Structure Solution of the Fluorescent Protein Cerulean Using MeshAndCollect
* These authors contributed equally
In This Article
Summary
We present the use of the MeshAndCollect protocol to obtain a complete diffraction data set, for use in subsequent structure determination, composed of partial diffraction data sets collected from many small crystals of the fluorescent protein Cerulean.
Abstract
X-ray crystallography is the major technique used to obtain high resolution information concerning the 3-dimensional structures of biological macromolecules. Until recently, a major requirement has been the availability of relatively large, well diffracting crystals, which are often challenging to obtain. However, the advent of serial crystallography and a renaissance in multi-crystal data collection methods has meant that the availability of large crystals need no longer be a limiting factor. Here, we illustrate the use of the automated MeshAndCollect protocol, which first identifies the positions of many small crystals mounted on the same sample holder and then directs the collection from the crystals of a series of partial diffraction data sets for subsequent merging and use in structure determination. MeshAndCollect can be applied to any type of micro-crystals, even if weakly diffracting. As an example, we present here the use of the technique to solve the crystal structure of the Cyan Fluorescent Protein (CFP) Cerulean.
Introduction
Macromolecular X-ray crystallography (MX) is, by far, the most used method for gaining atomic resolution insight into the three-dimensional structures of biological macromolecules. However, a major bottle necks is the requirement for relatively large, well diffracting crystals.
Often, and particularly when crystallizing membrane proteins, only very small crystals of a few microns in the largest dimension can be obtained. Radiation damage effects limit the resolution of a complete diffraction data set that can be collected from a single micro crystal2, and very often, it is necessary to improve the signal to noise ratio and hence data set resolution, by merging several partial diffraction data sets from different, but isomorphic crystals. The increases in flux density of X-ray beams at synchrotron sources and elsewhere (e.g. X-ray free-electron lasers (X-FELs)), have meant that useful partial diffraction data sets can be collected from even very small crystals of biological macromolecules. This, in turn, has led to the development of new techniques for the collection and merging of partial diffraction data sets collected from many different crystals in order to produce a complete data set for structure solution. Such techniques are commonly referred to as serial crystallography (SX)3,4,5,6,7,8. A prototypical example of SX is the use of injector devices to introduce a narrow stream of a crystal slurry into the X-ray beam3,4,5. A diffraction pattern is recorded every time a crystal is exposed to X-rays leading to the collection, from many thousands of individual crystals, of 'still' diffraction images, information which is then merged to produce a complete data set. However, a considerable disadvantage of this type of serial data collection is that the processing of still images can be problematic. The data quality is considerably improved if crystals can be rotated and/or several diffraction images are collected from the same crystal during serial crystallography experiments6.
MeshAndCollect1 was developed with the aim of combining SX with 'standard' MX rotation data collection and allows, in an automatic fashion, experimenters to collect partial diffraction data sets from numerous crystals of the same macromolecular target mounted on the same or different sample holders. A complete diffraction data set is then obtained by merging the most isomorphous of the partial data sets collected. MeshAndCollect is compatible with any state-of-the-art synchrotron X-ray beamline for MX (ideally an insertion device facility with a relatively small (20 µm or less) beam size at the sample position). In addition to the compilation of complete data sets from a series of small, well-diffracting crystals, the method is also very suitable for the initial experimental assessment of the diffraction quality of micro-crystals and for the processing of opaque samples, e.g., in meso grown microcrystals of membrane proteins9.
At the start of a MeshAndCollect experiment, the positions, in two dimensions, of each of the many crystal contained in a single sample holder are determined using a low dose X-ray scan. The diffraction images collected during this scan are automatically analyzed by the program DOZOR1, which sorts the positions of the crystals on the sample holder according to their respective diffraction strength. Positions for the collection of partial data sets are assigned automatically based on a diffraction strength cut-off and, in the last step, small wedges of diffraction data, typically ±5° of rotation, are collected from each chosen position. Experience has shown that this rotation range provides a sufficient amount of reflections per crystal for partial data set scaling purposes, while at the same time, reducing possible crystal centering issues and the chance of exposing multiple crystals in a particularly crowded support1. The individual diffraction data wedges (partial data sets) are then processed either manually or using automated data processing pipelines10,11,12,13. For downstream structure determination it is then necessary to find the best combination of partial data sets to be merged14,15,16 after which the resulting complete data set can be treated in the same way as one originating from a single crystal experiment.
As an example of MeshAndCollect in practice, we present here the solution of the crystal structure of the Cyan Fluorescent Protein (CFP) Cerulean, using a diffraction data set constructed from the combination of partial data sets collected from a series of microcrystals mounted on the same sample support. Cerulean has been engineered from the Green Fluorescent Protein (GFP) from the jellyfish Aequorea victoria17, whose fluorescent chromophore is autocatalytically formed from the cyclisation of three consecutive amino acid residues. Cerulean is obtained from GFP by mutating the first and second residues of the chromophore, a serine and a tyrosine, to threonine (S65T) and tryptophan (Y66W) respectively and adapting the chromophore environment with further mutations (Y145A, N146I, H148D, M153T and V163A) to produce a significant, yet suboptimal fluorescence level of QY = 0.4918,19,20. The suboptimal fluorescent properties of Cerulean have been proposed to be linked to complex protein dynamics involving the imperfect stabilization of one of the eleven β-strands of the protein21 and to the accommodation of two different chromophore isomers depending on the pH and irradiation conditions22. We chose to work with Cerulean as a model protein illustrating the use of the MeshAndCollect protocol due to the relatively ease of tuning crystal size depending on the crystallization. The structure of Cerulean is very similar to that of its parent protein GFP, as it is constituted of a β-barrel formed of eleven β-strands surrounding an α-helix, which bears the chromophore.
Protocol
1. Expression and Purification of Cerulean
Note: This is based on the protocol published by Lelimousin et al.21
- Express His-tagged Cerulean in Escherichia coli BL21 cells grown at 37 °C in 4 L of auto inducible medium23 until OD600=1 and then incubate overnight at 27 °C.
- Harvest the bacterial cells at 5,000 x g and lyse the cells via sonication (40%, 5 min, 10 s pulse, 10 s pause) in 200 mL of buffer comprised of 20 mM Tris pH 8.0, 500 mM NaCl and 1x EDTA-free protease inhibitors.
- Load the supernatant on a His-trap Ni-NTA column and elute Cerulean with 100 mM imidazole in the same buffer conditions.
- Pool the bright yellow colored fractions. The protein is intrinsically colored, hence the Cerulean-containing fractions are easily distinguishable.
- Purify the protein (4 mL) on an S75 column in 20 mM Tris pH 8.0.
- Pool the bright yellow fractions and concentrate the protein solution to 15 mg/mL.
2. Crystallization
- Use the hanging drop vapor diffusion technique24 at 20 °C in Linbro plates. Fill the wells with 1 mL of a precipitant solution consisting of 100 mM HEPES at pH 6.75, 12% PEG8000 and 100 mM MgCl2. For the hanging drops, mix 1 µL of protein concentrated to 15 mg/mL with 1 µL of precipitant solution. Crystals should appear in 24 h.
- Harvest the crystals obtained and transfer them to 100 µL of a seeding buffer comprised of 0.1 M HEPES pH 6.75, 22% PEG 8000.
- Grind the crystals with a 0.1 mL tissue grinder and dilute in seeding buffer (ratio 1:100).
- Digest an aliquot of the protein stock solution (15 mg/mL) with trypsin (0.5 mg/mL in the same buffer) for 1 h (1:10 (v/v)).
- Mix the digested protein solution with 10% of seed-containing buffer (v/v).
- Grow crystals (10*10*20 µm3) in 0.1 M HEPES pH 7, 14% PEG 8000, 0.1 MgCl2 in 1-1.5 μL hanging drops using the vapor diffusion method.
3. Crystal Mounting
- Use a suitable loop, e.g., a mesh loop 700 square holes of 25 µm each mounted on a SPINE standard sample holder25. Transfer crystals from the crystallization drop (Step 2.6) into 1 µL of cryoprotectant solution (the well precipitant solution mixed with glycerol (20% v/v final)).
- Mount the protein crystal slurry onto a mesh loop by moving the loop under the crystals and lifting them out of the drop. Ideally the crystals should be in the size range of 5 µm – 30 µm in maximum dimension with no overlap between crystals mounted in the loop.
- Wick off excess liquid by touching the mount quickly with filter paper. Sediment the crystals so that they sit in the plane of the loop surrounded by as little bulk liquid as possible.
- Plunge the mount into a unipuck full of liquid nitrogen. Store the puck at 100 K in a suitable storage container until beam time is available.
4. Offline preparation of the synchrotron experiment
Note: Request synchrotron beam time as early as possible and follow the online guidelines for available access types and on how to submit an application for a given synchrotron. The ESRF guidelines can be found at http://www.esrf.eu/UsersAndScience/UserGuide/Applying. If a member of an ESRF Block Allocation Group (BAG), an application for each specific project is not required. In this case experimenters should approach their BAG Responsible concerning the scheduling of beam time.
- After the proposal is accepted and an invitation for the experiment is received, have all participants complete safety training. Fill in the “A-form” (via the ESRF user portal, http://www.esrf.eu/UsersAndScience/UserGuide/Preparing/new-a-form) with the required safety information on the samples. Contact the local contact person to discuss the experiment. Once your A-form is submitted and validated it will give you the experiment number and password.
- Connect to extended ISPyB26 (http://www.esrf.fr/) and choose MX.
- Log in with the experiment number and the password from the A-form.
- Select Shipment | Add New and fill in the requested information.
- Select Add Parcel and fill in the relevant data. Select Add Container, choose a unipuck and fill in the information required, including the positions of the sample holders in the puck.
5. Loading of the Sample onto a Beamline
- In the experimental hutch, load the puck into the sample changer (SC) dewar and note its position.
- Interlock the experimental cabin and enter the control hutch.
- Log in to the ISPyB (https://esrf.fr/). Select Prepare Experiment, find the shipment, select Next and indicate the beamline and the puck position in the SC.
- Log in into the beamline control software, here MXCuBE227,28 with the experimental number and password provided on the A-form.
- Press Sync to synchronize the beamline control software with the ISPyB database.
- Use the beamline control software, to mount the sample holder onto the goniometer. In MXCuBE2, right click a position in the sample changer area and select Mount Sample.
- Taking advantage of the MK3 mini-kappa goniometer29 installed at most of the ESRF MX beamlines, use MXCuBE2’s “visual realignment” workflow30 to align the plane of the sample holder with the rotation axis of the goniometer.
- Select the Centre button, then 3-click center on the middle of the edge of the tip of the loop. Save the centered position by selecting Save.
- Click again on the Centre button, then 3-click center the middle of the start of the stem of the loop. Save the second position as well by clicking on Save.
- Select one of the saved centered positions by clicking on it.
- Under Advanced, add the workflow Visual Reorientation to the MxCuBE2 data collection queue.
- Launch the workflow by clicking on Collect Queue.
- After the workflow aligns the plane of the sample holder with the rotation axis of the goniometer, center the sample holder again, this time somewhere in the middle of the mesh.
- Orient the sample holder so that the face of the mesh is perpendicular to the X-ray beam direction by rotating the omega axis using MXCuBE2.
- In MXCuBE2, select the beam size required for the scanning of the sample holder (only for beamlines with variable beam size).
- Click on the aperture drop down menu in the beamline control software and select a value, e.g., 10 µm.
- Define a mesh for the mesh scan.
- Click on the mesh tool icon in MXCuBE2. The mesh tool window will appear.
- In the sample view of MXCuBE2, draw the mesh by left clicking and dragging the mouse over the area containing crystals on the sample holder.
- To save the mesh click on the Plus button in the mesh tool window (mesh becomes green).
6. Prepare and Execute the MeshAndCollect Workflow
- In the Resolution field of MXCuBE2, enter the resolution (dmin) at which diffraction images should be collected, e.g., here 1.8 Å.
- Select MeshAndCollect in the Advanced data collection tab, add it to the queue and click Collect the Queue.
- In the parameter window which appears, use the beamline dependent default parameters. In the experiment described here defaults parameters are 0.037 s exposure time per mesh scan point, 100% transmission (leading in this case to 4 x 1011 ph/s), 1° oscillation per mesh scan line.
- Click Continue. The mesh scan runs and the diffraction images collected at each grid point are analyzed and ranked according to diffraction strength with the software DOZOR1. This process runs in the background.
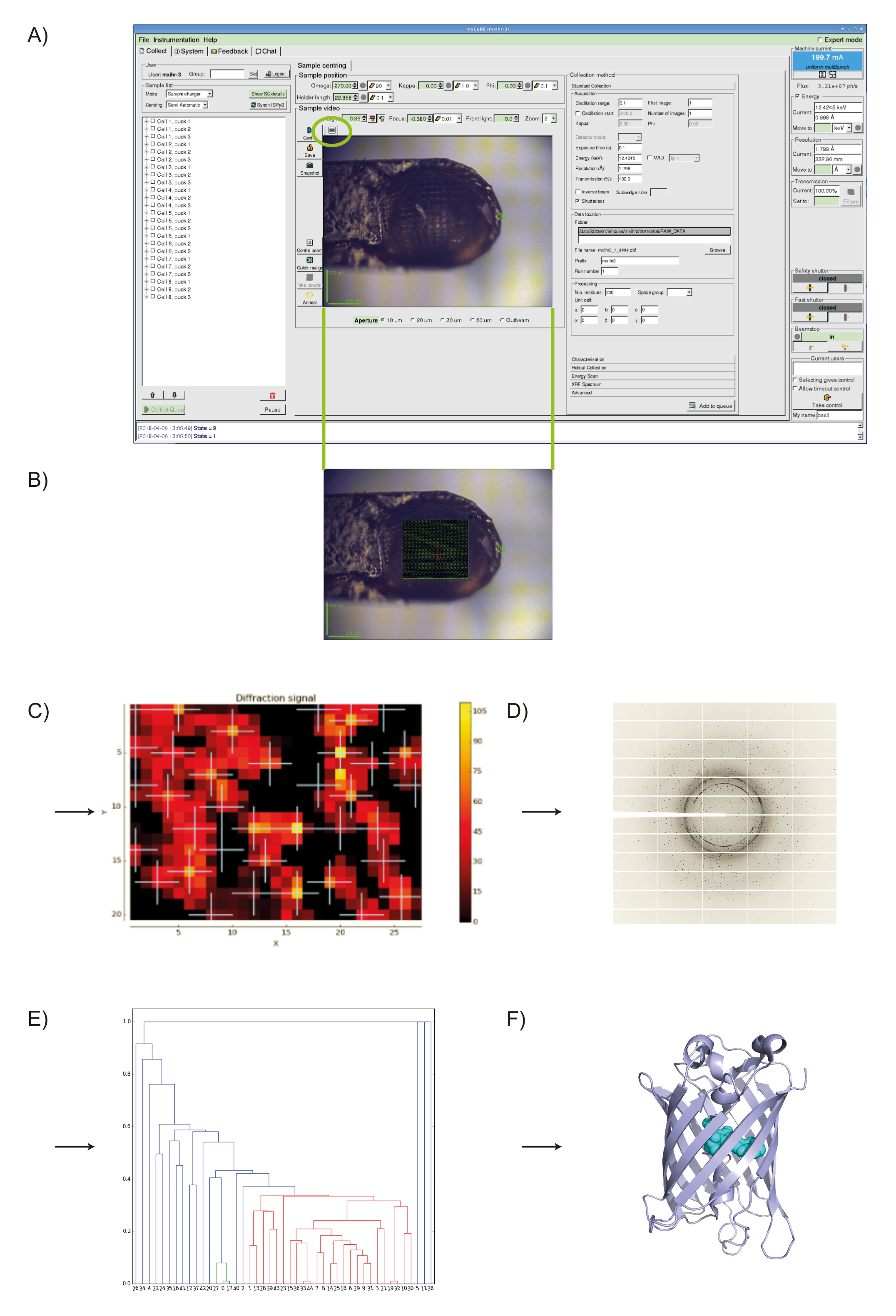
- After the DOZOR analysis a heat map is generated and the order for subsequent partial data collections is assigned automatically based on diffraction strength (see Figure 1).
Note: The results of this step can also be inspected in ISPyB. For the collection of partial data sets a new tab with settings pops up in the beamline control software, select suitable values for rotation range (i.e., 0.1°), number of images (i.e., 100), exposure time, resolution, transmission, inverse beam etc. Ideally the dose for each wedge to be collected should be below the Garman limit (30 MGy). The approximate exposure time per image is 0.037 s to 0.1 s in the described experimental conditions. - Click Continue to launch the partial data collections.
7. Data Processing
Note: The partial data sets are integrated with a suitable program (XDS10). For this a Python script will be used that recognizes each individual data set, integrates it and makes sure that indexing between the different partial data sets is consistent.
- Open the folder containing the images: /data/visitor/mxXXXX/beamline_name/date/RAW_DATA/Cerulean.
- Make a safety copy of the process subfolder that can be found in the folder where the partial data sets are collected.
- On the Linux terminal, use the command cp –r process process_backup.
- Navigate into the process folder and launch the processing script.
- On the Linux terminal, type the command cd process and hit enter.
- Type procMultiCrystalData and hit enter.
Note: The script will ask for a space group and cell parameters (this information is optional), enter those according to the instructions. After a last user confirmation, the script will run automatically.
8. Merging of Data Sets
Note: After all partial data sets are integrated the best combination of them are merged to produce the final data set for use in structure determination and refinement. Different aims of this merging process can be to obtain full completeness (highly recommended), high multiplicity or the best data statistics (high <I/σ(I)>, low R-factors, etc.). The latter can sometimes be at the expense of completeness and/or multiplicity so this option should be chosen with care.
- Merge the partial data sets using the program ccCluster14. It uses Hierarchical Cluster Analysis (HCA) to determine possible combinations of isomorphous partial data sets ().
- Type ccCluster in the Unix terminal to open its graphical user interface (GUI).
Note: In the ccCluster GUI a dendrogram is drawn. This gives a suggestion as to which partial data sets might be best merged based on isomorphism between them. - Click on a node that corresponds to a value of about 0.4 on the vertical axis. Generally, higher values will include more partial data sets but lead to worse merging statistics as partial data sets will be less isomorphous.
- Click on MERGE DATA. The selected cluster will be processed in the background and the estimated merging statistics will appear in a new tab in the GUI. This step can be repeated for different combinations of data sets. For a good combination of partial data sets the completeness should be close to 100%, the <I/σ(I)> values high (10 or higher in the lowest resolution shell) and the R-meas31 values low (around 5% in the low resolution shell).
- Type ccCluster in the Unix terminal to open its graphical user interface (GUI).
- For each combination selected use the generated input script to merge the partial data sets chosen into a single mtz file (i.e., pointless32).
- Definitively scale and merge the intensity data in this file using a scaling program (i.e., aimless32) and, as with a file originating from a single crystal data collection, use the output for subsequent phasing and structure solution33.
Representative Results
MeshAndCollect, as implemented in MXCuBE2 (see Figure 1A), was used for the collection of partial diffraction data sets from small crystals of Cerulean located on the same sample holder in which visual identification of crystals was difficult. To screen the sample holder, we drew a grid over the center of the meshloop (see Figure 1B) and based on the DOZOR score heat map (see Figures 1C, 1D) 85 partial diffraction data sets were automatically collected. These were individually integrated then merged (see above) to produce a data set with 99.8% completeness at dmin = 1.7Å (see Table 1). Half-set correlation (CC1/2)34 in the highest resolution shell was 60% ( = 4.7). As expected, the crystal structure of Cerulean could be straightforwardly solved by molecular replacement33 using the data set generated. After refinement, we obtained an Rwork of 22.8% and an Rfree of 25.4%. Superposition with the previously determined structure (PDB entry 2WSO21) shows a global rmsd on Cα positions of 0.1 Å.
| Statistics of the merged data set | |
| Clustering Threshold | 0.35 |
| Number of partial datasets | 25 |
| Space Group | P212121 |
| Unit Cell (a, b, c) | 50.98, 62.76, 69.50 |
| Resolution Range | 46.58-1.70 (1.73-1.70) |
| Rmerge (all I+ and I-) | 0.133 (0.743) |
| Rmeas (all I+ & I-) | 0.142 (0.813) |
| Rpim (all I+ & I-) | 0.047 (0.318) |
| Observations total/unique | 220693/25129 |
| Mean((I)/sd(I)) | 13.8 (4.7) |
| Mn(I) half-set correlation CC(1/2) | 0.994 (0.602) |
| Completeness | 99.8 (99.5) |
| Multiplicity | 8.8 (6.5) |
| Final Rcryst | 22.8 |
| Final Rfree | 25.4 |
Table 1: Statistics of the merged data set indicating the high quality of the data collected.

Figure 1: Using MeshAndCollect to collect a series of partial data sets from a series of small crystals contained in the same sample holder. A) User-interface of MXCuBE2. The green oval over the on-axis viewer field indicates the grid tool. B) With it a grid is drawn onto the image of sample holder in the life image field. C) Heat map of the DOZOR scores. D) Example of a diffraction image. E) Dendrogram after hierarchical cluster analysis. Data sets in red were used for merging. F) Overall structure of Cerulean. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The success of an MX experiment usually depends on the existence of relatively large, well diffracting crystals. For projects where optimization from small crystal showers to larger crystals fails, MeshAndCollect provides a possibility to obtain a complete diffraction dataset for structure solution via the combination of isomorphous partial data sets collected from a series of small crystals. The method is compatible with synchrotron beamlines for MX, ideally with a high photon flux and a small beam diameter, equipped with a state of the art diffractometer device and a fast-readout detector. On such an end station, the data collection part of such an experiment will take about 20 minutes, depending on the number of partial data sets to be collected and the number of crystal-containing sample holders to be analyzed.
The most important prerequisite for the success of a MeshAndCollect experiment is the existence of a sufficient number (at least 50, 100 ideally) of diffracting positions on the sample holder. From experience, the minimum size of the crystals to be analyzed should be about 5 µm in the smallest dimension. The method is compatible with any kind of standard cryo-cooling compatible sample holders with the best results being achieved using mesh mounts that are rigid and straight.
At the ESRF, MeshAndCollect is implemented in a user-friendly manner in a Passerelle (http://isencia.be/passerelle-edm-en) workflow30 available from the MXCuBE2 beamline control software. A major advantage of MeshAndCollect compared to other SX methods is that the data collected can be processed by standard programs and automated pipelines used for single crystal MX.
As our example shows, MeshAndCollect is very easy to apply and leads to a series of partial diffraction data sets, usually collected from small crystals, which can be merged to produce a complete data set for use in structure solution. Moreover, MeshAndCollect has the potential to open up the sampling space of protein crystallography as it provides a way to collect usable data from crystallization trials where the last optimization step, the production of large crystals, is unsuccessful.
In the light of the current developments towards brighter X-ray sources (e.g., Extremely Brilliant Source (EBS) project/ESRF35) it is foreseeable that due to increased radiation damage, the type of multi-crystal data collection facilitated by MeshAndCollect will become the standard method of data collection, rather than an exception – as is currently the case - at synchrotron-based MX beamlines.
Acknowledgements
We thank the ESRF for providing beam time through its in-house research program.
Materials
| Name | Company | Catalog Number | Comments |
| Beamline | ESRF ID 23-1 | ||
| Concentrators: Amicon Ultra-4 Ultracel -30K | Merck Millipore | UFC803024 | |
| Crystallization plates XDXm with sealant | Hampton Research | HR3-306 | |
| EDTA- free protease inhibitors | Roche | 4,693,159,001 | |
| Escherichia coli BL21 (DE3) | Life Technologies Thermo Fisher Scientific | C600003 | |
| glycerol | VWR Chemicals Prolabo | 14388.29T | |
| HEPES | Euromedex | 10-110-C | |
| His-trap HP | GE healthcare | 17-5247-01 | |
| imidazole | Sigma-Aldrich | 56750-500G | |
| MgCl2 | Sigma-Aldrich | 13452-1KG | |
| MicroMeshes 700/25 | MiTeGen | SKU: M3-L18SP-25L | |
| NaCl | Fisher Chemical | S/3160/60 | |
| PEG8000 | Sigma-Aldrich | P5413-500G | |
| Sonicator vibra cell 75/15 | SONICS | ||
| Superdex 75 10/300 -GL | GE healthcare | 17-5174-01 | |
| Tris base | Euromedex | 26-128-3094-B | |
| Trypsin | Sigma-Aldrich | T9201-1G | |
| Unipuck | Molecular Dimensions | MD7-601 | |
| Name | Company | Catalog Number | Comments |
| Programs | |||
| ISPyB | ESRF | Solange Delagenière, Patrice Brenchereau, Ludovic Launer, Alun W. Ashton, Ricardo Leal, Stéphanie Veyrier, José Gabadinho, Elspeth J. Gordon, Samuel D. Jones, Karl Erik Levik, Seán M. McSweeney, Stéphanie Monaco, Max Nanao, Darren Spruce, Olof Svensson, Martin A. Walsh, Gordon A. Leonard; ISPyB: an information management system for synchrotron macromolecular crystallography, Bioinformatics, Volume 27, Issue 22, 15 November 2011, Pages 3186–3192, https://doi.org/10.1093/bioinformatics/btr535 | local development |
| aimless | MRC Laboratory of Molecular Biology | Evans, P.R., Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallographica Section D Biological Crystallography. 69 (7), 1204–1214, doi: 10.1107/S0907444913000061 (2013). | |
| ccCluster | ESRF | Santoni, G., Zander, U., Mueller-Dieckmann, C., Leonard, G., Popov, A. Hierarchical clustering for multiple-crystal macromolecular crystallography experiments: the ccCluster program. Journal of Applied Crystallography. 50 (6), 1844–1851, doi: 10.1107/S1600576717015229 (2017). | local development |
| DOZOR | ESRF | Bourenkov and Popov, unpublished | local development |
| MeshAndCollect workflow | ESRF | Zander, U. et al. MeshAndCollect: an automated multi-crystal data-collection workflow for synchrotron macromolecular crystallography beamlines. Acta Crystallographica Section D Biological Crystallography. 71 (11), 2328–2343, doi: 10.1107/S1399004715017927 (2015). | local development |
| MXCuBE2 | ESRF | Gabadinho, J. et al. MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700–707, doi: 10.1107/S0909049510020005 (2010). De Santis, D., Leonard, G. Notiziario Neutroni e Luce di Sincrotrone,Consiglio Nazionale delle Ricerche. (19), 24–226 (2014). | local development |
| XDS | Max-Planck-Institut für Medizinische Forschung | Kabsch, W. XDS. Acta Crystallographica Section D Biological Crystallography. 66 (2), 125–132, doi: 10.1107/S0907444909047337 (2010) |
References
- Zander, U., et al. MeshAndCollect: an automated multi-crystal data-collection workflow for synchrotron macromolecular crystallography beamlines. Acta Crystallographica Section D Biological Crystallography. 71 (11), 2328-2343 (2015).
- Henderson, R. Cryo-Protection of Protein Crystals against Radiation Damage in Electron and X-Ray Diffraction. Proceedings of the Royal Society B: Biological Sciences. 241 (1300), 6-8 (1990).
- Chapman, H. N., et al. Femtosecond X-ray protein nanocrystallography. Nature. 470 (7332), 73-77 (2011).
- Schlichting, I. Serial femtosecond crystallography: the first five years. IUCrJ. 2 (2), 246-255 (2015).
- Stellato, F., et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ. 1 (4), 204-212 (2014).
- Gati, C., et al. Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ. 1 (2), 87-94 (2014).
- Coquelle, N., et al. Raster-scanning serial protein crystallography using micro- and nano-focused synchrotron beams. Acta Crystallographica Section D Biological Crystallography. 71 (5), 1184-1196 (2015).
- Diederichs, K., Wang, M. Serial Synchrotron X-Ray Crystallography (SSX). Protein Crystallography. 1607, 239-272 (2017).
- Borshchevskiy, V. I., Round, E. S., Popov, A. N., Büldt, G., Gordeliy, V. I. X-ray-Radiation-Induced Changes in Bacteriorhodopsin Structure. Journal of Molecular Biology. 409 (5), 813-825 (2011).
- Kabsch, W. XDS. Acta Crystallographica Section D Biological Crystallography. 66 (2), 125-132 (2010).
- Winter, G., et al. DIALS implementation and evaluation of a new integration package. Acta Crystallographica Section D Structural Biology. 74 (2), 85-97 (2018).
- Winter, G. xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography. 43 (1), 186-190 (2010).
- Monaco, S., et al. Automatic processing of macromolecular crystallography X-ray diffraction data at the ESRF. Journal of Applied Crystallography. 46 (3), 804-810 (2013).
- Santoni, G., Zander, U., Mueller-Dieckmann, C., Leonard, G., Popov, A. Hierarchical clustering for multiple-crystal macromolecular crystallography experiments: the ccCluster program. Journal of Applied Crystallography. 50 (6), 1844-1851 (2017).
- Zander, U., et al. Merging of synchrotron serial crystallographic data by a genetic algorithm. Acta Crystallographica Section D Structural Biology. 72 (9), 1026-1035 (2016).
- Foadi, J., et al. Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 69 (8), 1617-1632 (2013).
- Tsien, R. Y. The Green Fluorescent Protein. Annual Review of Biochemistry. 67 (1), 509-544 (1998).
- Heim, R., Prasher, D., Tsien, R. Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc Natl Acad Sci U S A. 91 (26), 12501-12504 (1994).
- Cubitt, A. B., Woollenweber, L. A., Heim, R. Chapter 2: Understanding Structure-Function Relationships in the Aequorea victoria Green Fluorescent Protein. Methods in Cell Biology. 58, 19-30 (1998).
- Rizzo, M. A., Springer, G. H., Granada, B., Piston, D. W. An improved cyan fluorescent protein variant useful for FRET. Nature Biotechnology. 22 (4), 445-449 (2004).
- Lelimousin, M., et al. Intrinsic Dynamics in ECFP and Cerulean Control Fluorescence Quantum Yield. Biochemistry. 48 (42), 10038-10046 (2009).
- Gotthard, G., von Stetten, D., Clavel, D., Noirclerc-Savoye, M., Royant, A. Chromophore Isomer Stabilization Is Critical to the Efficient Fluorescence of Cyan Fluorescent Proteins. Biochemistry. 56 (49), 6418-6422 (2017).
- Studier, F. W. Protein production by auto-induction in high density shaking cultures. Protein Expression and Purification. 41 (1), 207-234 (2005).
- Rhodes, G. . Crystallography made crystal clear: a guide for users of macromolecular models. , (2006).
- Cipriani, F., et al. Automation of sample mounting for macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 62 (10), 1251-1259 (2006).
- Delageniere, S., et al. ISPyB: an information management system for synchrotron macromolecular crystallography. Bioinformatics. 27 (22), 3186-3192 (2011).
- Gabadinho, J., et al. MxCuBE a synchrotron beamline control environment customized for macromolecular crystallography experiments. Journal of Synchrotron Radiation. 17 (5), 700-707 (2010).
- De Santis, D., Leonard, G. Notiziario Neutroni e Luce di Sincrotrone. Consiglio Nazionale delle Ricerche. , (2014).
- Brockhauser, S., Ravelli, R. B. G., McCarthy, A. A. The use of a mini-κ goniometer head in macromolecular crystallography diffraction experiments. Acta Crystallographica Section D Biological Crystallography. 69 (7), 1241-1251 (2013).
- Brockhauser, S., et al. The use of workflows in the design and implementation of complex experiments in macromolecular crystallography. Acta Crystallographica Section D Biological Crystallography. 68 (8), 975-984 (2012).
- Diederichs, K., Karplus, P. A. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nature Structural Biology. 4, 269 (1997).
- Evans, P. R., Murshudov, G. N. How good are my data and what is the resolution?. Acta Crystallographica Section D Biological Crystallography. 69 (7), 1204-1214 (2013).
- Taylor, G. L. Introduction to phasing. Acta Crystallographica Section D Biological Crystallography. 66 (4), 325-338 (2010).
- Karplus, P. A., Diederichs, K. Linking Crystallographic Model and Data Quality. Science. 336 (6084), 1030-1033 (2012).
- Dimper, R., Reichert, H., Raimondi, P., Ortiz, L. S., Sette, F., Susini, J. ESRF upgrade programme phase II (2015 - 2022). The orange book. , (2015).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved