sodium borohydride (NaBH4). Adding nickel chloride (NiCl2) to the horseradish peroxidase (HRP) reaction to obtain a black pigment reaction product also increases the contrast.
The following protocol describes the procedures used to ascertain excellent tissue preservation and highly detailed neuronal 3D reconstructions following intracellular recordings in vitro. The description of the slice preparation, intracellular paired recordings using sharp electrodes and subsequent histological procedures used in our laboratory have been previously reported28. Although the protocol applies to the cells filled intracellularly with biocytin in 450–500 μm thick slices, the same protocol may be used following whole-cell recordings. However, the use of thinner slices will result in less complete reconstructions of the cells.
Protocol
All procedures used throughout this study were carried out according to the British Home Office regulations with regard to the Animal Scientific Procedures Act 1986.
1. Determination of Calcium Binding Protein or Protein Content of Interneurons and Biocytin Visualization Following Electrophysiological Recordings and Biocytin Filling
Note: At the end of the electrophysiological recordings, slices that contain biocytin-filled cells are fixed overnight prior to histological procedures. The fixative solution will be replaced with a single change of 0.1 M phosphate buffer the next morning, if the rest of the procedure is carried out on another day, in order to prevent tissue damage. The fixation solution (4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M phosphate buffer (PB)) must be made fresh on the day of the recordings for best results.
- At the end of the electrophysiological recording, carefully pick up the slice containing the recorded cell(s) with a paintbrush and place it in a pot containing artificial cerebral spinal fluid (ACSF).
NOTE: Details of the protocol used for slice preparation and intracellular recordings using sharp electrodes have been described previously28. - In a fume hood, carefully pick up the brain slice with a paintbrush and roll it onto a small piece of fine quality filter paper. Place another piece of moistened filter paper onto the slice and place the two pieces of wet filter paper in a small plastic pot containing 5-10 mL of fixative solution and store in the fridge overnight at 4 °C.
- Prepare a solution of gelatin in distilled water. Place 20 mL of distilled water in a beaker on a hot plate and heat it to 60 °C. Add progressively 2.4 g of gelatin to the water, wait until dissolved and allow it to cool to 35–40 °C before use to prevent tissue damage.
- Replace the fixative solution with 2 mL of 0.1 M PB. Place the slice in a Petri dish and cut away any excess tissue with a scalpel blade. Place the trimmed tissue in a Petri dish (diameter of 9 cm, height of 1.4 cm), ensuring that it lies flat with no folds or creases, and remove the excess buffer using a dry paintbrush.
- Cover the tissue with the warm gelatin solution and place the Petri dish onto a frozen block to quickly cool the solution.
- Using an angle-poise lamp to provide contrast, look through the side of the dish and keep the slice flat using light pressure from a fine paintbrush until the gelatin begins to set.
NOTE: The gelatin can be re-melted if the tissue does not lie flat. Any imperfections on the surface of the gelatin caused by the removal of the paintbrush as it solidifies may be removed by melting the surface layer gently by moving the bulb of the lamp close to the surface for a few seconds. - Move the dish of setting gelatin to the fridge and leave at 4 °C for 30-60 min.
- In a fume hood, cut out a small block (~ 1 x 1 cm) containing the gelatin-embedded tissue of the dish using a scalpel blade, lift the block using a small spatula and carefully place it in the same but fresh fixative solution used to fix the slices for at least 30 min at 4 °C.
- Wash the gelatin block in 5 mL of 0.1 M PB three times, dry it using a piece of paper tissue and stick the block side up (i.e., with the tissue at the top) onto a vibratome chuck using superglue.
- Remove excess glue with a piece of filter paper and use a scalpel blade to cut the corners of the block off, leaving a diamond shape.
- Section the slice at 50 µm thickness using a vibratome and place each section carefully in a glass vial containing 10% sucrose.
- Carefully pick up a section from the vial, place it flat into a Petri dish lid. Using a dissecting microscope and a fresh scalpel blade, remove the gelatin from around the section and return the section to a vial containing 2 mL of fresh 10% sucrose
NOTE: It is crucial to remove as much gelatin as possible at this stage to reduce tissue shrinkage during the dehydration step (Step 1.37). - Cryo-protect the sections in 0.1 M PB-based sucrose-glycerol solution at room temperature by incubating them for 10 min in 10% sucrose solution, 20 min in 20% sucrose-6% glycerol solution twice and finally 30 min in 30% sucrose-12% glycerol solution twice under constant agitation.
- Place the sections flat onto a small rectangle of tin foil using a paintbrush. Remove any excess liquid from the sections and carefully fold the tin foil into a parcel.
- Hold the parcel close to the surface of liquid nitrogen without touching the surface for 30 s and then allow the sections to thaw completely for approximately 30 s. Repeat the freeze-thaw another two times.
- Remove all sections with a paintbrush and place them in a glass vial containing 2 mL of 0.1 M PB under constant agitation to wash off excess sucrose.
- Remove the PB with a Pasteur pipette and incubate the sections in 2 mL of 1% aqueous H2O2 for 30 min. Wash the sections in 2 mL of 0.1 M PB 3x 5 min.
- Remove the 0.1 M PB with a Pasteur pipette and add 1 % sodium borohydride (NaBH4) in 0.1 M PB.
NOTE: Do not cap the vial, as NaBH4 solution gives off hydrogen gas. - Remove the sodium borohydride with a Pasteur pipette and wash the sections thoroughly in 2 mL of 0.1 M PB 5x 5 min.
- Replace the 0.1 M PB with 10% normal goat serum (NGS) in 0.1 M PB for 30 min.
- Remove the goat serum and incubate the sections overnight at 4 °C in a mixture of mouse monoclonal and rabbit polyclonal antibodies made up in ABC solution.
NOTE: A list of primary antibodies used in previous studies is displayed in Table 1 in Botcher et al. (2014)29. - Incubate the sections for 2 h in the dark in a mixture of fluorescently-labelled secondary antibodies (Table of Materials).
- Mount the sections onto the slides in mounting medium and cover with a coverslip.
- Take images of fluorescence labelling at 40X magnification (Figure 1Bb).
- After the fluorescence imaging, place the slide into a glass Petri dish containing PBS and carefully remove the coverslip. Then wash the sections off the slide using gentle pulses of PBS from a Pasteur pipette. Place the sections into a clean glass vial containing PBS.
- Perform the avidin-HRP reaction by firstly incubating the sections in ABC for at least 2 h to amplify the HRP reaction product.
- Prepare the 3,5 diaminobenzidine (DAB) solution by adding one tablet to 5 mL of distilled water.
- Wash the sections with PBS three times for 10 min and then with Tris buffer twice for 10 min. Remove the Tris buffer after the last wash.
- Quickly add one drop of 8 % NiCl2 solution to the DAB solution, pipette the solution in and out to mix and quickly add 1 mL of this solution over the sections. Incubate the sections in the DAB/NiCl2 solution for 15 min.
- Add 10 µL of 1% H2O2 to the DAB solution. Allow the reaction to proceed in the dark under constant agitation for about 1 to 2 min and monitor the labelling of the filled cells with a dissecting microscope.
- Stop the reaction by removing the DAB/NiCl2/H2O2 solution and wash the sections with Tris buffer twice for 5 min.
- In a fume hood, place a small circle of filter paper into a Petri dish and dampen it with 0.1 M PB. Lift the sections one at a time from the glass vial using a paintbrush, and place them carefully flat upon the paper.
- Cover the sections with another moistened circle of filter paper and remove excess buffer by gently touching tissue paper to the surface.
- Apply 8–9 drops of 1% osmium tetroxide in 0.1 M PB to the top paper, cover the dish and retain in the fume hood for at least 30 min, but no more than 1 h.
- Open the Petri dish and lift the top filter paper. Lift the sections carefully one at a time with a paintbrush, place them in a glass vial and rinse them in distilled water twice.
- Dispose of osmium tetroxide waste appropriately. Rinse all disposable equipment and place them in appropriate bins.
- Place each section flat onto a glass slide and coverslip the sections. Transfer the slide into a Petri dish, place an empty glass vial over the coverslip to retain it in place and cover with 50% alcohol. After 15 min, remove the slide from the solution and remove the sections from the slide. Place the sections back on the slide and then place the slide in 70% alcohol for 15 min. Repeat the same process with 95% and finally 100% alcohol solution.
- Following the dehydration step, transfer the sections to a glass vial containing 100% alcohol on a shaker in a fume hood. Replace the alcohol solution with propylene oxide (C3H6O) and wash three times for 5 min. Following the last wash, keep ~2 mL of propylene oxide in the vial and add resin (1:1 ratio). Ensure that the resin is dissolved and keep the sections under constant agitation for 30 min.
- Place each section in an aluminum planchette containing epoxy resin using a wooden stick and incubate overnight.
NOTE: Do not leave the sections in the resin longer than 24 h to avoid the risk of damaging the sections. - Place the planchette over a hot plate for approximately 10 min. Pick up each section with a wooden stick and place them on a clean slide. Keep the orientation of each section consistent using a dissecting microscope. Place a coverslip over the sections. Place the slide in the oven for 48 h at 56 °C for curing.
2. 3D Neuronal Reconstructions
NOTE: Neurolucida software is used. Instructions provided below only apply to a specific neurone reconstruction system (Table of Materials). The sections obtained from the cutting are matched prior to the reconstructions using a microscope.
- Place a slide on the stage and secure with stage clip and open the neuron reconstruction software. Click on Acquire tab and select Live image.
- Measure the thickness of each section using 100X oil objective lens. Make a note of the value on the z-meter at the top and bottom of each section and calculate the section thickness as the difference of the two values.
- Use a low-magnification objective to focus on the Home section containing the cell body. Then use a 100x objective, focus on the cell body and click in the center to mark the reference point.
- From the Trace tab, select Serial Section Manager. Then select Create New Section (+ icon) in the Serial Section Manager window. Enter the number of sections. Name each section in the correct Z order and enter the cut thickness measured in Step 2.2.
- To trace the soma in 3D using 100X objective, select Contour from the Trace tab and select the Cell Body contour. Use the joystick to move the focus to the very top of the cell body. Place the points by clicking around the perimeter of the part that is currently in focus. Right-click and select Close Contour to finish this first outline. Repeat this process at different z positions until the bottom of the cell body is reached (Figure 2).
NOTE: Select 3D visualize in Trace tab to visualize the cell body in 3D. - To trace the cell body in 2D using 100X objective, select Cell Body from the Neuron menu in the Trace tab. Focus on the middle of the cell body. Place the points by clicking around the perimeter of the cell body. To complete the cell body, right-click and select Finish Cell Body.
- To trace the dendritic arbor, select Dendrite or Apical Dendrite in the Neuron menu. First trace a short, initial segment for each dendrite using the mouse scroll wheel to adjust the diameter of the cursor to match the diameter of the dendrite. Trace along each dendrites using the joystick to move across the section and the mouse scroll wheel to adjust the diameter of the tracing.
- Check the alignment of the tracing with the live microscope image and adjust if necessary, especially after moving using the joystick. In the Trace tab, select Align Tracing, click on the tracing and then click on the correct location.
- When a node in the tree is reached, right-click and select Bifurcating Node or Trifurcating Node from the drop-down menu.
- When the end of a branch has been reached, select an ending from the drop-down menu in the Neuron menu. Select the correct ending type, i.e., High ending, normal ending or low ending to facilitate matching across sections.
- Reduce the magnification on the microscope once all dendrites in the current section have been traced, select the Joy Free tab and move to a section that matches immediately above or below the completed section.
- To identify matching points between the dendrites in a section that matches the completed section, click on the Move tab and select Match points. Select the number of points needed to be matched (three or more points is preferred) and then press OK. Click on the ending of a completed branch and then click on the branch. Repeat this for each match point. Repeat this process at 100X magnification to ensure accurate matching.
- Add matching branches to the previous section directly by right-clicking on the ending. Select Add to Ending when the branch lines up with the completed traced branch and trace as previously described.
- Once all the dendrites of each section have been traced, trace the axon using the same process by selecting Axon from the Neuron menu.
- Go to the Home section, re-align the reconstruction with the live microscope image and select Contours from the Trace tab. Select a pre-defined contour/layer border/region border and click to trace along a contour. Select End Open Contour. Trace all desired layers and region outlines.
- Use 3D visualize in Trace tab to visualize the reconstructions in 3D.
- To record videos of 3D reconstructions (Video 1), open the 3D reconstruction and open Create movies. Set a file destination and desired rotation speed. It is recommended to set the rotation to 270°. Select Start recording, record for a few seconds, then click Auto-rotate. Select Stop recording when required. Edit the video with a video editing software (Table of Materials).
- To export the files into Tiff or JPEG files, select File | Export | Export tracing as image. Choose a µm/pixel ratio and select Fit. Select a background color and select File. Name the file, select Tiff or JPEG and press Save. To export the files into vector files, select File | Export | Export tracing as vector files.
- Use a neuron reconstruction analysis software to perform morphometric analyses.
- To analyze dendritic trees and obtain a dendrogram, open the file in Neurolucida Explorer. Select Analyze | Structure and select Dendrogram from the drop-down menu (Figure 3).
- To perform a comprehensive morphometric analysis of the 3D reconstructions (branch complexity, volumes of branches and somas, surface area), open the file in the software. In the View tab, click Select all. Select the Analyze tab. Then select Structure and Branch Structure Analysis.
3. Trouble-shooting
- Change camera settings. For best results, set the exposure time to less than 100 ms and gain and offset as close to 0 as possible.
- If the neuron reconstruction software does not automatically display the traced contours as a 3D cell body in 3D visualize in Trace tab, select Select objects in Trace tab, hold down the Ctrl key on the keyboard and click on all the drawn contours, right click and then select Set cell body on the drop-down menu.
- To adjust the z-parameters of an individual branch, select the tree (Select objects in Trace tab) and set the z-adjustment of all points via the right-click menu drop down. Select Modify z position, then select Shift z value and enter the required value.
- Realign the reconstructions using 'Go To' in the ' Move' tab when it is necessary to move a large distance over the tracing.
- Add additional nodes at any point during the reconstruction process. Select the branch where the node is to be placed into (Select Object in Trace tab and click on the branch), right-click, and select Insert Node into Selected Tree. Then click on the right location on the branch.
- When reconstructing a complex neurone, trace matching branches individually and later splice them for easier identification of branches. Trace the branches in the new section individually and then attach these branches to the matching branches on the previous section by hovering over the ending of the branch to be spliced, right-click and select Splice, then hovering over the ending that the branch is to be spliced to and click.
- If a branch is traced under the wrong tree type, select the tree, right-click and select Change Tree Type, and select the correct tree type.
- If a point is incorrectly placed, perform Ctrl + Z, or click the Undo button in the Trace menu to remove the last point. However, this procedure will not work if the joystick is used to move across the section before attempting to remove a point. In this case, the point may be removed when the branch is ended. Select the branch (Press Select Object and click on the branch), hover over the point to be removed, right-click and select remove point.
- If unsure whether two branches are matched, either 1) use 3D Visualize in Trace tab to check whether the branches match in the z-plane, or 2) use Show Flanking feature in the Serial Section Manager to display sections immediately above and below the current section in grey.
- If a section is flipped, go to Tools in the Trace tab. Select Adjust XY Scaling and change X-axis to -1. Then select Correct Z-shrinkage and change z-axis to -1. Then trace branches as usual. Remember to reverse these changes when moving to a section which had the original orientation. Changing the x- and z-axis will not change branch ending labels, so take care when splicing that the correct endings are being used.
- Correct Z-shrinkage can be corrected, if there is a significant difference between the section cut thickness and the mounted thickness measured. In the Trace tab, select Tools, then Correct Z-shrinkage, and enter the shrinkage factor for the z-axis as a decimal representation of the cut thickness divided by the mounted thickness.
Representative Results
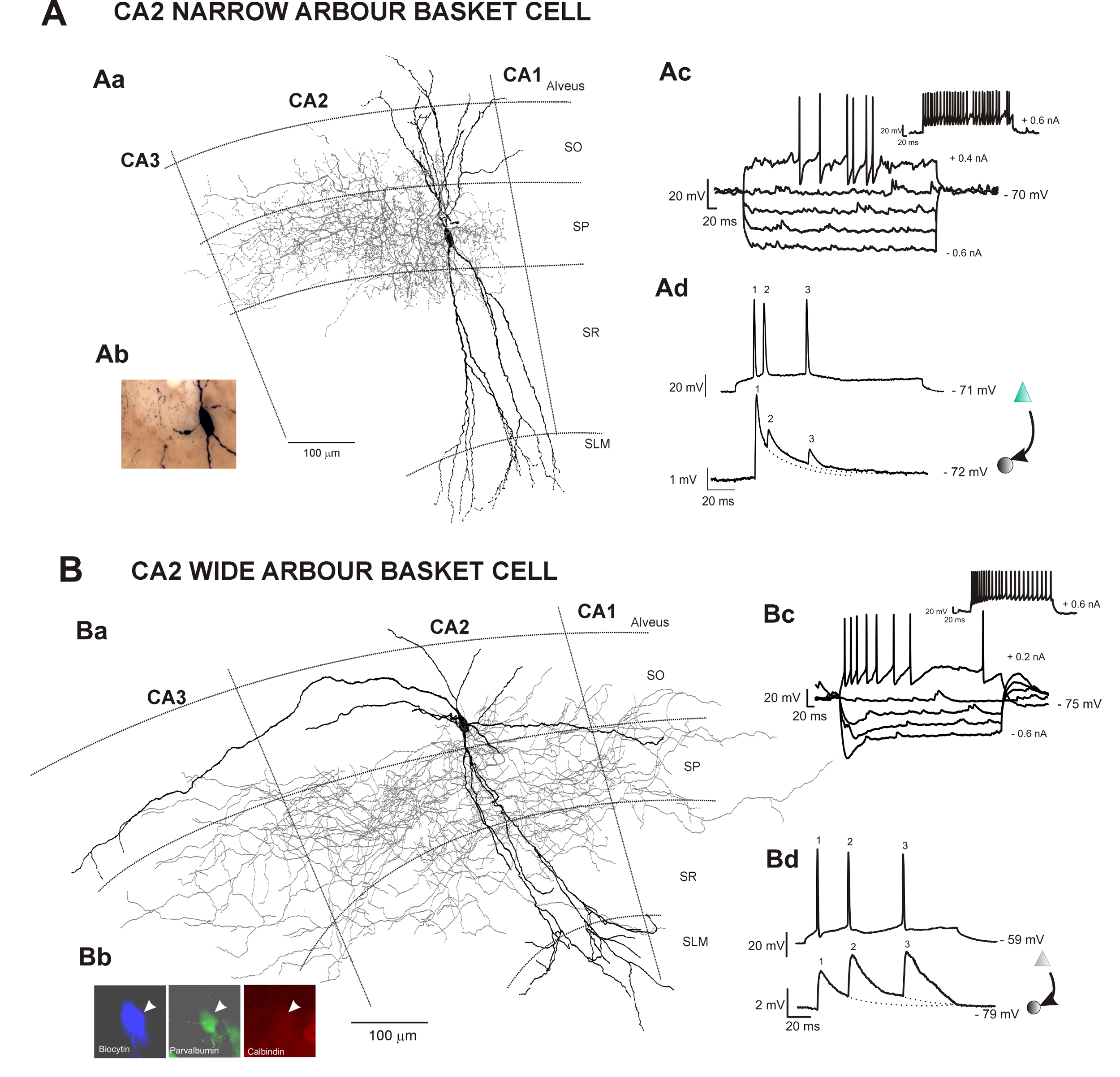
Neurons in hippocampal CA2 were filled with biocytin following electrophysiological recordings (Figures 1Ac, Ad and 1Bc, Bd). The slices were fixed overnight following the recordings and the neurochemistry and morphological characterization of neurons were revealed following the protocol described here.
The calcium binding protein or protein content of filled interneurons was determined by incubating the slices first with primary antibodies and then with fluorescently labelled secondary antibodies. The firing characteristics of the interneurons during the recordings will dictate the choice of primary antibodies used. Avidin-AMCA was used to visualize the biocytin-filled interneuron and anti-mouse fluorescein isothiocyanate (FITC) and goat anti-rabbit Texas Red (TR) were used to characterize the interneuronal neurochemistry (Figure 1Bb).
Following the fluorescence visualization, an HRP protocol was used to reveal the biocytin (Figure 1Ab). The fine detailed anatomy of CA2 interneurons was then drawn in 3D using a neuron reconstruction software (Figure 2 and Figure 3a). A video of a 3D reconstruction of a basket cell recorded and filled in CA2 is displayed in Video. Neuronal reconstructions were considered as complete if both the dendritic and axonal arbors were confined within the depth of the 450-500 μm slice. Poor axonal arbor staining was assessed by the presence of truncated branches with open endings at the top or the bottom of the slice or by a staining that was limited to the axon initial segment and very proximal branches. Figure 4 represents the examples of a good and a poor HRP staining following biocytin visualization.
Morphometric analysis of 3D reconstructions (Figure 3) can be carried out to demonstrate branch complexity, soma surface area and the surface area and volume of the dendritic and axonal arbors.

Figure 1: Neuronal reconstructions of two types of basket cells recorded and filled in the hippocampal CA2 region and correlated electrophysiological data obtained following intracellular recordings in vitro. This figure has been modified from previous studies23,24. SO Stratum Oriens, SP Stratum Pyramidale, SR Stratum Radiatum, SLM Stratum Lacunosum Moleculare. (A) Aa: Reconstruction of a CA2 basket cell with restricted dendritic and axonal arbor using a drawing tube (1000X). The dendrites are in black, and the axon is in red. Ab: Image of the biocytin-filled basket cell following the avidin-HRP protocol described here. Ac: Representative trace of voltage responses to hyperpolarizing and depolarizing current injection of a CA2 basket cell with restricted dendritic and axonal arbor. Ad: Example of a CA2 pyramid to narrow arbor basket cell connections recorded using sharp electrodes. Composite excitatory post-synaptic potential (EPSP) averages show brief train depression apparent during responses to trains of three spikes. (B) Ba: 2D reconstruction of a CA2 basket cell with wide dendritic and axonal arbors using a drawing tube (1000X). The dendritic tree of this basket cell (in black) extended radially through all layers of the CA2 region and horizontally in SO and SP of the CA2 and CA3 regions. One horizontal dendrite also reached the CA1 region. The axon (in red) extended to the CA3 and CA1 regions. Bb: The biocytin-filled (AMCA staining) basket cell was PV-immunopositive (FITC staining) and CB-immunonegative (Texas-Red staining). Bc: Representative trace of voltage responses to hyperpolarizing and depolarizing current injection of a CA2 basket cell with wide dendritic and axonal arbor. Bd: Composite EPSP averages show brief train facilitation apparent during responses to trains of three spikes. Examples of other types of interneurons recorded in CA2 can be found in previous studies25,30. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: 3D cell body reconstruction. (A) 3D tracing of the cell body. View of the different contours traced at different z positions whilst focusing through the cell body. (B) 3D view of the different contours. (C) 3D view of the cell body at a different angle. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Morphometry analyses of 3D neuronal reconstructions. (A) 3D reconstruction of a CA2 narrow arbor basket cell. Each dendritic branch is represented by a color (green, blue, red and pink) and axon is in light blue. (B) Dendrogram of the basket cell representing the number of dendritic branches and length of each segment contained within spheres concentric with the soma and at 100 μm intervals from the soma. The colors on the dendrogram correspond to those of the dendrites in A. (C) Example of morphometric analysis performed on CA2 pyramidal cells (adapted from a previous study23). The number of dendritic branches was plotted against the distance from the soma of CA2 pyramidal cells. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Examples of good (A) and poor (B) HRP staining. (A) Biocytin recovery revealed a very well filled interneuron in CA2. The cell body (CB) is darkly stained and has clear outlines. The dendrites are beaded and displayed some spines (represented by red stars). The axonal arbor (A) is dense and presents small boutons. (B) Example of a poor dendritic and axonal staining of 2 pyramidal cells in CA2. The staining of the CB is faint with no clear outline. Poor staining is often associated with shorter electrophysiological recordings resulting in the presence of very few biocytin-filled branches. Please click here to view a larger version of this figure.
{kind=link}
Video 1: 3D neuronal reconstruction of a CA2 basket cell with restricted dendritic arbor (also referred to as CA2 narrow arbor basket cell) with its soma in stratum pyramidale, dendrites spanning all layers and axon in CA2 stratum pyramidale and adjacent stratum oriens and radiatum. Very few branches reached the proximal CA3 stratum oriens and CA3 stratum pyramidale. This cell was filled with biocytin following electrophysiological recordings and sections were processed with avidin-HRP following the protocol described here. Due to slicing, only the axon within the depth of the slice was recovered, though the dendrites are intact. Dendrites are in dark pink and axon in white. Layer and region boundaries have been added at the beginning of the video. 3D reconstruction by Georgia Economides- 3D video by Svenja Falk. The video was recorded with a neuron reconstruction software as stated in Step 2.17 and edited with a video editing software. Link to the video:30. Please click here to download this video.
| Solutions used | Composition/Instructions |
| Fixation solution | 4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M phosphate buffer (PB) |
| 0.1M Phosphate Buffer pH 7.6 | Add 100 mL of stock 1 M Phosphate Buffer to 900 mL of distilled water |
| Phosphate buffered saline (PBS) pH 7.4/7.5 | Add 10 mL of 0.1M phosphate buffer, 0.2 g of KCl and 8.76 g of NaCl to 990 mL of distilled water |
| TRIS buffer pH 7.5 | Dissolve 5.72 g of Tris Hydrochloride and 1.66 g of Tris Base in 50 mL of distilled water. Then make up to 1 L with distilled water. |
| Buffered glutaraldehyde and paraformaldehyde fixative solution | 4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M Phosphate buffer. |
| ABC solution | Solution to be made at least 30 min before use from the ABC kit. Add 1 drop of solution A and 1 drop of solution B to 2.5 mL of PBS. |
| Durcupan epoxy resin: | To make 20 pots: 20 g of component A, 20 g of component B, 0.6 g of component C and 0.4 g of component D- |
| Protect the balance from spills by covering the plate with a circle of filter paper. Carefully weigh the reagents into a tripour beaker in the proportions stated above. Mix thoroughly by vigorously stirring using two wooden sticks for at least 5 min. The mixture should become a uniform density dark brown colour. Place the beaker into the oven at ~50 °C for a maximum of 10 min to remove as many air bubbles as possible. NOTE: The resin will start to cure if you leave the beaker in the oven longer than 10 min. Decant the resin out into plastic pots or 5 mL syringes, date them and store in the -20 °C freezer ready for use. |
Table 1: Table of solutions.
Discussion
Electrophysiological recordings in vitro (Figure 1 Ac,d and Bc,d) combined with histochemical and immunohistochemical procedures enable the detailed morphology, calcium binding protein content and identity of adult cortical interneurons recorded to be revealed. In the CA2 region, this technique allowed the study of the local circuitry for the first time and revealed subclasses of interneurons that had not been previously described in CA1 or CA3: wide dendritic and axonal arbor basket cells (Figure 1B), bistratified cells and SP-SR interneurons.
The protocol described here has been optimized to preserve the ultrastructure of the neurons and obtain excellent recovery of both dendritic (including spines) and axonal arbors. Critical steps includes the use of the double fixation technique to enhance contrast for light microscopy26 and the addition of glutaraldehyde and picric acid solution to the fixative solution to enhance antibody penetration and preserve the neuronal ultrastructure27. Gentle freeze-thaw permeabilization gives better preservation of fine structure, while osmication and resin embedding reduce z-plane shrinkage28. In addition, the visualization of very fine structures (fine axons with small boutons for example) is improved by incubating the sections with H2O2 and NaBH4 to reduce background staining. Contrast can also be increased with the addition of NiCl2 to the HRP reaction.
The histological procedure detailed here offers excellent results in terms of reproducibility and reliability. However, the duration of the electrophysiological recordings will determine the quality of the biocytin/fluorescence staining, with shorter recordings usually associated with poor axonal staining. The choice of recording protocols (intracellular recordings using sharp electrodes vs. whole-cell patch clamping) may also influence biocytin retention and preservation of fine anatomy.
While the difficulties encountered in preserving fine structure during histological processing described here and the time taken to reconstruct at 100X magnification (1-4weeks depending on the complexity of the axon) are appreciated, this method gives an accurate representation of dendritic and axonal diameters. The use of less demanding protocols to reveal biocytin-labelling is understandable, however, these often preclude clear visualization of fine axonal branches. Detergents, to promote entry of Avidin-HRP to reveal the biocytin and antibodies, are often necessary in thick sections, but can disrupt fine structure. Neuroscientists search constantly for semiautomatic methods of reconstruction, but, for now and for axons especially, biocytin-HRP with manual reconstruction remains the gold standard31.
Highly detailed neuronal reconstructions, especially accurate drawings of axonal boutons and nodes, the presence or absence of myelin and more generally the drawing of complete axonal arbor, with the representation of accurate axon-diameter changes along its length, provide further information for accurate identification of a distinct type of interneurone. Although many interneurons may not fit exactly into a specific class, the technique described above provides correlated data on neuronal electrophysiological properties, the short-term plasticity associated with a specific type of connection and detailed neuronal reconstructions, allowing the wiring diagram, in the CA2 region, for example, to be studied in detail.
Fine, detailed structure is often simplified in computational models. While understandable, this results in the loss of the information that could prove critical in the future. Analysis of detailed 3D reconstructions with parallel synaptic data will allow the addition of further criteria for interneuronal classification. Data can be deposited in public repositories and used by modellers to explore the outcome of sporadic changes in axon diameter and myelination on action potential propagation computationally.
Acknowledgements
This research has received funding from Novartis Pharma (Basel) and the Medical Research Council awarded to Prof Alex Thomson, the Biotechnology and Biological Sciences Research Council (BBSRC- BB/G008639/1), the Physiological Society, the European Union's Horizon 2020 Framework Programme for Research and Innovation under the Specific Grant Agreement No. 720270 (Human Brain Project SGA1) and under the Specific Grant Agreement No. 785907 (Human Brain Project SGA2). We are extremely grateful to Prof Alex Thomson for setting up the protocol in other cortical regions, securing funding and for her continuous support for this project. Contributions made by lab-members who helped optimising the recovery protocol and reconstructed CA2 neurones are gratefully acknowledged: J. Deuchars, H. Pawelzik, D. I. Hughes, A. P. Bannister, K. Eastlake, H. Trigg, N. A. Botcher.
Materials
| Name | Company | Catalog Number | Comments |
| Avidin-7-amino-4-methylcoumarin-3-acetic acid (Avidin-AMCA) antibody- 20.8 mg/mL | Vector laboratories | A2008 | |
| Biocytin ≥98% (TLC) | Sigma | B4261 | |
| 3,5 diaminobenzidine (DAB) tablet, To prepare 5 mL | Sigma | D4293 | |
| Durcupan epoxy resin A | Sigma | 44611 | |
| Durcupan epoxy resin B | Sigma | 44612 | |
| Durcupan epoxy resin C | Sigma | 44613 | |
| Durcupan epoxy resin D | Sigma | 44614 | |
| Ethanol, puriss. p.a., absolute, ≥99.8% | Sigma | 32221 | |
| Gelatin | Sigma | 48723 | |
| Glutaraldehyde solution, grade I, 25 % in H2O | Sigma | G5882 | |
| Glycerol, ≥99% | Sigma | G5516 | |
| Goat serum | Sigma | G9023 | |
| Goat anti-mouse fluorescein isothiocyanate (FITC)- 14.3 mg/mL | Sigma | F2653 | |
| Goat anti-rabbit Texas Red (TR)- 3.3 mg/mL | Invitrogen | T2767 | |
| Hydrogen peroxide, 30% solution | Sigma | H-1009 | |
| Immersion oil, viscosity 1.250 cSt (lit.) | Sigma | I0890 | |
| Nickel chloride (NiCl2 . 6H2O) | Sigma | N5756 | |
| Osmium tetroxide, for electron microscopy, 4% in H2O | Sigma | 75632 | |
| Paraformaldehyde, reagent grade, crystalline | Sigma | P6148 | |
| Picric acid, moistened with water, ≥98% | Sigma | 197378 | |
| Phosphate buffer 1 M | Sigma | P3619 | |
| Propylene oxide (C3H6O), 99% | Alfa Aesar | 30765 | |
| Sodium tetrahydroborate | VWR | 27885.134 | |
| Sucrose | Fisher scientific | S/8600/53 | |
| Trizma Hydrochloride, ≥99.0% | Sigma | T5941 | |
| Trizma base, ≥99.9% | Sigma | T6066 | |
| Vectashield Antifade Mounting Medium, , refractive index 1.45 | Vector laboratories | H-1000 | |
| Vectastain Elite ABC HRP kit | Vector laboratories | PK6100 | |
| Equipment used | |||
| Vibratome | Agar Scientific | ||
| C4A Cupped aluminium planchettes | GA-MA & ASSOCIATES, INC. | ||
| Leica DMR microscope | Leica Microsystems | ||
| X-Cite 120PC Q fluorescence light source | Excelitas Technologies | ||
| Leica DFC450 digital microscope camera | Leica Microsystems | ||
| Rapidograph technical drawing pen 0.18mm | London graphics centre | ||
| 0.18mm rapidograph nib | London graphics centre | ||
| Rapidograph technical drawing pen 0.25mm | London graphics centre | ||
| 0.25mm rapidograph nib | London graphics centre | ||
| Neurolucida system including PC workstation, stage, camera and joystic for XYZ stage control | Microbrighfield (MBF) Bioscience | ||
| Neurolucida software version 2017 | Microbrighfield (MBF) Bioscience | ||
| CorelDRAW graphics Suite X5 | Corel | ||
| Video editing software | Adobe Premiere Pro | ||
| Glass vials 14 mL | Fisher scientific |
References
- Klausberger, T., Somogyi, P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 321, 53-57 (2008).
- DeFelipe, J., et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nature Reviews Neuroscience. 14 (3), 202-216 (2013).
- Bezaire, M. J., Soltesz, I. Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity. Hippocampus. 23, 751-785 (2013).
- Katona, L., et al. Behavior-dependent activity patterns of GABAergic long-range projecting neurons in the rat hippocampus. Hippocampus. 27, 359-377 (2017).
- Ali, A. B., Thomson, A. M. Facilitating pyramid to horizontal oriens-alveus interneurone inputs: dual intracellular recordings in slices of rat hippocampus. Journal of Physiology. 507, 185-199 (1998).
- Ali, A. B., Thomson, A. M. Synaptic alpha 5 subunit-containing GABAA receptors mediate IPSPs elicited by dendrite-preferring cells in rat neocortex. Cerebral Cortex. 18, 1260-1271 (2008).
- Ali, A. B., Bannister, A. P., Thomson, A. M. Robust correlations between action potential duration and the properties of synaptic connections in layer 4 interneurones in neocortical slices from juvenile rats and adult rat and cat. Journal of Physiology. 580, 149-169 (2007).
- Pawelzik, H., Bannister, A. P., Deuchars, J., Ilia, M., Thomson, A. M. Modulation of bistratified cell IPSPs and basket cell IPSPs by pentobarbitone sodium, diazepam and Zn2+: dual recordings in slices of adult rat hippocampus. European Journal of Neuroscience. 11, 3552-3564 (1999).
- Pawelzik, H., Hughes, D. I., Thomson, A. M. Physiological and morphological diversity of immunocytochemically defined parvalbumin-and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. Journal of Comparative Neurology. 443, 346-367 (2002).
- Thomson, A. M., Lamy, C. Functional maps of neocortical local circuitry. Frontiers of Neuroscience. 1, 19-42 (2007).
- Kepecs, A., Fishell, G. Interneuron cell types are fit to function. Nature. 505 (7483), 318-326 (2014).
- Wilent, W. B., Nitz, D. A. Discrete Place Fields of Hippocampal Formation Interneurons. Journal of Neurophysiology. 97, 4152-4161 (2007).
- Hirsch, J. A., Martinez, L. M. Laminar processing in the visual cortical column. Current Opinion in Neurobiology. 16 (4), 377-384 (2006).
- Kay, K., Sosa, M., Chung, J. E., Karlsson, M. P., Larkin, M. C., Frank, L. M. A hippocampal network for spatial coding during immobility and sleep. Nature. 531, 185-190 (2016).
- Yu, J. Y., et al. Distinct hippocampal-cortical memory representations for experiences associated with movement versus immobility. eLife. 6, e27621 (2017).
- Markram, H., et al. Reconstruction and Simulation of Neocortical Microcircuitry. Cell. 163, 456-492 (2015).
- Van Geit, W., et al. BluePyOpt: Leveraging Open Source Software and Cloud Infrastructure to Optimise Model Parameters in Neuroscience. Frontiers in Neuroinformatics. 10, 17 (2016).
- Gal, E., et al. Rich cell-type-specific network topology in neocortical microcircuitry. Nature Neuroscience. 20, 1004-1013 (2017).
- Thomson, A. M., West, D. C., Wang, Y., Bannister, A. P. Synaptic connections and small circuits involving excitatory and inhibitory neurons in layers 2-5 of adult rat and cat neocortex: triple intracellular recordings and biocytin labeling in vitro. Cerebral Cortex. 12, 936-953 (2002).
- Mercer, A., West, D. C., Morris, O. T., Kirchhecker, S., Kerkhoff, J. E., Thomson, A. M. Excitatory connections made by presynaptic cortico-cortical pyramidal cells in layer 6 of the neocortex. Cerebral Cortex. 15, 1485-1496 (2005).
- West, D. C., Mercer, A., Kirchhecker, S., Morris, O. T., Thomson, A. M. Layer 6 cortico-thalamic pyramidal cells preferentially innervate interneurons and generate facilitating EPSPs. Cerebral Cortex. 16, 200-211 (2006).
- Bannister, A. P., Thomson, A. M. Dynamic properties of excitatory synaptic connections involving layer 4 pyramidal cells in adult rat and cat neocortex. Cerebral Cortex. 17, 2190-2203 (2007).
- Mercer, A., Trigg, H. L., Thomson, A. M. Characterization of neurons in the CA2 subfield of the adult rat hippocampus. Journal of Neuroscience. 27 (7), 7329-7338 (2007).
- Mercer, A., Eastlake, K., Trigg, H. L., Thomson, A. M. Local circuitry involving parvalbumin-positive basket cells in the CA2 region of the hippocampus. Hippocampus. 22 (1), 43-56 (2012).
- Mercer, A., Botcher, N. A., Eastlake, K., Thomson, A. M. SP-SR interneurones: a novel class of neurones of the CA2 region of the hippocampus. Hippocampus. 22 (8), 1758-1769 (2012).
- Sabatini, D. D., Bensch, K., Barrnett, R. J. Cytochemistry and electron microscopy: The Preservation of Cellular Ultrastructure and Enzymatic Activity by Aldehyde Fixation. Journal of Cell Biology. 17, 19-58 (1963).
- Somogyi, P., Takagi, H. A note on the use of picric acid-paraformaldehyde-glutaraldehyde fixative for correlated light and electron microscopic immunocytochemistry. Neuroscience. 7, 1779-1783 (1982).
- Hughes, D. I., Bannister, A. P., Pawelzik, H., Thomson, A. M. Double immunofluorescence, peroxidase labelling and ultrastructural analysis of interneurones following prolonged electrophysiological recordings in vitro. Journal of Neuroscience Methods. 101, 107-116 (2000).
- Botcher, N. A., Falck, J. E., Thomson, A. M., Mercer, A. Distributions of interneurones in the CA2 region of the rat hippocampus. Frontiers in Neuroanatomy. 8, 104 (2014).
- Mercer, A., Thomson, A. M. Cornu Ammonis Regions-Antecedents of Cortical Layers?. Frontiers in Neuroanatomy. 11, 83 (2017).
- Blackman, A. V., Grabuschnig, S., Legenstein, R., Sjöström, P. J. A comparison of manual neuronal reconstruction from biocytin histology or 2-photon imaging: morphometry and computer modeling. Frontiers in Neuroanatomy. 8, 65 (2014).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved