Gene-therapy Inspired Polycation Coating for Protection of DNA Origami Nanostructures
In This Article
Summary
Here, a protocol for the protection of DNA origami nanostructures in Mg-depleted and nuclease-rich media using natural cationic polysaccharide chitosan and synthetic linear polyethyleneimine (LPEI) coatings is presented.
Abstract
DNA origami nanostructures hold an immense potential to be used for biological and medical applications. However, low-salt conditions and nucleases in physiological fluids induce denaturation and degradation of self-assembled DNA nanostructures. In non-viral gene delivery, enzymatic degradation of DNA is overcome by the encapsulation of the negatively charged DNA in a cationic shell. Herein, inspired by gene delivery advancements, a simple, one-step and robust methodology is presented for the stabilization of DNA origami nanostructures by coating them with chitosan and linear polyethyleneimine. The polycation coating efficiently protects DNA origami nanostructures in Mg-depleted and nuclease-rich media. This method also preserves the full addressability of enzyme- and aptamer-based functionalization of DNA nanostructures.
Introduction
DNA is a versatile building block for the programmable self-assembly of nanoscale structures1. The most popular method for creating DNA nanostructures is the DNA origami technique, which is based on the self-assembly of a long circular, single stranded DNA scaffold with the aid of hundreds of shorter shape-determining synthetic staple strands2. Today, the creation of DNA nanostructures in almost any geometry and morphology is readily feasible. DNA nanostructures can be site-specifically functionalized with high precision3,4 and can be programmed to undergo allosteric conformational changes5,6. Hence, using DNA as a building material offers the unique opportunity to create programmable and responsive custom-designed nanostructures for applications in biosensing, diagnostics and drug delivery. However, DNA nanostructures are susceptible to digestion by exo- and endo-nucleases, and lattice-based 3D DNA origami nanostructures generally require high salinity buffers (e.g., 5-20 mM Mg+2) to maintain their integrity7,8.
The rapid degradation of DNA by nucleases present in blood and the extracellular matrix is a major obstacle to the efficient in vivo delivery of genetic products into cells9. To overcome this limitation, in non-viral gene delivery, DNA is mixed with a cationic polymer at a defined N/P charge ratio (ratio of amines in polycation to the phosphates in DNA)10. The complex of DNA with a cationic polymer, known as polyplex, protects DNA from nuclease-mediated degradation and enhances its cellular uptake11. Inspired by gene delivery advancements, oligolysine12, oligolysine-polyethylene glycol (PEG) copolymers13, poly (2-dimethylamino-ethylmethacrylate) (PDMAEMA)-based polymers14, and virus capsid proteins15 have been used for stabilizing DNA origami nanostructures.
Recently, we reported a method for the protection of DNA origami nanostructures in Mg-depleted and nuclease-rich media using the natural cationic polysaccharide chitosan and the synthetic linear polyethyleneimine (LPEI) was reported16. This article is an adaptation of our earlier work and describes the detailed protocol for the preparation of polyplexes, their characterization, testing the stability of naked and protected nanostructures in low-salt and nuclease-rich media and examining the addressability of enzyme- and aptamer-functionalized DNA origami nanostructures upon polycation coating.
Protocol
1. Preparing Polycation Stock Solution
- LPEI stock solution
- Add 10 mg of LPEI into a glass beaker containing <10 mL of H2O.
- To completely dissolve LPEI, add HCl (32%) till reaching pH 2.0.
- Adjust the pH to 7.0 by adding NaOH (10 M).
- Adjust the volume to the final concentration of 1 mg/mL.
- Filtrate the LPEI stock solution through a 0.22 µm membrane.
- Chitosan stock solution
- Dissolve 16.6 mg of chitosan oligosaccharide lactate (Mw ~ 5 kDa, 60% oligosaccharide composition, deacetylated from chitin by 90%) in 1 mL of acetic acid (10%) aqueous media to prepare a stock solution with a concentration of 10 mg/mL.
2. Purification of DNA Origami Nanostructures
- Mix 100 µL of the DNA origami sample (in 5 mM Tris, 1 mM EDTA, 5 mM NaCl buffer (denoted as TB) including 18 mM MgCl2) with the equivalent volume of 22 mM MgCl2 supplemented TB (100 µL) in a 1.5 mL tube.
- Add 200 µL of purification buffer (15% (w/v) PEG 8,000, 5 mM Tris, 1 mM EDTA and 505 mM NaCl). Mix gently by tube inversion.
- Spin the sample at 16,000 × g at room temperature (r.t.) for 25 min.
- Discard the supernatant carefully and resuspend the pellet in 16 mM MgCl2 supplemented TB buffer. The supernatant and the pellet contain excess staple strands and precipitated DNA origami, respectively.
- Incubate the sample for one day at r.t. at 650 rpm in a thermomixer. This step is for complete resuspension of DNA origami.
3. Agarose Gel Electrophoresis (AGE)
- Add 1.6 g of agarose and 80 mL of 0.5x TAE buffer (20 mM Tris, 0.5 mM EDTA, 10 mM acetic acid, pH 8) into a glass beaker to prepare a 2% agarose gel. Microwave for 5 min. Swirl the contents of the beaker for 1 min in a water bath. Add 352 µL of MgCl2 (2.5 M) to prepare the gel with 11 mM MgCl2. Pre-stain the gel with 10 µL of DNA gel stain.
- Pour the gel solution into a dry gel box and insert a gel electrophoresis comb. Let the solution solidify at r.t. for 15-30 min.
- Supplement the running buffer (0.5x TAE buffer) with 11 mM MgCl2.
- Mix 10-20 µL of the sample with 20% of 6x loading buffer (30% (v/v) glycerol, 0.25% (w/v) bromophenol blue, 0.25% (w/v) xylene cyanol FF) and load it into the agarose gel wells.
- Run the gel at 70 V at r.t. for 2.5- 3 h.
- Visualize the gel with a UV scanner.
4. Negative Stain Transmission Electron Microscopy (nsTEM)
- Apply 5 µL of the diluted nanostructure or the polyplex on a glow-discharged carbon-coated Cu400 TEM grid. Let it adsorb for ca. 1 min.
- Drain excess liquid from the edge of the grid by a piece of filter paper.
- Apply 5 µL of a 2% uranyl acetate aqueous solution and wait for 1 min.
- Use the filter paper edge to completely drain excess liquid from the edge of the grid.
- Let the grid dry completely before injecting it into the TEM.
5. Polyplex Formation
NOTE: Illustrative example for preparing a polyplex comprising DNA origami and chitosan at N/P 0.01-8 ratios.
- Calculate the number of amines per polycation using the formula-1 (Table 1).
- Measure the concentration of purified DNA origami using an ultra violet spectrophotometer at 260 nm. Use 2 µL of TB buffer as the blank to calibrate the machine. Calculate the "nmol of phosphate" using formula-2.
- Prepare 15 µL of the DNA origami structure including 1 nmol of phosphate.
- Use formula-3 to calculate the volume of chitosan, which is needed for preparing the polyplex at N/P 0.01-8. For example, to prepare the polyplex at N/P 8, 1.8 µL of chitosan (0.8 mg/mL) is needed (N/P is 8, nmol of phosphate is 1, molecular weight of chitosan is 5,000 g/mol, concentration of chitosan stock solution is 0.8 mg/mL and the number of amines per chitosan is 28). Add 13.2 µL of TB to 1.8 µL of chitosan (0.8 mg/mL). Add 15 µL of chitosan solution (0.8 mg/mL) to 15 µL of the DNA origami from step 5.3 to get a DNA origami-chitosan polyplex with NP 8. The polyplex is formed spontaneously.
- Repeat step 5.4 to prepare polyplexes at different N/P ratios.
- Perform an AGE as described in Step 3 (Figure 1A). Include the naked DNA origami as control.
- Image the polyplex as described in Step 4 (Figure 2).
Formula-1) number of amine groups per polycation

Formula-2) mole of phosphate
=
Formula-3) Volume (µL) of polycations
=
6. Decapsulation with Dextran Sulphate
- Calculate the number of sulphate groups per dextran sulphate using formula-4 (Table 2).
- Calculate the volume of dextran sulphate needed for the decomplexation of the polyplex at a predefined A/P ratio using formula-5. The A/P charge ratio is the ratio between the sulphate groups of the polyanion (A) to the phosphate groups (P) of the DNA origami. An excess amount of dextran sulphate is needed for an efficient decomplexation of the polyplexes (e.g. 500-1,000). For example, to decomplex the polyplex prepared in section 5 (polyplex contains 1 nmol of phosphate) at A/P 1,000, 3.6 µL of dextran sulphate 40 kDa (50 mg/mL) is needed (A/P is 1,000, nmol of phosphate is 1, the molecular weight of dextran sulphate is 40,000 g/mol, the concentration of dextran sulphate stock solution is 50 mg/mL and number of sulphate is 220). Add 3.6 µL of dextran sulphate (50 mg/mL) to the polyplex from step 5.4 and mix well.
- If working with a chitosan polyplex, add NaOH to facilitate the decomplexation process. Add NaOH for a final concentration of 10 mM. Skip this step for LPEI polyplexes.
- Perform an AGE as described in Step 3 (Figure 1B). Include a naked DNA origami and polyplex as control.
Formula-4) number of sulphate groups per polyanion=

Formula-5) Volume (µL) of dextran sulphate
=
7. Stability towards Mg Depletion
- Wash 20 µL of a DNA origami or the corresponding polyplex (including 2 nmol phosphate) with 4x 600 µL of Mg-zero buffer (5 mM Tris, 1 mM EDTA and 30 mM NaCl) using the ultrafiltration column (MWCO ~ 3 kDa). Spin each wash at 14,000 × g at r.t. for 3-5 min.
- Collect the remaining solution (80 µL) and incubate at 37 °C for one day at 650 rpm in a thermomixer.
- If testing LPEI polyplex, add 2.5 µL of MgCl2 (500 mM) for a final concentration of 16 mM MgCl2. Then, add 7 µL of dextran sulphate-40 kDa (50 mg/mL) (equivalent to A/P 1,000) to unravel the core DNA nanostructures (decapsulation, see Step 6).
NOTE: Skip this step if working with chitosan polyplex or naked DNA origami. - Follow nsTEM imaging as described in Step 3 (Figure 2).
8. DNase I Titration Assay of Naked DNA Origami Nanostructures
- Mix 3 µL of naked DNA origami including 1 nmol phosphate with 2 µL of 10x DNase I buffer (100 mM Tris-HCl, 25 mM MgCl2, 5 mM CaCl2, pH 7.6), 1 µL of MgCl2 (250 mM), 12 µL of water in 0.2 mL PCR tubes. Add 2 µL of DNase I (10x). Adjust the stock concentration of the DNase I (10x) to get final DNase I concentrations of 0.25-2 U/mL.
- Incubate the samples at 37 °C for 1-2 h in a thermocycler.
- Immerse the samples in ice and deactivate the nucleolytic activity by adding 2 µL of 500 mM dithiothreitol (DTT) and 2.5 µL of EGTA (33 mM).
- Analyze the samples using an AGE as described in Step 3.
9. Stability of Polyplexes towards DNase I
- Mix 4 µL of a polyplex (including 1 nmol of phosphate, prepared at different N/P ratios) with 1.5 µL of a DNase I buffer (10x), 8 µL of TB and 1.5 µL of DNase I (100 U/mL) in a 0.2 mL PCR tube.
- Incubate the sample for 24 h at 37 °C in a thermocycler.
- Add 2 µL of proteinase K (20 mg/mL) to digest the DNase I. Incubate for 30 min at 37 °C in a thermocycler.
NOTE: Instead of enzymatic digestion, chemical deactivation of DNase I by adding 1.5 µL of DTT (500 mM) and 2 µL of EGTA (33 mM) can be performed. - Add 3.6 µL of dextran sulphate-40 kDa (50 mg/mL) (equivalent to A/P 1,000) to unravel the core DNA nanostructures.
- Perform AGE as described in Step 3 (Figure 3A, Figure 3C). Include fresh DNA origami as control for measuring and comparing the band intensities on the gel.
- Excise the band on the AGE and extract the DNA origami by a squeeze freeze extraction column based on the protocol provided by the supplier.
- Follow nsTEM imaging as described in Step 4 (Figure 2).
10. Stability towards Fetal Bovine Serum (FBS)
- Mix 4 µL of naked DNA origami or its corresponding polyplex (including 1 nmol of phosphate) with 17 µL of TB + 10% FBS in 0.2 mL PCR tubes. Use freshly thawed FBS.
- Incubate the sample at 37 ˚C in a thermal cycler for 24 h.
- Immerse the sample in an ice bath.
- If testing the polyplex, perform the decapsulation process by adding 3.6 µL of dextran sulphate-40 kDa (50 mg/mL). Additionally, add NaOH if working with chitosan polyplexes (see step 6.3)
- Perform an AGE as described in Step 3. Include the fresh DNA origami as control for measuring and comparing the band intensities on the gel.
- Excise the band on the AGE and extract the DNA origami by a squeeze freeze extraction column.
- Add 2 µL of proteinase K (20 mg/mL) to the extracted DNA origami solution (20 µL) in a 0.2 mL PCR tube. Incubate for 30 min at 37 °C in a thermocycler to digest serum proteins bound to the nanostructures.
- Wash the sample with 5x 500 µL of TB including 16 mM MgCl2 using the ultracentrifugation column (MWCO ~ 100 kDa) to wash proteinase K (MW ~ 28.9 kDa) away. Spin each wash at 14,000 × g at r.t. for 3-5 min.
- Perform nsTEM imaging as described in step 4 (Figure 3).
NOTE: The gel extracted sample from step 10.6 might be directly used for nsTEM imaging. However, the imaging could be very challenging due to the attachment of serum proteins to the DNA origami nanostructures. Therefore, proteinase K treatment (steps 10.7-10.9) is recommended to facilitate the imaging process.
11. Testing the Addressability of Horseradish Peroxidase Enzyme (HRP)-functionalized DNA Origami upon Coating with Polycations
- Purify the self-assembled biotinylated-NR (Biotin-NR) as described in Step 2.
- Incubate 200 µL of purified Biotin-NR (36 nM, in 16 mM supplemented TB buffer) with 200 µL of HRP-conjugated streptavidin (540 nM, in TB buffer). Add 4.8 µL of MgCl2 (500 mM) to a final concentration of 14 mM. Incubate at r.t. for 12 h at 650 rpm in a thermomixer
- Purify HRP-functionalized NR (HRP-NR) by a PEG purification method (as described in Step 2) to remove the unbound HRP enzymes. Resuspend the precipitated HRP-NR in TB including 16 mM MgCl2.
- Mix 6.5 µL of purified HRP-NR (36 nM) with 6.5 µL of polycations of variant concentrations to prepare the polyplex at N/P ratios of 1, 2, 4, 10 and 20 (using formula 3).
- Repeat steps 11.2-11.4 for non-biotinylated NR. As the HRP enzyme may bind non-specifically to the DNA origami, the non-biotinylated DNA origami, which is treated with HRP enzymes and PEG purified (similar to Biotin-NR), will serve as the control in the assay.
- Mix 6.5 µL of distilled-water with 6.5 µL of polycations of variant concentrations corresponding to the defined N/P ratios of 1, 2, 4, 10 and 20. Theses samples serve as the blank group.
- Add the prepared solution in steps 11.4-11.6 (12.5 µL) to a 96-well plate including 72.5 µL of HEPES buffer (25 mM HEPES, 20 mM KCl, 200 mM NaCl, 0.05% Triton X-100, 1% DMSO, pH 7.4) and mix well.
- Add 10 µL of 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) (15 mM).
- Add 5 µL of H2O2 (12 mM) and mix well by pipetting up and down. H2O2 initiates the reaction.
- Measure the absorbance at 421 nm over 4 h with a multimode plate reader (Figure 4A).
12. Testing the Addressability of Hemin-binding Aptamers (HBA)-functionalized DNA Origami upon Coating with Polycations
- Purify the HBA functionalized-NR (HBA-NR) as described in Step 2. Resuspend the precipitated HBA-NR in TB supplemented with 6 mM MgCl2 (higher salt concentration leads to the aggregation of HBA-NR).
- Mix 10 µL of HBA-NR (40 nM) with 10 µL of polycations at varying concentration to prepare polyplex of defined N/P ratios of 1, 2, 10 and 20.
- Mix 10 µL of naked-NR (40 nM) or distilled-water with 10 µL of polycations at varying concentrations corresponding to N/P ratios of 1, 2, 10 and 20.
- Add the 20 µL prepared solution from steps 12.2 and 12.3 to the 96-well plate including 60 µL of HEPES buffer (25 mM HEPES, 20 mM KCl, 200 mM NaCl, 0.05% Triton X-100, 1% DMSO, pH 7.4) and mix well. Include at least one blank sample replacing the 20 µL polyplex or polycation solution (from steps 12.2 and 12.3) with water.
- Add 5 µL of hemin (0.04 mM) followed by 10 µL of ATBS (15 mM). Place the plate on an orbital shaker for 5 min.
- Add 5 µL of H2O2 (12 mM) and mix well by pipetting up and down.
- Measure the absorbance at 421 nmover 30 min with the multimode plate reader (Figure 4B).
Representative Results
Three DNA origami nanostructures of different configurations were designed, including a nanorod (NR), a nanobottle (NB) and a wireframe nanostructure (WN) (the 3D models of the nanostructures are illustrated in Figure 2). The NR and the NB were designed based on a square and honeycomb lattice, respectively, using caDNAno17 and the WN was created using the Daedalus software18. A comprehensive protocol for the design and self-assembly of DNA nanostructures has been published already19,20,21,22. The shape-specific staple strands were ordered, self-assembled and further purified based on a protocol described by Stahl et al.23 (step 2).
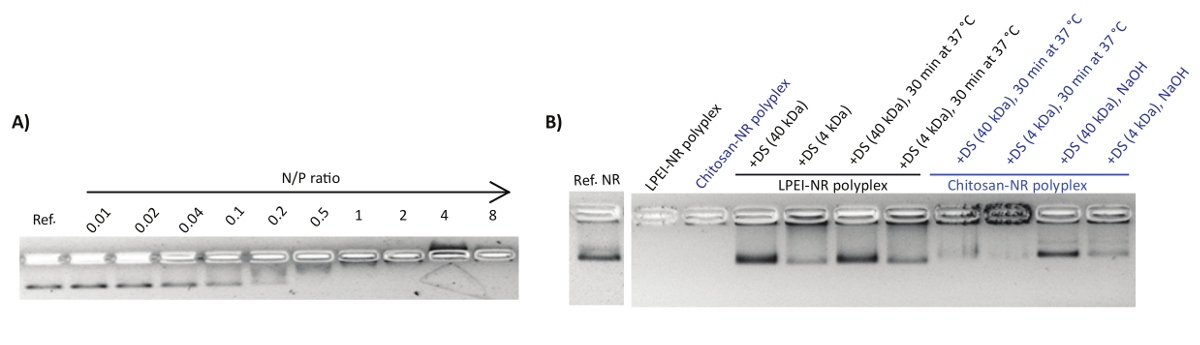
Polyplexes were characterized by the gel retardation assay and nsTEM imaging. Upon mixing the DNA origami with the polycations, a shift in the naked nanostructure band towards the cathode was observed (Figure 1A). This can be attributed to the counterbalancing of the negative charge of phosphate groups upon binding to polycations and the overall size increase of the complex. Adding an extra amount of polyanions such as dextran sulphate can reverse the polyplex formation. In this regard, dextran sulphate of higher molecular weight (MW ~ 40 kDa) showed to be more efficient for the decomplexation compared to lower molecular weight dextran sulphate (MW ~ 4 kDa) (Figure 1B).
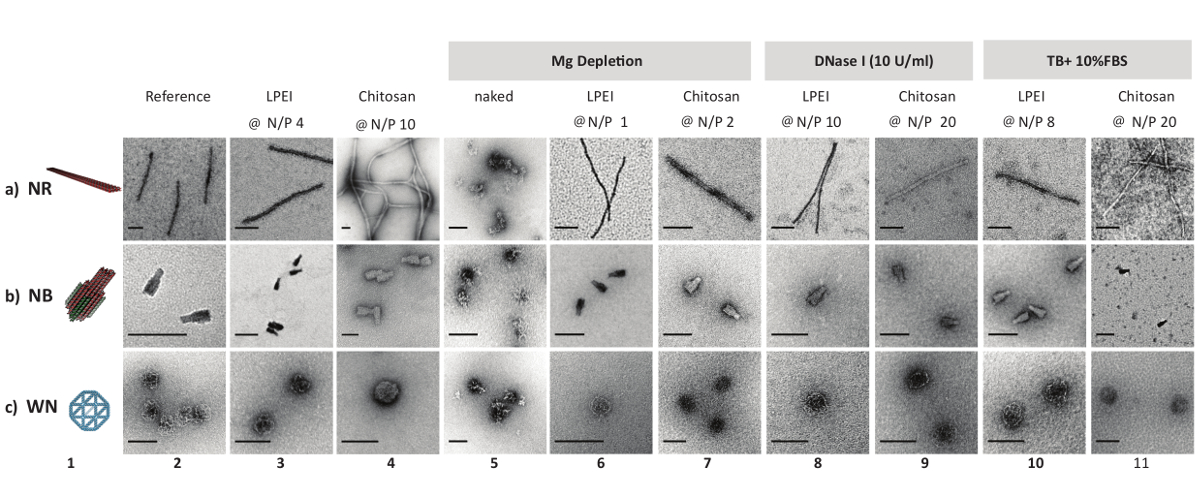
While imaging LPEI polyplexes by uranyl acetate negative stain TEM, it was difficult to image any particles. It was hypothesized that LPEI might hinder uranyl acetate from binding to the phosphate backbone due to tight shielding of DNA origami. Therefore, prior to nsTEM imaging, an excess amount of dextran sulphate was added to LEPI polyplexes. The unraveled DNA origami nanostructures showed no sign of defect nor decomposition. In contrary, chitosan polyplexes were successfully imaged with no need for polyanion treatment (Figure 2).
All naked DNA nanostructures (irrespective of their different configurations) were denatured completely after one day of incubation at 37 °C in Mg-zero buffer, as confirmed by nsTEM imaging. In contrast, nanostructures coated with LPEI or chitosan at N/P≥1 remained intact. Similarly, DNase I titration assays showed the susceptibility of unprotected nanostructures towards enzymatic digestion. While naked nanostructures were completely digested in the presence of 1 U/mL DNase I after 2 h, the LEPI or chitosan encapsulated DNA origami stayed intact in Mg-zero buffer supplemented with 10 U/mL DNase I for a minimum of one day (Figure 2). Higher stability towards nucleolytic digestion was achieved by increasing the N/P ratio of the polyplexes. Additionally, LPEI protects the DNA nanostructures more efficiently compared to chitosan (Figure 3A, Figure 3C), probably due to the higher charge density of LEPI, and thus, higher binding affinity towards the DNA24. No difference was observed while using LPEI of different molecular weight (Figure3B).
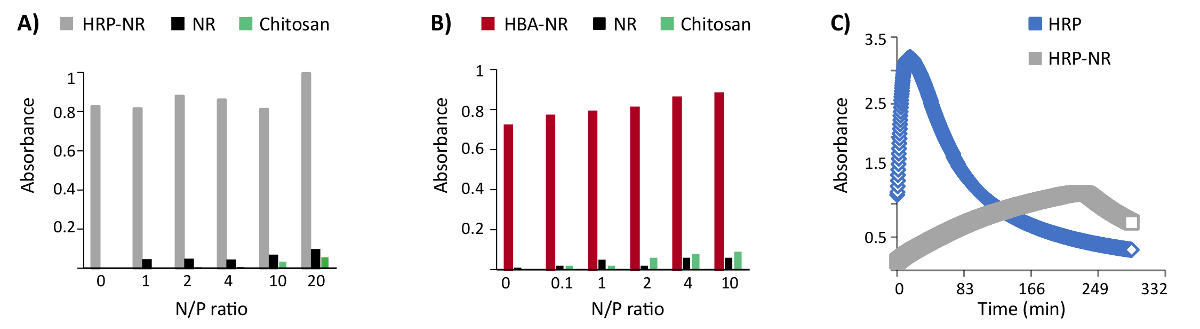
The addressability of functional groups in DNA nanostructures after encapsulation in a polymer shell is an important feature. Furthermore, the compatibility of polycation coating with the horseradish peroxidase enzyme (HRP) and hemin-binding aptamers (HBA) functionalized DNA origami was examined. Three biotinylated staple strands were protruded from the surface of the NR and then functionalized with HRP conjugated streptavidin (three enzymes per DNA origami). The catalytically highly active (Kd: 439 nM) HBA aptamer PS2.M (5'-GTGGGTAGGGCGGGTGG-3') was chosen for these experiments25. Up to 24 staple strands were protruded from the NR surface with a 5-nm length linker (5'-AAAAGAAAAGAAAAA-3') followed by the PS2.M sequence (24 aptamers per DNA origami). No remarkable hindrance of enzymatic or aptamer activity was observed upon coating HRP- or HBA-functionalized DNA origami with LPEI and chitosan at variant N/P ratios (Figure 4A-B)16. However, the kinetic of HRP enzyme has been dramatically changed after binding to the DNA origami (Figure 4C).

Figure 1: Representative AGE images of polyplex formation and decapsulation. (A) The electrophoretic mobility shift assay for NB mixed with chitosan at N/P ratios of 0.01-8. Each lane contains 1 nmol NB. The first lane is the reference NB. (B) Decapsulation of polyplexes by polyanionic dextran sulphate (DS). This figure has been modified from a previously published figure16. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Negative stain TEM micrographs of DNA origami and polyplexes. 3D model and nsTEM micrographs of naked DNA origami nanostructures (columns 1 and 2). nsTEM micrographs of LPEI polyplexes (after decapsulation) and chitosan polyplexes (columns 3 and 4). nsTEM images of naked and protected DNA origami (polyplex) subjected to Mg depletion (columns 5, 6 and 7), enzymatic degradation (columns 8 and 9), serum digestion (columns 10 and 11) for one day at 37 °C. LPEI-5 kDa was used in this assay. Naked DNA nanostructures were degraded beyond detection on an AGE in the presence of 1 U/mL DNase I or in TB + 10% FBS after 2 h of incubation at 37 °C. Scale bars: 100 nm. This figure has been modified from a previously published figure16. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative results of DNase I protection assays. (A) The DNase I protection assay for polyplexes of WN with LPEI-5 kDa, LPEI-10 kDa and LPEI-25 kDa prepared at N/P ratio of 2, 4, 8 and 10. The samples were subjected to 10 U/mL DNase I for 24 h at 37 °C. The last lane is the control WN. The polyplexes were decapsulated prior to loading into the gel. (B) The normalized mean band intensity for polyplexes of DNA origamis with different LPEI extracted from AGE image-A. Y axis represents normalized mean band intensity. (C) The DNase I protection assay of chitosan-WN polyplexes prepared at N/P ratios of 1, 2, 4, 10 and 20. The samples were subjected to 10 U/mL DNase I for 24 h at 37 °C. Lane 6 is the control polyplex (N/P 20). The last lane is the control WN. All chitosan polyplexes were decapsulated prior loading into the gel. This figure has been modified from a previously published figure16. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative results of colorimetric enzyme- and aptamer-functionalized DNA origami assays. (A) The colorimetric assay of enzyme-functionalized nanostructures (HRP-NR) coated with chitosan 3.7 h after the reaction start. (B) Colorimetric assay of aptamer-functionalized nanostructures (HBA-NR) coated with chitosan, 6 min after the reaction initiation. Y axis represents the normalized absorbance. (C) Monitoring the colorimetric assay of HRP and HRP-NR over time. This figure has been modified from a previously published figure16. Please click here to view a larger version of this figure.
{kind=link}
| Cationic polymers | Chitosan | LPEI-5 kDa | LPEI-10 kDa | LPEI-25 kDa |
| Molecular weight of polymer (kDa) | 5 | 5 | 10 | 25 |
| Molecualr weight of monomer (g/mol) | 161 | 43 | 43 | 43 |
| Number of amine per monomer | 1 | 1 | 1 | 1 |
| Number of amine per polymer | 28 | 116 | 232 | 581 |
Table 1. Calculating the number of amine groups per polycation.
| Molecular weight of dextran sulphate (kDa) | 4 | 40 |
| Molecular weight of monomer (g/mol) | 366 | 366 |
| Number of sulphate per monomer | 2 | 2 |
| Number of sulphate per polymer | 22 | 220 |
Table 2. Calculating the number of sulphate groups per polyanion.
Discussion
In the self-assembly of DNA origami, the staple strands are typically added in 5-10 excess ratio to the scaffold. These excess staple strands also bind to the polycation and form polyplexes in the monometer range which are further difficult to be separated from DNA origami polyplexes. Hence, a critical step in polyplex formation is to remove the excess staple strands and use well-purified DNA origami nanostructures.
Other methods have been developed for stabilizing DNA nanostructures such as the cyclization of DNA strands via a click reaction26. As alkyne- and azide-modified staple strands are required for the formation of interlocked single-stranded rings, this technique is limited for the stabilization of small nanostructures such a DNA catenane, and it is not readily scalable for larger DNA origami structure such as those used in this protocol. Recently, Gerling et al.27 reported a method for creating covalent cyclobutene pyrimidine dimer (CPD) bonds between neighboring thymidines within DNA nanostructures using ultraviolet irradiation. Although this method is site-selective and scalable, its efficiency in protecting DNA origami is much lower than polycation coating. For example, CPD-stabilized DNA origami objects endure only 0.4 U/mL DNase I for 1 h, while polyplexes were stable in the presence of 10 U/mL DNase I for at least one day.
In brief, gene-therapy inspired chitosan and LPEI coating is a low-cost, one-step and efficient method to address the long-term stability of DNA origami nanostructures in Mg-depleted and nuclease-rich media. The reversibility of the polyplex formation facilitates its application in multi-step diagnostic tests where the protection of DNA origami against nucleolytic degradation or salt-depletion is crucial in one step but may cause problems in other steps such as DNA amplification. Additionally, polycation coating is a potential approach for enhancing the cellular uptake of DNA origami nanostructures for drug-delivery applications28.
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 686647. TEM images were recorded on a Morgagni operated at 80 kV at the EM Facility of the Vienna Biocenter Core Facilities GmbH (VBCF). We would like to thank Tadija Kekic for assistance in the graphical design and Elisa De LIano for designing the wireframe nanostructure.
Materials
| Name | Company | Catalog Number | Comments |
| 10× DNase I reaction buffer | New England Biolab (NEB) | B0303 | |
| ABTS (2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt) | Alfa Aesar | J65535 | |
| Amicon ultracentrifugation columns | Merck Millipore | UCF500308, UCF510024 | |
| Chitosan oligosaccharide lactate (Mn ~ 4000-6000, > 90% deacetylated, 60%. composition oligosaccharide | Sigma-Aldrich | 523682 | |
| Design-specific staple strands | Integrate DNA Technologies (IDT) | ||
| Dextran sulfate sodium salt (Mr ~ 4 kDa) | Sigma-Aldrich | 75027 | |
| Dextran sulfate sodium salt (Mr ~ 40 kDa) | Sigma-Aldrich | 42867 | |
| DNA gel loading dye (6×) | ThermoFischer sceintific | R0611 | |
| DNase I (RNase free) | New England Biolab (NEB) | M0303 | |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) | Sigma-Aldrich | E3889 | |
| Ethylenediaminetetraacetic acid (EDTA) BioUltra, anhydrous, ≥99% (titration) | Sigma-Aldrich | EDS | |
| Freeze'N Squeeze DNA Gel Extraction Spin Columns | Biorad | 7326165 | |
| Hemin | Sigma-Aldrich | H9039 | |
| HEPES | Sigma-Aldrich | H3375 | |
| Hydrogen peroixde slution (32 wt%) | Sigma-Aldrich | 216763 | |
| LPEI-25 kDa | Polysciences, Inc | 23966-1 | |
| NuPAGE Sample Reducing Agent (500 mM dithiothreitol (DTT)) | ThermoFischer sceintific | NP0004 | |
| Polyethylene glycol (PEG, MW ~ 8000 Da) | Carl Roth | O263.1 | |
| Polyethylenimine, linear, average Mn 10,000, PDI ≤1.2 | Sigma-Aldrich | 765090 | |
| Polyethylenimine, linear, average Mn 5,000, PDI ≤1.3 | Sigma-Aldrich | 764582 | |
| Proteinase K solution (20 mg/ml) | ThermoFischer sceintific | AM2548 | |
| Streptavidin-conjugated HRP | ThermoFischer sceintific | N100 | |
| SYBER safe DNA gel stain | ThermoFischer sceintific | S33102 |
References
- Jones, M. R., Seeman, N. C., Mirkin, C. A. Nanomaterials. Programmable materials and the nature of the DNA bond. Science. 347 (6224), 1260901 (2015).
- Rothemund, P. W. Folding DNA to create nanoscale shapes and patterns. Nature. 440 (7082), 297-302 (2006).
- Takabayashi, S., Klein, W. P., Onodera, C., Rapp, B., Flores-Estrada, J., Lindau, E., Snowball, L., Sam, J. T., Padilla, J. E., Lee, J., Knowlton, W. B., Graugnard, E., Yurke, B., Kuang, W., Hughes, W. L. High precision and high yield fabrication of dense nanoparticle arrays onto DNA origami at statistically independent binding sites. Nanoscale. 6 (22), 13928-13938 (2014).
- Kuzyk, A., Schreiber, R., Zhang, H., Govorov, A. O., Liedl, T., Liu, N. Reconfigurable 3D plasmonic metamolecules. Nature Material. 13 (9), 862-866 (2014).
- Douglas, S. M., Bachelet, I., Church, G. M. A logic-gated nanorobot for targeted transport of molecular payloads. Science. 335 (6070), 831-834 (2012).
- Choi, Y., Choi, H., Lee, A. C., Lee, H., Kwon, S. A Reconfigurable DNA Accordion Rack. Angewandte Chemie International Edition. 57 (11), 2811-2815 (2018).
- Kielar, C., Xin, Y., Shen, B., Kostiainen, M. A., Grundmeier, G., Linko, V., Keller, A. On the stability of DNA origami nanostructures in low-magnesium buffers. Angewandte Chemie International Edition. 57 (30), 9470-9474 (2018).
- Hahn, J., Wickham, S. F. J., Shih, W. M., Perrault, S. D. Addressing the instability of DNA nanostructures in tissue culture. ACS Nano. 8 (9), 8765-8775 (2014).
- Al-Dosari, M. S., Gao, X. Nonviral Gene Delivery: Principle, limitations, and recent progress. The AAPS Journal. 11 (4), 671 (2009).
- Ibraheem, D., Elaissari, A., Fessi, H. Gene therapy and DNA delivery systems. International Journal of Pharmaceutics. 459 (1-2), 70-83 (2014).
- Pack, D. W., Hoffman, A. S., Pun, S., Stayton, P. S. Design and development of polymers for gene delivery. Nature Reviews Drug Discovery. 4 (7), 581-593 (2005).
- Ponnuswamy, N., Bastings, M. M. C., Nathwani, B., Ryu, J. H., et al. Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation. Nature Communication. 8, 15654 (2017).
- Agarwal, N. P., Matthies, M., Gür, F. N., Osada, K., Schmidt, T. L. Block copolymer micellization as a protection strategy for DNA origami. Angewandte Chemie International Edition. 56 (20), 5460-5464 (2017).
- Kiviaho, J. K., Linko, V., Ora, A., Tiainen, T., Järvihaavisto, E., Mikkilä, J., Tenhu, H., Nonappac, M. A., Kostiainen, Cationic polymers for DNA origami coating - examining their binding efficiency and tuning the enzymatic reaction rates. Nanoscale. 8 (22), 11674-11680 (2016).
- Perrault, S. D., Shih, W. M. Virus-inspired membrane encapsulation of DNA nanostructures to achieve in vivo stability. ACS Nano. 8 (5), 5132-5140 (2014).
- Ahmadi, Y., De Llano, E., Barišić, I. (Poly)cation-induced protection of conventional and wireframe DNA origami nanostructures. Nanoscale. 10 (16), 7494-7504 (2018).
- Douglas, S. M., Marblestone, A. H., Teerapittayanon, S., Vazquez, A., Church, G. M., Shih, W. M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Research. 37 (15), 5001-5006 (2009).
- Veneziano, R., Ratanalert, S., Zhang, K., Zhang, F., Yan, H., Chiu, W., Bathe, M. Designer nanoscale DNA assemblies programmed from the top down. Science. 352 (6293), 1534 (2016).
- Castro, C. E., Kilchherr, F., Kim, D. N., Shiao, E. L., Wauer, T., Wortmann, P., Bathe, M., Dietz, H. A primer to scaffolded DNA origami. Nature Methods. 8 (3), 221-229 (2011).
- Amir, Y., Abu-Horowitz, A., Bachelet, I. Folding and Characterization of a Bio-responsive Robot from DNA Origami. Journal of Visualized Experiment. (106), 51272 (2015).
- Ben-Ishay, E., Abu-Horowitz, A., Bachelet, I. Designing a bio-responsive robot from DNA origami. Journal of Visualized Experiment. (77), 50268 (2013).
- Wei, B., Vhudzijena, M. K., Robaszewski, J., Yin, P. Self-assembly of complex two-dimensional shapes from single-stranded DNA tiles. Journal of Visualized Experiment. (99), 52486 (2015).
- Stahl, E., Martin, T. G., Praetorius, F., Dietz, H. Facile and scalable preparation of pure and dense DNA origami solutions. Angewandte Chemie International Edition. 53 (47), 12735-12740 (2014).
- Grigsby, C. L., Leong, K. W. Balancing protection and release of DNA: tools to address a bottleneck of non-viral gene delivery. Journal of the Royal Society Interface. 7 (1), 67-82 (2010).
- Li, T., Dong, S., Wang, E. G-quadruplex aptamers with peroxidase-like DNAzyme functions: which is the best and how does it work. Chemistry - An Asian Journal. 4 (6), 918-922 (2009).
- Cassinelli, V. 1., Oberleitner, B., Sobotta, J., Nickels, P., Grossi, G., Kempter, S., Frischmuth, T., Liedl, T., Manetto, A. One-Step Formation of "Chain-Armor"-Stabilized DNA Nanostructures. Angewandte Chemie International Edition. 54 (27), 7795-7798 (2015).
- Gerling, T., Kube, M., Kick, B., Dietz, H. Sequence-programmable covalent bonding of designed DNA assemblies. Science Advances. , 1157 (2018).
- Xia, T., Kovochich, M., Liong, M., Meng, H., Kabehie, S., George, S., Zink, J. I., Nel, A. E. Polyethyleneimine Coating Enhances the Cellular Uptake of Mesoporous Silica Nanoparticles and Allows Safe Delivery of siRNA and DNA Constructs. ACS Nano. 3 (10), 3273-3286 (2009).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved