Absorbent Microbiopsy Sampling and RNA Extraction for Minimally Invasive, Simultaneous Blood and Skin Analysis
In This Article
Summary
In this article, we demonstrate how the absorbent microbiopsy technique is performed and how the sample can be used for RNA extraction for simple and simultaneous sampling of skin and blood in a minimally invasive manner.
Abstract
Conventional skin biopsy limits the clinical research that involves cosmetically sensitive areas or pediatric applications due to its invasiveness. Here, we describe the protocol for using an absorbent microneedle-based device, absorbent microbiopsy, for minimally invasive sampling of skin and blood mixture. Our goal is to help facilitate rapid progress in clinical research, the establishment of biomarkers for skin disease and reducing the risk for clinical research participants. In contrast to conventional skin biopsy techniques, the absorbent microbiopsy can be performed within seconds and does not require intensive training due to its simple design. In this report, we describe the use of absorbent microbiopsy, including loading and application, on a volunteer. Then, we show how to isolate RNA from the absorbed sample. Finally, we demonstrate the use of quantitative reverse transcription PCR (RT-qPCR) to quantify mRNA expression levels of both blood (CD3E and CD19) and skin (KRT14 and TYR). The methods that we describe utilize off the shelf kits and reagents. This protocol offers a minimally invasive approach for simultaneous sampling of skin and blood within the same absorbent microbiopsy matrix. We have found human ethics committees, clinicians and volunteers to be supportive of this approach to dermatological research.
Introduction
Skin biopsy is one of the most essential techniques in dermatology for skin sampling and subsequent diagnosis of skin diseases through histopathological assessment. The biopsy technique involves a medical professional using a blade or punch biopsy to remove the suspicious lesion on patient's skin for examination1. Though the technique is effective, it is highly invasive and limits clinical research as the end point usually involves molecular biology techniques2,3. Molecular analysis of skin diseases has the potential to provide highly specific biological information that histopathological analysis cannot, thus facilitating drug discovery and disease diagnosis4,5. Besides, the sample demand in most molecular techniques is comparatively small and may lead to a reduction in animal use and permit a larger number of replicates. Therefore, there is a clear need for an alternative technique that enables molecular analysis in clinical research and lowers risk for participants.
To address such a need in the field, our group has developed a novel microneedle-based diagnostic platform, absorbent microbiopsy, that enables the collection of a tiny amount of skin mixed with blood in a simple and minimally invasive manner6. The purpose of this publication is to describe the absorbent microbiopsy as a sampling tool to facilitate molecular analysis through RNA extraction in clinical research.
Previously, we have described the first version of microbiopsy, skin microbiopsy, which consists of a microneedle made of a three-layer steel plate design to extract tiny pieces of skin tissues7. The novelty of this device comes from the multiple contact points from the microneedle that permits efficient tissue extraction3. In contrast, a circular skin punch biopsy provides only one contact point and simply tears the skin without capturing any sample in some cases. Based on the skin microbiopsy, we recently developed the absorbent microbiopsy which has both blood and skin sampling capabilities. The device has been shown to be feasible for use in resource-poor areas in a recent epidemiological study6.
Due to its simple design, the absorbent microbiopsy can be performed within a few seconds and does not require extensive training. Additionally, local anesthetic is not needed, and the application site does not cause scarring. The present protocol enables researchers or medical professionals without relevant sampling training to obtain targeted skin data for molecular analysis. We expect microsampling devices to become routine in skin research in the future.
Although the microbiopsy has been reported in other skin disease studies that involved molecular diagnostic techniques6,8,9,10, such as human papilloma virus DNA detection, this protocol is the first to demonstrate the details of the sample extraction and processing techniques for the absorbent microbiopsy. Further, this is the first report describing the relative gene expression profiling of skin and blood cells in microbiopsy samples.
Protocol
The study was approved by Metro South Human Research Ethics Committee and the University of Queensland Human Research Ethics Committee (HREC-13-QPAH-551 and UQ2013001551) and the University of South Australia Human Ethics Committee (200607).
1. Absorbent Microbiopsy Fabrication
- Laser cut microneedle parts from 50 µm medical-grade stainless steel sheet and absorbent layer (filter paper), respectively (Figure 1).
Note: Other parts, including the plunger and the case, of the device are fabricated with rapid-prototyping techniques, such as 3D printing. - Soak all the parts, except for the absorbent layer, in 70% ethanol (EtOH) for 5 min and air dry them for 30 min afterwards.
- Assemble the three-layer microneedle, with the absorbent layer in the middle (Figure 1a).
- Insert the assembled microneedle into the slit of plunger.
- Put on the spring and insert the plunger into the case (Figure 1c). Avoid the microneedle touching anything in the assembling procedure to ensure the needle quality.
- Seal the assembled absorbent microbiopsy in a package for γ-radiation sterilization before using.
2. Sampling with the Absorbent Microbiopsy
- Wear disposable gloves, and spray hands and tools, such as forceps, with 70% EtOH.
- Sanitize the application site with an alcohol wipe.
- Remove the microbiopsy from the γ-radiation sterilized package without touching the microneedle in the process.
- Load the device by pulling the plunger to compress the spring until there is a ‘click’ sound and the plunger locks in place.
- Aim the device at the skin to be sampled, ensuring that the sampling area is in a fixed position.
- Ensure that the device is approximately perpendicular to the skin, then apply ≥1 kg of force to the device onto the skin.
Note: Applying force downwards into the skin is required to ensure sufficient penetration of the microneedle. - Press the trigger and hold the device in place for a minimum of 10 s for the blood sample to be absorbed.
Note: If the device is released before 10 s, there is less sample captured.
3. RNA Extraction
Note: The RNA extraction procedures were modified from manufacturers’ protocol for optimal isolation of RNA from microbiopsied sample. All the reagents and column used in RNA extraction are included in the kit unless indicated otherwise.
- Pull out the plunger and absorbent microneedle from the case by hands gently. Ensure that the tip of the microneedle does not come into contact with anything during removal.
- Remove the absorbent microneedle from the plunger. Hold onto the two holes on the microneedle with a pair of sterile forceps and pull it out.
- Put the intact microneedle in a 2 mL microcentrifuge tube (not from the RNA isolation kit; see Table of Materials) prefilled with 50 µL of Extraction Buffer, with the needle side facing down towards the bottom. To minimize sample loss, leave the tube on dry ice for 5 min before processing the sample.
- Vortex briefly (3 - 5 s) to remove some of the sampled materials from the tip.
- Incubate the microneedle in the tube on a rocker for 30 min at 42 °C.
Note: The buffer solution will be absorbed into the absorbent layer immediately. - Place the top part of the microneedle (the opposite side from the needle) level with the top edge of the 2 mL microfuge tube using sterile forceps.
- Close the lid of the tube such that the top of the microneedle is held in place between the cap and tube.
- Spin the tube at 16,000 x g or at maximum speed for 30 s to remove the sampled materials from the absorbent layer of the device.
- Remove the microneedle carefully with forceps without dipping back into the buffer solution. Keep the tube and discard the microneedle.
- Centrifuge the samples at 3,000 x g for 2 min. Pipette the supernatant containing the extracted RNA carefully into a new microcentrifuge tube.
- Add 250 µL of Conditioning Buffer onto the purification column filter membrane and incubate for 5 min at room temperature.
- Centrifuge the column in the collection tube at 16,000 x g or at maximum speed for 1 min.
- Add 50 µL of 70% EtOH into the collected sample from Step 3.9. Mix well by gently pipetting up and down.
- Transfer the mixture (~100 µL) into the preconditioned purification column.
- Centrifuge for 2 min at 100 x g immediately, followed by a centrifugation at 16,000 x g for 30 s to remove the flow-through.
- Add 100 µL of Wash Buffer 1 (W1) into the purification column and centrifuge for 1 min at 8,000 x g.
- Prepare DNase solution by adding 5 µL of DNase I Stock Solution to 35 µL of Buffer RDD in a separate microcentrifuge tube for each sample. Mix by gently inverting.
Note: The DNase treatment is to be performed on the purification column, instead of in the PCR plate afterwards, because of the quantity of the sample. - Add 40 µL of DNase incubation mix directly into the purification column membrane. Incubate at room temperature for 15 min.
- Add 40 µL of W1 into the purification column membrane. Centrifuge at 8,000 x g for 15 s.
- Pipette 100 µL of Wash Buffer 2 (W2) into the purification column after the DNase treatment and centrifuge for 1 min at 8,000 x g.
- Add another 100 µL of W2 into the purification column and centrifuge for 2 min at 16,000 x g. Check the purification column for any residual wash buffer. Re-centrifuge at 16,000 x g for 1 min if any wash buffer remains.
- Transfer the purification column to a new 0.5 mL microcentrifuge tube provided in the kit.
- Pipette 11 µL of RNase-free water (not provided in the RNA isolation kit), instead of Elution Buffer (EB), directly onto the membrane of the purification column. Gently touch the tip of the pipette to the surface of the membrane while dispensing the RNase-free water to ensure maximum absorption of RNase-free water into the membrane. Incubate at room temperature for 2 min.
- Centrifuge the column for 1 min at 1,000 x g to distribute the RNase-free water in the column, then centrifuge for 16,000 x g at 1 min to elute RNA.
Note: RNA quantification is not recommended due to the small quantity of the sample. - Stop point #1: Store the total RNA sample at -80 °C if cDNA synthesis is not performed immediately.
Note: Avoid freezing if not necessary given the low concentration of RNA sample.
4. cDNA Synthesis
- Thaw the frozen sample on ice (from Step 3.25).
- Synthesize cDNA from total RNA with cDNA synthesis kit according to the procedure below.
- Prepare the reaction mix: 11 µL of total RNA, 4 µL of 5x TransAmp Buffer, 1 µL of reverse transcriptase, 4 µL of DNase/RNase-free water. Mix gently by pipetting.
- Run reverse transcription on a thermal cycler apparatus: 25 °C for 10 min, 42 °C for 15 min, followed by a final reverse transcriptase inactivation step at 85 °C for 5 min.
- Stop point #2: Store the reverse transcribed sample at -80 °C until use.
5. qPCR Reaction and Data Analysis
Note: The primers for qPCR reactions were designed to span intron boundaries to avoid the amplification of genomic DNA using NCBI Primer BLAST (www.ncbi.nlm.nih.gov/tools/primer-blast/). The reference gene used in this study is RPLP0 (see the Discussion section for more information).
- Thaw the frozen sample on ice (from Step 4.5).
- Perform qPCR with the qPCR master mix kit according to the procedure below.
- Prepare master mix containing the followings per reaction: 5 µL of 2x qPCR reaction mix, 0.4 µL of 10 µM forward primer, 0.4 µL of 10 µM reverse primer, 2.2 µL of DNase-free water. Mix gently by pipetting. Set up the samples in triplicates.
- Pipette 8 µL of the master mix into each well of a 384-well PCR plate first and then 2 µL of cDNA from each sample (a total of 10 µL per well).
Note: No template control (NTC) and no reverse transcriptase control (-RT) are included as negative controls. - Seal the plate with adhesive film, and centrifuge briefly.
- Run the amplification reaction on a real-time thermal cycler: 95 °C for 2 min (polymerase activation), followed by 40 cycles of 95 °C for 5 s (denaturation), 60 °C for 10 s (annealing) and 72 °C for 10 s (extension).
- Calculate the RNA concentration of samples by interpolating from a standard curve prepared from templates with known concentrations.
- Create a New Experiment in the software.
- Click Define and Set Up Standards to set up serial dilution for plates.
- Select and arrange the wells for standards and samples. Click Apply.
- Click Analyze in the Result tab to assess the standard curve. Confirm the slope, R2 value, amplification efficiency and error meet the experimental criteria.
- Check the quantities of the unknown samples visually on the standard curve based on their Ct values.
Note: The construction of standard curve, including the choice of dilution factor, is performed according to published protocols11,12,13.
- Perform gene expression analysis of mRNA by using the ΔCt method (Ct is normalized to a refence gene).
- Subtract the Ct value of target gene from the Ct value of reference gene to obtain the ΔCt value.
- Average the ΔCt values of technical replicates.
- Plot and analyze the gene expression with statistics software11,14 (see Table of Materials).
Note: Alternatively, the gene expression analysis is performed according to published protocols13.
Representative Results
We have previously reported that the microneedle of the skin microbiopsy penetrates the skin approximately 500 µm deep7. The microneedle design of skin microbiopsy is highly similar to the one on absorbent microbiopsy (Figure 2a). While the absorbent microbiopsy consisted of an absorbent layer in the middle for blood absorption, the skin microbiopsy contained a channel for mechanical capturing of skin tissues. The use of absorbent layer also led to a slight difference in the microneedle dimension (absorbent: 1.50 x 0.50 x 0.21 mm vs skin: 1.50 x 0.50 x 0.15 mm).
Figure 2b shows the application sites 5 min after absorbent and skin microbiopsies were applied on the left volar arm of male volunteer. Due to the similarity between the two microneedle designs, the application sites from both devices were comparable, with minor erythema. Both application sites were not noticeable after 48 h with no scars left behind. This supports the hypothesis that this minimally invasive device has the potential to help screen multiple application sites or to be performed on a regular basis.
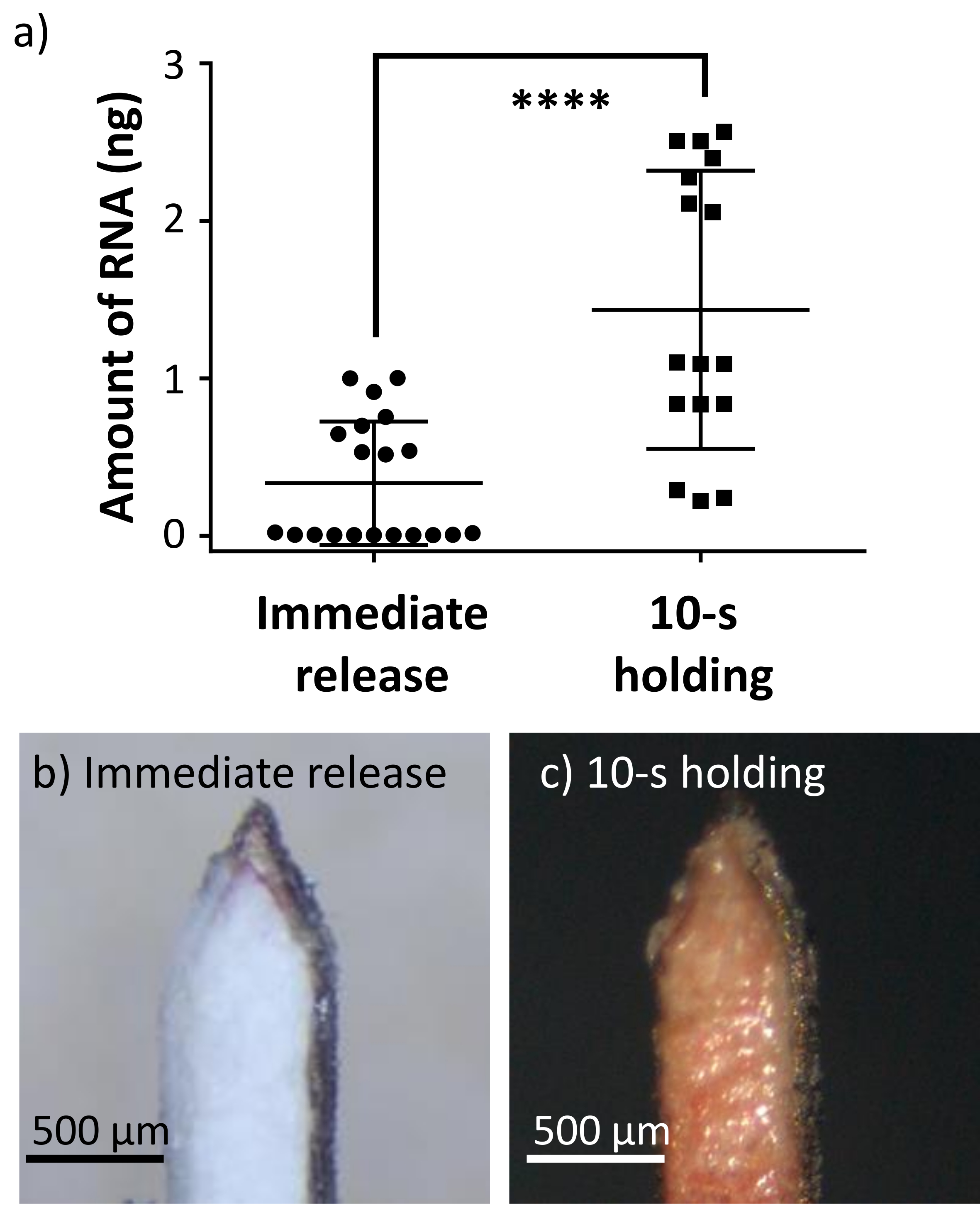
Figure 2c shows a representative picture of the absorbent layer of the absorbent microbiopsy after being applied to the volar arm of a male volunteer. As shown in the figure, a few tiny pieces of skin were captured near the tip of the microneedle, and some blood was absorbed into the filter paper. This indicates that the device penetrated the skin and captured both skin and blood simultaneously within the same absorbent microbiopsy matrix. Figure 2d shows a post-sampling skin microbiopsy, the previous generation of microbiopsy, for comparison. The channel of the skin microbiopsy captured a small piece of skin, but the amount of blood on the microneedle was small relative to the absorbent microbiopsy. Both application sites were not noticeable within 48 h. In the experiment, the absorbent microbiopsy was applied with a 10-s holding time post-application, while the skin microbiopsy was immediately released after applying due to the difference in design.
As shown in Figure 3a, two groups of absorbent microbiopsy were involved in this experiment based on the application protocol: 'Immediate release' and '10-s holding'. For the 'Immediate release' group, the device was applied using the same original skin microbiopsy protocol, with the device being removed from the application site immediately after application. For the '10-s holding' group, the device was held in place after the application for 10 s to improve sample collection. The two groups of absorbent microbiopsy were set up to demonstrate how the application approach could affect the sample amount. The 10-s holding time was chosen as a clinically reasonable time to demonstrate that the application time affects the amount of recoverable sample.
The amount of RNA recovered from the two absorbent microbiopsy groups were 0.33 ± 0.39 ng for 'Immediate release' and 1.43 ± 0.88 ng for '10-s holding' (Figure 3a, n = 20), suggesting a 4-fold increase with the extra holding time. This indicates that applying the absorbent device with the 10-s holding time resulted in more RNA extracted. The difference may be due to the presence of increased blood sample in the absorbent layer (Figure 3c). Indeed, the 'Immediate release' group (Figure 3b) failed to collect a similar amount of blood with the absorbent layer as compared to the '10-s holding' group (Figure 3c). It should also be noted that most immediately released microbiopsies were close to the x-axis, suggesting they were negative events or displayed a very low amount of RNA. Therefore, the result validated the hypothesis that the holding time would have an impact on the performance of the device as it might take time for the blood to be absorbed by the absorbent layer.
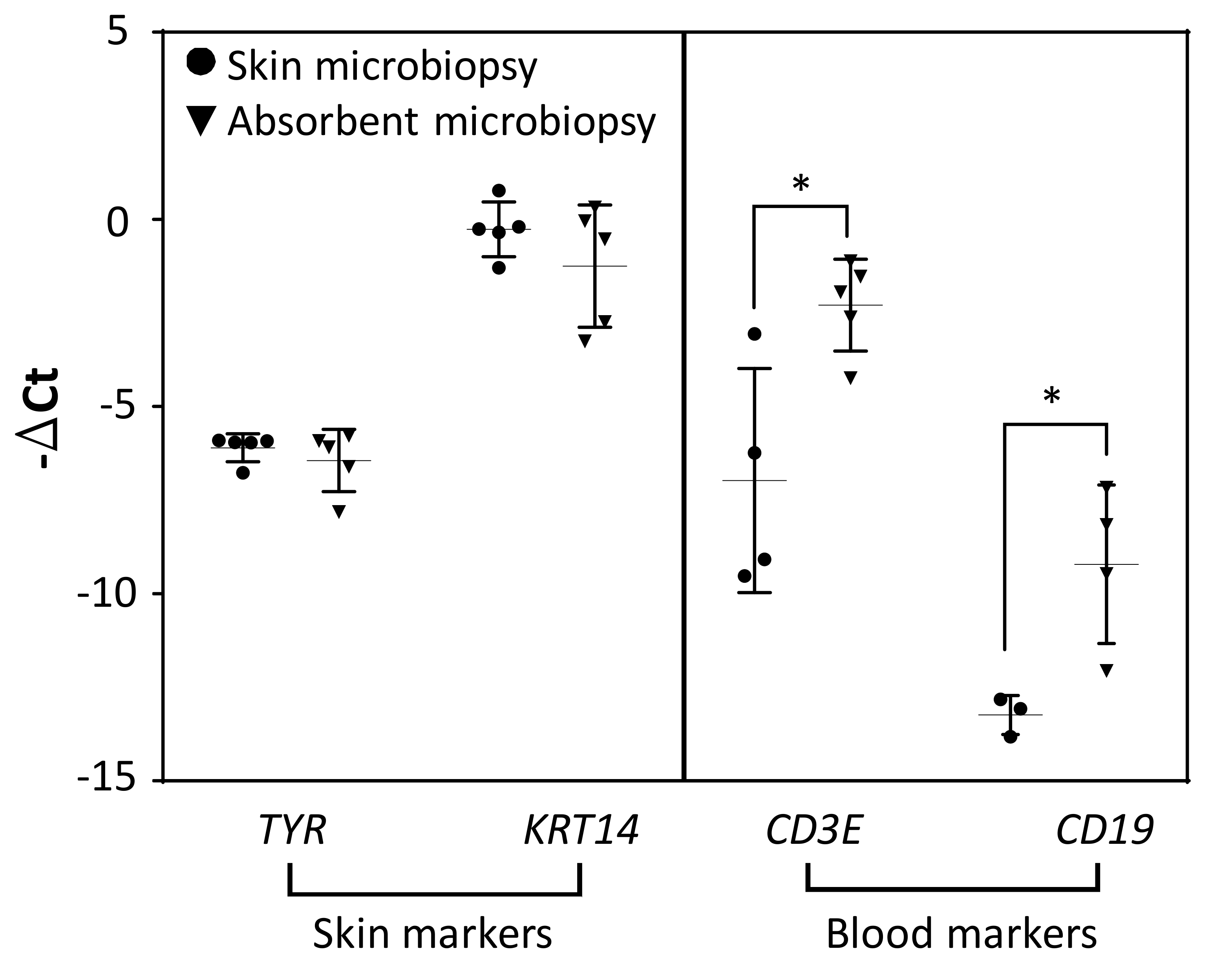
Since the device was designed for blood and skin sampling, qPCR was used to detect the expression of skin and blood biomarkers for both devices for comparison. We used tyrosinase, TYR, as a biomarker for melanocytes and KRT14 as a biomarker for keratinocytes in the viable epidermis. Skin microbiopsy samples were included in the experiment for comparison. As shown in Figure 4, though skin and absorbent microbiopsies were applied differently due to the difference in design, the expression levels for both skin biomarkers TYR and KRT14 were comparable for both devices as indicated by the data. White blood cell biomarkers (CD3E, T cells and CD19, B cells) were found to be more prevalent in absorbent microbiopsy samples than in skin microbiopsy samples. This result suggests that the absorbent microbiopsy performed better in blood collection but still maintained the capacity for capturing skin as compared to the skin microbiopsy (n = 5).

Figure 1. The fabrication of absorbent microbiopsy. (a) The three-layer microneedle. (b) All parts of the device. (c) The assembled absorbent microbiopsy. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. The absorbent microbiopsy was able to capture skin and blood samples simultaneously without leaving a scar on the left volar arm of male volunteer. (a) A comparison between the microneedles of absorbent and skin microbiopsies. (b) Application sites left by absorbent and skin microbiopsies 5 min after application. (c,d) Microneedles of absorbent and skin microbiopsies after application. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. The absorbent microbiopsy captured more blood and RNA sample when applied for 10-s. (a) The 10-s post-application holding time resulted in a higher amount of RNA than releasing the device immediately (n = 20). Error bars represent S.D. from the mean (****p<0.0001). (b,c) Representative pictures of the absorbent microbiopsies after application with 'Immediate release' and '10-s holding' approaches. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. The comparison of mRNA expression levels between skin and absorbent microbiopsies (n = 5). The gene expression had been normalized with reference gene RPLP0. Error bars represent S.D. from the mean (*p<0.05). Please click here to view a larger version of this figure.
{kind=link}
Discussion
These results demonstrate that the absorbent microbiopsy can be used as a tool for simple and minimally invasive sampling of skin and blood mixture for molecular characterization. Device application in accordance with our protocol is essential for obtaining reliable results as shown by the difference in RNA amount recovered with different application protocols (Figure 3). Once the sample is extracted, the subsequent sample processing step for RNA extraction is highly similar to established protocols15,16. Besides from the initial steps in RNA extraction that are modified for the absorbent microbiopsy, another key change is the use of RNase-free water to improve the results for downstream applications. Moreover, it is worth mentioning that the reference gene used in this study is RPLP0. RPLP0, whose function is well known for different cell and tissue types17, has been reported as a suitable reference gene for use in non-melanoma skin cancers and precancerous lesions18.
One of the main limitations of the device is the removal of the microbiopsy from the device, which is time-consuming and potentially increases the chance of sample loss, especially for sensitive samples like RNA. Nevertheless, the issue can be overcome by pre-cooling all the sterile processing tools, such as the 2 mL microcentrifuge tubes, on dry ice.
The use of the absorbent microbiopsy is simple and does not require intensive training. Conventional biopsy is not necessary as microbiopsy permits molecular characterization with established molecular diagnosis techniques, such as RT-qPCR. To further quantify and demonstrate the sampling ability of absorbent microbiopsy, previous literature that involved RNA extraction from human skin tissue was investigated. From a typical 3.0 or 4.0 mm skin punch biopsy, three studies have reported the extracted RNA amounts ranged from 50 to 200 ng per mg of skin tissue19,20,21. Comparing with the 1.43 ng of RNA that was recovered from the absorbent microbiopsy on average (Figure 3), the weight of sampled skin tissues is expected to range from 0.29-115 µg based on the same RNA-to-tissue ratio from skin punch biopsy studies.
This protocol is not without potential pitfalls, though some of the problems can be easily overcome. For example, the RNA extraction data suggested a considerable variation even with the 10-s holding time (Figure 3). To address the problem, one potential solution involves optimizing the application protocol. Parameters such as force applied to the skin and holding time should be tested and optimized to reduce the variations in sample extraction. The other potential issue is the removal of microbiopsy from the device, which might impact the sample integrity and recovery. Although removing the microbiopsy for RNA extraction is an effective approach, the entire procedure is tedious, and the sample might be exposed to contamination in the process. Therefore, the modification of the sample processing protocol is highly desired in order to ensure sample integrity and prevent sample loss.
Once the above two issues are addressed, it is expected that the device will become a standard tool for clinical research. It is important to note that the device captures both skin and blood sample simultaneously and this should be taken into consideration when analyzing gene expression data. In conclusion, this protocol describes the details of performing absorbent microbiopsy as an easy and minimally invasive tool for combined skin and blood sampling and the subsequent sample processing for relative gene expression profiling.
Acknowledgements
We would like to acknowledge NHMRC Fellowships APP1109749 and APP1111216, University of Queensland Centennial Scholarship and International Postgraduate Research Scholarship.
Materials
| Name | Company | Catalog Number | Comments |
| Absorbent microbiopsy fabrication and sampling | |||
| Absorbent Microbiopsy | Trajan Scientific and Medical | N/A | https://www.trajanscimed.com/ |
| Whatman filter paper, Grade 1 | Sigma Aldrich | WHA1001325 | N/A |
| RNA extraction | |||
| PicoPure RNA Isolation Kit | ThermoFisher | KIT0214 | Including all buffer soltuions described in protocol |
| UltraPure DNase/RNase-Free Distilled Water | ThermoFisher | 10977015 | Improving RNA quality in RNA elution step |
| 2.0 mL Microcentrifuge Tube, Sterile | Thomas Scientific | 1226S74 | N/A |
| Microcentrifuge 5415R | Eppendorf | Z605212 | N/A |
| cDNA synthesis | |||
| SensiFAST cDNA Synthesis Kit | Bioline | BIO-65053 | N/A |
| CFX96 Touch Real-Time PCR Detection System | Bio-Rad | 1855196 | N/A |
| qPCR reaction and data analysis | |||
| SensiFAST SYBR Lo-ROX Kit | Bioline | BIO-94005 | Including reagents described in qPCR reaction steps |
| MicroAmp Optical Adhesive Film | ThermoFisher Scinentific | 4311971 | N/A |
| MicroAmp Optical 384-Well Reaction Plate | ThermoFisher Scinentific | 4343370 | N/A |
| QuantStudio 6 Flex Real-Time PCR System | ThermoFisher Scinentific | 4485694 | N/A |
| GraphPad Prism (v6.04) | GraphPad | N/A; Windows version | Plotting and statistical analysis in qPCR data analysis steps |
| PCR primers | |||
| RPLP0 F | Sigma Aldrich | N/A | ATC AAC GGG TAC AAA CGA GTC |
| RPLP0 R | Sigma Aldrich | N/A | CAG ATG GAT CAG CCA AGA AGG |
| TYR F | Sigma Aldrich | N/A | TCA GCA CCC CAC AAA TCC TAA |
| TYR R | Sigma Aldrich | N/A | AAT CGG CTA CAG ACA ATC TGC |
| KRT14 F | Sigma Aldrich | N/A | CCT CCT CCC AGT TCT CCT |
| KRT14 R | Sigma Aldrich | N/A | ACA CCA CCT TGC CAT CG |
| CD3E F | Sigma Aldrich | N/A | CAA AGG GGA CAA AAC AAG GAG |
| CD3E R | Sigma Aldrich | N/A | GTT CTC CAG AGG GTC AGA TG |
| CD19 F | Sigma Aldrich | N/A | TTC TGC CTG TGT TCC CTT G |
| CD19 R | Sigma Aldrich | N/A | GCG TCA CTT TGA AGA ATC TCC T |
References
- Wang, C. Y., Maibach, H. I. Why minimally invasive skin sampling techniques? A bright scientific future. Cutaneous and Ocular Toxicology. 30, 1-6 (2011).
- Kirstein, O. D., et al. Minimally invasive microbiopsies: a novel sampling method for identifying asymptomatic, potentially infectious carriers of Leishmania donovani. International journal for parasitology. 47, 609-616 (2017).
- Lin, L. L. . The skin microbiopsy. , (2015).
- Dhingra, N., et al. Molecular profiling of contact dermatitis skin identifies allergen-dependent differences in immune response. Journal of Allergy and Clinical Immunology. 134, 362-372 (2014).
- Chapman, P. B., et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. New England Journal of Medicine. 364, 2507-2516 (2011).
- Kirstein, O. D., et al. Minimally invasive microbiopsies: a novel sampling method for identifying asymptomatic, potentially infectious carriers of Leishmania donovani. International journal for parasitology. 47, 609-616 (2017).
- Lin, L. L., et al. Microbiopsy engineered for minimally invasive and suture-free sub-millimetre skin sampling. F1000Research. 2, (2013).
- Tom, L. N., et al. Skin microbiopsy for HPV DNA detection in cutaneous warts. Journal of the European Academy of Dermatology and Venereology. 30, e216-e217 (2016).
- Sobarun, P., et al. Microbiopsy Biomarker Profiling in a Superficial Melanoma Resembling a Pigmented Basal Cell Carcinoma. JAMA Dermatology. 153, 334 (2017).
- Banan, P., Lin, L. L., Lambie, D., Prow, T., Soyer, H. P. Effects of Ex Vivo Skin Microbiopsy on Histopathologic Diagnosis in Melanocytic Skin Lesions. JAMA Dermatology. 149, 1107 (2013).
- . GraphPad - FAQ 1753 - Prism 3 -- Calculating "Unknown" Concentrations using a Standard Curve Available from: https://www.graphpad.com/support/faq/prism-3-calculating-unknown-concentrations-using-a-standard-curve/ (2018)
- Kubista, M., et al. The real-time polymerase chain reaction. Molecular Aspects of Medicine. 27, 95-125 (2006).
- Goni, R., García, P., Foissac, S. . The qPCR data statistical analysis. , 1-9 (2009).
- . . GraphPad Statistics Guide. , (1995).
- Goldstein, N. B., et al. Isolating RNA from precursor and mature melanocytes from human vitiligo and normal skin using laser capture microdissection. Experimental dermatology. 25, 805-811 (2016).
- Shearstone, J. R., Allaire, N. E., Campos-Rivera, J., Rao, S., Perrin, S. Accurate and precise transcriptional profiles from 50 pg of total RNA or 100 flow-sorted primary lymphocytes. Genomics. 88, 111-121 (2006).
- Bär, M., Bär, D., Lehmann, B. Selection and Validation of Candidate Housekeeping Genes for Studies of Human Keratinocytes-Review and Recommendations. Journal of Investigative Dermatology. 129, 535-537 (2009).
- Hoang, V. L. T., et al. RNA-seq reveals more consistent reference genes for gene expression studies in human non-melanoma skin cancers. PeerJ. 5, e3631 (2017).
- Döbbeling, U. Simultaneous RNA and DNA Extraction from Biopsy Material, Culture Cells, Plants, and Bacteria. Nucleic Acid Protocols Handbook, The. , 53-56 (2000).
- Bruning, O., et al. RNA isolation for transcriptomics of human and mouse small skin biopsies. BMC Research Notes. 4, 438 (2011).
- Berglund, S. R., et al. Optimized Methodology for Sequential Extraction of RNA and Protein from Small Human Skin Biopsies. Journal of Investigative Dermatology. 127, 349-353 (2007).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved