Combined Size and Density Fractionation of Soils for Investigations of Organo-Mineral Interactions
In This Article
Summary
Combined size and density fractionation (CSDF) is a method to physically separate soil into fractions differing in texture (particle size) and mineralogy (density). The purpose is to isolate fractions with different reactivities towards soil organic matter (SOM), in order to better understand organo-mineral interactions and SOM dynamics.
Abstract
Combined size and density fractionation (CSDF) is a method used to physically separate soil into fractions differing in particle size and mineralogy. CSDF relies on sequential density separation and sedimentation steps to isolate (1) the free light fraction (uncomplexed organic matter), (2) the occluded light fraction (uncomplexed organic matter trapped in soil aggregates) and (3) a variable number of heavy fractions (soil minerals and their associated organic matter) differing in composition. Provided that the parameters of the CSDF (dispersion energy, density cut-offs, sedimentation time) are properly selected, the method yields heavy fractions of relatively homogeneous mineral composition. Each of these fractions is expected to have a different complexing ability towards organic matter, rendering this a useful method to isolate and study the nature of organo-mineral interactions. Combining density and particle size separation brings an improved resolution compared to simple size or density fractionation methods, allowing the separation of heavy components according to both mineralogy and size (related to surface area) criteria. As is the case for all physical fractionation methods, it may be considered as less disruptive or aggressive than chemically-based extraction methods. However, CSDF is a time-consuming method and furthermore, the quantity of material obtained in some fractions can be limiting for subsequent analysis. Following CSDF, the fractions may be analyzed for mineralogical composition, soil organic carbon concentration and organic matter chemistry. The method provides quantitative information about organic carbon distribution within a soil sample and brings light to the sorptive capacity of the different, naturally-occurring mineral phases, thus providing mechanistic information about the preferential nature of organo-mineral interactions in soils (i.e., which minerals, what type of organic matter).
Introduction
Soil is a complex system which contains elements of geological and biological origin. The study of their inter-relation is a cornerstone of our understanding of ecosystem function1. In particular, organo-mineral interactions are thought to play a key role in soil organic matter (SOM) dynamics2. Unravelling SOM dynamics is presently a very active research area for several reasons. A soil with high SOM stocks will tend to show good intrinsic fertility and may also constitute an environmentally valuable carbon sequestration opportunity3,4.
Organic matter in soil is highly heterogeneous, with some components turning over in the space within a few hours while others may persist for thousands of years5. The determinants of this heterogeneity remain a controversial topic, but association with the mineral matrix is thought to be particularly important6,7, especially for subsoil horizons8. As a result, mineral phases known to closely associate with organic components are receiving increasing interest9,10,11.
Soils contain a wide range of minerals with qualitatively and quantitatively varying sorptive potential towards SOM. Minerals with large specific surface areas and/or highly reactive surfaces have been shown to have a high sorption capacity for organic compounds4,12. In soils, secondary minerals such as high-activity phyllosilicates (e.g., smectites), iron oxyhydroxides and poorly crystalline aluminosilicates have all been shown to engage significantly in the sorptive preservation of some organic compounds13,14,15,16,17. Separating soil into fractions differing in mineralogy could thus help isolate organic matter pools with relative functional homogeneity.
The aim of this paper is to present a methodology to isolate organo-mineral complexes according to composition, which then facilitates the study of their properties. The method combines size and density fractionation to physically separate bulk soil into a sequence of fractions of different composition. Combined size and density fractionation (CSDF) integrates two effective physical fractionation approaches (particle size separation and density separation). The combination of these two approaches brings improved resolution to our understanding of organo-mineral associations in soil.
There are many different approaches (chemical, physical and / or biochemical) that can be used to specify fractions in a bulk soil sample18,19. Simple density fractionation is a physical separation which has been widely used by soil scientists to study SOM dynamics (see for instance Grunwald et al., 2017 and references therein)20. In its classical form, simple density fractionation separates materials lighter than a given cutoff (generally 1.6 to 1.85 g·cm-3) - the light fraction (LF) from heavier materials - the heavy fraction (hF). The LF is sometimes further split into free light fraction (fLF) and occluded light fraction (oLF)21.
In many soils, the largest SOM pool is found in the hF22. SOM in the hF is generally thought to be more stable than that in the LF23, yet it has been shown to retain a high compositional and probably, functional heterogeneity18. This points to the need to further separate the hF into more homogeneous subfractions, with the view of isolating pools of SOM with distinct biogeochemical properties (such as residence time or functionality). Sequential density fractionation, as described by Sollins et al. (2009)24, has indeed proved to be a successful method; yet a separation done solely on the basis of density runs the risk of overlooking differences arising from variation in grain size and thus specific surface area. For instance, kaolinite has approximately the same density as quartz but may be separated on the basis of its size mode (Table 1). CSDF includes consideration of grain size and improves the resolution of the fractionation.
SOM fractionation based on physical, chemical or biochemical properties has a long history. Physical methods such as CSDF are based on physical attributes of soil components, such as size (of particles or aggregates) or density. Chemical methods include selective extractions of specific compounds or classes of compounds, as well as chemical oxidation. Biochemical methods rely on microbial oxidation under various experimental conditions. Chemical and biochemical methods are based on different principles and have different objectives compared to physical methods but are nevertheless briefly reviewed below.
The alkaline extraction (with sodium hydroxide for example) ranks among the earliest methods used to chemically isolate the organic component of soils6. Examples of more modern, chemical methods for SOM fractionation include i) alkaline extraction with Na-pyrophosphate aimed at isolating SOM bound to minerals; ii) acid hydrolysis (HCl) aimed at quantifying old, persistent SOM; and iii) selective oxidation of SOM with chemical agents aimed at attacking free or labile SOM2. While these methods can be useful to gain insight into functionally different organic matter pool, they suffer from several limitations. First, the extractions can be imperfect or incomplete. For example, the classical alkaline method extracts only 50-70% of soil organic carbon (SOC)6. Second, fractionation products may not be representative of the SOM found in situ and may be difficult to categorize5. Third, these chemical methods only offer limited insight into the organo-mineral interaction since many of them do not preserve the original association between organics and minerals.
Biochemical extraction including incubations experiments are used mainly to study labile and reactive SOM (see Strosser32 for a review of biochemical methods). Incubation experiments can be thought of as a measure of biochemical oxygen demand and is intuitively well-suited to the determination of bioavailable organic substrates. However, the need for long incubation times in conditions that differ from the field (temperature, humidity, physical disturbance, absence of new inputs) makes the extrapolation to in-situ SOM dynamics delicate.
Compared to chemical or biochemical methods which are believed to be either transformative or destructive, physical fractionation techniques can be considered as more preservative22 (with the important exception of soluble organic compounds, which are lost during the procedure). At their best, physical soil fractions can be thought of as a 'snapshot' of solid-phase soil components as present in the field and could thus relate more directly to SOM dynamics in situ33. Moreover, the non-destructive nature of the technique means that the fractions can be subsequently characterized using a variety of analyses or further fractionated according to chemical or biochemical methods.
Physical fractionation of soils is not a recent idea. Scientific literature about physical separation techniques dates back to the mid-20 century. Applications of density fractionation were reported as early as 196534, 35. During the same period and in the following decades, publications about the dynamics of SOM and its interaction with minerals were already becoming widespread amongst soil scientists36,37,38,39.
Separation based on density, aggregate size or particle size are the most common physical separation methods used currently. One of the main challenges of physical separation is the isolation of homogenous functional SOM pools, as defined by turn-over rate, size or other indicator of function. Combining separation methods or criteria, as in CSDF, may help bring functional resolution to soil fractions; indeed, these methods seem to be used more and more in combination18,40,41,42,43. By combining sequential density separation, able to yield fractions with different organic matter content and mineralogical composition, with size separation, which accounts for differences attributable to specific surface area, CSDF holds the promise of yielding insight into the diversity and function of organo-mineral associations in soil.
CSDF aims to physically fractionate bulk soil samples into fractions of relative mineralogical and textural homogeneity. The density and particle size cut-offs, as well as the dispersion energy used here have been selected based on our soil type, but these parameters may be adapted depending on the samples to be fractioned and the purpose of the study. In this example, we have chosen to use one dispersion step, two density and one size cut-offs, resulting in the separation of the bulk soil into 6 fractions (Table 2). Figure 1 gives a conceptual overview of the method. The materials being fractioned here are tropical soils, but the method can be applied to any soil type as well as sediments. CSDF is generally used as a preparatory step before further analyses, even though the distribution of materials among fractions can be very informative in and of itself. When applied to soils, CSDF yields fractions differing in (1) mineral composition (mineralogy and texture) and (2) SOM concentration and composition.
Protocol
1. Sample Preparation
- Air-dry and sieve 20 g or more of bulk soils to 2 mm.
- Determine the residual moisture content of bulk soil (mass of water still present in air-dried soil) for correction of initial fractionation mass44.
2. Dense Solution Preparation
- Add sodium polytungstate (SPT) in portions to deionized (DI) water warmed on a hot stir plate (~60 °C) to obtain dense solutions of the desired densities (i.e., 1.62 and 2.78 g·cm-3). Use Table 3 to approximate proportions of water and SPT to use. Set a vigorous agitation to help dissolve the SPT.
CAUTION: SPT is corrosive, and it is harmful if swallowed. It causes serious eye damage. Wear protective gloves/eyewear. Harmful to aquatic life with long lasting effects. Avoid release to the environment. - Check the final density with a hydrometer once the solution is back to room temperature (between 20-25 °C). Adjust the density by adding SPT or water.
NOTE: It is easiest to prepare SPT at a slightly greater concentration (+ 0.02 – 0.05 g·cm-3) than needed then dilute it to the required density. - Store SPT at room temperature in plastic containers. SPT is known to salvage calcium from glassware and precipitate insoluble Ca metatungstate. SPT can also react with low-quality stainless steel and ceramics. Avoid storing SPT for several months since crystals will start nucleating at local supersaturated points.
3. Light Fraction (LF) Separation
NOTE: The LFs are isolated by flotation on a solution with the density of 1.62 g·cm-3 prior to ultrasonic dispersion (fLF) and following dispersion at 280 J·mL-1 (oLF).
- fLF Separation
- Weigh 5-8 g of air-dried, sieved soil in a 50 mL conical centrifuge tube. Record the mass of soil with four significant figures.
NOTE: The amount of soil that can be added to each tube depends on the soil texture. Generally, smaller amounts should be used for clay soils and higher amounts may be used for sandy soils. In order to get sufficient amounts of material in each fraction, it is possible to use several tubes for the same soil (we generally use two) or larger (250 mL) conical bottles. A pilot fractionation run can help get an idea of the distribution of soil among fractions and adjust starting soil masses. - Using a graduated cylinder, add 35-40 mL of SPT (1.62 g·cm-3). Aim for a soil/solution ratio of 1:5 or lower.
NOTE: To prevent aggregate slaking, it is possible to saturate the soil by capillary rise prior to adding the solution42,45. - Balance the total mass of tubes with lids in view of centrifugation. Equalize tube mass using SPT solution. Install the tubes on a shaking table for 10 min at 200 rpm.
NOTE: In soils with poor aggregate stability, this step may be replaced by a gentle inversion by hand6 or skipped altogether42. - Set the tubes upright and let the content settle for at least 30 min. Centrifuge for 90 min at 2,500 x g in a swinging-bucket centrifuge to afford a clear separation between the fLF and the pellet (‘sinking’ material).
NOTE: Centrifugation times can be reduced for sandy or well-flocculated samples. It is not recommended to centrifuge at speeds nearing the machine’s maximum capabilities; doing so risks damaging rotor axis. Bear in mind that most centrifuges are not designed to handle solutions of density greatly exceeding that of water. - Remove the tubes from the centrifuge very carefully, avoiding any shock. Set upright.
NOTE: At this point, it is possible to check the density of the SPT by pipetting 5 mL of the clear supernatant between the floating material and the pellet and weighing it. This is particularly important if the soil was not dry. - Pour all floating and suspended materials (the fLF) into a 250 mL polycarbonate centrifuge bottle. Ensure that the pellet remains firmly lodged at the bottom of the tube.
- Rinse the material adhering to the wall of the tube into the same polycarbonate bottle. Use a squirt bottle filled with DI water, holding the tube almost upside down above the polycarbonate bottle. Be careful not to disrupt the pellet.
NOTE: For some samples, the pellet may not adhere strongly to the bottom of the tube, making complete recovery of the fLF difficult. In such cases, aspiration with a pump or a high-volume pipette can help.
- Weigh 5-8 g of air-dried, sieved soil in a 50 mL conical centrifuge tube. Record the mass of soil with four significant figures.
- Washing the fLs by Ultrafiltration
NOTE: LFs are rinsed by ultrafiltration, since the density of some organics can be lower than that of water, precluding washing by centrifugation. A limitation of ultrafiltration is that sub-micrometric, dispersed colloids may not be retained. Commonly used filter pore sizes range from 0.2 to 1.2 μm. Filters with larger pores may not retain colloids as effectively but are less prone to clogging. Here, we used 0.45 μm filters.- Setup a vacuum ultrafiltration line with 0.45 μm filters. Slightly moisten the filters and apply vacuum before tightening the funnels to avoid tears.
- Slowly pour the content of the polycarbonate bottle into the funnel of a vacuum filtration unit. Do not allow the solution to pool over a depth greater than ~ 1 cm over the filter. Rinse out any residue left in the polycarbonate bottle into the filtration unit.
- Once all the solution has passed through the filter, recover it in a plastic jar labelled “used SPT” for recycling. Subsequent rinses can be discarded in the sink.
- Add more than 10 mL of DI water to the filtering unit at least 3 times to ensure that all traces of SPT are removed. Make sure to rinse the side of the filtration funnel. Keep track of the washes by adding a mark on the funnel when adding water.
NOTE: Complete removal of SPT can be checked by measuring the electrical conductivity of the last rinse. The conductivity should be ≤ 5 μS·cm-1 46.
- Recovering and Storing the fLF
- Release the vacuum on the filtration line.
- Remove the funnel from the filtration unit and recover the material adhering to the sides into a labelled aluminum boat using a squirt bottle filled with DI water. Carefully lift the filter with tweezers and rinse the material present on it into the same aluminum boat using a squirt bottle filled with DI water.
- Dry the boat contents at a maximum of 65 °C to constant weight (at least 48 h after complete evaporation). Cool in a desiccator containing fresh desiccant for at least 30 min.
NOTE: The use of a freeze-drier is another possibility. - Gently scrape the material off the aluminum boat with a plastic spatula. If the sample is strongly adhering to the aluminum boat, it is possible to use a metal spatula. Be careful not to scratch into the aluminum boat.
- Record the mass of the fLF with four significant figures, then place the sample in a storage vial. Avoid plastic vials because of static electricity which makes small organic particles adhere to the sides.
- oLF Release by Sonication
NOTE: The goal is to destroy stable macro-aggregates and large micro-aggregates by sonication to free occluded particulate organic matter, while minimizing extraction or redistribution of mineral-sorbed polymers. A sonication energy of 280 J·mL-1 was selected here. See the section on Aggregate disruption strategy in the discussion in order to help with the choice of sonication energy.- Calculate the amount of time necessary to reach the sonication target energy of 280 J·mL-1 using a low amplitude (e.g., 30 %) and 20 KHz frequency.
NOTE: Each sonifier should be regularly calibrated to relate sonication time to energy output; see North (1976)47 to help with calibration. - Add 35-40 mL of SPT (1.62 g·cm-3) to the centrifuge tube containing the pellet from the fLF extraction. Re-suspend the pellet by tapping the tube upside down and vortexing.
- Insert the ultrasonic probe at 2 cm below the surface of the solution and place the tube in iced water to prevent bulk solution heating.
- Sonicate the sample using the necessary time to reach the target energy of 280 J·mL-1. Always prefer low amplitude and longer sonication time. High amplitudes make light materials such as organic particles slide up the probe and spatter out of the tube, resulting in a loss of sample.
- Calculate the amount of time necessary to reach the sonication target energy of 280 J·mL-1 using a low amplitude (e.g., 30 %) and 20 KHz frequency.
- oLF Separation
- Balance the total mass of tubes with lids in view of centrifugation. Equalize tube mass using 1.62 g·cm-3 SPT solution.
- Set the tubes upright and let the content settle for at least 30 min. Centrifuge for 90 min at 2,500 x g in the swinging-bucket centrifuge.
- Pour all floating and suspended materials (the oLF) into a 250 mL polycarbonate centrifuge bottle. Ensure that the pellet remains firmly lodged at the bottom of the tube.
- Rinse the occluded light organic material adhering to the wall of the tube into the same polycarbonate bottle. Use a squirt bottle filled with DI water, holding the tube almost upside down above the polycarbonate bottle. Be careful not to disrupt the pellet adhering to the bottom.
- Washing the oLF by Ultrafiltration
- Setup a vacuum ultrafiltration line with 0.45 μm filters. Slightly moisten the filters and apply vacuum before tightening the funnels to avoid tears.
- Pour a small amount of suspension from the polycarbonate bottle into the funnel of a vacuum filtration unit. If the solution does not go through easily, add DI water to dilute the contents of the polycarbonate bottle.
NOTE: Samples high in organics (colored) will be difficult to filter. If the filter shows signs of clogging, set up a second filtration unit or try a pressure filtration system. Another possibility is to centrifuge diluted contents of polycarbonate bottles; the supernatant should go through the filter more easily as it contains fewer colloids and suspended particles than the original suspension. The settled material can then be re-suspended in water and rinsed by centrifugation. Eventually, all material needs to be rinsed at least 3 times and recovered. - Recover the first SPT filtrate in a plastic jar labelled “used SPT” for recycling. Subsequent rinses can be discarded in the sink.
- Rinse the material on the filtration unit three times with >10 mL of DI water.
- Collect the oLF in an aluminum boat and dry at a maximum of 65 °C to constant weight. Store in the same way as the fLF (see step 3.3).
4. Separation of the hFs According to Particle Size
NOTE: The next step is to fractionate the residue from step 3 (the hF) according to particle size. The size cut-off here is set at 8 µm and produces a clay + fine silt fraction (< 8 µm) and a coarse silt + sand fraction (> 8 µm). The grouping of clay and fine silt in the finer fraction reflects the documented affinity of both clay and fine silt for soil organic matter sorption33,48. Particle size separation here is done by sedimentation (based on Stokes’ law49). For cut-offs at 50 µm or larger, the separation can be simply effected by wet sieving without risking too much abrasion or disruption of organo-mineral complexes.
- Removing Residual SPT from the hF
- Add 35-40 mL of DI water to the tube containing the residue from step 4. Re-suspend the residue by tapping the tube upside down and vortexing. Centrifuge for 20 min at 2,500 x g.
- Pour out the supernatant. If it looks cloudy or milky, collect it and centrifuge it again to settle the colloids. Combine the new pellet with the one resulting from the first centrifugation.
NOTE: More than one rinse may be necessary if there was a significant amount of SPT remaining in the residue from step 3 (e.g., incomplete decantation of the supernatant). The density of the suspension should be <1.1 g·cm-3 prior to sedimentation.
- Dispersing the hF and Sedimentation
NOTE: Prior to sedimentation, it is important to have a well-dispersed hF. We use sonication rather than chemical dispersants, which would be difficult to rinse out. Stokes’ law requires a constant temperature (e.g., 20 °C). Starting with a cold suspension prior to sonication is an effective way to prevent over-heating.- Add refrigerated DI water to the tube containing the pellet (to the 40 mL mark). Place in the fridge for 30 min.
- Sonicate at 75 J·mL-1. Calculate the required sonication time as in 3.4.1.
- Rinse the ultrasonic probe with DI water to recover all materials. Using DI water, adjust the volume to the 50 mL mark in the tubes. Check the temperature of the suspension (it should match the room temperature used in calculation of sedimentation time).
- Homogenize tube content by inverting by hand. Leave the content to sediment for the time required to allow material (< 8 µm) to settle. Based on Stokes’ law for a mean particle travel distance of 7 cm at 20 °C, sedimentation time is 20 min.
- Pipet out the supernatant (down to 10 mL mark) into a 50 mL centrifuge tube.
- Repeat the sedimentation step 3 times (steps 4.2.1 – 4.2.5). Collect each supernatant in a new centrifuge tube.
- Collection of the Two Size Fractions
- Set the tubes containing the supernatant to evaporate in the oven at 45 °C. Do not allow them to dry. Once the volume is sufficiently reduced, consolidate the three supernatants into one 50 mL centrifuge tube, which now contains the < 8 µm fraction.
- Place the tubes containing the < 8 µm fraction as well as those containing the > 8 µm fraction (sediment from step 5.2) back into the oven at 45 °C to evaporate the remaining excess liquid. Do not allow the contents to dry completely.
NOTE: Check the oven frequently and remove the samples as soon as there is no more pooled water in the tubes. Desiccation can result in strong aggregation, especially in the fine fractions.
5. Separation of the Two Size Fractions According to Density
NOTE: Density fractionation is applied here to both particle size fractions. The objective was to separate silicates from oxides. We thus opted for a density cut-off of at 2.78 g·cm-3. Optional, additional separations may be performed. For instance, a 2.4 g·cm-3 solution would allow for the separation of high-activity clays from kaolinite and primary silicates. Keep in mind than high organic matter loading will decrease the theoretical density of mineral particles43.
- Density Fractionation
- Add 35 – 40 mL of SPT with the density of 2.78 g·cm-3 to each size fraction. Balance the tubes mass in view of centrifugation using SPT solution.
- Re-suspend the material by tapping the tube upside down and vortexing. Agitate for minimum 4 h on a shaking table at 200 rpm.
NOTE: The < 8 µm fraction is difficult to properly re-suspend. If the clods persist after 4 h, set on the shaking table overnight. Sonication in very dense SPT solution is not recommended. Due to the high density and viscosity, the sonication energy does not propagate well. - Set the tubes upright and let the content settle for at least 30 min. Centrifuge for 90 min at 2,500 x g.
- Pour all floating and suspended materials (<2.78 g·cm-3 fractions) into a 250 mL polycarbonate centrifuge bottle. Ensure that the pellet (> 2.78 g·cm-3 fractions) remains firmly lodged at the bottom of the tube.
- Washing the Four hFs by Centrifugation
NOTE: Following density separation, we have obtained four hFs (hF1 to hF4; Table 2). Each needs to be rinsed by centrifugation to remove SPT. Ultrafiltration is not recommended for hFs (and not possible for the fine fractions, hF3 and hF4).- Transfer the > 2.78 g·cm-3 pellets into 250 mL polycarbonate bottles using a squirt bottle filled with DI water.
- Add DI water to the polycarbonate bottles to lower the density, without exceeding the 200 mL mark. Do this for all fractions.
- Weigh the bottles and the lids and equalize masses using DI water in view of centrifugation. Check that the maximum load of the rotor is not exceeded.
- Place on vortex to re-suspend the material. Centrifuge for 20 min at 5,000 x g.
- Decant the supernatant into “used SPT” jar for recycling. Following rinses may be discarded in the sink.
NOTE: Be careful not to lose fine particles while decanting. Stop the decanting as soon as the supernatant shows traces of suspended particles (swirls, flakes or cloudy appearance). - Re-suspend the pellet in DI water (wash 1), not exceeding the 200 mL mark.
NOTE: It is important to ensure proper re-suspension so that SPT may not remain stuck between particles. Fine particles adhering to the side of the bottle are most easily suspended using a water jet from a squirt bottle. The pellet can be re-suspended by tapping the bottle upside down and vortexing. If the sample remains pelleted, use the shaking table. A low-energy sonication step (e.g., 75 J·mL-1) is also possible. - Repeat the washing procedure (steps 5.2.3 – 5.2.6) two more times (wash 2 and wash 3).
- Check the density of the last supernatant by weighing a volume of 10 mL; its density should be < 1.01 g·cm-3. If the supernatant is denser than this, perform a 4th wash.
- Perform a final wash using 0.01 M NaCl as a rinse solution to prevent dispersion of colloids. Fully decant the last rinse solution.
- Collect hFs (pellet + fine particles adhering to the side of the bottles) in an aluminum boat and dry at a maximum of 105 °C to constant weight. Store in the same way as the LFs.
6. Recycling of SPT
NOTE: The SPT solution can be recycled in view of re-use by passing it through a column containing activated charcoal and a cation exchange resin50. Activated charcoal retains organics while the sodium-saturated cation exchange resin removes calcium and other cations from the solution and replaces them with sodium. We digest the SPT in hydrogen peroxide prior to passing it through the column to ensure quantitative removal of dissolved organics.
- Setting up and Changing the Recycling Column
- Drill 5-8 holes of approximately 4-5 mm diameter at the base of a 1,000 mL polycarbonate graduated cylinder.
- Cut 2 circular 11 μm-pore-size filters to size and place them at the bottom of the cylinder. Overlay the filters with 3-5 cm of glass wool. Add 8-10 cm of activated charcoal.
NOTE: Because the majority of organics are removed by the H2O2 digestion, the amount of activated charcoal in the column may be reduced compared to the recommendation of Six50. - Cover with 3-5 cm of glass wool. Add 8-10 cm of new or regenerated cationic exchange resin. Finish with 3-5 cm of glass wool.
- Clamp the column up onto a metal stand. Place large plastic funnels fitted with 2.5 μm-pore-size filters at the top and bottom of the column. The bottom funnel will be connected to tubing leading to a plastic bottle.
- Rinse the column by passing 2 L of DI water through the column before first use.
- Replace the activated charcoal and regenerate the cation exchange resin after approximately 10 to 15 L of SPT have passed through.
- SPT Cleaning
- Place the used SPT solution on a hot plate with magnetic stirring. Add 15 mL of 35% H2O2 for each liter of used SPT solution and digest for 24 h at approximately 60 °C.
- Remove the SPT from the hot plate and let the solution sit at room temperature in plasticware for a minimum of 7 days.
- Percolate through the recycling column. Change the filters in the funnels every day.
- Rinse the column with approximately 2 x 200 mL of DI water to recover all SPT.
- Evaporation
- Place the recycled SPT on a hot plate with magnetic stirring. Add 15 mL of 70% ethanol for each liter of SPT solution to neutralize any remaining oxidizing capacity. Excess ethanol will readily volatilize from the solution during evaporation.
- Evaporate the solution on low heat to desired density.
- Cool the solution and check the density. Store in plastic bottles.
Representative Results
Samples are tropical soils originating from the Albertine rift valley in Uganda. They consist of profiles from 3 cultivated sites receiving no external inputs such as fertilizer or phytosanitary products. These samples were chosen to represent a large spectrum of mineralogy. Preliminary analyses showed that site 1 was least weathered and richest in primary silicates (feldspars). Site 2 showed signs of more advanced weathering with a high content of secondary clays such as kaolinite and a relative enrichment in quartz. Site 3 was highly weathered with signs of desilicification and residual accumulation of iron oxides and oxyhydroxides. Site 3 contained an extremely high total iron concentration (34%, expressed as Fe2O3 oxide) due to the presence of plinthic material (iron-rich induration51,52). For each profile, two horizons were sampled: topsoil (A) and subsoil (B). CSDF was performed on these 6 samples in four replicates.
The first step in evaluating the effectiveness of the fractionation procedure is to look at recovery rates, (i.e., whether the initial material is quantitatively retrieved at the end of the experiment). We assessed recovery rates based on the whole soil and SOC contents.
Overall, whole soil recovery rates were considered to be very good, with 16 out of 20 replicates having recovery rates of more than 90% and 4 replicates showing recovery rates between 85-90% (Table 4). The cause of incomplete recovery was most likely a loss of dissolved and colloidal substances during rinsing. Two replicates showed a slight gain of mass (in the order of 1%) that could possibly be caused by SPT residues or weighing imprecisions. It should be noted that reasonable mass imbalances (<10-15%) are common and do not generally compromise the validity of the fractionation.
SOC recovery was generally within the range of reports from other studies53,54 and remarkably constant considering the large variation in initial SOC content (Table 5). Most samples showed a SOC recovery rate of 80-85%. Losses are attributable to the flushing of soluble C, which is an unavoidable feature of the method; however, soluble organic C can easily be quantified using a separate extraction in water, salt or chemical dispersant55. A small loss of dispersed organic colloids during fractionation is also likely. One site showed a slight gain of carbon which can probably be attributed to analytical error, as the absolute value for the difference was small (3 mg).
The method reproducibility may be verified by analyzing dispersion between replicates. We assessed the standard error of the mean (SEM) as well as the coefficient of variation (CV) of fraction mass between replicates.
Standard errors of the mean were small (Table 6), being generally 1 to 2 orders of magnitude smaller than mean values. This shows that working in 4 replicates allowed us to reliably estimate the central tendency for distribution of materials between fractions.
Coefficients of variation ranged from 2 to 70% (Table 7). All CVs greater than 35% occurred for fractions with small amounts of material (<0.25 g). These high values are simply due to the fact that division by a small mean yields high CVs. A few hF1 and hF3 fractions (coarse and fine silicates) showed relatively high CVs, between 20-35%, yet comprised large amounts of materials (1 - 4 g). This may reflect the high potential for human errors during several sensitive steps (i.e., (1) the separation of floating and suspended materials from the pellet in dense solutions, (2) sedimentation to isolate particle size fractions, (3) sample rinsing and recovery). This result confirms the need to work in several replicates to obtain robust results. It is also recommended that the whole process be handled by the same person, who becomes an expert in performing manipulations in a reproducible way and will acutely notice any details that might be different from previous batches.
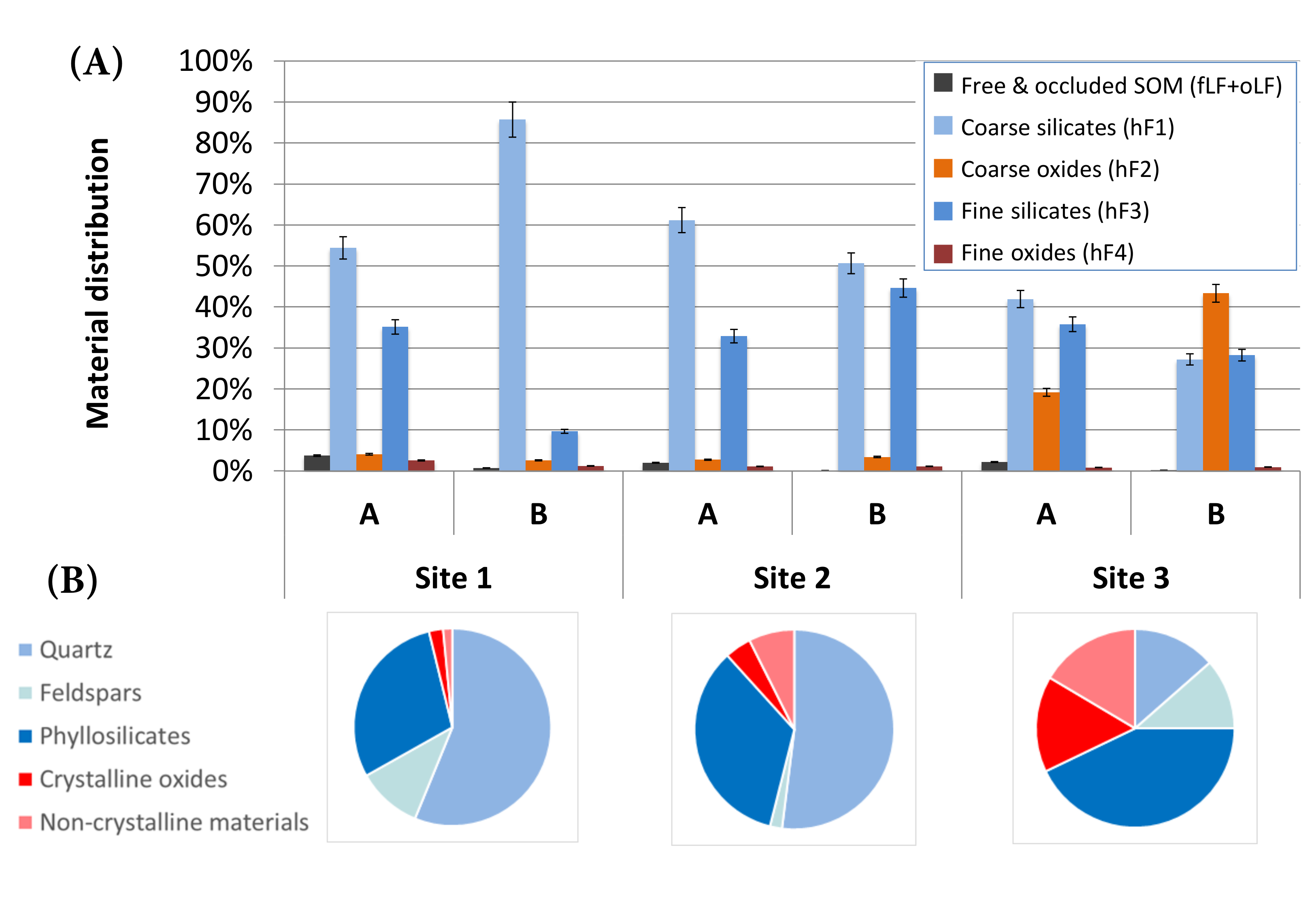
The distribution of material mass between fractions showed strong differences between sites, as could be expected given the initial differences in mineralogy (Figure 2). At site 1, dominated by primary silicates such as quartz and feldspars, most of the material was recovered in hF1 (designed to concentrate coarse silicates). Site 2 showed a greater percentage of phyllosilicates (mostly kaolinite) during mineralogical analysis; accordingly, hF3 (designed to concentrate fine silicates) had more materials than at site 1. Finally, site 3 was the richest in oxides and also showed the greatest amount of material in hF2, designed to concentrate coarse oxides. This indicates that the method was successful in physically fractionating bulk samples into their main mineralogical components.
The amount of materials recovered in coarse (hF1 + hF2) versus fine (hF3 + hF4) fractions was compared to what was expected based on particle-size distribution determined by laser granulometry (Table 8). Agreement was good (< 10%) for three samples. The three other samples showed an excess of material in the order of 20% in the coarse fractions. The large amount of oxides in the soils (particularly at site 3) may be partially responsible for this difference. Oxide grains have a greater density compared to silicates and will sediment faster. Other factors could include incomplete dispersion or partial flocculation of samples during sedimentation, since we did not use chemical dispersion, and removal of some fine materials in the light fractions (fLF and oLF). Finally, laser granulometry is based on volume estimates under the assumption of particle sphericity, while sedimentation yields mass-based estimates. These contrasting principles of measurement are likely to give somewhat diverging results.
CSDF isolates fractions of relative mineralogical homogeneity and their associated organic matter (organo-mineral complexes). It is most useful as a preparation step prior to subsequent geochemical, biochemical and mineralogical analyses. Arguably, the most powerful experiments will aim to characterize both the organic matter and the minerals in each fraction. This will provide direct evidence for the nature of organo-mineral association in soils.
Sample analyses could include determination of SOM quantity (e.g., elemental analysis of organic C and total nitrogen) and quality (e.g., differential Fourier-transform infrared spectroscopy, pyrolysis gas chromatography mass spectrometry, or thermal analysis such as Rock-Eval pyrolysis56,57,58). When looking at the mineral partner, useful analyses could include particle size analysis, quantification of reactive aluminum and iron phases59, X-ray diffraction (XRD) on powdered samples for bulk mineralogy or on oriented slides for clay mineralogy60.
Techniques able to yield simultaneous information on both the organic and inorganic components could be of particular interest. Elemental mapping by secondary ion mass spectrometry (SIMS) or electron microscopy coupled with X-ray microanalysis (WDS or EDS, wavelength or energy dispersive X-ray spectroscopy) can allow for the co-localization of C, N and elements associated with reactive mineral phases such as Fe, Al, Mn or Ca. X-ray photoelectron spectroscopy (XPS) can reveal the chemical composition of SOM and the surface elemental composition of each fraction61.

Figure 1: Flowchart. Fractionation steps and cut-offs used in the method are presented here. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Distribution of materials between fractions as a function of soil mineralogy for two horizons (A and B) at three sites. (A) Bar chart showing the repartition of materials between fractions. Bars represent the mean and error bars represent the standard error of the mean of four replicates. For each sample, the five bars sum to 100%. (B) Bulk sample mineralogy as assessed by powder X-ray diffraction. Please click here to view a larger version of this figure.
{kind=link}
| Soil component | Class | Density [g cm-3] | Size distribution | Source |
| Organic | Organic matter | 1.00-1.50 | Variable | Multiple sources. See Rühlmann et al. (2006)25 for a review |
| Imogolite | Poorly crystalline phase | 1.70-2.33 | Clay | Wada and Wada (1977)26 |
| Allophane | Poorly crystalline phase | 1.84-2.35 | Clay | Wada and Wada (1977)26 ; Wilson (2013)27 |
| Opal | Poorly crystalline phase | 1.90-2.30 | Variable | Hudson Institute of Mineralogy (2017)28 |
| Montmorillonite | Clay mineral | 2.30-2.35 | Clay | Wada and Wada (1977)26; Wilson (2013)27 |
| Vermiculite | Clay mineral | 2.30-2.50 | Clay | Wilson (2013)27 |
| Gibbsite | Al oxide | 2.34-2.42 | Variable | Hudson Institute of Mineralogy (2017)28 |
| K-feldspars | Primary Si-rich silicates | 2.54-2.57 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Albite | Primary Si-rich silicates | 2.60-2.62 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Kaolinite | Clay mineral | 2.60-2.68 | Clay and silt | Klein and Philpotts (2017)29; Wilson (2013)27 |
| Quartz | Primary Si-rich silicates | 2.63-2.66 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Calcite | Carbonate | 2.71 | Variable | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Anorthite | Primary Si-rich silicates | 2.74-2.76 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Illite | Fine-grained mica | 2.75-2.80 | Clay | Wilson (2013)27; Hudson Institute of Mineralogy (2017)28 |
| Muscovite | Mica | 2.77-2.88 | Variable | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Biotite | Mica | 2.78-3.20 | Variable | Klein and Philpotts (2017)29; Skopp (2000)30 |
| Dolomite | Carbonate | 2.84-2.86 | Variable | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Amphiboles | Primary ferromagnesian silicates | 3.00-3.40 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Pyroxenes | Primary ferromagnesian silicates | 3.20-3.60 | Silt and sand | Klein and Philpotts (2017)29; Hudson Institute of Mineralogy (2017)28 |
| Goethite | Fe oxide | 3.30-4.37 | Variable | Hiemstra and van Riemskijk (2009)31; Klein and Philpotts (2017)29 |
| Ferrihydrite | Fe oxide | 3.50-3.90 | Clay | Hiemstra and van Riemskijk (2009)31 |
| Lepidocrocite | Fe oxide | 4.00-4.13 | Variable | Hiemstra and van Riemskijk (2009)31; Hudson Institute of Mineralogy (2017)28 |
| Hematite | Fe oxide | 4.80-5.30 | Variable | Klein and Philpotts (2017)29; Skopp (2000)30 |
Table 1: Main soil components in order of increasing density. Their prevalence in the main textural classes (clay fraction, 0-2 µm; silt fraction, 2-50 µm; sand fraction, 50-2000 µm) for moderately weathered soils is also indicated.
| Name of fraction | Abbreviation | Cut-offs |
| Free organics | fLF | < 1.62 g cm-3 (before sonication) |
| Occluded organics | oLF | < 1.62 g cm-3 (after sonication) |
| Coarse silicates | hF1 | > 8 µm, 1.62 g cm-3< hF1 < 2.78 g cm-3 |
| Coarse oxides | hF2 | > 8 µm, > 2.78 g cm-3 |
| Fine silicates | hF3 | < 8 µm, 1.62 g cm-3< hF3 < 2.78 g cm-3 |
| Fine oxides | hF4 | < 8 µm, > 2.78 g cm-3 |
Table 2: List of fractions resulting from CSDF using one sonication, two density and one size separation steps.
| Solution volume [mL] | Desired density [g cm-3] | Mass SPT [g] | Volume H2O [mL] |
| 1000 | 1.6 | 741 | 856 |
| 1000 | 1.8 | 990 | 810 |
| 1000 | 2 | 1250 | 750 |
| 1000 | 2.2 | 1490 | 715 |
| 1000 | 2.4 | 1803 | 595 |
| 1000 | 2.6 | 2052 | 545 |
| 1000 | 2.8 | 2297 | 504 |
| 1000 | 3 | 2552 | 450 |
Table 3: Guide to the preparation of common SPT solutions.
| Site | Horizon | Replicate | Starting mass [g] | Final mass [g] | Difference [g] | Difference [%] |
| 1 | A | 1 | 10.110 | 9.531 | 0.579 | 5.73 |
| 2 | 10.057 | 9.354 | 0.703 | 6.99 | ||
| 3 | 10.010 | 8.589 | 1.421 | 14.19 | ||
| 4 | 10.043 | 10.197 | -0.154 | -1.53 | ||
| B | 1 | 10.054 | 9.891 | 0.163 | 1.62 | |
| 2 | 10.069 | 9.746 | 0.323 | 3.21 | ||
| 3 | 10.058 | 9.699 | 0.359 | 3.57 | ||
| 4 | 10.059 | 9.782 | 0.277 | 2.76 | ||
| 2 | A | 1 | 10.130 | 9.252 | 0.878 | 8.67 |
| 2 | 10.182 | 9.246 | 0.936 | 9.20 | ||
| 3 | 10.053 | 9.372 | 0.681 | 6.77 | ||
| 4 | 10.031 | 9.577 | 0.454 | 4.53 | ||
| B | 1 | 10.123 | 8.824 | 1.299 | 12.83 | |
| 2 | 10.052 | 8.938 | 1.114 | 11.08 | ||
| 3 | 10.029 | 9.006 | 1.023 | 10.20 | ||
| 4 | 10.086 | 9.118 | 0.968 | 9.60 | ||
| 3 | A | 1 | 10.020 | 9.187 | 0.833 | 8.32 |
| 2 | 10.060 | 9.139 | 0.921 | 9.15 | ||
| 3 | 10.069 | 9.386 | 0.683 | 6.79 | ||
| 4 | 10.049 | 9.638 | 0.411 | 4.09 | ||
| B | 1 | 10.071 | 9.207 | 0.864 | 8.58 | |
| 2 | 10.065 | 9.314 | 0.751 | 7.46 | ||
| 3 | 10.155 | 10.241 | -0.086 | -0.85 | ||
| 4 | 10.046 | 9.549 | 0.497 | 4.95 |
Table 4: Recovery rate of whole soil, showing the starting mass at the beginning of the fractionation procedure and final mass calculated as the sum of all fractions. Differences are expressed as % starting mass.
| Site | Horizon | Starting SOC mass | Final SOC mass | Difference | Difference |
| [g] | [g] | [g] | [%] | ||
| 1 | A | 0.50 | 0.41 | 0.09 | 18.07 |
| B | 0.026 | 0.029 | -0.003 | -10.63 | |
| 2 | A | 0.34 | 0.27 | 0.07 | 20.19 |
| B | 0.07 | 0.06 | 0.01 | 12.33 | |
| 3 | A | 1.08 | 0.94 | 0.14 | 12.56 |
| B | 0.31 | 0.27 | 0.05 | 14.51 |
Table 5: Recovery rate of soil organic carbon. Initial SOC content was calculated as the product of organic C concentration measured by elemental analysis and initial sample mass. Final SOC content was calculated as the product of organic C concentration and each fraction mass, summed for all fractions. Differences are expressed as % starting mass.
| Site | Horizon | fLF 1.62 g cm-3 | oLF 1.62 g cm-3 | hF1 > 8 µm < 2.78 g cm-3 | hF2 > 8 µm > 2.78 g cm-3 | hF3 < 8 µm < 2.78 g cm-3 | hF4 < 8 µm > 2.78 g cm-3 |
| 1 | A | 0.0794 ± 0.0052 | 0.1093 ± 0.0076 | 5.2188 ± 0.3079 | 0.3925 ± 0.0416 | 3.3699 ± 0.1504 | 0.2478 ± 0.0689 |
| B | 0.0044 ± 0.0005 | 0.0074 ± 0.0011 | 8.4351 ± 0.16 | 0.2569 ± 0.0301 | 0.9528 ± 0.1013 | 0.1226 ± 0.0124 | |
| 2 | A | 0.066 ± 0.011 | 0.1353 ± 0.0152 | 5.722 ± 0.1033 | 0.2575 ± 0.008 | 3.0761 ± 0.1464 | 0.1047 ± 0.0364 |
| B | 0.0024 ± 0.0002 | 0.0165 ± 0.0022 | 4.5416 ± 0.0387 | 0.3082 ± 0.0072 | 4.0005 ± 0.0547 | 0.1025 ± 0.0268 | |
| 3 | A | 0.2107 ± 0.0099 | 0.1489 ± 0.0223 | 3.8507 ± 0.6801 | 1.762 ± 0.0923 | 3.2862 ± 0.4892 | 0.0792 ± 0.0165 |
| B | 0.0305 ± 0.0035 | 0.0433 ± 0.0065 | 2.5929 ± 0.376 | 4.1277 ± 0.1025 | 2.6909 ± 0.13 | 0.0927 ± 0.0087 |
Table 6: Mean value and SEM for fraction mass (g). Each cell represents a mean of 4 replicates.
| Site | Horizon | fLF < 1.62 g cm-3 | oLF < 1.62 g cm-3 | hF1 > 8 µm < 2.78 g cm-3 | hF2 > 8 µm > 2.78 g cm-3 | hF3 < 8 µm < 2.78 g cm-3 | hF4 < 8 µm > 2.78 g cm-3 |
| 1 | A | 0.13 | 0.14 | 0.12 | 0.21 | 0.09 | 0.56 |
| B | 0.22 | 0.29 | 0.04 | 0.23 | 0.21 | 0.20 | |
| 2 | A | 0.33 | 0.22 | 0.04 | 0.06 | 0.10 | 0.70 |
| B | 0.17 | 0.26 | 0.02 | 0.05 | 0.03 | 0.52 | |
| 3 | A | 0.09 | 0.30 | 0.35 | 0.10 | 0.30 | 0.42 |
| B | 0.23 | 0.30 | 0.29 | 0.05 | 0.10 | 0.19 |
Table 7: Coefficients of variation of fraction mass for 4 replicates.
| Site | Horizon | Texture < 8 µm % | Texture > 8 µm % | Materials in fine fractions (hF3 + hF4) % | Materials in coarse fractions (hF1 + hF2) % | Coarse fraction 'excess' % |
| 1 | A | 48 | 52 | 39 | 61 | 9 |
| B | 31 | 69 | 11 | 89 | 20 | |
| 2 | A | 43 | 57 | 35 | 65 | 8 |
| B | 46 | 54 | 46 | 54 | 1 | |
| 3 | A | 58 | 42 | 37 | 63 | 21 |
| B | 52 | 48 | 29 | 71 | 23 |
Table 8: Comparison between soil's texture determined by laser granulometry and the distribution of particles in size fractions. The last column shows the excess of materials in the coarse fractions compared to what was expected based on textural analysis.
Discussion
The success of CSDF experiments hinges on the selection of appropriate parameters for the method, so that fractions of relatively homogeneous composition may be isolated. Key considerations in the selection of fractionation parameters are discussed below.
The fLF represents organic matter for which interaction with minerals is minimal. Extraction of this fraction is delicate, since the mixing of soil with the dense solution may already break-up some macroaggregates. There are, however, indications that the organic matter present in macroaggregates may be more similar to the fLF stricto sensu than to the oLF released by high-energy sonication18. Some authors have even proposed a low-energy sonication step to isolate the pool of free and weakly mineral-interacting organic matter, termed 'intra-aggregate particulate organic matter', iPOM54.
For the release of occluded organic matter, different techniques exist to disrupt soil aggregates. The most widespread are sonication, agitation with glass beads and the use of chemical dispersants33,62,63. Sonication was chosen here because the output energy can be finely controlled and is believed to distribute more or less uniformly in the sample. By precluding the need to use chemical dispersants, sonication may be considered as relatively preservative towards organo-mineral complexes22, 33. The dispersion step, however, remains one of the most delicate operations. On the one hand, a weak dispersion will leave the aggregates intact and may lead to an over-estimate of hF SOC; on the other hand, a highly vigorous dispersion step could cause re-distribution of SOC across the fractions by partial destruction of organo-mineral complexes. Weak organic-sand associations may be particularly vulnerable to this process. Since occlusion within aggregates and surface sorption are processes occurring along a continuum2, no perfect solution exists. Therefore, the energy level of sonication needs to be adjusted thoughtfully according to the soil properties. Kaiser and Berhe64 have published a very helpful review that proposes a strategy to minimize artifacts caused by ultrasound when dispersing soils.
Reported sonication energies range from 60 to 5,000 J·mL-1. Several research groups have reported that 100 J·mL-1 could be sufficient to destroy macroaggregates and effectively disperse sandy soils, while 500 J·mL-1 would destroy large microaggregates and provide a reasonable dispersion of reactive soils63,65,66,67,68. In physical fractionation schemes, complete dispersion of silt and clay-sized aggregates may not be necessary, since the protection mechanism is likely to become indistinguishable from sorptive stabilization in these size ranges. A reasonable objective of dispersion prior to size or density fractionation may be to disrupt macro- (> 250 µm) and large micro- (> 53 µm) aggregates. Energies of 100 J·mL-1 (sandy soils) to 200 J·mL-1 (loamy soils) may be appropriate choices. An energy of 200 J·mL-1 may already extract a portion of microbial metabolites (supposedly mineral-associated)69, thus the use of higher sonication energies should be subject to caution. However, mineralogically reactive soils with cemented aggregates could require up to 500 J·mL-1 to disperse. It is essential that the dispersion energy be adjusted to match each soil type as well as study objectives. Finally, it is important to remember that even after supposedly complete ultrasonic dispersion, clay-sized microaggregates are likely to persist70.
A difficulty with harmonizing physical fractionation techniques resides in the heterogeneity found in soils, in particular in their mineral composition. The choice of dense solutions should be made on the basis of known or inferred soil mineralogy, with the ultimate goal to isolate fractions which are as homogeneous as possible.
In the article, the dense solution used was SPT - pH 371, 72. The low pH minimizes losses of soluble organic compounds. However, density fractionation may be performed with different dense solutions. Historically, organic liquids were used (tetrabromoethane, tetrachloromethane), but were gradually abandoned at the profit of inorganic salts (sodium iodide, SPT) because of the toxicity of halogenated hydrocarbons and the inherent contamination of soil organics. Nowadays, SPT is the preferred solution because its density can be adjusted between 1.0 to 3.1 g·cm-3, it can be recycled and has a low toxicity (unless ingested)22, 50. Main manufacturers offer a range of SPT grades differing in the level of carbon and nitrogen contamination. For density fractionation of soils, the purest grade is recommended, particularly if the fractions are to be analyzed for isotopic composition.
A solution of density 1.6 g·cm-3 has classically been used to separate light organic from mineral-associated fractions - see for example Golchin et al.21. While some authors have suggested that a density of 1 g·cm-3 (water) could be sufficient to extract most of the light fraction73, 74, others have proposed higher density cut-offs such as 1.62 or 1.65 g·cm-3 based on the idea that some organic components could show densities up to 1.60 g·cm-3 33,75,76. Densities as high as 1.85 g·cm-3 have even been employed50. When selecting a density to separate light from heavy fractions, it should be noted that no perfect solution exists. Indeed, lower densities risk attributing some 'light' organics to the heavy fractions, while higher densities risk including some minerals into the light fractions. This last effect can be detected when observing the carbon content of the light fractions, with a % SOC lower than 40-45% indicating some degree of mineral contamination.
For heavy fractions, preliminary analysis such as XRD can provide insight into the mineralogy of the bulk sample60 and help define density cut-offs capable of distinguishing between the main mineral components of a soil, keeping in mind that high organic loadings will lower the density of a mineral compared to its theoretical value. Similarly, for particle-size separation, a textural analysis77,78 can help set appropriate limits. Particle-size separation is a particularly attractive addition to simple density fractionation whenever sequential density fractionation is difficult. This is the case for instance for soils containing large amounts of oxyhydroxides and low-activity clays, which result in sample dispersion and prevent clear separations in heavy liquids. A particle-size separation step is also indicated to separate minerals of similar densities but different sizes (e.g., quartz and illite).
Free calcium ions will react with SPT to form insoluble Ca metatungstate. The procedure is thus inappropriate for alkaline soils containing large amounts of poorly crystalline, pedogenic carbonates. Small amounts of low-reactivity carbonates do not interfere with the fractionation as long as the samples are not left in contact with SPT for too long. Ca metatungstate precipitates will lead to an over-estimate of fraction masses. If LFs are run on an elemental analyzer for C concentration, the problem will be discovered but the fractionation will be compromised.
In addition to these technical difficulties, the fundamental limitation of CSDF (or of any physical fractionation scheme) stems from the fact that reactive minerals in soils rarely occur as discrete separates, but instead as coatings and cements. The occurrence of highly sorptive but very thin coatings on otherwise unreactive minerals (such as quartz) can lead to a biased view of organo-mineral associations. Caution is thus required when interpreting results, particularly for soils whose reactivity is dominated by poorly crystalline and oxide phases. Further characterization of fractions can help alleviate such ambiguities. Nevertheless, detailed physical fractionation methods such as CSDF have an unmatched ability to gain insight into the composition of naturally-occurring organo-mineral complexes. Such insight is expected to yield new understanding of the biogeochemistry of the largest pool of organic matter in soils, the mineral-associated one.
Acknowledgements
The development of this method was supported by the Fond d'Investissement (FINV) of the Faculty of Geosciences at the University of Lausanne. We acknowledge the Uganda National Council for Science and Technology and Uganda Wildlife Authority for granting us permission to collect research samples. The authors further wish to thank Prof. Thierry Adatte for CHN and XRD analyses. We are grateful to Prof. Erika Marin-Spiotta for providing initial training in classical density fractionation. We also thank laboratory manager Laetitia Monbaron for her assistance in securing supplies and equipment.
Materials
| Name | Company | Catalog Number | Comments |
| Fractionation | |||
| Sodium polytungstate | Sometu | SPT 0 (low C and N) is recommended. Lower grade polytungstate may contaminate samples. | |
| Hydrometers (1-1.5, 1.5-2, 2-2.5, 2.5-3 g.cm-3) | Allafrance | Calibrated at 20 °C, e.g. 3050FG250/20-qp | |
| Vortex mixer | Fisher | Fixed speed standard vortex mixer, e.g. 02-215-410 | |
| Sonifier | VWR | Qsonica LLC - Q500 system with standard probe 4220 | |
| Sonifier stand | VWR | Large clamp stand | |
| Sonifier enclosure | VWR | Soundproof cabinet (optional) | |
| Swinging-bucket centrifuge | Beckman | Able to achieve speeds of 4000 g or more, fitted with rotor accommodating 50 mL Falcon tubes | |
| High-speed centrifuge with fixed angle rotor | Beckman | Able to achieve speeds of 7500 g or more, fitted with rotor accommodating 250 mL bottles | |
| 50 mL centrifuge Falcon tubes | Corning | e.g. 352070 | |
| 250 mL centrifuge bottles | Beckman | Polycarbonate bottles (e.g. 352070) are recommended because they are clearer than other plastics. | |
| Vaccum filtration units | Semadeni | Polusulfone reusable units, e.g. 3029 | |
| Polypropylene hose | Semadeni | To connect the filtration unit to vaccuum source | |
| Ultrafiltration disks, 0.45 µm pore size | Millipore | e.g. HAWP04700 | |
| Dessicator cabinet | Fisher scientific | 3 shelves, e.g. 305317-0120 | |
| Drierite absorbent indicating | Millipore | Blue drierite, e.g. 10276750 | |
| Scintillation vials | Fisher scientific | HDPE - separated cap 20mL, e.g. 12341599 | |
| 150 mL aluminium boats (smooth sides) | Fisher scientific | Any model. | |
| Laboratory oven | Fisher scientific | Any model. | |
| Recycling SPT column | |||
| Cation exchange resin | Sigma-Aldrich | Dowex® Marathon™ C sodium form, strongly acidic, 20-50 mesh | |
| Activated charcoal | Sigma-Aldrich | Darco S-51, 4-12 mesh | |
| Glass wool | Fisher scientific | Pyrex | |
| Filter paper, 2.5 µm pore size | Sigma-Aldrich | Whatman grade 42, e.g. WHA1442150 | |
| Hydrogen peroxide | Sigma-Aldrich | Reagent grade. | |
| Ethanol | Sigma-Aldrich | Reagent grade. | |
| Polycarbonate 1000mL graduated cylinder | Semadeni | Any model. | |
| Stand and clamp | Sigma-Aldrich | Size L - 2-prong | |
| Polypropylene hose | Semadeni | Any model. | |
| Polypropylene hose clamp | Semadeni | Any model. | |
| Polypropylene funnels | Semadeni | Any model. | |
| Polypropylene bottle (1L, 2L) | Semadeni | Any model. | |
| Heating plate | Fisher scientific | Any model. |
References
- Adhikari, K., Hartemink, A. E. Linking soils to ecosystem services — A global review. Geoderma. 262 (Supplement C), 101-111 (2016).
- Kögel-Knabner, I., et al. Organo-mineral associations in temperate soils: Integrating biology, mineralogy, and organic matter chemistry. Journal of Plant Nutrition and Soil Science. 171 (1), 61-82 (2008).
- Hansen, J., et al. Young people’s burden: requirement of negative CO2 emissions. Earth System Dynamics. 8 (3), 577-616 (2017).
- Lützow, M. v., et al. Stabilization of organic matter in temperate soils: mechanisms and their relevance under different soil conditions - a review. European Journal of Soil Science. 57 (4), 426-445 (2006).
- Schmidt, M. W. I., et al. Persistence of soil organic matter as an ecosystem property. Nature. 478 (7367), (2011).
- Lehmann, J., Kleber, M. The contentious nature of soil organic matter. Nature. 528 (7580), 60 (2015).
- Rowley, M. C., Grand, S., Verrecchia, &. #. 2. 0. 1. ;. P. Calcium-mediated stabilisation of soil organic carbon. Biogeochemistry. 137 (1-2), 27-49 (2018).
- Matteodo, M., et al. Decoupling of topsoil and subsoil controls on organic matter dynamics in the Swiss Alps. Geoderma. 330, 41-51 (2018).
- Coward, E. K., Thompson, A. T., Plante, A. F. Iron-mediated mineralogical control of organic matter accumulation in tropical soils. Geoderma. 306 (Supplement C), 206-216 (2017).
- Torres-Sallan, G., et al. Clay illuviation provides a long-term sink for C sequestration in subsoils. Scientific Reports. 7, 45635 (2017).
- Rasmussen, C., et al. Beyond clay: towards an improved set of variables for predicting soil organic matter content. Biogeochemistry. 137 (3), 297-306 (2018).
- Khomo, L., Trumbore, S., Bern, C. R., Chadwick, O. A. Timescales of carbon turnover in soils with mixed crystalline mineralogies. SOIL. 3 (1), 17-30 (2017).
- Basile-Doelsch, I., et al. Mineralogical control of organic carbon dynamics in a volcanic ash soil on La Réunion. European Journal of Soil Science. 56 (6), 689-703 (2005).
- Basile-Doelsch, I., Brun, T., Borschneck, D., Masion, A., Marol, C., Balesdent, J. Effect of landuse on organic matter stabilized in organomineral complexes: A study combining density fractionation, mineralogy and δ13C. Geoderma. 151 (3), 77-86 (2009).
- Kleber, M., Mikutta, R., Torn, M. S., Jahn, R. Poorly crystalline mineral phases protect organic matter in acid subsoil horizons. European Journal of Soil Science. 56 (6), 717-725 (2005).
- Krull, E. S., Baldock, J. A., Skjemstad, J. O. Importance of mechanisms and processes of the stabilisation of soil organic matter for modelling carbon turnover. Functional Plant Biology. 30 (2), 207-222 (2003).
- Sposito, G., Skipper, N. T., Sutton, R., Park, S., Soper, A. K., Greathouse, J. A. Surface geochemistry of the clay minerals. Proceedings of the National Academy of Sciences of the United States of America. 96 (7), 3358-3364 (1999).
- Poeplau, C., et al. Isolating organic carbon fractions with varying turnover rates in temperate agricultural soils - A comprehensive method comparison. Soil Biology and Biochemistry. 125, 10-26 (2018).
- Poeplau, C., et al. Reproducibility of a soil organic carbon fractionation method to derive RothC carbon pools. European Journal of Soil Science. 64 (6), 735-746 (2013).
- Grunwald, D., et al. Influence of elevated soil temperature and biochar application on organic matter associated with aggregate-size and density fractions in an arable soil. Agriculture, Ecosystems & Environment. 241 (Supplement C), 79-87 (2017).
- Golchin, A., Oades, J., Skjemstad, J., Clarke, P. Study of free and occluded particulate organic matter in soils by solid state 13C Cp/MAS NMR spectroscopy and scanning electron microscopy). Australian Journal of Soil Research. 32 (2), 285 (1994).
- Lützow, M. v., et al. SOM fractionation methods: Relevance to functional pools and to stabilization mechanisms. Soil Biology and Biochemistry. 39 (9), 2183-2207 (2007).
- Christensen, B. T. Physical fractionation of soil and structural and functional complexity in organic matter turnover. European Journal of Soil Science. 52 (3), 345-353 (2001).
- Sollins, P., et al. Sequential density fractionation across soils of contrasting mineralogy: evidence for both microbial- and mineral-controlled soil organic matter stabilization. Biogeochemistry. 96 (1-3), 209-231 (2009).
- Rühlmann, J., Körschens, M., Graefe, J. A new approach to calculate the particle density of soils considering properties of the soil organic matter and the mineral matrix. Geoderma. 130 (3-4), 272-283 (2006).
- Wada, S. I., Wada, K. Density and structure of allophane. Clay Minerals. 12, 289-298 (1977).
- Wilson, M. J., Deer, W. A., Howie, R. A., Zussman, J. Sheet Silicates: Clay Minerals, 2nd edition. Rock-Forming Minerals. 3C, (2013).
- . mindat.org Available from: https://www.mindat.org/ (2017)
- Klein, C., Philpotts, A. . Earth Materials: Introduction to Mineralogy and Petrology. , (2017).
- Skopp, J. M., Huang, P. M., Li, Y., Sumner, M. E. Physical properties of primary particles. Handbook of soil science: properties and processes. , 1.1-1.10 (2012).
- Hiemstra, T., Van Riemsdijk, W. H. A surface structural model for ferrihydrite I: Sites related to primary charge, molar mass, and mass density. Geochimica et Cosmochimica Acta. 73 (15), 4423-4436 (2009).
- Strosser, E. Methods for determination of labile soil organic matter: An overview. Journal of Agrobiology. 27 (2), (2010).
- Christensen, B. T. Physical fractionation of soil and organic matter in primary particle size and density separates. Advances in Soil Science. , 1-90 (1992).
- Greenland, D. J. Interaction between clays and organic compounds in soils. Part I. Mechanisms of interaction between clays and defined organic compounds. Soils Fertility. 28, 415-425 (1965).
- Greenland, D. J. Interaction between clays and organic compounds in soils. Part II. Adsorption of soil organic compounds and its effect on soil properties. Soils Fertility. 28, 521-532 (1965).
- Allison, F. E., Sherman, M. S., Pinck, L. A. Maintenance of soil organic matter: I. Inorganic soil colloid as a factor in retention of carbon during formation of humus. Soil Science. 68 (6), 463 (1949).
- Anderson, D. W. The effect of parent material and soil development on nutrient cycling in temperate ecosystems. Biogeochemistry. 5 (1), 71-97 (1988).
- Oades, J. M. The retention of organic matter in soils. Biogeochemistry. 5 (1), 35-70 (1988).
- Tiessen, H., Stewart, J. W. B., Hunt, H. W. Concepts of soil organic matter transformations in relation to organo-mineral particle size fractions. Plant and Soil. 76 (1-3), 287-295 (1984).
- Trumbore, S. E., Zheng, S. Comparison of fractionation methods for soil organic matter 14C analysis. Radiocarbon. 38 (2), 219-229 (1996).
- Moni, C., Derrien, D., Hatton, P. -. J., Zeller, B., Kleber, M. Density fractions versus size separates: does physical fractionation isolate functional soil compartments?. Biogeosciences. 9 (12), 5181-5197 (2012).
- Mueller, C. W., et al. Initial differentiation of vertical soil organic matter distribution and composition under juvenile beech (Fagus sylvatica L.) trees. Plant and soil. 323 (1-2), 111-123 (2009).
- Schweizer, S. A., Hoeschen, C., Schlüter, S., Kögel-Knabner, I., Mueller, C. W. Rapid soil formation after glacial retreat shaped by spatial patterns of organic matter accrual in microaggregates. Global Change Biology. 24 (4), 1637-1650 (2018).
- Kalra, Y. P., Maynard, D. G. . Methods manual for forest soil and plant analysis. , (1991).
- Mueller, C. W., Rethemeyer, J., Kao-Kniffin, J., Löppmann, S., Hinkel, K. M., Bockheim, J. G. Large amounts of labile organic carbon in permafrost soils of northern Alaska. Global Change Biology. 21, 2804-2817 (2015).
- Höfle, S., Rethemeyer, J., Mueller, C., John, S. Organic matter composition and stabilization in a polygonal tundra soil of the Lena Delta. Biogeosciences. 10, (2013).
- North, P. F. Towards an absolute measurement of soil structural stability using ultrasound. Journal of Soil Science. 27 (4), 451-459 (1976).
- Grand, S., Lavkulich, L. M. Depth distribution and predictors of soil organic carbon in Podzols of a forested watershed in Southwestern Canada. Soil Science. 176, 164-174 (2011).
- Laird, D. A., Dowdy, R. H., Amonette, J. E., Zelazny, L. W. Preconcentration techniques in soil mineralogical analyses. Quantitative Methods in Soil Mineralogy. , (1994).
- Six, J. Recycling of sodium polytungstate used in soil organic matter studies. Soil Biology and Biochemistry. 31 (8), 1193-1196 (1999).
- dos Anjos, L. H. C., Franzmeier, D. P., Schulze, D. G. Formation of soils with plinthite on a toposequence in Maranhaño State, Brazil. Geoderma. 64 (3), 257-279 (1995).
- . Keys to Soil Taxonomy | NRCS Soils Available from: https://www.nrcs.usda.gov/wps/portal/nrcs/detail/soils/survey/class/taxonomy/?cid=nrcs142p2_053580 (2014)
- Baldock, J. A., et al. Aspects of the chemical structure of soil organic materials as revealed by solid-state 13C NMR spectroscopy. Biogeochemistry. 16, 1-42 (1992).
- Vos, C., Jaconi, A., Jacobs, A., Don, A. Hot regions of labile and stable soil organic carbon in Germany - Spatial variability and driving factors. SOIL. 4, 153-167 (2018).
- Magid, J., Gorissen, A., Giller, K. E. In search of the elusive "active" fraction of soil organic matter: three size-density fractionation methods for tracing the fate of homogeneously 14C-labelled plant materials. Soil Biology and Biochemistry. 28 (1), 89-99 (1996).
- Albrecht, R., Sebag, D., Verrecchia, E. Organic matter decomposition: bridging the gap between Rock-Eval pyrolysis and chemical characterization (CPMAS 13C NMR). Biogeochemistry. 122 (1), 101-111 (2015).
- Disnar, J. R., Guillet, B., Keravis, D., Di-Giovanni, C., Sebag, D. Soil organic matter (SOM) characterization by Rock-Eval pyrolysis: scope and limitations. Organic Geochemistry. 34 (3), 327-343 (2003).
- Sebag, D., Disnar, J. R., Guillet, B., Di Giovanni, C., Verrecchia, E. P., Durand, A. Monitoring organic matter dynamics in soil profiles by ‘Rock-Eval pyrolysis’: bulk characterization and quantification of degradation. European Journal of Soil Science. 57 (3), 344-355 (2006).
- Shang, C., Zelazny, L. W., Ulery, A. L., Richard Drees, L., Ulery, A. L., Drees, L. R. Selective dissolution techniques for mineral analysis of soils and sediments. Methods of Soil Analysis, part 5. Mineralogical Methods. , (2008).
- Harris, W., Norman White, G., Ulery, A. L., Richard Drees, L., Ulery, A. L., Drees, L. R. X-ray diffraction techniques for soil mineral identification. Methods of Soil Analysis, part 5. Mineralogical Methods. , (2008).
- Jones, E., Singh, B. Organo-mineral interactions in contrasting soils under natural vegetation. Frontiers in Environmental Science. 2, 2 (2014).
- Amelung, W., Zech, W. Minimisation of organic matter disruption during particle-size fractionation of grassland epipedons. Geoderma. 92 (1), 73-85 (1999).
- Schmidt, M. W. I., Rumpel, C., Kögel-Knabner, I. Particle size fractionation of soil containing coal and combusted particles. European Journal of Soil Science. 50 (3), 515-522 (1999).
- Kaiser, M., Berhe, A. A. How does sonication affect the mineral and organic constituents of soil aggregates?—A review. Journal of Plant Nutrition and Soil Science. 177 (4), 479-495 (2014).
- Asano, M., Wagai, R. Evidence of aggregate hierarchy at micro- to submicron scales in an allophanic Andisol. Geoderma. 216, 62-74 (2014).
- Heckman, K., et al. Sorptive fractionation of organic matter and formation of organo-hydroxy-aluminum complexes during litter biodegradation in the presence of gibbsite. Geochimica et Cosmochimica Acta. , 667-683 (2013).
- Schrumpf, M., Kaiser, K., Guggenberger, G., Persson, T., Kögel-Knabner, I., Schulze, E. -. D. Storage and stability of organic carbon in soils as related to depth, occlusion within aggregates, and attachment to minerals. Biogeosciences. 10, 1675-1691 (2013).
- Rumpel, C., Eusterhues, K., Kögel-Knabner, I. Location and chemical composition of stabilized organic carbon in topsoil and subsoil horizons of two acid forest soils. Soil Biology and Biochemistry. 36, 177-190 (2004).
- Höfle, S., Rethemeyer, J., Mueller, C., John, S. Organic matter composition and stabilization in a polygonal tundra soil of the Lena Delta. Biogeosciences. 10, (2013).
- Chenu, C., Plante, A. F. Clay-sized organo-mineral complexes in a cultivation chronosequence: revisiting the concept of the ‘primary organo-mineral complex. European Journal of Soil Science. 57 (4), 596-607 (2006).
- Gregory, M. R., Johnston, K. A. A nontoxic substitute for hazardous heavy liquids—aqueous sodium polytungstate (3Na2WO4.9WO3.H2O) solution (Note). New Zealand Journal of Geology and Geophysics. 30 (3), 317-320 (1987).
- Munsterman, D., Kerstholt, S. Sodium polytungstate, a new non-toxic alternative to bromoform in heavy liquid separation. Review of Palaeobotany and Palynology. 91 (1), 417-422 (1996).
- Spielvogel, S., Prietzel, J., Kögel-Knabner, I. Changes of lignin phenols and neutral sugars in different soil types of a high-elevation forest ecosystem 25 years after forest dieback. Soil Biology and Biochemistry. 39 (2), 655-668 (2007).
- Kaiser, M., Wirth, S., Ellerbrock, R. H., Sommer, M. Microbial respiration activities related to sequentially separated, particulate and water-soluble organic matter fractions from arable and forest topsoils. Soil biology and Biochemistry. 42 (3), 418-428 (2010).
- Sollins, P., et al. Organic C and N stabilization in a forest soil: Evidence from sequential density fractionation. Soil Biology and Biochemistry. 38 (11), 3313-3324 (2006).
- Wander, M., Magdoff, F., Weil, R. Soil organic matter fractions and their relevance to soil function. Soil Organic Matter in Sustainable Agriculture. , (2004).
- Gee, G. W., Or, D., Dane, J. H., Topp, C. G. 2.4 Particle-size analysis. Methods of Soil Analysis: Part 4 Physical Methods. , (2002).
- Sheldrick, B. H., Wang, C., Carter, M. R., Gregorich, E. G. Particle size distribution. Soil Sampling and Methods of Analysis. , (1993).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved