Using Human Induced Pluripotent Stem Cell-derived Intestinal Organoids to Study and Modify Epithelial Cell Protection Against Salmonella and Other Pathogens
In This Article
Summary
Human induced pluripotent stem cell (hiPSC)-derived intestinal organoids offer exciting opportunities to model enteric diseases in vitro. We demonstrate the differentiation of hiPSCs into intestinal organoids (iHOs), the stimulation of these iHOs with cytokines, and the microinjection of Salmonella Typhimurium into the iHO lumen, enabling the study of an epithelial invasion by this pathogen.
Abstract
The intestinal ‘organoid’ (iHO) system, wherein 3-D structures representative of the epithelial lining of the human gut can be produced from human induced pluripotent stem cells (hiPSCs) and maintained in culture, provides an exciting opportunity to facilitate the modeling of the epithelial response to enteric infections. In vivo, intestinal epithelial cells (IECs) play a key role in regulating intestinal homeostasis and may directly inhibit pathogens, although the mechanisms by which this occurs are not fully elucidated. The cytokine interleukin-22 (IL-22) has been shown to play a role in the maintenance and defense of the gut epithelial barrier, including inducing a release of antimicrobial peptides and chemokines in response to infection.
We describe the differentiation of healthy control hiPSCs into iHOs via the addition of specific cytokine combinations to their culture medium before embedding them into a basement membrane matrix-based prointestinal culture system. Once embedded, the iHOs are grown in media supplemented with Noggin, R-spondin-1, epidermal growth factor (EGF), CHIR99021, prostaglandin E2, and Y-27632 dihydrochloride monohydrate. Weekly passages by manual disruption of the iHO ultrastructure lead to the formation of budded iHOs, with some exhibiting a crypt/villus structure. All iHOs demonstrate a differentiated epithelium consisting of goblet cells, enteroendocrine cells, Paneth cells, and polarized enterocytes, which can be confirmed via immunostaining for specific markers of each cell subset, transmission electron microscopy (TEM), and quantitative PCR (qPCR). To model infection, Salmonella enterica serovar Typhimurium SL1344 are microinjected into the lumen of the iHOs and incubated for 90 min at 37 °C, and a modified gentamicin protection assay is performed to identify the levels of intracellular bacterial invasion. Some iHOs are also pretreated with recombinant human IL-22 (rhIL-22) prior to infection to establish whether this cytokine is protective against Salmonella infection.
Introduction
In recent years, the study of host-pathogen interactions has been enhanced by the development of ‘organoid’ models, wherein 3-D representations of the intestinal epithelium can be produced from various progenitors. ‘Primary’ organoids can be generated directly from intestinal stem cells harvested from intestinal biopsies. In addition, intestinal organoids can be generated from hiPSCs. The same can be said of numerous tissues, with gastric1, liver2, pancreatic3,4, brain5,6, lung7, and prostate8 organoids used by many researchers to model disease. There are numerous exciting applications of the organoid system, including modeling cancer9 and drug screening10, but here we focus on the use of iHOs as an infection model, using S. enterica serovar Typhimurium (S. Typhimurium) as an exemplary pathogen and pretreatment with IL-22 as a therapy.
In this study, the hiPSCs used to generate iHO are ‘Kolf2’ iPSCs, generated from a healthy individual and available from the Human Induced Pluripotent Stem Cells Initiative Consortium (HipSci; www.hipsci.org), an open-access reference panel of characterized hiPSC lines11. One advantage of using hiPSCs as progenitors for organoids is that there are now extensive banks of healthy donor iPSC lines available, meaning that results can be validated in a number of cell lines with different genetic backgrounds. In addition, should researchers wish to look at specific disease-associated single nucleotide polymorphisms (SNPs), it is possible to use CRISPR/Cas9 to engineer mutations in a healthy cell line, thereby producing both a mutant line and retaining the isogenic control line for comparison12. In our experience, hiPSC-derived intestinal organoids are larger in size than their primary counterparts and more consistent in culture, making for a less technically challenging microinjection and potentially allowing a more diverse range of pathogens to be studied. iHOs can be cryogenically preserved, and we have propagated iHO cultures for up to a year to produce material for experimentation.
In vivo, IECs play a key role in regulating intestinal homeostasis and may directly inhibit pathogens, although the mechanisms by which this occurs are not well understood. The cytokine IL-22 is known to have a role in the maintenance of the gut epithelial barrier13 and is involved in the induction and secretion of antimicrobial peptides and chemokines in response to infection14. It is produced by activated T cells (particularly, Th17 cells) as well as by natural killer (NK) cells and binds to a heterodimeric receptor composed of IL-22R1 and IL-10R2 subunits15. The receptor for IL-22 is expressed basally on IECs, meaning that in the iHO model, it is possible to pretreat the organoids with rhIL-22 simply by its addition to culture medium16. One disadvantage of the organoid system is that it lacks the associated immune response normally delivered by other immune cell types; however, models are emerging that attempt to coculture organoids with intestinal lymphocytes to better represent this17,18.
The use of the microinjection system is key to simulating infections in the iHO model, since this allows the direct delivery of pathogens to the apical surface of the epithelium, as would occur in the case of in vivo infection. The addition of phenol red to the bacterial solution injected into the iHO marks the ones that have been infected, thus avoiding repeated injections of the same iHO. Organoids as vessels for infection modeling are growing in use, with pathogens such as Helicobacter pylori,19 the Norovirus,20 the Rotavirus,21 Shiga toxin-producing Escherichia coli22, Cryptosporidium23, and the Zika virus24 having been shown to survive and replicate within these systems. This technology could be applied to a wider range of pathogens, particularly to organisms that are difficult to culture, such as protozoa, or human restricted pathogens, in order to obtain direct information about the human epithelial response to infection.
Protocol
1. Culturing and Passaging of Induced Pluripotent Stem Cells
NOTE: All methods described here use commercially available human cell lines. All tissue culture work detailed below should be done in a Class II laminar flow hood. iPSCs are routinely maintained in stem cell culture medium (see the Table of Materials), as per the manufacturer’s instructions, which allows a weekend-free culture of iPSCs. iPSCs can be adapted from other iPSC culture systems with relative ease.
- Passage the cells once the colonies cover approximately 80%–90% of the plate surface.
- Prepare plates for the passage 1 h prior to use by adding vitronectin 10 μg/mL diluted in Dulbecco’s phosphate-buffered saline (DPBS; without calcium [Ca] or magnesium [Mg]) to tissue-culture-treated plates. The volumes for coating are dependent on the plate size and can be found in the manufacturer’s instructions. During this time, warm stem cell culture medium to room temperature (RT).
- Remove the media from the iPSCs ready for passage and wash the cells 2x with DPBS (without Ca or Mg).
- Add EDTA solution (see the Table of Materials) to the plates, making sure the entire surface is coated and incubate at RT for 5–8 min. When holes start to appear in the center of the iPSC colonies, aspirate and discard the EDTA solution.
- Add stem cell culture medium to the wells; dislodge the iPSCs by gently washing media over the plate surface a few times. The iPSCs are passaged as clumps and not as single cells, so make sure that the EDTA solution is not left on for too long. Move any dislodged iPSCs to a 15 mL conical tube.
- Aspirate the vitronectin from the precoated plates and replace it with stem cell culture medium. Invert the iPSC suspension a number of times to make sure the iPSCs have not settled at the bottom of the conical tube, and add the appropriate volume of suspension to give a 1:10 dilution of cells on a new plate. Split ratios may be adjusted depending on the iPSC growth rate, which may vary between iPSC lines.
- Rock the plate to disperse the iPSCs over the surface and place it in an incubator at 37 °C/5% CO2. Feed the iPSCs the day after passaging.
2. Differentiation from iPSCs to the Hindgut
- On Day 0, split the iPSCs on a 10 cm tissue-culture-treated dish, precoated with vitronectin as described in step 1.2, into 10 mL of stem cell culture medium supplemented with activin A (10 ng/mL) + basic fibroblast growth factor (bFGF; 12 ng/mL). Add the growth factors to the media directly before use; do this in all subsequent steps.
- On Day 1, change the media (10 mL of stem cell culture medium with activin A [10 ng/mL] + bFGF [12 ng/mL]).
- On Day 2, begin the differentiation by changing the media to 10 mL of stem cell culture medium supplemented with the following growth factors: activin A (100 ng/mL), bFGF (100 ng/mL), bone morphogenetic protein 4 (BMP-4; 10 ng/mL), phosphoinositol 3-kinase inhibitor LY294002 (10 μM), and GSK3 inhibitor CHIR99021 (3 μM).
- On Day 3, change the media to 10 mL of stem cell culture medium supplemented with activin A (100 ng/mL), bFGF (100 ng/mL), BMP-4 (10 ng/mL), and LY294002 (10 μM). Endoderm specification induced by this media should result in visible changes to the iPSC colony morphology over the next 24 h.
- On Day 4, change the media to 10 mL of RPMI/B27 media supplemented with activin A (100 ng/mL) and bFGF (100 ng/mL).
NOTE: RPMI/B27 media contains 500 mL of RPMI medium with L-glutamine supplement (see Table 1), 10 mL of B27 supplement (50x, serum free), and 5 mL of nonessential amino acids. Optional: add 5 mL of penicillin-streptomycin (10,000 U/mL). Filter-sterilize before use. - On Day 5, change the media to 10 mL of RPMI/B-27 media supplemented with activin A (50 ng/mL).
- On Day 6, to begin patterning the posterior endoderm to the hindgut, change the media to 10 mL of RPMI/B27 media supplemented with CHIR99021 (6 μM) + retinoic acid (3 μM).
- On Days 7, 8, and 9, repeat step 2.7. During these steps, visible 3-D structures of the hindgut should become apparent, covering the surface of the plate.
- On Day 10, embed the resulting hindgut in basement membrane matrix (see the Table of Materials).
3. Embedding of the Hindgut in Basement Membrane Matrix
- Make up iHO base growth media (500 mL of advanced Dulbecco’s modified Eagle’s medium [DMEM]/F12, 10 mL of B27 supplement [50x, serum free], 5 mL of N2 supplement [100x, serum free], 5 mL of 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and 5 mL of nonessential amino acids [100x]. Optional: add 5 mL of penicillin-streptomycin [10,000 U/mL]; filter-sterilize before use. See Table 1).
- Remove the media from the hindgut plate, and wash the plate 1x with DPBS (without Ca or Mg). Add 5 mL of collagenase solution to the plate and incubate at 37 °C for 5 min.
- Produce collagenase solution by adding 500 mg of collagenase IV powder to 400 mL of advanced DMEM/F12. Following this, add 100 mL of serum replacement (see the Table of Materials), 5 mL of L-glutamine (200 mM), and 3.5 μL of 2-mercaptoethanol, and swirl the solution to mix. Filter-sterilize the solution once the collagenase powder is fully dissolved.
NOTE: This can be stored at -20 °C for up to 6 months in smaller aliquots.
- Produce collagenase solution by adding 500 mg of collagenase IV powder to 400 mL of advanced DMEM/F12. Following this, add 100 mL of serum replacement (see the Table of Materials), 5 mL of L-glutamine (200 mM), and 3.5 μL of 2-mercaptoethanol, and swirl the solution to mix. Filter-sterilize the solution once the collagenase powder is fully dissolved.
- Inactivate the collagenase by adding 5 mL of iHO base growth media to the plate and scrape off the hindgut cells using a cell scraper, collecting the hindgut suspension in a 15 mL conical tube.
- Centrifuge at 240 x g for 1 min and pipette off the supernatant.
- Add 10 mL of media, break up the hindgut into smaller pieces by gently pipetting, and centrifuge again at 95 x g for 1 min.
- Wash the cells 2x in iHO base growth media by repeating step 3.5. Resuspend the cells in a small volume of base growth medium (~300–500 μL) and add around 100 μL of this solution to 1.5 mL of basement membrane matrix. The matrix must remain on ice during this time as it will begin to solidify rapidly at RT.
- Set up a 24-well plate on a plate heater at 37 °C and spot out 60 μL into one well of the 24-well plate. Allow it to set briefly and check the density under a microscope.
- If required, add more hindgut solution to the basement membrane matrix in increments until the desired concentration is achieved, and spot out the solution into the remaining wells.
- Incubate at 37 °C for 10 min; then, add 800 μL of iHO base growth media containing growth factors to each well of the 24-well plate at the following concentrations (see Table 1): 500 ng/mL R-spondin-1, 100 ng/mL Noggin, 100 ng/mL epidermal growth factor (EGF), 3 μM CHIR99021, 2.5 μM prostaglandin E2, and 10 μM Y-27632 dihydrochloride monohydrate (ROCK inhibitor).
- Change the iHO base growth media every 2–3 days, or immediately if the media begins to discolor. After the initial seeding into basement membrane matrix, allow the iHOs to develop for 7 days before splitting them. By day 3–4, distinct spheres should be visible in the culture.
- For media change alone, omit Y-27632, as this is only required when splitting/seeding.
4. Maintenance and Passage of iHOs
- To allow gradual thawing, put out the required volume of basement membrane matrix in a covered ice bucket at 4 °C overnight, 24 h prior to splitting.
- Remove the media from the iHOs and replace it with 500 μL of cell-lifting solution (see the Table of Materials) per well. Incubate at 4 °C for 40–50 min, at which point the iHOs should be floating in the solution.
- Optional: use an in-hood imaging system to select only iHOs with the desired morphology (see the Table of Materials).
- Gently pipette the iHO/cell lifting solution suspension into 15 mL conical tubes, trying not to break up the iHOs. Allow the IHOs to settle for 3–5 min and remove the supernatant and single cells.
- Resuspend the iHOs in 5 mL of iHO base growth medium and pipette them gently to wash. Centrifuge at 95 x g for 2 min.
- Set up a 24-well plate on a plate heater at 37 °C within the hood.
- Remove the supernatant and resuspend the iHOs in ~300–500 μL of base growth media, Using a P1000 pipette to break up the iHOs into smaller chunks. Note that the force that needs to be applied will vary dependent on the iHO line and maturation state, so start gently, increasing the force if required.
- Place ~100 μL of the iHOs (the volume is dependent on the density of the solution) into 1.5 mL of basement membrane matrix and pipette briefly to mix.
- Spot out 1 x 60 μL of basement membrane matrix into one well of the 24-well plate, leave it to solidify for ~30 s, and then check the density under the microscope. If the density is too low, add more iHOs to the matrix.
- Repeat step 4.8 until the correct density is acquired, and then spot out the rest of the matrix into a 24-well plate.
- Place it in an incubator at 37 °C for 10 min and, then, overlay it with 800 μL of base growth medium with growth factors, as described in step 3.7.
- To prepare the iHOs for an invasion assay experiment (outlined below), passage the iHOs 4–5 days prior to the experiment as described in steps 4.1–4.10, but place 120 μL of the matrix iHO solution generated in step 4.7 into 5 mm glass-bottomed microinjection dishes.
- Rather than leaving the iHO suspension in a droplet as with routine passaging, spread the droplet over the bottom of the dish to create a thin layer of matrix. Cover it with 2.5 mL of base growth medium plus growth factors.
NOTE: If antibiotics have been used in culture medium, these must be removed and replaced with nonantibiotic supplemented media for microinjection experiments.
- Rather than leaving the iHO suspension in a droplet as with routine passaging, spread the droplet over the bottom of the dish to create a thin layer of matrix. Cover it with 2.5 mL of base growth medium plus growth factors.
5. Prestimulation of iHOs with rhIL-22
- Aspirate the media from the iHOs and replace it with fresh base growth media (which must not contain antibiotics) 18 h prior to the invasion assay.
- Add rhIL-22 to culture media to a final concentration of 100 ng/mL.
6. Microinjection of iHOs and Intracellular Invasion Assays
- On the day prior to the experiment, set up S. Typhimurium SL1344 culture in 10 mL of Luria-Bertani broth and incubate at 37 °C overnight with shaking.
- On the day of the experiment, if a microscope with an enclosed heat chamber is available, turn it on and allow the temperature to reach 37 °C prior to starting the assay.
- Dilute overnight bacterial cultures in DPBS (containing Ca and Mg) to an optical density of 2 at 600 nm (OD600) and, then, mix it 1:1 with phenol red.
- Load the microinjection dish containing iHOs onto the microscope stage, remove the lid, and bring the iHOs into focus, ready for the injection to begin.
- Turn the injector and the arm control stations on. Ensure the injector is set to a pressure of 600 kPa and an injection time of 0.5 s. If it is not already backed away from the microscope stage, rotate the injection arm to make sure it is.
- Set up a 6 μm microinjection drill tip by removing the wrapping and the plastic cylinder from the needle. Remove the grip head from the injecting arm.
- Load the drill tip with 10 μL of the inoculum, gripping the drill tip gently at its blunt end. Place the drill tip into the grip head and reattach it to the microinjection arm.
- Gently move the arm into position so that the needle is situated 1–2 cm above the microinjection dish. Use the arm control to position the needle tip in the center of the dish and lower it until it is just over the surface of the media.
- Program the arm control station to return the needle to this point after all injections.
- Focus the microscope on the iHOs and select the target to inject. Position the needle just above and to the right of the iHO to be injected and move the needle downward and laterally into the iHO lumen.
- Press the inject button on the microinjector; the phenol-stained bacterial mixture will emerge from the needle. Inject each iHO 3x. Inject at least 30 iHOs per condition.
NOTE: Due to heterogeneity in iHO size and structure within a culture, it is necessary to inject a large number of iHOs to control for variation. - When all required iHOs are injected, remove the microinjection plate from the stage, replace the lid, and incubate the plate at 37 °C for 90 min.
- After 90 min, aspirate the growth media and replace it with 3 mL of cell lifting solution; incubate at 4 °C for 45 min.
- Gently move the iHOs/cell lifting solution to a 15 mL conical tube containing 5 mL of DPBS. Ensure that all injected iHOs have been removed from the plate (rinse the plate with 1 mL of the DPBS if required). Centrifuge at 370 x g for 3 min.
- Remove the supernatant and resuspend the iHOs in base growth media containing gentamicin at 0.1 mg/mL (add 1 mL of media; then, use a P1000 pipette ~50x to break up the iHOs, and add 4 mL of further media).
- Incubate at 37 °C for 1 h to kill extracellular bacteria.
- Centrifuge the iHOs at 370 x g for 3 min and aspirate the supernatant, leaving as little as possible. Wash the iHOs 1x with DPBS and centrifuge again.
NOTE: This step removes gentamicin, which is important as any remaining gentamicin may kill intracellular bacteria once the cells are lysed. - Resuspend the iHOs in 500 μL of lysis buffer (see the Table of Materials) and manually dissociate the organoids by pipetting ~50x. Leave this mixture for 5 min at RT.
- Serially dilute the resulting solution 10-fold in DPBS to generate 10-1, 10-2, and 10-3 concentrations. Pipette 3 x 20 μL droplets of the neat and diluted solutions onto prewarmed LB agar plates.
- Incubate overnight at 37 °C and perform colony counting and calculation of the colony-forming units (CFU). Colony counts will reflect the numbers of intracellular bacteria which were released during the cell lysing process.
7. Cell Freezing and Recovery
NOTE: As noted earlier, it is possible to cryogenically preserve iHOs and reconstitute them when desired. The freezing and thawing processes are outlined below.
- Select the wells of iHOs you wish to freeze. Add cell lifting solution to the wells and incubate for 40–50 min at 4 °C. The iHOs should be floating in the solution.
- Gently pipette the iHOs into a 15 mL conical tube and allow them to settle. Remove the media and wash the iHOs 1x with base growth media (no growth factors).
- Centrifuge at 95 x g for 2 min, remove the supernatant, and replace it with an appropriate volume of cell freezing medium (see the Table of Materials; use the medium as per the manufacturer’s instructions), decanting the iHOs into cryogenic vials.
- Store the vials in a -80 °C freezer overnight to allow more gradual freezing; then, transfer them to liquid nitrogen storage.
- To reconstitute the iHOs, rapidly defrost a cryogenic vial at 37 °C using a water/bead bath; then, gently pipette its contents into 10 mL of base growth media (no growth factors). Allow the iHOs to settle and replace the media with ~300 μL of fresh base growth media.
- Do NOT manually dissociate iHOs. Add iHOs to basement membrane matrix and plate them out as described in section 3.
Representative Results
Following the commencement of the differentiation process, the cells should pass through the stage of definitive endoderm formation followed by hindgut patterning prior to embedding into basement membrane matrix. Spheroids will form and cultures with large amounts of contaminating material will clear over a period of several weeks as the iHOs mature. Exemplar images of the differentiation, embedding, and passaging process are shown in Figure 1.
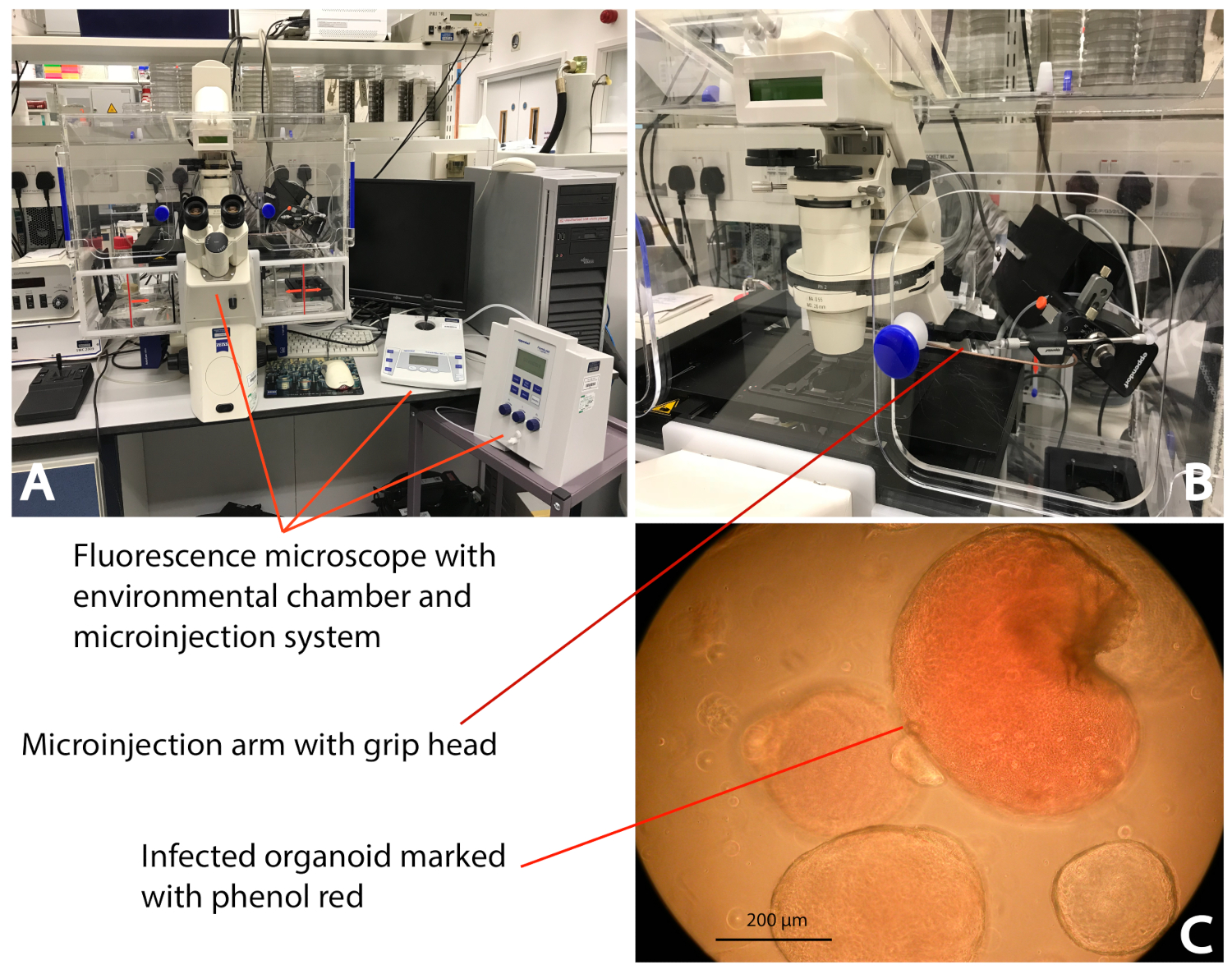
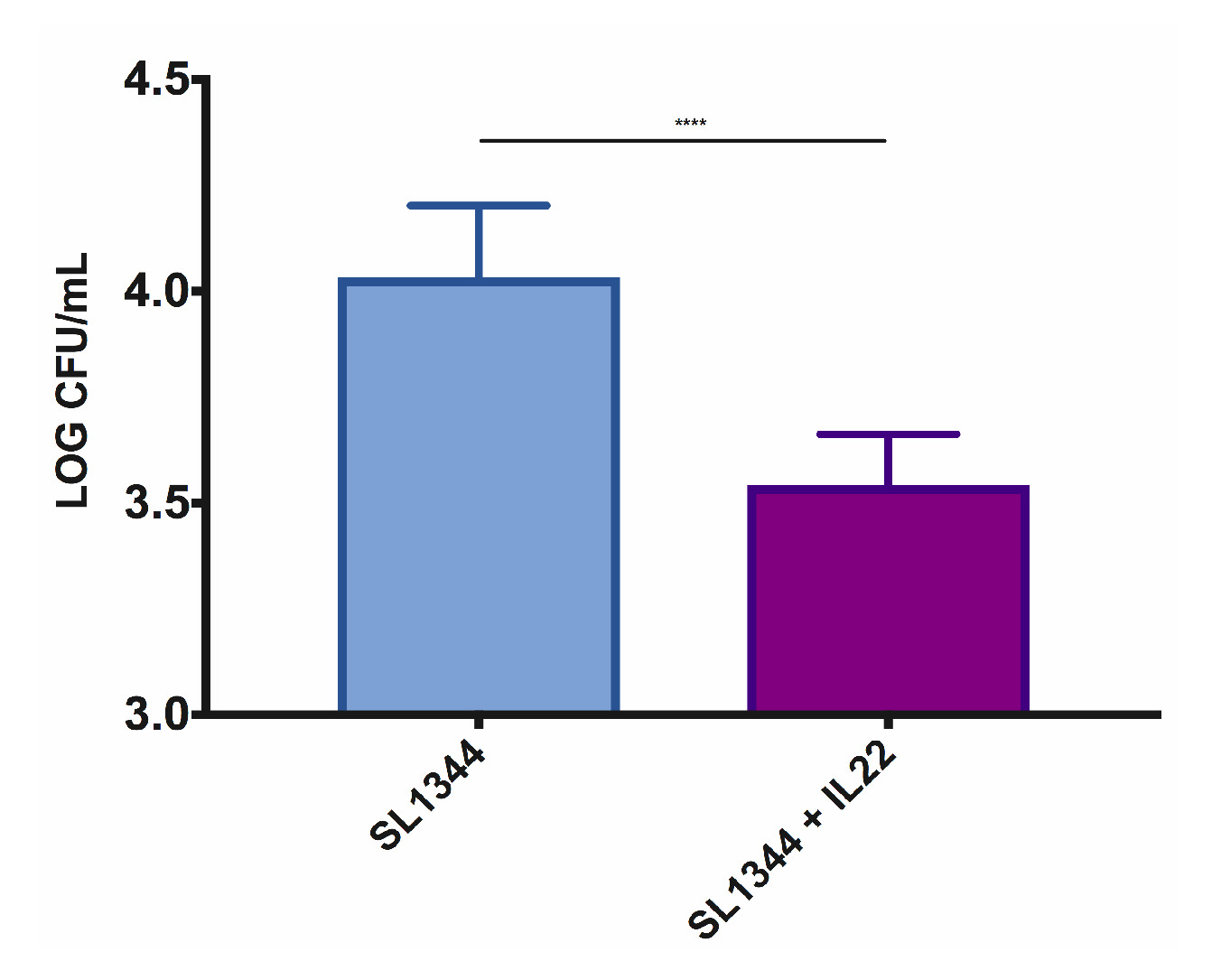
The setup of the microinjection system is as demonstrated in Figure 2. The iHOs are microinjected with the phenol red/bacterial solution and retain their red color, allowing the identification of infected iHOs to prevent duplicate injections. Counts of plated intracellular bacteria are performed following the modified gentamicin protection assay; prestimulation with rhIL-22 restricts the S. Typhimurium infection, with fewer intracellular bacteria being observed following rhIL-22 treatment (Figure 3). We also routinely process infected iHOs for immunostaining, or TEM, in order to facilitate the visualization of host IEC-bacterial interactions (Figure 4).

Figure 1: Directed differentiation of iPSCs to iHOs. Representative sequence of differentiation from iPSCs to iHOs, demonstrating the morphological changes observed and the growth factors required to drive these changes. Definitive endoderm is formed at day 4 of the differentiation, following exposure to specific concentrations of combinations of activin A, FGF, BMP-4, LY294002, and CHIR99021. After 8 days, the patterning of this definitive endoderm with specific concentrations of CHIR99021 and retinoic acid results in hindgut formation. Postembedding, spheroid formation is observed. After sustained passaging using a supporting basement membrane matrix overlaid with medium supplemented with prointestinal proliferation factors R-spondin 1, Noggin, EGF, CHIR99021, and prostaglandin E2, the spheroids progress into budded iHOs. (Images were taken at 4x–10x magnification). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Microinjection of an iHO with S. Typhimurium. (A) The microinjection system (see the Table of Materials) enclosed in the environmental chamber; this allows the injection of the iHOs in a controlled environment (37 °C/5% CO2). (B) The bacterial inoculum is delivered directly into the iHO lumen using a microcapillary attached to the microinjection system. (C) By mixing bacterial inoculums with phenol red, it is clear which iHOs have been infected, thus avoiding duplicate injections of the same iHO. The images were taken at 10x magnification. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Pretreatment of iHOs derived from the Kolf2 cell line with rhIL-22 restricts the S. Typhimurium SL1344 invasion into intestinal epithelial cells. For gentamicin protection assays, iHOs were treated with 100 ng/mL rhIL-22 18 h prior to infection, or left untreated, and incubated for 90 min postinfection. The data are means of three technical replicates for three biological replicates ± SEM. For significance testing, Mann-Whitney U tests were used; ****p < 0.0001. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Interaction of IECs with S. Typhimurium SL1344. These panels demonstrate iHOs injected with S. Typhimurium SL1344 and incubated for 3 h, prior to fixation and processing for (A) immunofluorescence or (B) transmission electron microscopy. In panel A, bacteria are seen within the iHO lumen and interacting with the epithelium. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI) dilactate (blue), cell membranes with phalloidin (red), and bacteria with CSA-1 (green). The images are taken at 20x magnification. Panel B demonstrates three different intracellular processing pathways of Salmonella following invasion; bacteria are seen (a) within a Salmonella-containing vacuole, (b) free within the cytoplasm, and (c) undergoing autophagy. Please click here to view a larger version of this figure.
{kind=link}
| RPMI/B27 medium | |
| Component: | Amount: |
| RPMI 1640 media with glutamine supplement | 500 mL |
| B27 serum-free supplement 50x | 10 mL |
| iHO base growth medium | |
| Component: | Amount: |
| Advanced DMEM/F12 | 500 mL |
| B27 serum-free supplement 50x | 10 mL |
| N2 serum-free supplement 100x | 5 mL |
| HEPES 1 M | 5 mL |
| L-glutamine 200 mM | 5 mL |
| Growth factors for iHO base growth medium | |
| Component: | Amount: |
| Recombinant human R-spondin1 | 500 ng/mL |
| Recombinant human Noggin | 100 ng/mL |
| Epidermal growth factor (EGF) | 100 ng/mL |
| Prostaglandin E2 | 2.5 µM |
| CHIR99021 | 3 µM |
| Y-27632 dihydrochloride monohydrate | 10 µM |
Table 1: Media recipes.
Discussion
This protocol outlines the differentiation of hiPSCs into iHOs and their utility as a model in which to simulate enteric infections. Below, we outline the critical steps in the protocol and any modifications or improvements we have made.
This protocol streamlines the differentiation process of hiPSCs compared to previously published work25. Previously used methods required the transfer of hiPSCs from other hiPSC culture systems (e.g., feeder-dependent hiPSC culture) to chemically-defined medium–polyvinyl alcohol (CDM-PVA). This transfer to CDM-PVA typically takes 2–3 weeks and requires daily feeding of the hiPSCs. This protocol was also not consistently effective, with some differentiations failing; therefore, we trialed differentiation using the same growth factors but starting with hiPSCs grown in stem cell culture medium (rather than CDM-PVA) and replacement of CDM-PVA with stem cell culture medium during differentiation days 0–3. This has been successful for the five independent hiPSC lines trialed thus far, making the differentiation process much more rapid and efficient. This also allows weekend-free culturing of hiPSCs prior to differentiation, allowing more flexibility in the hiPSC culture. iHO lines produced by this method have been phenotyped for markers of intestinal epithelium as we have previously described for the hiPSC lines Kolf2, Yemz1, and Lise116 and appear phenotypically indistinguishable from iHOs produced using the previous protocol.
Following seeding, iHOs require at least 1 month of routine passaging, with splitting every 4–7 days to facilitate maturation. Note that there will be some variation in iHO development depending on the iPSC line used and the density of the initial culture. During the first few passages, there will be visibly contaminating cells which are not iHOs. These will eventually die, leaving a clean culture of spherical and, after approximately 4 weeks, budded iHOs. In addition, an in-hood imaging system can be used to select and passage only iHOs with the desired morphology. As iHOs mature, they will require splitting every 6–7 days, dependent on the growth rate and density. If any of the following occur, iHOs should be split prior to this point: the luminal cavities of the iHOs start to fill up with dead cells, the basement membrane matrix starts to disintegrate, the iHOs start to grow out of the basement membrane matrix, or the culture is too dense and the media starts to go yellow very quickly.
Once the iHO culture is established, if at any time the appearance of the iHOs changes or is different than expected (for example, the cultures remain spherical, rather than budding), phenotyping via immunohistochemistry and qPCR for cell markers ought to be repeated to ensure that the differentiation of the cell types within the iHOs (e.g., goblet cells, Paneth cells) remains intact. If the iHOs are no longer differentiating, then they should be discarded and redifferentiated or an earlier passage of the iHOs should be thawed and reconstituted. If the iHOs cease to differentiate, the potential causes are the age of the culture (if it is over 6 months old), the activity of the growth factors (ensure that these are reconstituted as per the manufacturers’ instructions and kept frozen in small aliquots to avoid multiple freeze-thaw cycles), too frequent or violent passages (in general, passaging should only occur once a week, and if the iHOs are manually dissociated too vigorously on a regular basis, they will cease to fully differentiate).
We established via RNA sequencing that IL-22 stimulation 18 h prior to infection upregulates antimicrobial genes and those involved in the barrier defense phenotype. Prior to the use of new iHsO for assays involving prestimulation with rhIL-22 (or an alternative cytokine if the system is being used for this), it is advisable to check the activity of genes known to be upregulated by the cytokine (in the case of IL-22, we used DUOX2 and LCN2) via qPCR after stimulation of the iHOs, to ensure receptor expression and intact signalling. Prior to the first use of IL-22, we also carried out immunohistochemistry to locate the IL-22 receptor on iHOs to establish that the expression of the IL-22 receptor was basal, meaning that prestimulation could be achieved simply by adding rhIL-22 to the iHO culture medium. However, if a receptor is apically expressed, this protocol may have to be adapted to deliver ligands apically.
Pitfalls regarding the microinjection system are generally related to the delicacy of the needles required for the injection. Here, we use commercially available drill tips with a 6 μm lumen. It is possible to pull the injection needles from glass capillaries26 although this may be less uniform, leading to leakages from the needle tip or inconsistent volumes being injected into the iHOs. It is important to be sure that the injection has taken place into the iHO lumen, which is one reason for the use of phenol red as a dye; the iHOs will visibly expand and hold the red inoculum, allowing certainty about which iHOs have been injected. Occasionally needles will clog with debris from the iHO wall; if this is the case, remove the needle tip from inside the iHO and press the clean button on the microinjection system. This will produce a brief period of higher air pressure which should clear the blockage. It will also induce some leakage of the bacterial inoculum onto the plate; therefore, if this occurs, the clean action should be repeated on all plates to ensure equality of bacterial inoculum per plate. One large advantage of the hiPSC-derived iHOs is their size. Intestinal organoids from mice and primary human organoids are much smaller (measuring up to ~100 μm and 100–300 μm, respectively27, versus 250–1500 μm for hiPSC-derived iHOs), meaning that injections of large amounts of organoids will be slower. This allows larger-scale injection experiments to be trialed in the hiPSC-derived iHOs. It is also possible to study the luminal contents of the iHOs by harvesting them postinfection and manually dissociating the iHOs into DPBS, releasing their luminal contents. For microinjection, we recommend using a high concentration of bacteria. We found that lower concentrations were not sufficient to generate a response from the IECs comprising the iHOs. Additionally, it was difficult to subsequently locate internalized bacteria using microscopy. Inoculums may have to be optimized for different bacterial strains.
In summary, hiPSC-derived iHOs provide a promising model for directly dissecting the epithelial response to enteric infections, whether by studying intracellular invasion counts, imaging, measuring cytokine levels in the iHO supernatants, or harvesting RNA to study transcriptional changes after exposure to pathogens. Their utility will be even more apparent in the future for establishing infection models for human-restricted pathogens and in exploiting the possibilities of using this technology to personalize research by studying specific disease-related genetic mutations and drug responses.
Disclosures
The authors have nothing to disclose.
Acknowledgements
This work was supported by funding from The Wellcome Trust, The Gates Foundation, and the Cambridge Biomedical Research Centre. E.A.L. is a clinical Ph.D. student supported by the Wellcome Trust.
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250-10ML | |
| 5 mm glass bottom injection dishes | MatTek corporation | P50G-0-14-F | |
| Advanced DMEM/F12 | Gibco | 12634010 | |
| Alexa fluor 647 phalloidin | Life Technologies | A22287 | Use at 1:1000 concentration |
| B27 serum-free supplement | Life Technologies | 17504044 | Stock concentration 50x, final concentration 1x |
| BMP-4 recombinant human protein | R&D | PHC9534 | Stock concentration 10 μg/mL, final concentration 10 ng/mL |
| Cell recovery solution (cell lifting solution) | BD | 354253 | |
| CHIR99021 | Abcam | ab120890-5mg | Stock concentration 3 mM, final concentration 3 µM |
| Collagenase, type IV powder | Life Technologies | 17104019 | Reconstitute at 0.1% |
| Corning cryogenic vials | Corning | 430487 | |
| Costar TC treated 24 well culture plates | Corning | CLS3527 | |
| DAPI dilactate | Sigma-Aldrich | D9564-10MG | Use at 10 nM concentration |
| Dulbecco’s PBS (No MgCl2 or CaCl2) | Life Technologies | 14190-144 | |
| Dulbecco’s PBS (with MgCl2 and CaCl2) | Sigma-Aldrich | D8662-100ML | |
| Epidermal growth factor | R&D | 236-EG-200 | Stock concentration 100 μg/mL, final concentration 100 ng/mL |
| Eppendorf TransferMan NK2 (microinjection system) | Eppendorf | 920000011 | |
| Eppendorf Femtojet express (microinjection system) | Eppendorf | 5248 000.017 | |
| Essential 8 Flex medium kit (stem cell culture medium) | Life Technologies | A2858501 | |
| EVOS XL imaging system (in-hood imaging system) | |||
| Gentamicin | Sigma-Aldrich | G1272-10ML | Stock concentration 10 mg/mL, final concentration 0.1 mg/mL |
| Goat anti-Salmonella, CSA-1 | Insight Biotechnology | 02-91-99 | Use at 1:20 concentration |
| HEPES 1 M | Life Technologies | 15630056 | |
| KnockOut Serum Replacement (setrum replacment) | Gibco | 10828010 | |
| GlutaMAX supplement (glutamine supplement | ThermoFisher | ||
| L-glutamine | Life Technologies | A2916801 | Stock concentration 200 mM, final concentration 2 mM |
| LY294002 | Promega UK | V1201 | Stock concentration 50 mM, final concentration 10μM |
| Matrigel, GFR, phenol free (basement membrane matrix) | Corning | 356231 | |
| MEM non-essential amino acids solution (100x) | Gibco | 11140035 | |
| N2 serum-free supplement | Life Technologies | 17502048 | Stock concentration 100x, final concentration 1x |
| Penicillin-streptomycin | Life Technologies | 15140163 | Stock concentration 10,000 U/mL, final concentration 100 U/mL |
| Phenol red | Sigma-Aldrich | P0290-100ML | |
| Piezo Drill Tip Mouse ICSI, 25° tip angle, 6 µm inner diameter | Eppendorf | 5195000087 | |
| Prostaglandin E2 | Sigma | P0409-1MG | Stock concentration 2.5 mM, final concentration 2.5 mM |
| Recombinant human FGF basic | R&D | 233-FB-025 | Stock concentration 100 μg/mL, final concentration 100 ng/mL |
| Recombinant human IL-22 | R&D | 6057-NG-100 | Stock concentration 100 μg/mL, final concentration 100 ng/mL |
| Recombinant human Noggin | R&D | 6057-NG-100 | Stock concentration 100 μg/mL, final concentration 100 ng/mL |
| Recombinant human R-spondin1 | R&D | 4645-RS-025 | Stock concentration 25 mg/mL, final concentration 500 ng/mL |
| Recombinant human/mouse/rat Activin A | R&D | 338-AC-050 | Stock concentration 100 μg/mL, final concentration 100 ng/mL |
| Recovery cell culture freezing medium (cell freezing medium) | Gibco | 12648010 | |
| Retinoic acid | Sigma-Aldrich | R2625-50MG | Stock concentration 3μM, final concentration 3mM |
| RPMI 1640 media with Glutamax supplement (RPMI Medium with L-glutamine supplement) | Life Technologies | 61870010 | |
| Triton X-100 (cell lysis buffer) | Sigma-Aldrich | RES9690T-A101X | Use at 1% concentration |
| Versene (EDTA solution) | Life Technologies | 15040066 | |
| Vitronectin XF | Stemcell Technologies | 7180 | |
| Y-27632 dihydrochloride monohydrate | Sigma-Aldrich | Y0503-1MG | Stock concentration 3 mM, final concentration 10 mM |
References
- Barker, N., et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 6 (1), 25-36 (2010).
- Huch, M., Boj, S. F., Clevers, H. Lgr5(+) liver stem cells, hepatic organoids and regenerative medicine. Regenerative Medicine. 8 (4), 385-387 (2013).
- Huch, M., et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. The EMBO Journal. 32 (20), 2708-2721 (2013).
- Boj, S. F., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 160 (1-2), 324-338 (2015).
- Lancaster, M. A., et al. Cerebral organoids model human brain development and microcephaly. Nature. 501 (7467), 373-379 (2013).
- Mariani, J., et al. Modeling human cortical development in vitro using induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 109 (31), 12770-12775 (2012).
- Dye, B. R., et al. In vitro generation of human pluripotent stem cell derived lung organoids. eLife. 4, (2015).
- Karthaus, W. R., et al. Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell. 159 (1), 163-175 (2014).
- Matano, M., et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nature Medicine. 21 (3), 256-262 (2015).
- Ogawa, M., et al. Directed differentiation of cholangiocytes from human pluripotent stem cells. Nature Biotechnology. 33 (8), 853-861 (2015).
- Leha, A., et al. A high-content platform to characterise human induced pluripotent stem cell lines. Methods. 96, 85-96 (2016).
- Drost, J., et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature. 521 (7550), 43-47 (2015).
- Schreiber, F., Arasteh, J. M., Lawley, T. D. Pathogen Resistance Mediated by IL-22 Signaling at the Epithelial-Microbiota Interface. Journal of Molecular Biology. 427 (23), 3676-3682 (2015).
- Sabat, R., Ouyang, W., Wolk, K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nature Reviews Drug Discovery. 13 (1), 21-38 (2014).
- Kotenko, S. V., et al. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta ) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. Journal of Biological Chemistry. 276 (4), 2725-2732 (2001).
- Forbester, J. L., et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proceedings of the National Academy of Sciences of the United States of America. 115 (40), 10118-10123 (2018).
- Nozaki, K., et al. Co-culture with intestinal epithelial organoids allows efficient expansion and motility analysis of intraepithelial lymphocytes. Journal of Gastroenterology. 51 (3), 206-213 (2016).
- Dijkstra, K. K., et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell. 174 (6), 1586-1598 (2018).
- Schlaermann, P., et al. A novel human gastric primary cell culture system for modelling Helicobacter pylori infection in vitro. Gut. 65 (2), 202-213 (2016).
- Ettayebi, K., et al. Replication of human noroviruses in stem cell-derived human enteroids. Science. 353 (6306), 1387-1393 (2016).
- Saxena, K., et al. Human Intestinal Enteroids: a New Model To Study Human Rotavirus Infection, Host Restriction, and Pathophysiology. Journal of Virology. 90 (1), 43-56 (2016).
- Karve, S. S., Pradhan, S., Ward, D. V., Weiss, A. A. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLOS ONE. 12 (6), e0178966 (2017).
- Heo, I., et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nature Microbiology. 3 (7), 814-823 (2018).
- Garcez, P. P., et al. Zika virus impairs growth in human neurospheres and brain organoids. Science. 352 (6287), 816-818 (2016).
- Forbester, J. L., Hannan, N., Vallier, L., Dougan, G. Derivation of Intestinal Organoids from Human Induced Pluripotent Stem Cells for Use as an Infection System. Methods in Molecular Biology. , (2016).
- Wilson, S. S., Tocchi, A., Holly, M. K., Parks, W. C., Smith, J. G. A small intestinal organoid model of non-invasive enteric pathogen-epithelial cell interactions. Mucosal Immunology. 8 (2), 352-361 (2015).
- Sato, T., et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 141 (5), 1762-1772 (2011).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved