Sequencing of mRNA from Whole Blood using Nanopore Sequencing
In This Article
Summary
Nanopore sequencing is a novel technology that allows cost-effective sequencing in remote locations and resource-poor settings. Here, we present a protocol for sequencing of mRNAs from whole blood that is compatible with such conditions.
Abstract
Sequencing in remote locations and resource-poor settings presents unique challenges. Nanopore sequencing can be successfully used under such conditions, and was deployed to West Africa during the recent Ebola virus epidemic, highlighting this possibility. In addition to its practical advantages (low cost, ease of equipment transport and use), this technology also provides fundamental advantages over second-generation sequencing approaches, particularly the very long read length, ability to directly sequence RNA, and real-time availability of data. Raw read accuracy is lower than with other sequencing platforms, which represents the main limitation of this technology; however, this can be partially mitigated by the high read depth generated. Here, we present a field-compatible protocol for sequencing of the mRNAs encoding for Niemann-Pick C1, which is the cellular receptor for ebolaviruses. This protocol encompasses extraction of RNA from animal blood samples, followed by RT-PCR for target enrichment, barcoding, library preparation, and the sequencing run itself, and can be easily adapted for use with other DNA or RNA targets.
Introduction
Sequencing is a powerful and important tool in biological and biomedical research. It allows analysis of genomes, genetic variations, and RNA expression profiles, and thus plays an important role in the investigation of human and animal diseases alike1,2. Sanger sequencing, one of the oldest methods available for DNA sequencing, is still routinely used to this day and has been a corner-stone of molecular biology. Over the past 50 years, this technology has been improved to achieve read-lengths of more than 1,000 nt and an accuracy as high as 99.999%1. However, Sanger sequencing also has limitations. Sequencing a larger set of samples or the analysis of whole genomes with this method is time consuming and expensive1,3. Second-generation (next-generation) DNA sequencing methods such as 454 pyrosequencing and Illumina technology have allowed us to significantly reduce the cost and workload required for sequencing in the last decade, and have led to a tremendous increase in the amount of biological sequence information available4. Nevertheless, individual sequencing runs using these second-generation technologies are expensive, and sequencing under field conditions is challenging, as the necessary equipment is bulky and fragile (similar to Sanger sequencing devices), and often has to be calibrated and serviced by specially trained personnel. Also, for many of the second-generation technologies read-lengths are rather limited, which often makes downstream bioinformatics analysis of these data challenging.
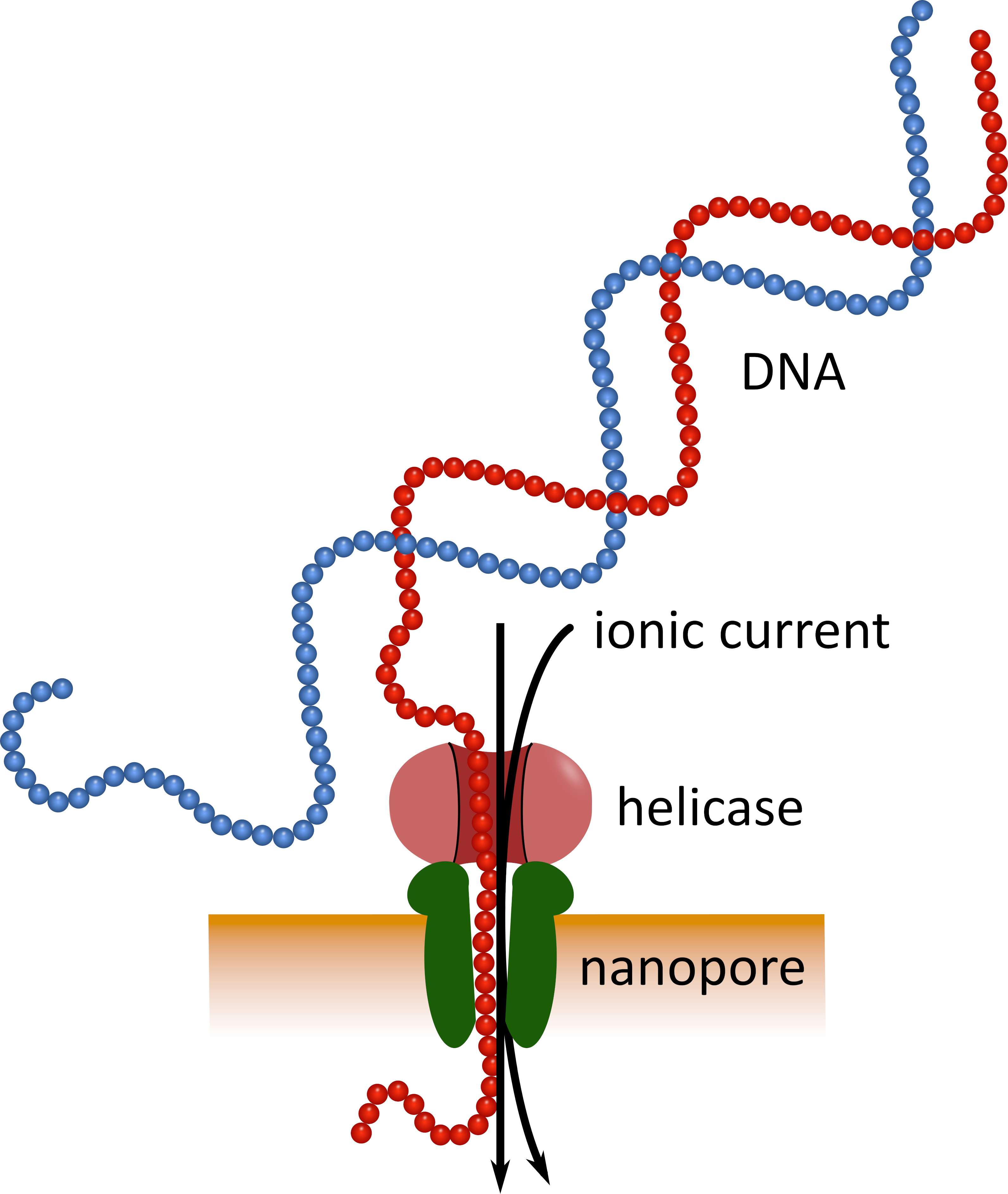
Third-generation sequencing using pocket-sized nanopore sequencing devices (see Table of Materials) can serve as an alternative to these established sequencing platforms. In these devices a single-stranded DNA or RNA molecule passes through a nanopore simultaneously with an ionic current that is then measured by a sensor (Figure 1). As the strand traverses the nanopore, the modulation of the current by the nucleotides present in the pore at any given time is detected, and computationally back-translated into the nucleotide sequence5. Because of this operational principle, nanopore sequencing allows both the generation of very long reads (close to 1 x 106 nucleotides6) and the analysis of sequencing data in real time. Barcoding is possible by attaching defined nucleotide sequences to the nucleic acids in a sample, which allows analysis of multiple samples in a single sequencing run, thus increasing sample throughput and lowering per sample costs. Due to their high portability and ease of use, nanopore sequencing devices have been used successfully in the field during the recent Ebola virus disease epidemic in West Africa, highlighting their suitability for rapid deployment into remote regions7,8.
Here, we describe a detailed field-compatible protocol for the sequencing of mRNA encoding for the Niemann-Pick C1 (NPC1) protein, which is the obligate entry receptor for filoviruses such as ebolaviruses, and has been shown to limit species susceptibility to these viruses9,10. The protocol encompasses extraction of whole RNA from blood samples, specific amplification of NPC1 mRNA by RT-PCR, barcoding of samples, library preparation and sequencing with a nanopore sequencing device. Data analysis cannot be discussed due to space limitations, although some basic directions are provided in the representative results; however, the interested reader is referred to a previous publication11 for a more detailed description of the workflow we used, as well as to publications by others12,13,14 for detailed information regarding the analysis tools used in this workflow.
Protocol
Samples were collected following the Njala University Institutional Review Board (NUIRB) protocol no. IRB00008861/FWA00018924.
1. RNA Extraction from Blood Samples
- Collect 3 mL of whole blood from the species to be analyzed into a blood collection tube prefilled with 6 mL of DNA/RNA stabilizing reagent (see Table of Materials) and mix by inverting 5 times. Store the blood sample for up to one month at 4 °C.
- Transfer the contents of the blood collection tube into a 50 mL collection tube, add 120 µL of proteinase K, and mix by vortexing for 5 s. Incubate the sample for 30 min at room temperature.
- Add 9 mL of isopropanol to the mixture and vortex for 5 s.
- Place a reservoir onto an RNA purification spin column (see Table of Materials) and place the assembly onto a vacuum manifold (see Table of Materials). Add the sample mixture into the reservoir. Apply a vacuum until all liquid has passed through the column.
- Alternatively, if no vacuum manifold is available, the blood can be passed through the column in 700 µL portions by repeated centrifugation for 30 s at 12,000 x g with the flow-through discarded between centrifugation steps. However, this will require approximately 26 centrifugation steps.
- Place the RNA purification spin column into a collection tube and add 400 µL DNA/RNA prep buffer (see Table of Materials). Centrifuge at 12,000 x g for 30 s and discard the flow-through.
- Add 400 µL of DNA/RNA wash buffer (see Table of Materials) to the column, centrifuge at 12,000 x g for 30 s and discard the flow-through.
- Mix 5 µL of DNase I (1 U/µL) (see Table of Materials) with 75 µL of DNA digestion buffer (see Table of Materials) and add the mixture onto the column. Incubate for 15 min at room temperature.
- Add 400 µL of DNA/RNA prep buffer to the column and centrifuge at 12,000 x g for 30 s. Discard the flow-through.
- Wash the column with 700 µL of DNA/RNA wash buffer and centrifuge at 12,000 x g for 30 s. Discard the flow-through.
- Repeat step 1.9 with 400 µL of DNA/RNA wash buffer and centrifuge at 12,000 x g for 2 min to remove all residual wash buffer and to dry the column. When removing the column from the collection tube, make sure not to wet the underside of the column with the buffer in the collection tube.
- Place the column into a new 1.5 mL microcentrifuge tube and add 70 µL of nuclease-free water. Incubate for 1 min at room temperature and centrifuge for 30 s at 12,000 x g. Store the RNA at -80 °C until further use (or use immediately).
- OPTIONAL: To quantify the RNA, take an aliquot and determine the concentration using a UV spectrophotometer (see Table of Materials).
2. Reverse Transcription of NPC1 mRNA into cDNA
- In a 0.2 mL reaction tube, add 8 µL of template RNA (1 pg to 2.5 µg of RNA) and 1 µL each of 10x DNase buffer and DNase enzyme (see Table of Materials). Incubate at 37 °C for 2 min. Subsequently, centrifuge the reaction briefly and place it on ice.
- Add 4 µL of reverse transcriptase master mix (see Table of Materials) and 6 µL of nuclease-free water to the reaction tube and mix gently. Incubate the reaction in a thermocycler for 10 min at 25 °C (for primer annealing), followed by 10 min at 50 °C (for reverse transcription of RNA). To inactivate the enzyme, incubate for 5 min at 85 °C.
- Transfer cDNA to a new 1.5 mL microcentrifuge tube and store at -80 °C until further use (or use immediately).
3. Amplification of the NPC1 Open Reading Frame
- Initial amplification step
- Set up a touchdown PCR15,16 to amplify the NPC1 cDNA with Primer Set 1 (see Table 1), using a hot start high fidelity DNA polymerase (see Table of Materials) with the appropriate reaction buffer in a 50 µL reaction volume with 1 µL template. If possible, set up the reaction on ice or in a 4 °C cool block.
- Incubate the reaction in a thermocycler with an initial denaturation step of 30 s at 98 °C, followed by 10 cycles with denaturation at 98 °C for 10 s, primer annealing for 20 s at 65 °C, lowering the temperature by 0.5 °C per cycle, and elongation for 1 min at 72 °C. Subsequently, run an additional 20 cycles with 10 s at 98 °C, 20 s at 60 °C, and 1 min at 72 °C, followed by a final elongation step of 5 min at 72 °C.
- PCR purification using magnetic beads
- Transfer 50 µL of PCR product into a 1.5 mL DNA-low binding reaction tube (see Table of Materials). Resuspend magnetic beads (see Table of Materials) thoroughly by vortexing and add 50 µL beads to the PCR reaction. Mix well. Incubate the sample on a rotating mixer (see Table of Materials) for 5 min at room temperature (at 15 rpm).
- Briefly spin down the sample and place the 1.5 mL microcentrifuge tube on a magnetic rack (see Table of Materials) to pellet the magnetic beads. Wait until the supernatant has been completely clarified before continuing with the next step.
- Aspirate the supernatant without disturbing the bead pellet and discard.
- Pipette 200 µL of 70% ethanol into the reaction tube and incubate for 30 s. Aspirate the ethanol without disturbing the pellet and discard. Repeat for a total of two washes. Make sure that no ethanol is left. It may be necessary to first aspirate with a larger pipette (e.g., 1000 µL), and then to remove any remaining ethanol droplets with a smaller pipette (e.g., 10 µL).
- Air-dry the pellet for 1 min at room temperature.
- Remove the reaction tube from the magnetic rack, resuspend the pellet in 30 µL of nuclease-free water and incubate for 2 min at room temperature.
- Place the reaction tube back on the magnetic rack and wait until the beads are completely pelleted.
- Remove the supernatant without disturbing the pellet and transfer it to a new 1.5 mL reaction tube.
- Addition of barcode adapters by nested PCR
- Set up a 50 µL touchdown PCR with hot start High-Fidelity DNA polymerase with 5x reaction buffer and Primer Set 2 (see Table 1). Primers in this set consist of a target sequence-specific region to allow binding of PCR products generated in step 3.2 (inside of the primer set 1 sequences), as well as an adapter sequence that is used as target in the subsequent barcoding PCR reaction (cf. section 4). If possible, set up the reaction on ice or in a 4 °C cool block. Use 1 µL of the purified PCR product prepared in section 3.2 as template.
- Incubate the reaction mix in a thermocycler using an initial denaturation step of 30 s at 98 °C, followed by 10 cycles with denaturation at 98 °C for 10 s, primer annealing for 20 s at 65 °C, lowering the temperature by 0.5 °C per cycle, and elongation for 1 min at 72 °C. Subsequently, incubate the reaction for a further 30 cycles for 10 s at 98 °C, 20 s at 71 °C, and 1 min at 72 °C, followed by a final elongation step of 5 min at 72 °C.
- Clean up the PCR product using magnetic beads as described in section 3.2.
4. Barcoding of NPC1 Amplicons
- For each PCR product generated in section 3, set up a barcoding PCR reaction in a 0.2 mL reaction tube using 50 µL of Taq DNA polymerase 2x master mix (see Table of Materials), 2 µL of one of the barcode primers from a PCR barcoding kit (see Table of Materials and Table 2), and 1 µL of purified PCR product from step 3.3.2 as template. Add 47 µL of nuclease free water to obtain a final volume of 100 µL.
- Incubate the reaction in a thermocycler at 95 °C for 3 min as an initial denaturation. Subsequently, run 15 cycles for 15 s at 95 °C, 15 s at 62 °C, and 1.5 min at 65 °C. As a final elongation, incubate the reaction at 65 °C for 5 min.
- Purify the PCR product as described under 3.2, but use 100 µL of magnetic beads and elute in 30 µL of nuclease-free water.
- If possible, quantify the sample using a UV spectrophotometer.
5. Library Preparation
- Combine an equal amount of barcoded DNA from each sample for a total of 1 µg of DNA in a volume of 45 µL (if necessary, add nuclease-free water) in a 0.2 mL reaction tube. If no UV spectrophotometer is available, use equal volumes of each sample. For dA-tailing, add 7 µL of End-prep reaction buffer (see Table of Materials), 3 µL of End-prep enzyme mix (see Table of Materials), and 5 µL of nuclease-free water. Mix gently by flicking the tube.
- Incubate the reaction for 5 min at 20 °C, followed by 5 min at 65 °C in a thermocycler.
- Purify the reaction product as described in section 3.2, but use 60 µL of magnetic beads and elute in 25 µL of nuclease-free water.
- OPTIONAL: Take 1 µL to quantify the concentration of the sample using a UV spectrophotometer. The total amount should be more than 700 ng.
- Combine 22.5 µL of purified DNA from step 5.3 with 2.5 µL of 1D2 adapter (see Table of Materials) and 25 µL of blunt/TA ligase master mix (see Table of Materials) in a new 1.5 mL DNA-low binding reaction tube, mix gently by flicking, and briefly spin down. Incubate for 10 min at room temperature.
- Purify the reaction product as described in section 3.2, but use 20 µL of magnetic beads, increase the incubation time for DNA binding to 10 min, perform two wash steps with 1 mL of ethanol each, and elute in 46 µL of nuclease-free water.
- Combine 45 µL of the reaction product from step 5.6 with 5 µL of barcode adapter mix (see Table of Materials) and 50 µL of blunt/TA ligase master mix in a DNA-low binding reaction tube. Mix gently by flicking and incubate for 10 min at room temperature.
- Purify the reaction product as described in section 3.2, but use 40 µL of magnetic beads, do two wash steps with 140 µL of ABB buffer (see Table of Materials) instead of ethanol, resuspend the beads by flicking and pellet on a magnetic rack. Elute in 15 µL of elution buffer (see Table of Materials). Increase the incubation times for the initial binding of DNA to the beads as well as for the elution step to 10 min. Store the resulting product on ice or at 4 °C until use.
6. Quality Check of Flow Cell
- Perform a quality check on the flow cell before use. To this end, connect the sequencing device to the host computer and open the software.
- Insert a flow cell (see Table of Materials) into the sequencing device and choose the flow cell type from the selector box and confirm by clicking Available.
- Click Check flow cell in the bottom of the screen and choose the correct flow cell type.
- Click Start test to start the quality check. A minimum of 800 active nanopores in total is required for the flow cell to be usable.
7. Loading the Flow Cell and Starting the Sequencing Run
- Open the priming port cover by sliding it in a clockwise direction. Set a P1000 pipette to 200 µL and insert the tip into the priming port. Adjust the pipette to 230 µL while keeping the tip in the priming port, to draw up 20-30 µL buffer and remove any air bubbles.
- In a new 1.5 mL DNA-low binding reaction tube prepare the priming mix by combining 576 µL of RBF buffer (see Table of Materials) with 624 µL of nuclease-free water.
- Carefully pipette 800 µL of the prepared priming mix into the priming port and wait 5 min. Lift the sample port cover, and pipette an additional 200 µL of the prepared priming mix into the priming port.
- Pipette 35 µL of RBF buffer into a new, clean 1.5 mL DNA low-binding reaction tube. Thoroughly mix LLB beads (see Table of Materials) by pipetting and add 25.5 µL of the beads to the RBF buffer. Add 2.5 µL nuclease-free water and 12 µL of DNA library from step 5.8 and mix by pipetting.
- Add 75 µL of the sample mixture in a slow dropwise fashion to the flow cell via the sample port.
- Replace the sample port cover, close the priming port, and close the lid of the sequencing device.
- Within the software, confirm that the flow cell is still available, open a new experiment, and set up the run parameters by selecting the kit used. Select live base-calling. Start the sequencing run by clicking Begin Experiment. Continue the sequencing run until sufficient experimental data is collected.
Representative Results
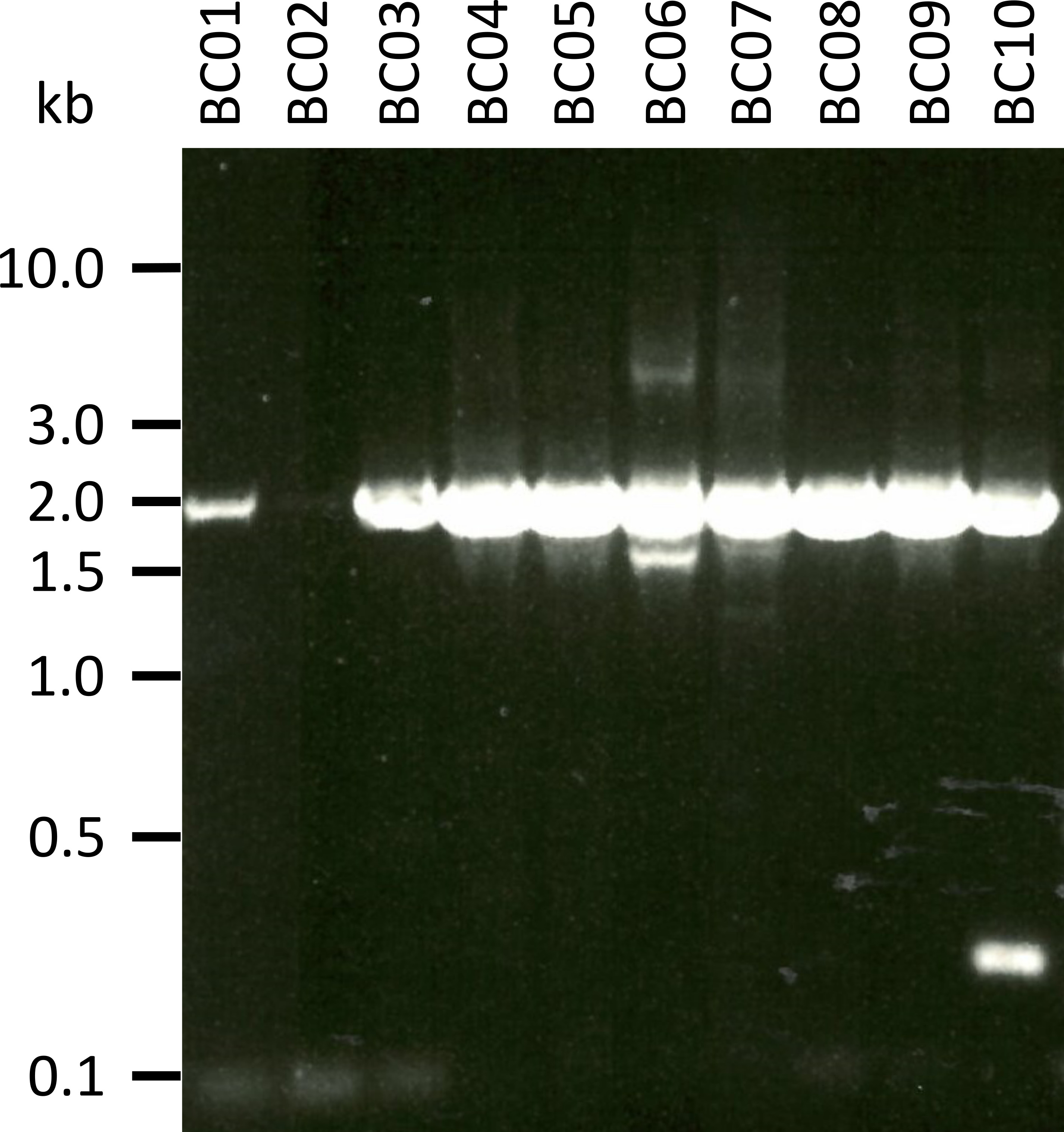
In a representative experiment to test the presented protocol we extracted the RNA from 10 different blood samples of five animal species (i.e., 2 individuals per species (goat, sheep, swine, dog, cattle)) (Table 3). RNA yields and quality following extraction can vary widely, in particular due to differences in sample handling and storage. In our representative experiment, we observed RNA concentrations between 43 ng and 543 ng per µL (Table 3). Also, after amplification by RT-PCR, gel analysis of the NPC-1 PCR-products showed various outcomes (Figure 2), with markedly weaker bands for samples BC01 and BC02 (both goat). These differences were most likely caused by differences in sample quality, although differences in PCR efficacy due to differences in primer binding to the NPC1 gene of different species cannot be excluded. However, these differences in yield and/or amplification efficiency did not markedly impact the overall sequencing outcome. Further, an additional non-specific PCR product occurred in sample BC10 (cattle). In contrast to Sanger sequencing, such non-specific products do not negatively influence the results of nanopore sequencing, as these reads are discarded during mapping of the obtained reads to a reference sequence as part of the data analysis.
Prior to each sequencing run, a quality check of the flow cell to be used is strongly recommended, with a minimum requirement of 800 total pores. In our representative experiment, this quality check returned 1,102 pores available for sequencing. Since the data are provided in real time and can be analyzed immediately, the length of a sequencing run can be adjusted for the individual application (i.e., until sufficient sequencing data is produced for the desired analysis). In our experiments, sequencing runs are typically performed overnight, and in the case of our representative experiment we obtained approximately 1.4 million reads during such a 14 h run.
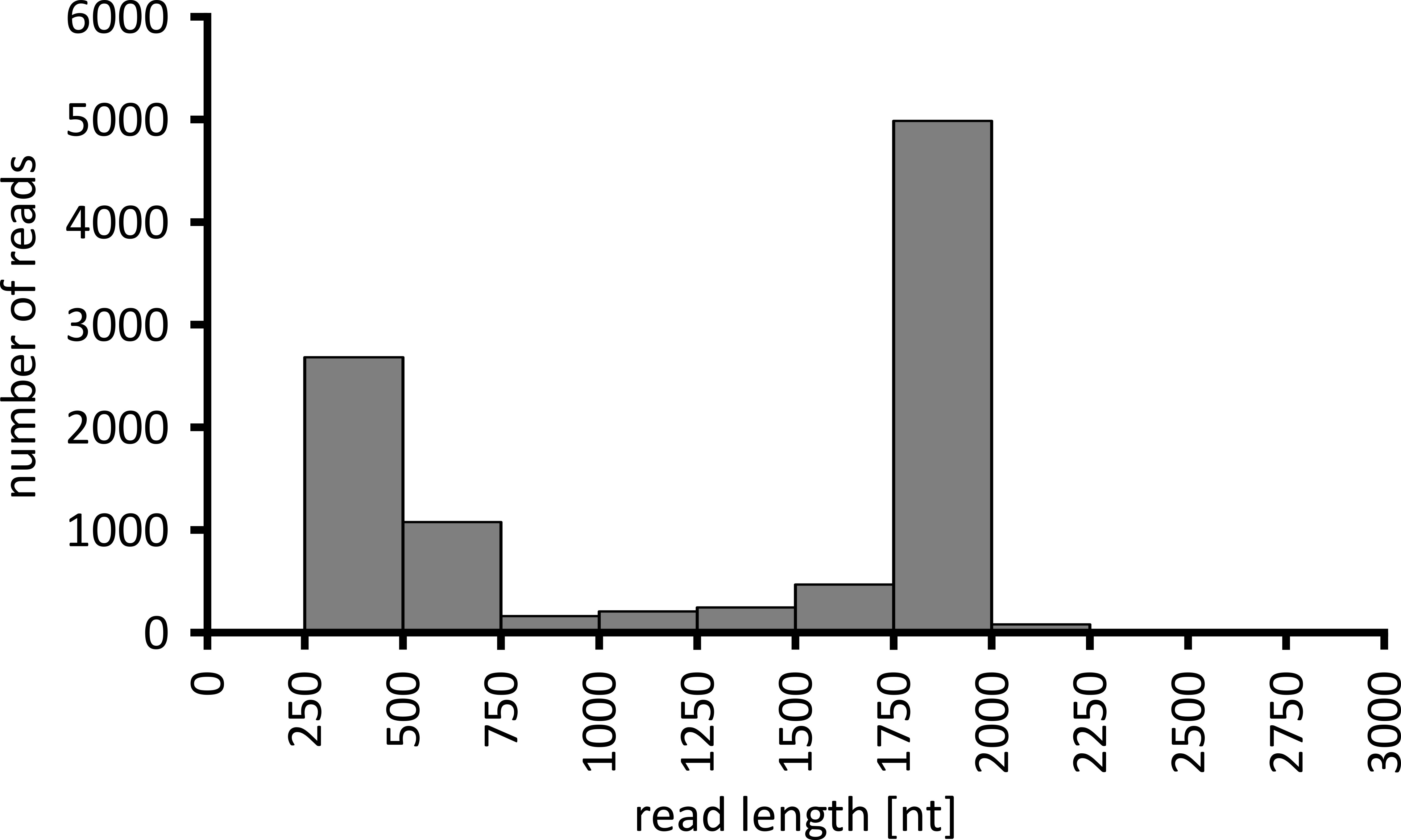
Depending on the type of data analysis to be performed, it can be advisable to process only a subset of the obtained reads. In the case of our representative experiment, a subset of 10,000 reads was selected for further analysis. To this end, the fastq files generated during the sequencing run were further processed in an Ubuntu 18.04 LTS environment, and demultiplexed using flexbar v3.0.3 with parameters optimized for demultiplexing of nanopore sequencing data (barcode-tail-length 300, barcode-error-rate 0.2, barcode-gap-penalty -1)12. After demultiplexing, read mapping and consensus generation can then be done using a number of different tools, but a detailed discussion of the bioinformatics aspect of nanopore sequencing goes beyond the scope of this manuscript. However, in the case of our representative results, read mapping to a reference sequence was performed using Geneious 10.2.3. Of the 10,000 reads analyzed, 5,457 showed a length between 1,750 and 2,000 nucleotides, which matches the expected sizes for the PCR fragments amplified as part of our workflow (1,769 nt, Figure 3). An additional peak in the length distribution of reads was observed between 250 to 500 nucleotides, which can be attributed to unspecific PCR products. Demultiplexing of reads allowed the assignment of 87.6% of the reads to one of the 10 barcodes/samples analyzed (Figure 4). The proportion of demultiplexed reads for each barcode ranged from 3.4% for barcode 1 to 16.9% for barcode 10; however, due to the overall large number of reads this still allowed meaningful consensus calling with a high read depth even for these lower abundance barcode datasets. Indeed, mapping of the sorted reads to a reference sequence of NPC1 resulted in between 31.7% (barcode 2) and 100% (barcode 7 and 8) of reads mapping to the reference, giving a read depth of more than 90 reads at any position for each sample. This is then more than adequate to allow confident consensus base-calling with a negligible error rate.

Figure 1: Schematic representation of DNA sequencing using nanopore technology. A single-stranded DNA molecule passes through a nanopore embedded in an electrically resistant membrane, with a helicase regulating the transition speed. An ionic current simultaneously passes through the pore and is continuously measured. Modulations of the current caused by the nucleotides present in the pore are detected and computationally back-translated into the nucleotide sequence of the DNA strand. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Amplification of PCR products of Niemann-Pick C1 from mRNA. mRNA was isolated from goat (BC01 and 02), sheep (BC03 and 04), swine (BC05 and 06), dog (BC07 and 08), and cattle (BC09 and 10). Nested PCR products were separated in a 0.8% agarose gel in 1x TAE buffer (prepared from 50x TAE buffer: 242.28 g of Tris base, 57.1 mL of glacial acetic acid, 100 mL of 0.5 M EDTA, dH2O to 1 L, pH adjusted to 8.0) for 45 min at 100 V and stained with Sybr Safe. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Read-length distribution of 10,000 reads from the representative experiment. The number of reads obtained having a given read length interval is indicated. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Distribution of reads after demultiplexing. The number and percentage of demultiplexed (grey) and mapped reads (black) for each barcode are shown. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Overview of primer sets used. Initial amplification of target sequences was performed with Primer Set 1. Primer Set 2 was then used for nested amplification and adapter addition. Adapters are indicated in red. Please click here to download this file.
Table 2: Overview of barcode sequences. Individual barcodes were used to identify each sequenced sample. Please click here to download this file.
Table 3: RNA concentrations obtained following extraction from blood samples sequenced in the representative experiment. The RNA concentrations of two individuals from each of five species are shown, and the ratios of the optical densities at 260/280 nm and 260/230 nm are indicated. Please click here to download this file.
Discussion
Over the last two decades, sequencing of biological samples has become an increasingly important aspect of studies in a wide range of subject areas. The development of second-generation sequencing systems based on the sequencing of a dense array of DNA features using iterative cycles of enzymatic manipulation and image-based data acquisition1 has dramatically increased throughput compared to the traditional Sanger sequencing technique, and allows analysis of multiple samples as well as various nucleic acid species in a given sample in parallel4. However, for most of the commonly used second-generation systems, only short reads are produced, and all platforms rely on sensitive, bulky, and expensive equipment3,4.
In contrast to second-generation sequencing platforms, the sequencing device used in this protocol is based on nanopore technology. Here a single-stranded nucleic acid molecule passes through a nanopore, resulting in modulation of an ionic current that is also flowing through the same nanopore, and which can be measured and back-translated to infer the sequence of the nucleic acid molecule. This third-generation sequencing approach imparts a number of advantages over other approaches. The main advantages that are directly related to the unique working principle of this technology are the extremely long read length produced (read lengths of up to 8.8 x 105 nucleotides have been reported6), the ability to sequence not only DNA but also RNA directly, which was recently demonstrated for a complete influenza virus genome17, and the ability to analyze data in real-time as they are being generated, which allows rapid metagenomics detection of pathogens within minutes18. Additional practical advantages are the extremely small size of the nanopore sequencing device, allowing its use in any laboratory or on field missions to remote locations19,20, and the low price in comparison to other sequencing platforms. In terms of running costs, currently a new flow cell is required for each sequencing run, which results in costs of about $1,100 per run for the flow cell and library preparation reagents. These costs can be reduced in some cases by washing and reusing the flow cell, or by barcoding and sequencing multiple samples in a single run. Also, a novel type of flow cell is currently being beta-tested by a small number of laboratories, which will require the use of a flow cell adaptor (called a “flongle”), and should significantly reduce flow cell price and thus running costs.
The major shortcoming of nanopore sequencing remains its accuracy, with single read accuracies in the range of 83 to 86% being reported6,21,22, and most of the inaccuracies being caused by insertion/deletions (indels)5,21. However, high read depth can compensate for these inaccuracies, and a recent study suggested based on theoretical considerations that a read depth of >10 might increase overall accuracy to >99.8%21. Nevertheless, further improvements in accuracy will be needed, particularly if analysis is to be performed on a single molecule level rather than on a consensus sequence level. The use of 1D2 technology as described in this protocol, which is based on the addition of the 1D2 and barcode adapters (cf. section 5.5) that result in both strands of a single DNA molecule being sequenced by the same nanopore, increases read accuracy since information from both DNA strands can be used for sequence determination. Further, a workaround strategy that can be pursued in order to combine the advantages of nanopore sequencing (particularly long read length) with the higher accuracy of other sequencing technologies is to use nanopore sequencing information as a scaffold, which is then polished using sequencing data from other platforms6.
The most critical factor for the success of the protocol presented here is sample quality, and particularly the amount and quality of the extracted RNA. Proper storage and prompt extraction of the RNA help in achieving an adequate RNA yield. The use of appropriate blood collection tubes allows the storage of blood samples for up to one month, but blood clotting can be an issue, particularly when samples are being stored at elevated temperatures, which can be the case under field conditions. The second critical step is the amplification of target sequences, and particular under field conditions PCR reactions often perform less well than under standard laboratory conditions7. To this end, careful primer design and optimization is paramount to achieve robust amplification. Additionally, nested PCR approaches and touchdown PCR, as used in this protocol, can increase both specificity and sensitivity of target gene amplification4,7. Indeed, in our experience in Liberia and Guinea with this technology nested protocols were required under field conditions with field samples even for primer sets which allowed amplification of targets from laboratory samples and under laboratory conditions with a single round of PCR (7 and unpublished results).
In contrast to these more critical steps, library preparation and the sequencing run itself are rather robust procedures. However, under field conditions practical issues such as the availability of certain pieces of equipment can be problematic. For example, a UV spectrophotometer is needed to determine DNA concentrations prior to library preparation of barcoded samples. However, should such a device not be available under field conditions, an equal volume of each sample can simply be combined to make up the 45 µL required for library preparation, with differences in sample input material then usually being mitigated by the large number of reads. Similarly, the need for internet connectivity for the sequencing run can be an issue, even though the base-calling no longer has to be performed online but can be done locally; however, this necessity can be removed under certain circumstances by the manufacturer if required.
In summary, the presented protocol allows relatively low-cost sequencing in locations with no access to traditional sequencing equipment, including in remote locations. It can easily be adapted to any target RNA or DNA, thus allowing researchers to answer numerous biological questions.
Acknowledgements
The authors thank Allison Groseth for critical reading of the manuscript. This work was financially supported by the German Federal Ministry of Food and Agriculture (BMEL) based on a decision of the Parliament of the Federal Republic of Germany through the Federal Office for Agriculture and Food (BLE).
Materials
| Name | Company | Catalog Number | Comments |
| 1D2 adapter, barcode adapter mix, ABB buffer, elution buffer, RBF buffer, LBB beads | Oxford Nanopore Technologies | SQK-LSK308 | 1D² Sequencing Kit |
| blood collection tube with DNA/RNA stabilizing reagent | Zymo Research | R1150 | DNA/RNA Shield - Blood Collection Tube |
| blunt/TA ligase master mix | New England Biolabs | M0367S | Blunt/TA Ligase Master Mix |
| DNA-low binding reaction tube | Eppendorf | 30108051 | DNA LoBind Tube |
| DNase buffer and DNase | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| flow cell | Oxford Nanopore Technologies | FLO-MIN105.24 | flow cell R9.4 |
| hot start high fidelity DNA polymerase | New England Biolabs | M0493L | Q5 Hot Start High-Fidelity DNA Polymerase (500 U) |
| magnetic beads | Beckman Coulter | A63881 | Agencourt AMPure XP beads |
| magnetic rack | ThermoFisher Scientific | 12321D | DynaMag-2 Magnet |
| nanopore sequencing device | Oxford Nanopore Technologies | - | MinION Mk 1B |
| PCR barcoding kit | Oxford Nanopore Technologies | EXP-PBC001 | PCR Barcoding Kit I (R9) |
| reverse transcriptase master mix | ThermoFisher Scientific | 11766050 | SuperScript™ IV VILO™ Master Mix with ezDNase™ Enzyme |
| RNA purification spin column, DNA/RNA prep buffer, DNA/RNA wash buffer, DNase I, DNA digestion buffer | Zymo Research | R1151 | Quick-DNA/RNA Blood Tube Kit |
| rotating mixer | ThermoFisher Scientific | 15920D | HulaMixer Sample Mixer |
| Taq DNA polymerase | New England Biolabs | M0287S | LongAmp Taq 2X Master Mix |
| Ultra II End-prep kit | New England Biolabs | E7546S | NEBNext Ultra II End-Repair/dA-tailing Modul |
| UV spectrophotometer | Implen | - | NanoPhotometer |
| vacuum manifold | Zymo Research | S7000 | EZ-Vac Vacuum Manifold |
References
- Shendure, J., Ji, H. Next-generation DNA sequencing. Nature Biotechnology. 26 (10), 1135-1145 (2008).
- Shendure, J., Lieberman Aiden, E. The expanding scope of DNA sequencing. Nature Biotechnology. 30 (11), 1084-1094 (2012).
- Liu, L., et al. Comparison of next-generation sequencing systems. Journal of Biomedicine and Biotechnology. 2012, 251364 (2012).
- Levy, S. E., Myers, R. M. Advancements in Next-Generation Sequencing. Annual Review of Genomics and Human Genetics. 17, 95-115 (2016).
- Lu, H., Giordano, F., Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics, Proteomics & Bioinformatics. 14 (5), 265-279 (2016).
- Jain, M., et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nature Biotechnology. 36 (4), 338-345 (2018).
- Hoenen, T., et al. Nanopore Sequencing as a Rapidly Deployable Ebola Outbreak Tool. Emerging Infectious Diseases. 22 (2), 331-334 (2016).
- Quick, J., et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
- Carette, J. E., et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 477 (7364), 340-343 (2011).
- Ndungo, E., et al. A Single Residue in Ebola Virus Receptor NPC1 Influences Cellular Host Range in Reptiles. mSphere. 1 (2), (2016).
- Martin, S., et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Medicine. 10 (1), 58 (2018).
- Roehr, J. T., Dieterich, C., Reinert, K. Flexbar 3.0 - SIMD and multicore parallelization. Bioinformatics. 33 (18), 2941-2942 (2017).
- Li, H., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Kielbasa, S. M., Wan, R., Sato, K., Horton, P., Frith, M. C. Adaptive seeds tame genomic sequence comparison. Genome Research. 21 (3), 487-493 (2011).
- Don, R. H., Cox, P. T., Wainwright, B. J., Baker, K., Mattick, J. S. Touchdown' PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 19 (14), 4008 (1991).
- Korbie, D. J., Mattick, J. S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nature Protocols. 3 (9), 1452-1456 (2008).
- Keller, M. W., et al. Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome. Scientific Reports. 8 (1), 14408 (2018).
- Greninger, A. L., et al. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Medicine. 7, 99 (2015).
- Castro-Wallace, S. L., et al. Nanopore DNA Sequencing and Genome Assembly on the International Space Station. Scientific Reports. 7 (1), 18022 (2017).
- Goordial, J., et al. In Situ Field Sequencing and Life Detection in Remote (79 degrees 26'N) Canadian High Arctic Permafrost Ice Wedge Microbial Communities. Frontiers in Microbiology. 8, 2594 (2017).
- Runtuwene, L. R., et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Scientific Reports. 8 (1), 8286 (2018).
- Rang, F. J., Kloosterman, W. P., de Ridder, J. From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biology. 19 (1), 90 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved