Production, Purification, and Quality Control for Adeno-associated Virus-based Vectors
In This Article
Summary
Here, we describe an efficient and reproducible strategy to produce, titer, and quality-control batches of adeno-associated virus vectors. It allows the user to obtain a vector preparation with high-titer (≥1 x 1013 vector genomes/mL) and a high purity, ready for in vitro or in vivo use.
Abstract
Gene delivery tools based on adeno-associated viruses (AAVs) are a popular choice for the delivery of transgenes to the central nervous system (CNS), including gene therapy applications. AAV vectors are non-replicating, able to infect both dividing and non-dividing cells and provide long-term transgene expression. Importantly, some serotypes, such as the newly described PHP.B, can cross the blood-brain-barrier (BBB) in animal models, following systemic delivery. AAV vectors can be efficiently produced in the laboratory. However, robust and reproducible protocols are required to obtain AAV vectors with sufficient purity levels and titer values high enough for in vivo applications. This protocol describes an efficient and reproducible strategy for AAV vector production, based on an iodixanol gradient purification strategy. The iodixanol purification method is suitable for obtaining batches of high-titer AAV vectors of high purity, when compared to other purification methods. Furthermore, the protocol is generally faster than other methods currently described. In addition, a quantitative polymerase chain reaction (qPCR)-based strategy is described for a fast and accurate determination of the vector titer, as well as a silver staining method to determine the purity of the vector batch. Finally, representative results of gene delivery to the CNS, following systemic administration of AAV-PHP.B, are presented. Such results should be possible in all labs using the protocols described in this article.
Introduction
Over the past 30 years, wild-type AAVs have been engineered to create recombinant AAV vectors, which have proven to be exceptionally useful tools for gene transfer into the CNS1,2,3,4,5,6 and gene therapy approaches to disease (including FDA- and EMA-approved therapies)4,7. Their suitability for use in the CNS largely derives from their ability to infect non-dividing, post-mitotic cells, typically found in the CNS8. However, AAV-based vectors also have the advantage of allowing a long-term expression of any given therapeutic transgene4,9 while eliciting a milder immune response compared to other viral vectors7,8,10,11,12.
The main elements of any AAV vector are the genome and the capsid. Wild-type AAVs are single-stranded (ss) DNA viruses with a genome of approximately 5 kilobases (kb)13. For the production of recombinant AAV vectors, the rep and cap genes (necessary for genome replication and the assembly of the viral capsid) are deleted from the genome of the wild-type AAV and provided in trans, leaving room for an expression cassette containing the transgene14,15. The inverted terminal repeat sequences (ITRs) of the original viral genome are the only retained elements in an AAV vector, as these are essential for replication and packaging3,10,14. AAV vectors can be engineered to enhance transgene expression; a mutation in one of the ITRs leads to the formation of a hairpin loop which effectively allows the generation of a complementary DNA strand3,7,15. The main advantage of this configuration, termed a self-complementary (sc) genome, is that it bypasses the need for second-strand synthesis typical in the conventional life cycle of AAVs, considerably increasing the speed and levels of transgene expression1. Nevertheless, using an scAAV genome reduces the cargo capacity of the vector to approximately 2.4 kb. This includes transgene sequences, as well as any regulatory sequences such as promoters or microRNA-binding sites, to limit expression to specific cell types16.
The AAV capsid determines the vector-host cell interaction and confers a degree of cell type or tissue tropism for an AAV serotype, which can also be exploited to limit transgene expression to specific locations. Several AAV serotypes are found in nature, whereas others have been produced in laboratories through recombinant approaches (i.e., PHP.B). In addition, some capsids also bestow other useful characteristics, such as the ability to cross the BBB, resulting in the delivery of transgenes throughout the CNS after systemic administration. This has been shown for AAV9, as well as for the recently described PHP.B capsid17. As a consequence, these serotypes are proving to be particularly relevant for new gene therapy approaches for neurodegenerative disorders1,17,18.
The aim of this protocol is to describe a cost-effective method for the small-scale production of AAV vectors with high titer and purity. Although the results presented here use the PHP.B capsid and a scAAV expression cassette, the protocol is suitable for the production of several AAV vector serotypes and genome configurations, allowing maximum experimental flexibility. However, the vector yield and final purity can vary depending on the chosen serotype.
The protocol itself is a variant of the classical tri-transfection method for viral vector production and incorporates the use of an iodixanol gradient for vector clean-up, which, in comparison to the classical use of cesium chloride (CsCl) gradients, has been reported to produce AAV vectors of higher purity in a more time-efficient manner19,20,21.
The transfection, purification and concentration steps are intended to be performed according to good laboratory practice (GLP) in a tissue culture laboratory rated for viral vector work. Each task needs to be performed in compliance with the relevant local and national legislation concerning viral vector production and use. Work must be carried out under a laminar flow hood and in sterile conditions. Inside the vector facility, it is recommended to wear a lab apron, in addition to the regular tissue culture lab coat. Moreover, a double pair of gloves, as well as plastic overshoes, should be worn at all times.
Before starting the vector production, ensure all necessary equipment and plasmids are available. 1) pCapsid plasmid contains the rep gene that encodes four non-structural proteins necessary for replication, namely Rep78, Rep68, Rep52, and Rep40, and the cap gene that encodes three structural capsid proteins, namely VP1, VP2, and VP3. 2) pHelper plasmid contains the genes E4, E2A, and VA from adenovirus, which facilitate AAV production in HEK293T cells. 3) pTransgene plasmid contains the transgene expression cassette, flanked by two ITRs. These plasmids can be synthesized de novo in the laboratory using sequences available online22. For plasmids made de novo, especially those containing novel transgenes, sequencing is required, to make sure that the transgene and ITRs are correct. Alternatively, premade plasmids can be directly acquired through online plasmid repositories. When necessary, plasmids can be amplified and purified using standard kits, according to the manufacturer's instructions23.
Vector titer and purity can adversely affect the transduction ability of the vector. Additional protocols are supplied to evaluate the quality of the produced vector. The final vectors will be useful for studies of the CNS cell function in both in vitro and in vivo applications.
Protocol
| Solution | Composition | |
| Cell culture and transfection | ||
| DMEM1 | DMEM 1x | |
| 1% FBS (v/v) | ||
| 1% GlutaMAX (v/v) | ||
| DMEM10 | DMEM 1x | |
| 10% FBS (v/v) | ||
| 1% GlutaMAX (v/v) | ||
| 150 mM NaCl | 4.380 g NaCl | |
| Up to 500 mL Ultrapure water | ||
| AAV purification and desalting | ||
| 5 M NaCl | 146.1 g NaCl | |
| Up to 500 mL Ultrapure water | ||
| 1 M Tris HCl (pH 8.5) | 12.11 g Tris base | CAUTION |
| Up to 100 mL Ultrapure water | ||
| Add 1 M HCl using a Pasteur pipette to reduce the pH to 8.5 | ||
| Lysis buffer | 15 mL of 5 M NaCl | |
| 25 mL of 1 M Tris HCl (pH 8.5) | CAUTION | |
| Up to 500 mL Ultrapure water | ||
| 10x Phosphate-buffered saline (PBS) | 80 g NaCl | |
| 2 g KCl | CAUTION | |
| 14.4 g Na2HPO4 | ||
| 2.4 g KH2PO4 | ||
| Up to 1 L ddwater | ||
| 1 M MgCl2 | 20.33 g MgCl2-6H2O | |
| Up to 100 mL Ultrapure water | ||
| 1 M KCl | 7.45 g KCl | CAUTION |
| Up to 100 mL Ultrapure water | ||
| 5x PBS Magnesium-Potassium (PBS-MK) stock solution | 250 mL of 10x PBS | |
| 2.5 mL of 1 M MgCl2 | ||
| 6.25 mL of 1 M KCl | CAUTION | |
| Up to 500 mL Ultrapure water H2O | ||
| 15% Iodixanol | 12.5 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5 M NaCl | ||
| 10 mL of 5x PBS-MK | ||
| 17.5 mL of Ultrapure water | ||
| 25% Iodixanol | 20.8 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5x PBS-MK | ||
| 19.2 mL of Ultrapure water | ||
| 100 µL of phenol red | ||
| 40% Iodixanol | 33.3 mL of Optiprep density gradient medium | CAUTION |
| 10 mL of 5x PBS-MK | ||
| 6.7 mL of Ultrapure water | ||
| 60% Iodixanol | 50 mL of Optiprep density gradient medium | CAUTION |
| 100 µL of phenol red | CAUTION | |
| AAV purity control | ||
| 10x Tris acetate EDTA (TEA) buffer | 44.8 g Tris base | CAUTION |
| 11.4 mL glacial acetic acid (17.4M) | ||
| 3.7 g EDTA | ||
| Up to 1 L Ultrapure water | ||
| Agarose gel | 0.8 g Ultrapure agarose | |
| Up to 100 mL 1x TEA buffer | ||
| Gel buffer | 181.7 g Tris base | CAUTION |
| 1.5 g SDS | CAUTION | |
| Adjust pH to 8.45 with 1 M HCl | CAUTION | |
| Up to 500 mL Ultrapure water | ||
| Cathode buffer 10x | 121.14 g Tris base | CAUTION |
| 179.2 g Tricine | CAUTION | |
| 1% SDS (w/w) | CAUTION | |
| Up to 1 L Ultrapure water | ||
| Anode buffer 10x | 242.3 g Tris base | CAUTION |
| Up to 1 L Ultrapure water | ||
| Adjust pH to 8.9 with 1 M HCl | CAUTION | |
| Sample buffer 5x | For 20 mL: | |
| 605 mg Tris base | CAUTION | |
| 4 g SDS | CAUTION | |
| 10 mg Serva Blue G | ||
| 12 g Glycerol | ||
| Adjust pH to 6.8 with 1 M HCl, aliquot and store at -20 °C | CAUTION | |
| Stacking gel | For 2 gels: | |
| 400 µL acrylamide | CAUTION | |
| 750 µL gel buffer | ||
| 1.85 mL Ultrapure water | ||
| 4 µL TEMED | CAUTION | |
| 20 µL 10% APS (v/v) | CAUTION | |
| Add TEMED and 10% APS immediately before pouring the gel. Use both chemicals under a chemical hood. | ||
| Resolving gel | For 2 gels: | |
| 3.32 mL acrylamide | CAUTION | |
| 3.35 mL gel buffer | ||
| 1.14 mL Ultrapure water | ||
| 2.12 mL 50% glycerol | ||
| 6 µL TEMED | CAUTION | |
| 50 µL 10% APS (w/v) | CAUTION | |
| Add TEMED and 10% APS immediately before pouring the gel. Use both chemicals under a chemical hood. | ||
| Water-saturated butanol | 10 mL n-butanol | CAUTION |
| 1 mL Ultrapure water | ||
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. | ||
Table 1: Composition of the required solutions.
1. Tri-transfection of HEK293T Cells
NOTE: Please refer to Table 1 for the composition of the buffers and solutions used in the protocol.
NOTE: Performance of this section of the protocol takes approximately 4 days.
- Thaw a vial of human embryonic kidney (HEK) 293T cells in a water bath set at 37 °C.
NOTE: Use only cells that have been passaged less than 20x to guarantee optimal transfection efficiency. - Seed HEK293T cells at a density of 2 x 103 to 6 x 103 cells/cm2 in DMEM10 in 15 cm diameter cell culture dishes.
- Grow the cells to 70–80% confluence in a standard incubator set at 37 °C, with 95% humidity and 5% CO2.
NOTE: The production of one batch of AAV vectors using this protocol requires 18 cell culture dishes (15 cm diameter). A cell confluence of 70% - 80% corresponds to 6 x 103 to 7 x 103 cells/cm2, maintained in 17–20 mL of DMEM10 culture medium. - Prepare polyethylenimine (PEI)/DNA mix at a concentration ratio of 1/3.5 (w/w).
- Prepare the DNA mix for 18 cell culture dishes in one 50 mL conical tube by mixing 360 µg of pΔF6, 180 µg of pCapsid, and 180 µg of pTransgene in 18 mL of 150 mM NaCl.
- Distribute the DNA mix over three 50 mL conical tubes (6 mL of DNA mix per conical tube).
- Prepare the PEI mix for six cell culture dishes in a new 50 mL conical tube by mixing 840 µg of PEI (1 µg/µL) in 6 mL of 150 mM NaCl.
- Prepare the PEI/DNA mix by adding 6 mL of the PEI mix (prepared in step 1.4.3), drop by drop, to one of the conical tubes containing the DNA mix (prepared in step 1.4.2) and incubate for 20 min at room temperature.
NOTE: After 20 min of incubation, the PEI/DNA mix will become slightly turbid.
- Take six cell culture dishes out of the incubator and completely aspirate the medium from each culture dish in a laminar flow hood. Remove the traces of the medium by rinsing the dishes with 5 mL of prewarmed Dulbecco’s phosphate-buffered saline (DPBS).
- Ensure the distribution of DPBS over the entire surface by gently tilting the dish.
- Gently aspirate the DPBS and add 12 mL of DMEM1 to each dish.
NOTE: Avoid detaching the cells by adding the medium slowly, from a pipette placed at the edge of the dish. - Mix the PEI/DNA by pipetting up and down 3x - 5x. Add 2 mL of the PEI/DNA mix to each of the six cell culture dishes in a drop-by-drop fashion, carefully distributing it over the entire surface. Once the mix is added to each dish, place the dishes back in the incubator. Repeat steps 1.4.3– 1.8 for the remaining culture dishes.
- Incubate the transfected cells for 5 h at 37 °C, with 95% humidity and 5% CO2.
- Add an additional 12 mL of DMEM10 to each culture dish without removing the pre-existing medium (total medium volume = 25 mL).
- Incubate the transfected cells up to 72 h post-transfection at 37 °C, with 95% humidity and 5% CO2.

Supplementary Figure 1: HEK 293T cell morphology visualized by phase contrast microscopy (left) with confirmation of GFP expression visualized by fluorescence imaging (right). (A) Successful transfection of HEK293T cells with a GFP-encoding pTransgene is confirmed by fluorescence imaging. (B) HEK293T cells treated solely with transfection reagents show no GFP expression. Please click here to view a larger version of this figure.
{kind=link}
- Harvest the medium and the cells 72 h post-transfection. Use a cell scraper to carefully detach the cells from the culture dish. Collect the medium and the cells in a 50 mL conical tube kept on ice.
NOTE: The contents of two cell culture dishes can be collected into a single 50 mL conical tube. At the end of this step, each of the nine tubes will contain approximately 50 mL of medium. - Centrifuge the conical tubes at 420 x g for 10 min at 4 °C with the acceleration and deceleration of the centrifuge set to maximum.
- Carefully discard the supernatant from each tube. Do not use pipettes to prevent the loss of material. Instead, gently pour the supernatant into a waste disposal container and place the tubes containing the cell pellets back on the ice.
- Resuspend each cell pellet in 2 mL of lysis buffer directly in the 50 mL conical tube by pipetting up and down 5–10x. Do not vortex. Pool the lysates from three tubes together.
NOTE: At this point, there will be three 50 mL conical tubes, each one containing 6 mL of the resuspended cells in lysis buffer.
2. AAV Vector Purification
NOTE: Performance of this protocol section takes approximately 1 day. Perform the following steps simultaneously on each of the three 50 mL conical tubes containing the cells resuspended in lysis buffer (see the previous note).
- Freeze and thaw the resuspended cells 3x to lyse them and release the AAV particles. Perform the freezing step by placing the tubes in a bucket containing dry ice mixed with ethanol. Perform thawing by immediately placing the cells in a water bath set at 37 °C.
- After the third thawing step, centrifuge at 1,167 x g for 15 min at 4 °C.
NOTE: A noticeable pellet composed of cell debris will be formed. When handling the tubes, avoid sudden movements as these can cause the detachment of the pellet into the supernatant, which will compromise the purity of the final vector. - Carefully transfer the supernatants to clean 50 mL conical tubes and then add nuclease to each tube, to a final concentration of 50 U/mL supernatant.
- Incubate for 30 min at 37 °C. Swirl the 50 mL conical tubes by hand every 10 min to ensure that the nuclease is thoroughly mixed with the supernatant.
- Clarify the supernatant by centrifugation at 13,490 x g for 20 min at 4 °C.
- Attach a 0.45 µm filter to a 10 mL syringe and place it on top of a clean 50 mL conical tube. Carefully remove the plunger and fill the syringe with the supernatant from step 2.5.
- Use the plunger to force the lysate through the filter. Use a new filter and syringe for each tube of supernatant obtained in step 2.5.
NOTE: The obtained fraction is known as ‘crude lysate’. - Prepare each of the 15%, 25%, 40%, and 60% iodixanol fractions in four separate 50 mL conical tubes, according to the instructions in Table 1.
- Prepare the iodixanol gradients in three ultracentrifugation tubes using the following order of iodixanol fractions: 8 mL of 15% iodixanol, 5.5 mL of 25% iodixanol, 5 mL of 40% iodixanol, and 4.5 mL of 60% iodixanol.
CAUTION: Iodixanol can cause irritation to eyes, skin, and the gastrointestinal and respiratory tracts. When handling iodixanol gradients, wear gloves and work under a laminar flow hood.- Pipette 8 mL of 15% iodixanol into each ultracentrifugation tube.
- Pipette 5.5 mL of 25% iodixanol solution into a clean 50 mL conical tube (Figure 1A).
- Use a non-graduated Pasteur pipette (as the neck of the ultracentrifugation tube is too narrow for conventional graduated pipettes) to carefully layer 5.5 mL of the 25% iodixanol solution below the 15% iodixanol solution (Figure 1B).
NOTE: This can be successfully achieved by adding the iodixanol in three steps since the Pasteur pipette can only hold around 2 mL. - Add the 40% and 60% iodixanol solutions as described in step 2.9.3. Do not disturb the different iodixanol interfaces during layering.
NOTE: The proper preparation of the different iodixanol fractions can be ensured by visual confirmation thanks to the phenol red added to the iodixanol fractions (see the instructions in Table 1). Since each fraction has a specific density, they will not intermix during the layering step, if it is performed properly (check Figure 1C).
- Layer the crude lysate on top of the 15% iodixanol gradient with a Pasteur pipette. Proceed drop by drop to avoid disturbing the interface between the crude lysate and the iodixanol solution.
- Fill the ultracentrifugation tube up with lysis buffer until the meniscus reaches the base of the tube neck to ensure the tube does not collapse under the very high forces generated during the ultracentrifugation (Figure 1C).
- Close the ultracentrifugation tubes using appropriate lids. Use a digital scale to make sure all three ultracentrifugation tubes have the same weight. Adjust the weight, if necessary, by adding more lysis buffer on top of the ‘crude lysate’.
NOTE: The weight difference between the ultracentrifugation tubes must be below 0.1 g to ensure the safe operation of the ultracentrifuge. Ultracentrifuges are potentially dangerous pieces of equipment and should only be used by properly trained personnel. - Centrifuge the tubes at 301,580 x g, using a fixed-angle titanium rotor, for 1 h and 40 min at 12 °C, using maximum speed of acceleration and deceleration.
- Carefully insert a stainless-steel blunt needle at the 40% and 60% iodixanol interface.
- Attach the needle to a 5 mL syringe (Figure 1E).
- Aspirate only the clear fraction, containing the vector particles.
NOTE: The total volume collected is approximately 2.5–3 mL. Avoid the collection of material from the 25–40% interface, since this will increase the level of contamination in the final vector batch, due to the presence of non-desirable proteins. - Process the collected fraction (desalting and concentration) or store it overnight at 4 °C.

Figure 1: Setup for iodixanol gradient purification and subsequent vector collection. (A) Before pipetting the different iodixanol gradients into the ultracentrifugation tube, pipette an adequate volume of each iodixanol solution into a separate conical tube. (B) Then use a Pasteur pipette to sequentially transfer each iodixanol solution to the ultracentrifugation tube: layers of an increasingly high iodixanol concentration should be added at the bottom of the tube underneath the previous layer(s). (C) Layer the crude vector lysate on top once the gradient has been prepared. This vector collection system does not use sharp needles, which present a risk of 'needlestick' injuries. (D) A stainless-steel 316-syringe needle is inserted through the iodixanol gradient up to the 40%/60% iodixanol interface. (E) Vector particles are found in the 40% iodixanol phase and are collected. Please click here to view a larger version of this figure.
{kind=link}
3. Desalting and Concentration of the AAV Vector
NOTE: Performance of this section of the protocol takes approximately 2 h.
- Prerinse the filter membrane of the centrifugal filter by adding 5 mL of 1x PBS-MK and centrifuge for 5 min at 4,000 x g.
- Add 5 mL of 1x PBS-MK to the collected AAV vector fraction, mix and then add the total volume to the filter. Upon addition of 1x PBS-MK, turbidity is observed. Centrifuge at 4,000 x g until the volume present on the cone-shaped filter has been reduced to 1 mL. Discard the flow-through accumulated in the outer collection tube.
- Add 13 mL of 1x PBS-MK to the cone-shaped filter and repeat the centrifugation step as many times as necessary (minimum 3x), until there is no more turbidity observed when fresh 1x PBS-MK is added.
- In the final wash step, add 13 mL of PBS + 0.01% (v/v) non-ionic surfactant and centrifuge at 4,000 x g until the volume is reduced to 300–350 µL.
- Collect the remaining fraction present on the filter by pipetting the whole volume into a sterile skirted microcentrifuge tube.
NOTE: This fraction contains the desalted and concentrated AAV vector. In the following steps of this protocol, this fraction is referred to as the ‘primary’ fraction. Make sure to handle the tube under the hood and store it at 4 °C. - Rinse the filter with 200 µL of 1x PBS-MK to collect the residual vector particles retained on the filter. Collect in a sterile microcentrifuge tube.
NOTE: This is a diluted vector stock, which may be suitable for some applications requiring lower vector titers. In future steps, this fraction is referred to as the ‘secondary’ fraction. - For short-term storage, store the vector fraction(s) at 4 °C (less than 2 weeks). Aliquot and store the vector at ‑20 °C when long-term storage is required.
- Perform quality control as described in section 4.
4. Titration of the Vector by Quantitative Polymerase Chain Reaction
NOTE: Performance of this section of the protocol takes approximately 3 h.
- Standard curve
- Linearize the transgene-expressing plasmid (pTransgene) used for the transfection of HEK-293T cells in section 1.
- Prepare the restriction digest mix in a 0.5 mL microcentrifuge tube (refer to Table 2 for the composition of the restriction digest mix).
NOTE: Adjust the composition of the restriction digest mix according to the enzyme used and the related guidelines from the manufacturer. - Incubate the restriction digest mix for 1 h at 37 °C.
- Check the efficiency of the restriction digestion by running the linearized plasmid on a 0.8% (w/v) agarose gel at 100 V for 1 h. Complete digestion of the plasmid should produce a single fragment of defined size.
- Purify the linear plasmid DNA using a PCR purification kit, following the manufacturer’s instructions24, and measure the DNA concentration with a spectrophotometer by measuring the absorption at 260 nm. After the titration, store the remaining aliquot of the linear plasmid at ‑20 °C for further use.
- Calculate the molecules (i.e. DNA copies) of the plasmid stock per microliter as follows.
- First, calculate the molecular weight of the plasmid. Assuming that the average weight of a DNA base pair (bp) is 650 Daltons (Da) and that one mole of a bp weighs 650 g, the molecular weight of any double-stranded DNA template can be estimated by taking the product of its length (in bp) and weight per base pair.
Plasmid molecular weight [g/mol] = plasmid size [bp] x 650 [Da/bp] - Next, calculate the number of moles of plasmid per microliter.
Moles of plasmid per microliter = plasmid concentration [g/µL] / plasmid molecular weight [g/mol]
NOTE: The inverse of the molecular weight is the number of moles of plasmid present in 1 g of the material. - Then, calculate the number of plasmid molecules per microliter, using Avogadro’s number (6.022 x 1023 molecules/mole).
Molecules of plasmid per microliter = moles of plasmid per microliter [moles/µL] x Avogadro’s number [molecules/mole] - Finally, dilute the plasmid stock (molecules/µL) to obtain a 100 µL solution with the desired concentration of 1 x 109 molecules (vector genomes (vg) per microliter).
Plasmid stock (100 µL) = (desired concentration [molecules/µL] x 100 µL) / plasmid molecules [molecules/µL]
- First, calculate the molecular weight of the plasmid. Assuming that the average weight of a DNA base pair (bp) is 650 Daltons (Da) and that one mole of a bp weighs 650 g, the molecular weight of any double-stranded DNA template can be estimated by taking the product of its length (in bp) and weight per base pair.
- Make serial dilutions of the plasmid stock (1 x 109 vg/µL) in triplicates:
10 µL of 1 x 109 vg/µL plasmid stock + 90 µL of H2O = 1 x 108 vg/µL solution
10 µL of 1 x 108 vg/µL dilution + 90 µL of H2O = 1 x 107 vg/µL solution
10 µL of 1 x 107 vg/µL dilution + 90 µL of H2O = 1 x 106 vg/µL solution
10 µL of 1 x 106 vg/µL dilution + 90 µL of H2O = 1 x 105 vg/µL solution
Continue to obtain a 1x101 vg/µL solution. - Keep the serial dilutions of the standard plasmid stock on ice until loading them on the qPCR plate (section 4.3).
| Component | Amount |
| 10x Restriction enzyme buffer | 5 µL |
| Restriction enzyme | 2,5 µL |
| Plasmid | 5 µg |
| H2O | up to 50 µL |
Table 2: Restriction digest mix composition.
Table 3: Stock plasmid volume calculator. Please click here to download this table.
- DNA extraction from the AAV vector
- Mix 2 µL of the AAV vector stock (the primary fraction from step 3.5) with 198 µL of DNase I buffer (1x) in strip tubes (PCR tubes) and add 2 µL of DNase I.
NOTE: DNase I will degrade any genetic material that is not contained inside a vector capsid (which would distort the qPCR results). This solution is referred to as dilution ‘dil1 x 10-2’. - Incubate for 30 min at 37 °C, followed by 10 min at 95 °C.
NOTE: The protocol can be stopped at this point and the material can be stored indefinitely at 4 °C, to avoid product deterioration. - Add 2 µL of proteinase K to the dil1 x 10-2 solution (step 4.2.1) and incubate for 60 min at 50 °C, followed by 20 min at 95 °C.
NOTE: This step will disassemble the AAV vector capsid and release the AAV vector genome into the solution. Add proteinase K in excess as the protein (capsid) content of the sample is not known. Note, it is essential to ensure that all proteinase K activity is removed by denaturation, prior to qPCR, to avoid issues with (partial) polymerase degradation influencing the final result. - Prepare 1:10 serial dilutions of the proteinase K treated dil1 x 10-2 solution (step 4.2.3) in 1.5 mL microcentrifuge tubes as follows:
10 µL of dil1 x 10-2 + 90 µL of H2O = dil1 x 10-3 dilution
10 µL of dil1 x 10-3 + 90 µL of H2O = dil1 x 10-4 dilution
10 µL of dil1 x 10-4 + 90 µL of H2O = dil1 x 10-5 dilution - Keep the serial dilutions of DNA extracted from the vector on ice until loading them on the qPCR plate (section 4.3).
- Mix 2 µL of the AAV vector stock (the primary fraction from step 3.5) with 198 µL of DNase I buffer (1x) in strip tubes (PCR tubes) and add 2 µL of DNase I.
- Titration by SYBR Green detection-based qPCR
- Prepare the qPCR master mix in a 1.5 mL microcentrifuge tube, for both the sample and the standards. Use 10 µL of SYBR Green Master Mix, 1 µL of forward primer (10 µM stock), 1 µL of reverse primer (10 µM stock), and 3 µL of H2O per reaction. See the protocol in Table 4 for primer sequences.
- Pipette the qPCR master mix up and down, but do not vortex.
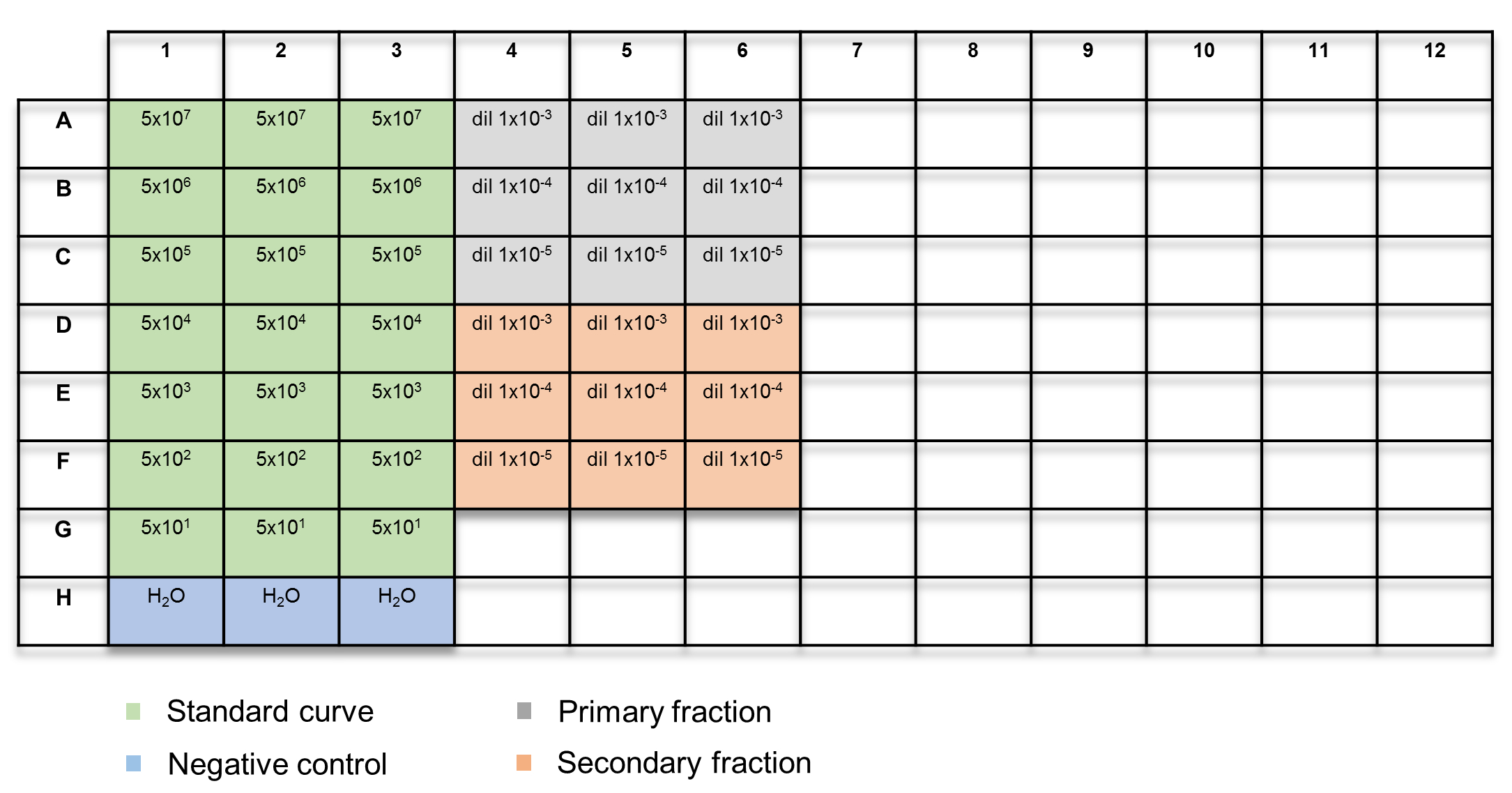
- Add 15 µL of the qPCR master mix, followed by either 5 µL of the standard curve prepared in section 4.1 or the DNA extracted from the AAV vector in section 4.2, into each well. Include three wells containing only the qPCR master mix, as a negative control. Refer to Figure 2 for the plate layout.
- Seal the plate with a sealing film and briefly centrifuge the qPCR plate at 1,500 x g for 30 s at 4 °C.
- Run the qPCR reaction on a plate-based real-time PCR amplification and detection instrument, using the conditions suggested in Table 5.
| Primer name | Sequence |
| Forward primer | 5’-CCCACTTGGCAGTACATCAA-3’ |
| Reverse primer | 5’-GCCAAGTAGGAAAGTCCCAT-3’ |
Table 4: Primer sequences designed against the CBA promoter.

Figure 2: Plate layout for qPCR-based vector titration. The samples are color-coded: green = standard curve; blue = H2O control; grey = primary fraction; orange = secondary fraction. Please click here to view a larger version of this figure.
{kind=link}
| Step | Time | Temperature | Cycles | Aim |
| Pre-incubation | 5 min | 95 °C | x1 | DNA denaturation and polymerase activation (hot-start reaction). |
| Amplification | 10 min | 95 °C | x1 | Amplification of the DNA. Settings may be optimized if alternative primers with different annealing temperature are used. |
| 10 s | 95 °C | x40 | ||
| 40 s | 60 °C | |||
| 1 s | 72 °C | |||
| Cooling | 10 s | 40 °C | x1 | Plate cooling. End of the PCR. |
Table 5: Thermal cycling protocol for SYBR green-based qPCR titration.
- Data analysis to determine the AAV vector titer
- Fill the spreadsheet data cells (Table 6A) with the Ct values obtained for the different dilutions of the standard sample prepared in section 4.1 to generate a standard curve.
NOTE: The equation of the standard curve will be shown (y = a ln(x) + b) together with the R2 efficiency (Table 6B). A qPCR must have an efficiency close to 100% and R2 close to 1.0 (≥ 0.96). - Use the calculated values of a and b to fill in the corresponding data cells in the spreadsheet (Table 6C).
- Complete the spreadsheet with the Ct values obtained for the different dilutions of the AAV sample prepared in section 4.2.
- Calculate the average AAV vector titer in vector genomes per microliter of the 'primary fraction' by using the following formula:

NOTE: The factor 400 is used for single stranded genomes, while sc genomes use a multiplication factor of 20025. In fact, as DNA quantities double during each qPCR cycle, sc DNA is detected 2x in comparison to an ss genome. The aim of the titration is to calculate the concentration in terms of vector genomes per microliter. A correction is necessary for running the qPCR when using 2 µL of starting material (step 4.2.1). dil represents the dilution factors from step 4.2.4. The titer of the 'secondary' AAV vector fraction (step 3.6) can be calculated simultaneously.
- Fill the spreadsheet data cells (Table 6A) with the Ct values obtained for the different dilutions of the standard sample prepared in section 4.1 to generate a standard curve.
Table 6: Template for qPCR data analysis. Please click here to download this file.
5. Purity Control by SDS-PAGE and Silver Staining
NOTE: Performance of this section of the protocol takes approximately 5 h.
- Use 70% (v/v) ethanol to thoroughly clean the glass plates used for casting the gels.
- Assemble the gel casting system. Ensure that the bottoms of the glass plates are not chipped, to prevent leakage of the acrylamide mixture when casting the gel.
- Prepare the stacking and the resolving gel solutions in two separate 50 mL conical tubes. Omit the tetramethylethylenediamine (TEMED) and the 10% (w/v) ammonium persulfate (APS), as they are responsible for polymerization of the gel. Add them immediately before casting the gel.
NOTE: The final percentage of acrylamide in the gel influences the separation profile of the proteins: typically, a 10% (v/v) final concentration of acrylamide will be sufficient to test the purity of an AAV vector preparation. - Mix gently by swirling the tube containing the gel components. Do not vortex, since excessive oxygenation can impair polymerization.
- Add TEMED and 10% (w/v) APS to the resolving gel solution. Mix and then pour into the gel holder until it reaches 1-2 cm below the top of the small glass plate.
- Place a layer of water-saturated butanol on the top of the acrylamide mixture. This will ensure the formation of a flat surface during polymerization. Do not leave the gel under alcohol for longer than 30 min as this will dehydrate the gel and impair its function.
CAUTION: Butanol is hazardous in case of skin contact. It also presents a fire risk. Handle with care. - Wait for the gel to polymerize and then pour off the butanol. Tip: Check to see whether the excess gel mix in the tube has solidified. Polymerization occurs in less than 20 min. Wash the surface of the gel with H2O and then dry it using paper towels, taking care not to disturb the surface of the gel.
- Add TEMED and 10% (w/v) APS to the stacking gel and pour the gel into the gel holder on top of the separating gel.
- Place the provided comb in the gel. Perform this action with one steady movement to avoid the formation of air bubbles inside the wells.
- Wait for at least 20 min for the gel to polymerize. Tip: Check if the excess gel mix in the conical tube has solidified.
- Prepare the two mixes (Table 7) for both the primary and the secondary AAV vector fractions (steps 3.5 and 3.6, respectively).
| High Amount | Low Amount | |
| AAV vector stock | 5 µL | 1 µL |
| 5x Sample buffer | 3 µL | 3 µL |
| H2O | 7 µL | 11 µL |
Table 7: Composition of the sample mixes required for silver staining.
- Fully denature the sample mixes by heating them for 5 min at 95 °C. Assemble the electrophoresis tank, paying attention to the electrode orientation.
- Fill the tank with 1x cathode buffer on the inside of the electrode assembly and 1x anode buffer on the outside.
NOTE: Anode buffer can be recycled from run-to-run. Fresh cathode buffer must always be used. - Remove the comb from the gel and clean the wells of the gel with 1x cathode buffer.
- Load 1 µL of protein ladder in a well. Load the sample mixes in different wells on the same gel (Figure 3). Run the gel at 50 V until the samples enter the resolving gel. Then increase the voltage to 100 V until the dye front reaches the lower limit of the gel.
- Extract the gel carefully from the glass plates and use the silver staining kit (according to the manufacturer’s instructions26) to visualize the viral proteins (VP1, VP2, and VP3 subunits) that comprise the AAV capsid, as well as to check for possible protein contamination (Figure 3).

Figure 3: Evaluation of vector purity using SDS-PAGE and silver staining. Using a Tricine-SDS gel, 5 µL of various vector preparations were separated. Proteins were subsequently detected by silver staining. Vectors are considered pure when VP1 (82 kDa), VP2 (67 kDa), and VP3 (60 kDa) are visible in a 1:1:10 ratio (lane 1), without excessive background (lane 2) or non-specific bands (lane 3). Please click here to view a larger version of this figure.
{kind=link}
Representative Results
AAV9 was considered, until recently, to be the most effective AAV vector serotype in crossing the BBB and transducing cells of the CNS, following peripheral administration. A significant advance in capsid design was achieved when Deverman et al. reported the use of a capsid selection method called Cre recombination-based AAV-targeted evolution (CREATE)17. Using this method, they identified a new capsid, named PHP.B, which they reported as able to transduce the majority of astrocytes and neurons in multiple CNS regions, following systemic injection17. At this point, it should be noted that even though PHP.B provides positive results in C57/Bl6 mice (which was the strain used in the initial isolation experiments), preliminary reports suggest its efficiency may vary in a strain-dependent manner. Further experiments will, no doubt, shed more light on this issue31.
However, despite these issues, PHP.B offers exciting possibilities for noninvasive gene manipulation in the CNS of mice, including proof-of-concept gene therapy experiments in disease models. As such, we chose to evaluate the efficiency of transgene expression using PHP.B versus AAV9, which has been the 'gold-standard' vector for CNS transduction following peripheral administration since 20092. To perform a direct comparison of both serotypes, under optimal conditions for transgene expression, we used an sc genome configuration32. Both vectors carried the transgene for green fluorescent protein (GFP) under the control of the ubiquitous chicken β-actin (CBA) promoter. Female C57/Bl6 mice at postnatal day 42 (approximately 20 g in weight) received a dose of 1 x 1012 vg per mouse of either scAAV2/PHP.B-CBA-GFP or scAAV2/9-CBA-GFP. Vector administration was performed via tail vein injection. The experiments were approved by the Ethical Committee of the KU Leuven.
Three weeks postinjection, the mice underwent transcardial perfusion with ice-cold PBS, followed by 4% (w/v) ice-cold paraformaldehyde (PFA). Their brains were harvested and underwent further postfixation by an overnight incubation in the same fixative, before transferring to 0.01% (w/v) Na-azide/PBS for storage until further analysis. Afterward, the brains were sectioned using a vibrating microtome, and immunohistochemistry was performed on 50 µm thick sections.
To evaluate the levels of transgene expression, sections were stained with primary antibodies against GFP (rabbit anti-GFP), with detection using secondary antibodies conjugated to a fluorescent dye (anti-rabbit Alexa Fluor 488) (Figure 4A, B). Fluorescence intensity measurements (in arbitrary units [au]) confirmed a significant increase in GFP expression when an sc genome and the PHP.B capsid were used relative to AAV9. Increases in GFP were observed in the cerebrum (2105 ± 161 vs. 1441 ± 99 au; p = 0.0032), the cerebellum (2601 ± 196 vs. 1737 ± 135 au; p = 0.0032), and the brainstem (3082 ± 319 vs. 2485 ± 88 au; p = 0.0038) (Figure 4C).

Figure 4: Systemic delivery of scAAV2/PHP.B-CBA-GFP leads to a high GFP expression in the CNS. scAAV2/PHP.B-CBA-GFP or scAAV2/9-CBA-GFP (1 x 1012 vg/mouse) was administered to 6-weeks-old C57/Bl6 mice via tail vein injection. GFP was detected using immunohistochemistry on coronal brain sections 3 weeks postinjection. (A) The cerebrum and (B) the cerebellum and brainstem are shown. Scale bars = 1 mm. (C) The quantification of relative fluorescence intensities was performed to determine the levels of GFP signal achieved with each vector (10 sections per mouse; three mice per vector group). A one-way ANOVA test was performed, followed by a two-tailed Student's t-test; the data are expressed as mean ± standard deviation; **p < 0.01; au. arbitrary units. pCapsid, used for AAV vector production, contains the gene rep from serotype 2 and the gene cap from serotype PHP.B or AAV9, accordingly. This figure has been modified from Rincon et al.32. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The production of recombinant AAV vectors described here uses materials and equipment common to most molecular biology labs and cell culture facilities. It allows the user to obtain pure, preclinical grade AAV vectors that can be used to target multiple cell and tissue types across a range of in vitro and in vivo applications. One of the greatest advantages of this protocol, compared to others (i.e., CsCl-based purification), is the shorter working time needed. Ready-to-use AAV vectors are obtainable in a maximum of 6 working days after the initial transfection of HEK293T cells.
Several factors can negatively influence the final yield or the quality of the AAV vector. Poor transfection efficiency is one of the main reasons for a low viral yield33. A major recommendation is the use of HEK293T cells that have not been passaged more than 20 times and do not have a cell confluence greater than 90% at the time of transfection21. In addition, the transfection method selected has a major impact on the results. This protocol is based on the use of PEI. PEI is a cationic polymer with the ability to deliver exogenous DNA to the cell nucleus through the generation of complexes of polymer and nucleic acid, known as polyplexes, which are taken up by the cell and trafficked via endosomes34. PEI-based transfection is easy and fast to perform, in contrast to other widely used methods, such as the co-precipitation of DNA with calcium phosphate35. Also, PEI-based transfection is much cheaper when compared to other newly introduced methods, such as the usage of cationic lipids and magnet-mediated transfection36.
The purification strategy plays a key role in the protocol. Compared to other methods, iodixanol-based purifications tend to contain a higher percentage of empty viral particles (20%)20. This is offset, to a degree, by the fact that iodixanol-based purification routinely results in AAV vector preparations with a particle-to-infectivity ratio of less than 100. This represents a significant improvement in comparison to conventional CsCl-based procedures, for which substantial loss of particle infectivity is reported37. Another common alternative method to purify AAV vectors is chromatography-based purification. However, this method has the major drawback that a specific column is required for each vector capsid used: for example, while AAV2 is classically isolated using heparin columns, this methodology does not work with AAV4 and AAV5, which do not possess heparin-binding sites on their capsids38. Considering that chromatography purification is also expensive, iodixanol-based purification is generally more suitable for laboratories that wish to produce high-quality batches of AAV vectors on a small scale33,39,40. However, to maximize the final yield and purity of the vector, extreme care is needed when making the iodixanol gradients. The various iodixanol fractions should be transferred to the ultracentrifugation tube using a sterile Pasteur pipette whose tip is touching the wall of the tube: iodixanol should be expelled from the pipette slowly and continuously. As the vector particles accumulate in the 40% iodixanol layer, care needs to be taken to ensure that the gradient interfaces do not mix20. Finally, the fraction containing the vector should be recovered by the insertion of a stainless-steel blunt needle with a gauge not larger than 20 G. To maximize vector recovery, the clear fraction should be retrieved in its entirety. During this step, timing is critical. To avoid compromising the purity of the preparation, it is essential to stop the collection before other (contaminating) phases of the gradient are collected.
Differences in the obtained vector titer can be also attributed to the intrinsic ability of the vector to produce packaged particles. A comparison between different AAV serotypes showed that some AAV vectors are more difficult to produce at a higher titer than others (e.g., AAV2)41. Precipitation of the vector, during the desalting step, can be a possible reason for a lower titer and easily prevented by avoiding overconcentration33. Moreover, it is also possible that the efficiency of iodixanol gradient-based purification slightly differs between serotypes and, therefore, discrepancies in the titer obtained with different serotypes can be observed41.
Finally, it needs to be pointed out that even though qPCR is a very accurate method for DNA quantification some inherent variability in the technique can be observed. Accuracy in the titration primarily depends on the precise pipetting and proper vortexing of all the solutions. To guarantee the most accurate titer reading, the qPCR can be independently repeated, and the obtained values averaged. The choice of primers presented in this protocol is based on the sequence of the CBA promoter located in the pTransgene plasmid used in our laboratory. The CBA promoter is a strong synthetic promoter that is widely used in the vector field to drive expression across multiple cell types. It incorporates multiple elements, including the cytomegalovirus (CMV) early enhancer element; the promoter, first exon, and the first intron of the CBA gene; and the splice acceptor of the rabbit β-globin gene. However, primers can be designed for virtually any element located within the expression cassette (including the promoter, transgene, and regulatory elements). The comparison of titer across batches is also possible, providing primers are used against regions common to the vectors in question.
In conclusion, this protocol can be used to produce AAV vectors with a variety of capsids, genome configurations, promoter types and transgene cargos. This will allow users to easily adapt the final characteristics of their vectors to best suit experimental needs. In the example presented in the representative results, the use of the PHP.B capsid, which efficiently crosses the BBB, gave highly efficient gene expression in the CNS, following tail vein injection32. The systemic administration of CNS penetrant vectors has considerable advantages in terms of possible side-effects2,17,32. A possible alternative to peripheral injection, while avoiding the caveats of invasive techniques, is intrathecal delivery, which consists of delivery of the AAV vector into the cerebrospinal fluid. This delivery route is proven to be effective, showing widespread expression of transgene across the CNS, less off-target effects in peripheral organs, and low levels of immune response42. However, intrathecal injections are much more challenging, as they require higher technical skills than tail vein injection.
Further capsid development to refine this technology will be driven by the opportunities for AAV vector use in gene therapy applications. Such approaches offer attractive possibilities to treat currently incurable CNS disorders, such as amyotrophic lateral sclerosis, Charcot-Marie-Tooth disease, Parkinson’s and Alzheimer’s disease18.
Disclosures
The authors have nothing to disclose.
Acknowledgements
M.R. is supported by a Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO) postdoctoral fellowship (133722/1204517N) and acknowledges the continuous support of the Fundación Cardiovascular de Colombia and the Administrative Department of Science, Technology and Innovation (Grant CT-FP44842-307-2016, project code 656671250485). M.M. is supported by an FWO doctoral fellowship (1S48018N). M.G.H. was supported by a VIB institutional grant and external support from the Thierry Latran Foundation (SOD-VIP), The Foundation for Alzheimer Research (SAO-FRA) (P#14006), the FWO (Grant 1513616N), and the European Research Council (ERC) (Starting Grant 281961 - AstroFunc; Proof of Concept Grant 713755 - AD-VIP). The authors acknowledge Jeason Haughton for his help with mouse husbandry, Stephanie Castaldo for her help performing the tail vein injections, and Caroline Eykens for providing the images of transfected HEK293T cells. M.G.H. acknowledges Michael Dunlop, Peter Hickman, and Dean Harrison.
Materials
| Name | Company | Catalog Number | Comments | |
| Plasmid production | ||||
| pTransgene plasmid | De novo design or obtained from a plasmid repository | N/A | See step 1 of main protocol for further details | |
| pCapsid | De novo design or obtained from a plasmid repository | N/A | See step 1 of main protocol for further details | |
| pHelper | Agilent | 240071 | ||
| Plasmid Plus maxi kit | Qiagen | 12963 | ||
| QIAquick PCR purification Kit | Qiagen | 28104 | ||
| AAV Helper-Free System | Agilent | 240071 | ||
| Cell culture and transfection | ||||
| Dulbecco’s Modified Eagle Medium (DMEM), high glucose, no glutamine | Life technologies | 11960-044 | Supplement DMEM with FBS (1% or 10% v/v) and GlutaMAX 200 mM (1% v/v) then filter sterilize the medium using a 0.22 mm filter | |
| Fetal bovine serum (FBS) | GIBCO | 10500-064 | ||
| GlutaMAX supplement | GIBCO | 35050038 | ||
| (200 mM) | ||||
| Corning bottle-top vacuum filter system | Sigma-Aldrich | CLS430769 | ||
| Dulbecco’s Phosphate Buffered Saline (DPBS) with no calcium and no magnesium | GIBCO | 14190094 | ||
| HEK293T cells | American Tissue Culture Collection | CRL3216 | Upon receipt, thaw the cells and culture as described in the protocol. After a minimal number of passages, freeze a subfraction for future in aliquots. Always use cells below passage number 20. Once cultured cells have been passaged more than 20 times, restart a culture from the stored aliquots | |
| Cell culture dishes | Greiner Cellstar | 639160 | 15 cm diameter culture dishes | |
| Cell scrapers | VWR | 10062-904 | ||
| Polyethylenimine (PEI) | Polyscience | 23966-2 | PEI in powder form is dissolved at 1 µg/µL in deionized water (ddwater) at pH=2 (use HCl). Prepare in a beaker and stir for 2-3 h. When dissolved, bring the pH back to 7 with NaOH. Filter sterilize and store the resuspended stock solution in 1 ml aliquots at -20 °C. PEI aliquots can freeze/thawed multiple times. | |
| Virkon solution | Fisher Scientific | NC9821357 | Disinfect any material that has been in contact with assembled viral particles with Virkon solution | |
| Mutexi long-sleeve aprons | Fisher Scientific | 11735423 | Wear an apron over the top of a regular lab coat | |
| Fisherbrand maximum protection disposable overshoes | Fisher Scientific | 15401952 | ||
| AAV Purification and desalting | ||||
| Optiprep density gradient medium | Sigma-Aldrich | D1556 | Optiprep is a 60% (w/v) solution of iodixanol in water (sterile). CAUTION. Use under a laminar flow hood. Wear gloves | |
| Phenol red | Sigma-Aldrich | P0290 | CAUTION. Use under a laminar flow hood | |
| Pasteur pipette | Sigma-Aldrich | Z627992 | Sterilize before use | |
| OptiSeal Polypropylene tubes | Beckman | 361625 | ||
| Benzonase (250 U/µL) | Sigma-Aldrich | E1014 | Supplied as a ready-to-use solution | |
| Acrodisc syringe filter | Pall corporation | 4614 | ||
| Omnifix syringe (5mL) | Braun | 4617053V | ||
| Blunt syringe needle | Sigma Aldrich | Z261378 | Stainless steel 316 syringe needle, pipetting blunt 90° tip gauge 16, L 4 in. Referred to in the text as a blunt-end needle | |
| Aqua Ecotainer | B. Braun | 0082479E | Sterile endotoxin-free water. Referred to as 'Ultrapure water' | |
| Amicon ultra-15 centrifugal filter unit | Millipore | UFC910024 | These filters concentrate the final product by collecting the viral particles in consecutive centrifugation steps | |
| Pluronic F68 (100X) | Thermo Fisher | 24040032 | Non-ionic surfactant. Dilute in sterile PBS to use at 0,01% (v/v) | |
| Fisherbrand Sterile Microcentrifuge Tubes with Screw Caps (2 mL) | Fisher Scientific | 02-681-374 | Use skirted tubes for easy handling | |
| AAV Titration | ||||
| Restriction enzyme: StuI (10 U/µL) | Promega | R6421 | ||
| DNAse I (1 U/µL) | Fisher scientific | EN0521 | ||
| Proteinase K | Sigma-Aldrich | 3115852001 | Reconstitute in ultrapure water and use at a final concentration of 10 mg/ml. Solution can be stored at -20°C | |
| EasyStrip Plus Tube Strips (with attached caps) | Fisher scientific | AB2000 | ||
| Eppendorf microtube 3810x | Sigma-Aldrich | Z606340-1000EA | ||
| LightCycler 480 SYBR Green I Master Mix | Roche | 4707516001 | ||
| LightCycler Multiwell Plates, 96 wells | Roche | 4729692001 | White polypropylene plate (with unique identifying barcode) | |
| Microseal 'A' PCR Plate and PCR Tube Sealing Film | Bio-Rad | msa5001 | ||
| AAV Purity control | ||||
| Ammonium persulfate (APS) | Sigma-Aldrich | A3678 | Reconstitute in ultrapure water to 10% (v/v). CAUTION. Use under laminar flow hood. Wear gloves | |
| Tetramethylethylenediamine (TEMED) | Sigma-Aldrich | T9281 | CAUTION. Use under a laminar flow hood. Wear gloves | |
| Tris Base ULTROL Grade | Merck | 648311 | CAUTION. Use under a laminar flow hood. Wear gloves | |
| UltraPure Agarose | Thermo Fisher | 16500-500 | ||
| Rotiphorese® Gel 30 (37,5:1) | Carl Roth | 3029.3 | Aqueous 30 % acrylamide and bisacrylamide stock solution at a ratio of 37.5:1. CAUTION. Use under laminar flow hood. Wear gloves | |
| Serva Blue G | Sigma-Aldrich | 6104-58-1 | ||
| Precision Plus prestained marker | Bio-Rad | 1610374edu | ||
| 1-Butanol | Sigma-Aldrich | B7906 | CAUTION. Use under a laminar flow hood. Wear gloves | |
| Immunohistochemistry | ||||
| Rabbit anti-GFP | Synaptic System | 132002 | 1:300 dilution | |
| Anti-rabbit Alexa Fluor 488 | Invitrogen | A21206 | 1:1000 dilution | |
| Equipment | Company | Catalog number | Comments | |
| Vector production lab | ||||
| Rotina 380 bench-top centrifuge * | Hettich | 1701 | ||
| Optima XPN 80 ultracentrifuge * | Beckmann Coulter | A95765 | ||
| Type 50.2 Ti fixed-angle titanium rotor * | Beckmann Coulter | 337901 | ||
| Entris digital scale * | Sartorius | 2202-1S | ||
| Warm water bath * | Set at 37°C | |||
| Ice bucket * | VWR | 10146-290 | Keep material used in the vector production lab separate from that used in standard lab areas | |
| Pipetboy pro * | Integra | 156,400 | ||
| Graduated pipettes: Cell star * | Greiner bio-one | 606180 | Capacity of 5 ml, 10 ml and 25 ml | |
| Graduated pipettes: Cell star * | Greiner bio-one | 607180 | Capacity of 5 ml, 10 ml and 25 ml | |
| Graduated pipettes: Cell star * | Greiner bio-one | 760180 | Capacity of 5 ml, 10 ml and 25 ml | |
| Co2 incubator CB150 * | Binder | 9040-0038 | Set at 37°C, 5% CO2 and 95% humidity | |
| Nuaire safety cabinet NU 437-400E * | Labexchange | 31324 | Clean all the surfaces with 70% ethanol and Virkon before and after use | |
| Conventional lab | ||||
| T100 thermal cycler * | Bio-Rad | 1861096 | ||
| LightCycler 480 Instrument II * | Roche | 5015278001 | ||
| ThermoMixer * | Eppendorf | C 5382000015 | ||
| Nanodrop * | ThermoFisher Scientific | ND 2000 | ||
| Mini-Protean Tetra Cell* | Bio-Rad | 1658001FC | For use with handmade or precast gels | |
| ProteoSilver silver stain kit | Sigma-Aldrich | PROTSIL1 | High sensitivity protein detection with low background | |
| Centrifuge 5804 R * | Eppendorf | B1_022628045 | High speed centrifuge for medium capacity needs (up to 250 ml) | |
| Graduated pipettes Cell star * | Greiner bio-one | 606180 | 5 ml, 10 ml and 25 ml | |
| Graduated pipettes Cell star * | Greiner bio-one | 607180 | 5 ml, 10 ml and 25 ml | |
| Graduated pipettes Cell star * | Greiner bio-one | 760180 | 5 ml, 10 ml and 25 ml | |
| Filter tips * | Greiner bio-one | 750257 | 2 µl, 20 µl, 200 µl | |
| Filter tips * | Greiner bio-one | 738257 | 2 µl, 20 µl, 200 µl | |
| Filter tips * | Greiner bio-one | 771257 | 2 µl, 20 µl, 200 µl | |
| Ice bucket with lid * | VWR | 10146-290 | ||
| Mini diaphragm vacuum pump, VP 86 * | VWR | 181-0065 | ||
| Pipetman P2, P20, P100, P200, P1000 | Gilson | F144801 | ||
| Pipetman P2, P20, P100, P200, P1000 | Gilson | F123600 | ||
| Pipetman P2, P20, P100, P200, P1000 | Gilson | F123615 | ||
| Pipetman P2, P20, P100, P200, P1000 | Gilson | F123601 | ||
| Pipetman P2, P20, P100, P200, P1000 | Gilson | F123602 | ||
| * Materials marked with an asterisk are expensive vpieces of equipment and are usually central infrastructure items shared between multiple labs. These items can also be replaced by equivalents if available. Note, when a different ultracentrifuge is used, care must be taken to select the correct rotor and centrifuge tubes. | ||||
References
- Daya, S., Berns, K. I. Gene Therapy Using Adeno-Associated Virus Vectors. Clinical Microbiology Reviews. 21 (4), 583-593 (2008).
- Duque, S., et al. Intravenous Administration of Self-complementary AAV9 Enables Transgene Delivery to Adult Motor Neurons. Molecular Therapy. 17 (7), 1187-1196 (2009).
- Bourdenx, M., Dutheil, N., Bezard, E., Dehay, B. Systemic gene delivery to the central nervous system using Adeno-associated virus. Frontiers in Molecular Neuroscience. 7, (2014).
- Kantor, B., McCown, T., Leone, P., Gray, S. J. Clinical Applications Involving CNS Gene Transfer. Advances in Genetics. 87, 71-124 (2014).
- Crystal, R. G. Adenovirus: The First Effective In Vivo Gene Delivery Vector. Human Gene Therapy. 25 (1), 3-11 (2014).
- Weinberg, M. S., Samulski, R. J., McCown, T. J. Adeno-associated virus (AAV) gene therapy for neurological disease. Neuropharmacology. 69, 82-88 (2013).
- Henckaerts, E., Linden, R. M. Adeno-associated virus: a key to the human genome?. Future Virology. 5 (5), 555-574 (2010).
- Howarth, J. L., Lee, Y. B., Uney, J. B. Using viral vectors as gene transfer tools (Cell Biology and Toxicology Special Issue: ETCS-UK 1 day meeting on genetic manipulation of cells). Cell Biology and Toxicology. 26 (1), 1-20 (2010).
- Nathwani, A. C., et al. Adenovirus-Associated Virus Vector-Mediated Gene Transfer in Hemophilia B. The New England Journal of Medicine. 365 (25), 2357-2365 (2011).
- Carter, B. J. Adeno-associated virus and the development of adeno-associated virus vectors: a historical perspective. Molecular Therapy. 10 (6), 981-989 (2004).
- Schambach, A., Zychlinski, D., Ehrnstroem, B., Baum, C. Biosafety Features of Lentiviral Vectors. Human Gene Therapy. 24 (2), 132-142 (2013).
- Van Vliet, K. M., Blouin, V., Brument, N., Agbandje-McKenna, M., Snyder, R. O. The Role of the Adeno-Associated Virus Capsid in Gene Transfer. Methods in Molecular Biology. 437, 51-91 (2008).
- Rincon, M. Y., VandenDriessche, T., Chuah, M. K. Gene therapy for cardiovascular disease: advances in vector development, targeting, and delivery for clinical translation. Cardiovascular Research. 108 (1), 4-20 (2015).
- Samulski, R. J., Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annual Review of Virology. 1 (1), 427-451 (2014).
- McCarty, D. M. Self-complementary AAV Vectors; Advances and Applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Choi, J. H., et al. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Molecular Brain. 7, 17 (2014).
- Deverman, B. E., et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nature Biotechnology. 34 (2), 204-209 (2016).
- Jackson, K. L., Dayton, R. D., Deverman, B. E., Klein, R. L. Better Targeting, Better Efficiency for Wide-Scale Neuronal Transduction with the Synapsin Promoter and AAV-PHP.B. Frontiers in Molecular Neuroscience. 9, (2016).
- Lock, M., et al. Versatile Manufacturing of Recombinant Adeno-Associated Viral Vectors at Scale. Human Gene Therapy. 21 (10), 1259-1271 (2010).
- Strobel, B., Miller, F. D., Rist, W., LamLa, T. Comparative Analysis of Cesium Chloride- and Iodixanol-Based Purification of Recombinant Adeno-Associated Viral Vectors for Preclinical Applications. Human Gene Therapy Methods. 26 (4), 147-157 (2015).
- Jungmann, A., Leuchs, B., Katus, H. A., Rommelaere, J., Müller, O. J. Protocol for efficient generation and characterization of adeno-associated viral (AAV) vectors. Human Gene Therapy Methods. , (2017).
- Tolmachov, O. Designing Plasmid Vectors. Methods in Molecular Biology. 542, 117-129 (2009).
- . QIAGEN Plasmid Purification Handbook Available from: https://www.qiagen.com/us/resources/download.aspx?id=46205595-0440-459e-9d93-50eb02e5707e&lang=en (2018)
- Lock, M., Alvira, M. R., Chen, S. J., Wilson, J. M. Absolute Determination of Single-Stranded and Self-Complementary Adeno-Associated Viral Vector Genome Titers by Droplet Digital PCR. Human Gene Therapy Methods. 25 (2), 115-125 (2014).
- . ProteoSilver Silver Stain Kit (PROTSIL1) - Bulletin, ProteoSilver Available from: https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Bulletin/protsil1bul.pdf (2018)
- Machholz, E., et al. Manual Restraint and Common Compound Administration Routes in Mice and Rats. Journal of Visualized Experiments. (67), e2771 (2012).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Puntel, M., et al. Gene transfer into rat brain using adenoviral vectors. Current Protocols in Neuroscience. 50 (1), (2010).
- Bachman, J. Immunohistochemistry on Freely Floating Fixed Tissue Sections. Methods in Enzymology. 533, 207-215 (2013).
- Hordeaux, J., et al. The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Molecular Therapy. 26 (3), 664-669 (2018).
- Rincon, M. Y., et al. Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Therapy. 25 (2), 83-92 (2018).
- Crosson, S. M., Dib, P., Smith, J. K., Zolotukhin, S. Helper-free Production of Laboratory Grade AAV and Purification by Iodixanol Density Gradient Centrifugation. Molecular Therapy. Methods & Clinical Development. 10, 1-7 (2018).
- Merdan, T., Kunath, K., Fischer, D., Kopecek, J., Kissel, T. Intracellular processing of poly(ethylene imine)/ribozyme complexes can be observed in living cells by using confocal laser scanning microscopy and inhibitor experiments. Pharmaceutical Research. 19 (2), 140-146 (2002).
- Meissner, P., et al. Transient gene expression: recombinant protein production with suspension-adapted HEK293-EBNA cells. Biotechnology and Bioengineering. 75 (2), 197-203 (2001).
- Reed, S. E., Staley, E. M., Mayginnes, J. P., Pintel, D. J., Tullis, G. E. Transfection of mammalian cells using linear polyethylenimine is a simple and effective means of producing recombinant adeno-associated virus vectors. Journal of Virological Methods. 138 (1-2), 85-98 (2006).
- Zolotukhin, S., et al. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Therapy. 6 (6), 973-985 (1999).
- Kaludov, N., Handelman, B., Chiorini, J. A. Scalable Purification of Adeno-Associated Virus Type 2, 4, or 5 Using Ion-Exchange Chromatography. Human Gene Therapy. 13 (10), 1235-1243 (2002).
- Nass, S. A., et al. Universal Method for the Purification of Recombinant AAV Vectors of Differing Serotypes. Molecular Therapy. Methods & Clinical Development. 9, 33-46 (2017).
- Qu, G., et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. Journal of Virological Methods. 140 (1-2), 183-192 (2007).
- Holehonnur, R., et al. Adeno-associated viral serotypes produce differing titers and differentially transduce neurons within the rat basal and lateral amygdala. BMC Neuroscience. 15, 28 (2014).
- Wang, H., et al. Widespread spinal cord transduction by intrathecal injection of rAAV delivers efficacious RNAi therapy for amyotrophic lateral sclerosis. Human Molecular Genetics. 23 (3), 668-681 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved