Single-Step Enrichment of a TAP-Tagged Histone Deacetylase of the Filamentous Fungus Aspergillus nidulans for Enzymatic Activity Assay
In This Article
Summary
Class 1 histone deacetylases (HDACs) like RpdA have gained importance as potential targets to treat fungal infections. Here we present a protocol for the specific enrichment of TAP-tagged RpdA combined with an HDAC activity assay that allows in vitro efficacy testing of histone deacetylase inhibitors.
Abstract
Class 1 histone deacetylases (HDACs) like RpdA have gained importance as potential targets for treatment of fungal infections and for genome mining of fungal secondary metabolites. Inhibitor screening, however, requires purified enzyme activities. Since class 1 deacetylases exert their function as multiprotein complexes, they are usually not active when expressed as single polypeptides in bacteria. Therefore, endogenous complexes need to be isolated, which, when conventional techniques like ion exchange and size exclusion chromatography are applied, is laborious and time consuming. Tandem affinity purification has been developed as a tool to enrich multiprotein complexes from cells and thus turned out to be ideal for the isolation of endogenous enzymes. Here we provide a detailed protocol for the single-step enrichment of active RpdA complexes via the first purification step of C-terminally TAP-tagged RpdA from Aspergillus nidulans. The purified complexes may then be used for the subsequent inhibitor screening applying a deacetylase assay. The protein enrichment together with the enzymatic activity assay can be completed within two days.
Introduction
Histone deacetylases (HDACs) are Zn2+-dependent hydrolytic enzymes capable of removing acetyl groups from lysine residues of histones and other proteins. Based on the sequence similarity, HDACs are grouped into several classes1. Recently, the fungal class 1 HDAC RpdA, an ortholog of baker's yeast (Saccharomyces cerevisiae) Rpd3p, was shown to be essential for the opportunistic fungal pathogen Aspergillus fumigatus2. Therefore, RpdA has gained importance as a potential target to treat fungal infections2. Assessment of deacetylase activity in vitro is important for the characterization of enzymatic properties and allows to determine the efficacy of novel substances for inhibitor development. Although recombinant expression of a codon-optimized version of human HDAC1 in Escherichia coli has been reported recently3, attempts to express full-length RpdA in this host failed4. Furthermore, since fungal class 1 HDACs such as RpdA and HosA exert their function as multiprotein complexes, it is favorable to use native endogenous complexes for enzymatic inhibitor studies. However, due to inhibiting factors and the presence of different HDACs in fungal lysates, catalytic activity measured in whole protein extracts is relatively low and cannot be assigned to individual enzymes. Moreover, previous studies in the filamentous fungus A. nidulans identified a class 2 HDAC, HdaA, as predominant deacetylase in chromatographic fractions of fungal extracts. Thus, multiple conventional chromatographic purification steps are needed to separate non-HdaA activity from fungal strains4. The introduction of the tandem affinity purification (TAP) strategy in A. nidulans5 has significantly eased the enrichment of specific deacetylase activities. The original TAP tag is composed of two protein A domains and a calmodulin binding peptide (CBP) separated by a tobacco etch virus protease (TEV) cleavage site6. This allows for native purification and elution of tagged proteins including their interaction partners7. When using the enriched proteins for activity assays, the mild elution under native conditions by protease cleavage is an important feature of the TAP tag purification. A GFP-tagged protein, for example, can be enriched by immobilized antibodies as well, however, cannot be eluted under native conditions.
Here we provide a detailed protocol for the single-step enrichment of active RpdA complexes via the first purification step of C-terminally TAP-tagged RpdA from A. nidulans (IgG separation and TEV cleavage) for subsequent inhibitor screening applying a histone deacetylase assay. As proved to be sufficient, affinity enrichment was restricted to just one purification step also because the enzymatic activity was significantly reduced after two-step TAP purification when compared to IgG purification alone.
Nevertheless, the introduced protocol should as well be applicable for the enrichment of other tagged enzymes involved in chromatin regulation such as acetyltransferases, methyltransferases, and demethylases. By appending the second purification step of the TAP protocol, proteins co-purified with the tagged baits can be considered as complex partners of (novel) enzymatic complexes.
Protocol
1. Cultivation of A. nidulans
- Inoculum preparation
NOTE: All steps apart from the incubation should be performed under a laminar flow cabinet.- Streak out TAP strain (e.g. TIB32.12) from the glycerol stock (-80 °C) onto glucose/xylose minimal medium (GXMM; per liter: 10.0 g of glucose, 0.5 g of xylose, 10 mL of 1 M di-ammonium tartrate solution, 20 mL of 50 × salt solution [per liter: 26.0 g KCl, 26.0 g MgSO4 · 7 H2O, 76.0 g KH2PO4, 1 mL of chloroform], 1 mL of 1,000 × Hutner’s trace elements8) including 1.5% (w/v) agar and the required supplements as described by Todd et al. 20079. Incubate for 2–3 days at 37 °C.
NOTE: TIB32.1 contains an rpdA::TAP fusion controlled by a xylose-inducible promoter, xylP(p)10. This is why xylose is included in the above recipe. Omit xylose when growing strains expressing constructs that are not under xylP(p) control. - Prepare a sterile 1.5 mL centrifuge tube with 1.5 mL of sterile conidial suspension solution (CSS; 0.9% (w/v) NaCl, 0.01% (v/v) polysorbate 80).
- Wet a disposable inoculation loop in CSS, scrape off conidia from the plate from step 1.1.1 and suspend in the tube from step 1.1.2.
- Transfer 150 µL each of conidial suspension to ten 25 cm2 cell culture flasks with vent cap containing GXMM agar including supplements and 200 µL of CSS.
- Spread the conidial suspension within the flasks using a sterile inoculation loop and incubate the flasks at 37 °C for 2–3 days.
- To harvest the conidia, use 10 mL of CSS per flask. Pour the solution into a flask, tightly close the flask with the provided screw cap and vigorously shake the flask.
- After wetting the fungal surface, completely scrape off remaining conidia with a sterile inoculation loop.
- Pass conidia through 40 µm cell strainers placed onto a sterile 50 mL centrifuge tube and collect the suspension of five flasks in one tube.
- Centrifuge the tube at 1,000 × g for 10 min.
- Decant the supernatant and resuspend the pellet in 10 mL of CSS per tube.
- Collect both suspensions in one tube, rinse the empty tube with 40 mL of CSS, add to the suspension, and centrifuge as described in step 1.1.9.
- Decant the supernatant and resuspend the conidial pellet in 4 mL of CSS.
- Prepare two serial 1:50 dilutions of the conidial suspension and determine the number of conidia in the resulting 1:2,500-diluted suspension with a counting chamber as described11.
- Streak out TAP strain (e.g. TIB32.12) from the glycerol stock (-80 °C) onto glucose/xylose minimal medium (GXMM; per liter: 10.0 g of glucose, 0.5 g of xylose, 10 mL of 1 M di-ammonium tartrate solution, 20 mL of 50 × salt solution [per liter: 26.0 g KCl, 26.0 g MgSO4 · 7 H2O, 76.0 g KH2PO4, 1 mL of chloroform], 1 mL of 1,000 × Hutner’s trace elements8) including 1.5% (w/v) agar and the required supplements as described by Todd et al. 20079. Incubate for 2–3 days at 37 °C.
- Growth and harvesting of mycelia
- While working under a laminar flow cabinet, inoculate 4–6 one-liter conical flasks each containing 250 mL of GXMM including appropriate supplements at a density of 5 × 106 conidia/mL and incubate at 180 rpm at 37 °C for 14–16 h.
- Place cheese cloth into a funnel on top of a flask and filter the mycelia through the cloth. Wash briefly with deionized water.
- Remove as much moisture as possible by squeezing the mycelia trapped in the cheese cloth between first the hands and then paper towels.
- Transfer the dried mycelia as flat sheets to a plastic beaker with a screw lid and flash freeze with liquid nitrogen. This ensures a high area/volume ratio for the following lyophilization process.
- Store the frozen mycelia at -80 °C prior to lyophilization.
NOTE: The protocol can be paused here. - Lyophilize the mycelia overnight.
- Stop the freeze-drying process when the temperature of mycelia remains constant (18–24 h). Remove the beakers and immediately seal with the provided screw caps.
NOTE: When tightly sealed, lyophilized mycelia can be stored for several weeks at RT.
2. Single-step enrichment of TAP-tagged HDAC (adapted from Bayram et al. 2012)12
- Preparation of buffers and solutions
NOTE: Add 2-mercaptoethanol (EtSH) and protease inhibitors to buffers directly prior to use. Filter all buffers used for chromatography through 0.22 µm nitrocellulose membranes to avoid introduction of impurities/contaminations to the chromatography resin. Instructions in the steps below refer to the preparation of 1 L of each buffer. Store buffers at 4 °C.- Extraction buffer (B250): 250 mM NaCl, 100 mM Tris-HCl pH 7.5 (RT), 0.1% (v/v) TX-100, 5 mM EtSH.
- Dissolve 12.35 g of Tris-HCl, 2.62 g of Tris (free base), and 13.2 g of NaCl in 800 mL of deionized water and add 10 mL of a 10% (v/v) TX-100 solution.
- Check pH at RT and adjust to pH 7.5 with either NaOH or HCl [5 M], if necessary.
- Make up to 1 L and filter through a 0.22 µm nitrocellulose membrane.
- Add 35 µL of EtSH per 100 mL of buffer (5 mM final concentration) directly prior to use.
- Washing buffer 250 (WB250): 250 mM NaCl, 40 mM Tris-HCl pH 8.0 (RT), 0.1% (v/v) TX-100, 5 mM EtSH.
- Prepare WB250 as described for B250 (2.1.1) but with 3.59 g of Tris-HCl, and 2.08 g of Tris (free base).
- Washing buffer 150 (WB150): 150 mM NaCl, 40 mM Tris-HCl pH 8.0 (RT), 0.1% (v/v) TX-100, 5 mM EtSH.
- Prepare WB150 as described for B250 (2.1.1) but with 3.59 g of Tris-HCl, 2.08 g of Tris (free base), and 8.77 g of NaCl.
- TEV equilibration buffer (TEB): 150 mM NaCl, 40 mM Tris-HCl pH 8.0 (RT), 0.5 mM EDTA, 0.1% (v/v) TX-100, 5 mM EtSH.
- Same as WB150 but add EDTA to 0.5 mM final concentration.
- TEV cleavage buffer (TCB): 150 mM NaCl, 40 mM Tris-HCl pH 8.0 (RT), 0.5 mM EDTA, 0.1% (v/v) TX-100, 10% (v/v) glycerol, 5 mM EtSH.
- Prepare as described in step 2.1.1 with 3.59 g of Tris-HCl, 2.08 g of Tris (free base), and 8.77 g of NaCl and add 100 mL of glycerol before adjusting the volume to 1 L.
- 50 × protease inhibitor cocktail: Dissolve 1 tablet of a commercially available cocktail of protease inhibitors in 1 mL of water and store at -20 °C.
- TBS-T: 50 mM Tris-HCl pH 7.6 (RT), 0.9% (w/v) NaCl, 0.1% (v/v) polysorbate 20.
- Prepare as described in step 2.1.1. Use 6.06 g of Tris-HCl, 1.40 g of Tris (free base), 9 g of NaCl, and 1 mL of polysorbate 20.
- 5 × LSB (Laemmli sample buffer): 315 mM Tris-HCl pH 6.8 (RT), 25% (v/v) EtSH, 50% (v/v) glycerol, 10% (w/v) SDS, 0.05% (w/v) bromophenol blue.
- Extraction buffer (B250): 250 mM NaCl, 100 mM Tris-HCl pH 7.5 (RT), 0.1% (v/v) TX-100, 5 mM EtSH.
- Preparation of protein extract
- Add 1.5 g of lyophilized mycelia and a grinding ball into the grinding jar of a ball mill.
NOTE: Fresh mycelia might be used as well. When doing so, dry the mycelia as thoroughly as possible before freezing and make sure to precool the grinding jars in liquid nitrogen for grinding fresh mycelia. - Grind mycelia to powder at 25 Hz for 30 s.
NOTE: In cases where no grinding machine is available, grind mycelia to a powder using a mortar and pestle, as described by Bayram et al.12. - Transfer mycelial powder to a 15 mL centrifuge tube.
- Tilt the centrifuge tube including ground mycelia to allow subsequent mixing of mycelia with buffer.
- Add 6 mL of ice-cold B250 including 1 × protease inhibitor cocktail per gram of mycelial powder and blend with a small spatula until complete homogenization of the crude extract is achieved. When using fresh mycelia, refer to Bayram et al.12 to achieve the right biomass to extraction buffer ratio.
- Keep the tube on ice for 5 min.
- Place the tube and a balance tube into a centrifuge and spin at 40,000 × g for ≥20 min at 4 °C. Perform equilibration of IgG resin (step 2.3) during the centrifugation.
- After centrifugation, remove 10 µL of the supernatant for SDS-PAGE analysis. Place the sample into a 1.5 mL tube containing 40 µL of water and 12.5 µL of 5 × LSB; referred to as “Ex” in Figure 1.
- Carefully remove the supernatant (cleared lysate) using a serological pipette and transfer onto the column containing the equilibrated IgG beads (step2.3) and tightly close using the provided end cap.
- Add 1.5 g of lyophilized mycelia and a grinding ball into the grinding jar of a ball mill.
- Equilibration of IgG resin (performed during step 2.2.7)
- Prepare a 10 mL disposable chromatography column and pipet 300 µL of well-resuspended IgG resin (50% slurry) into the column. Fill up the column to 10 mL with B250 and let the buffer flow through by gravity.
- Add 1 mL of B250 including 1 × protease inhibitor cocktail and let flow through. Plug the bottom of the column.
- Batch purification of TAP-tagged HDAC
- Incubate the chromatography column containing the equilibrated beads and the cleared lysate from step 2.2.9 on a rotary mixer at 10 rpm at 4 °C for 2–4 h.
- After the batch binding, remove the cap and open the column at the bottom to collect the flow-through.
- Take a sample for SDS-PAGE analysis as described in step 2.2.8; referred to as “FT” in Figure 1.
- Wash beads with 10 mL of WB250. First, use 1 mL of buffer to remove trapped beads from the column cap using a pipettor and transfer this suspension in one flush onto the settled resin to allow resuspension of the beads. Then fill up the column to the top, close using a stack cap and connect to a peristaltic pump. Adjust a flow rate of approx. 1–5 mL/min. Prevent the resin from running dry.
- Repeat step 2.4.4 three times for a total of four washes with WB250.
- Wash beads three times with 10 mL of TEB as described in step 2.4.4.
- Close the chromatography column at the bottom, resuspend IgG beads in 1 mL of TCB, and add 20 µL of 50 × protease inhibitor cocktail as well as 10 µL of TEV (~1 mg/mL stock, S219V mutant variant produced in-house).
- Cap the column and incubate on a rotary mixer at 10 rpm at 4 °C overnight to elute the protein complexes bound via the tagged HDAC.
NOTE: Alternatively perform TEV digest at elevated temperature (16–25 °C), which reduces the reaction time, however, is raising the risk of protein degradation. - On the next day open the column and collect the eluate in a 2 mL centrifuge tube. Use 0.7 mL of TCB to remove beads from the cap and rinse the wall of the column.
- Place the column onto the open 2 mL tube from the previous step which itself is placed within a 50 mL centrifuge tube.
- Transfer this assembly into a table top centrifuge and spin at 300 × g for 2 min. This is the TEV eluate.
NOTE: When performing tandem affinity purification, use the TEV eluate as input for the calmodulin affinity step. You also may split the TEV eluate and use one part for HDAC activity determination and the second part to complete the TAP purification. - Remove 50 µL from the TEV eluate and add 12.5 µL of 5 × LSB for SDS-PAGE (referred to as “TE” in Figures 1 and 2) and keep the remaining eluate on ice.
- To assess the efficacy of protease elution, add 2 mL of 5% (v/v) acetic acid and incubate at RT for 5 min. Again, use the first mL to resuspend the resin.
- Collect the acid eluate and again take 50 µL sample and add 12.5 µL of 5 × LSB (referred to as “AE” in Figure 1). LSB will turn yellow when added to the acidic solution. To neutralize the acid, add 10 M NaOH in steps of 1 µL and mix well until the color changes to blue again.
- Re-equilibrate the IgG resin with TBS-T to neutralize the acid. Store the resin in TBS-T/20% (v/v) ethanol at 4 °C for reuse with the same tagged protein.
- Storage of elution fractions
- Aliquot the eluate into ~100 µL fractions, to avoid multiple freeze-thaw cycles.
- Freeze aliquots in liquid nitrogen and keep at -80 °C.
NOTE: Samples stored this way will be stable for months without losing enzymatic activity.
3. Analysis of purification by SDS-PAGE and western blotting
- Use standard protocols for casting SDS-polyacrylamide gels or use precast gels13. Denature gel samples from the previous section at 95 °C for 5 min, centrifuge at ≥15,000 × g for 5 min, and load onto 12% gels. Recommended loading volumes are given in the legend of Figure 1. Electrophorese the samples in 1 × Tris-glycine SDS-PAGE running buffer at 180 V constant for 60–70 min, until the bromophenol blue marker of the SDS-PAGE loading buffer starts to migrate out of the gel.
- Use standard protocols for silver staining of the gels. For example, use the protocol of Blum et al. 198714 that is also compatible with MS analysis.
- Use standard protocols for western blotting15. For the generation of representative results below, a commercially available blotting system was used.
- Probe blots with commercially available anti-calmodulin binding protein (anti-CBP) antibody in 5% (w/v) milk powder in TBS-T at 4 °C overnight.
NOTE: Anti-CBP antibody is directed against the part of the TAP tag still present after TEV cleavage. - Use standard protocols for detection and development of blots. For example, anti-rabbit IgG-alkaline phosphatase conjugate and a BCIP/NBT color development substrate were used for the generation of representative results below.
4. Deacetylase assay using in vitro [3H] acetate-labeled chicken reticulocyte histones (adapted from Trojer et al. 2003)4
- Refer to the protocol of Kölle et al. 199816 for the preparation of [3H] acetate-labeled chicken reticulocyte histones.
- Per assay condition place three 1.5 mL centrifuge tubes on ice for measurements in triplicates. Also prepare three tubes for buffer-only background control.
- Put 25 µL of WB150 into each tube and add 25 µL of the TEV eluate from step 2.4.11 and keep on ice.
- Preheat a tube incubator to 25 °C.
- Use 15 s intervals (stop watch) for the addition of 10 µL of labeled histones [1.5 mg/mL] to each tube.
- Before starting the assay, take up 10 µL of the [3H] acetate-labeled chicken histones and start the stop watch.
- Five seconds after the start, add the labeled histones, close the tube tightly, vortex, and put it into the incubator.
- Use 15 s intervals for the addition of 10 µL of labeled histones and proceed as described in the previous step.
- After 60 min incubation add 50 µL of stop solution (1 M HCl/0.4 M acetic acid) to each tube in 15 s intervals; vortex immediately. After the addition of the acidic solution, the assay mix is stable and can be kept at RT until the next step.
- Add 800 µL of ethyl acetate to each tube to extract the released [3H] acetic acid.
- Tightly close the tubes and vortex each tube for 5 s.
- Place the tube into a microcentrifuge and spin at 10,000 × g for 10 min at RT.
- In the meantime, prepare one scintillation vial per assay sample and add 3 mL of scintillation cocktail for hydrophobic samples.
- After centrifugation (step 2.12), carefully transfer 600 µL of the upper organic phase to the prepared scintillation tubes and close the tubes tightly.
- Measure the radioactivity corresponding to the HDAC activity in a liquid scintillation counter.
Representative Results
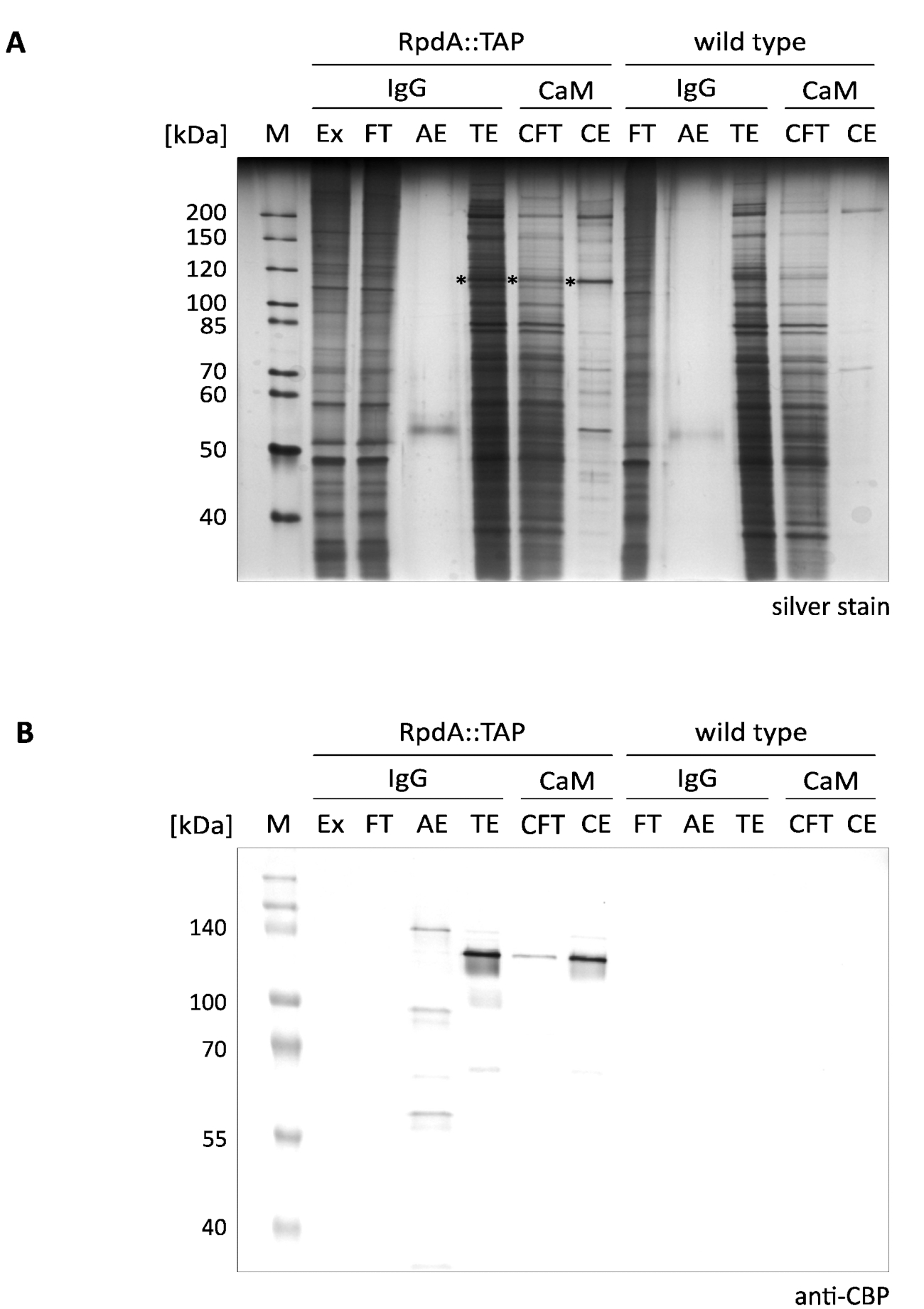
A typical outcome of the presented single-step enrichment of RpdA is shown in Figure 1 (referred to as "IgG"). For the sake of completeness, we also have included flow-through and elution fractions ("CFT" and "CE") illustrating the second purification step by a calmodulin resin ("CaM") as described12. The displayed silver-stained gel (A) clearly illustrates the efficacy of the first affinity step that is even further increased when performing the tandem purification. Most prominent proteins present in the protein extract and the flow-through, however, already are depleted in the TEV eluate (TE). It is important to notice that the TEV elution fractions are >100 × concentrated compared to extract and flow-through. The asterisks mark tagged RpdA (compare panel B). The calculated MW of RpdA including the CBP (RpdA::CBP) is 82 kDa, however, the protein migrates at a much higher apparent molecular weight of approx. 120 kDa. This phenomenon has been observed previously and can be assigned to the specific properties of its C-terminus4,17. The immunoblot (B) shows strong signals migrating at approx. 120 kDa corresponding to CBP-tagged full-length RpdA (RpdA::CBP) in the TEV eluate ("TE"), the calmodulin flow-through ("CFT"), and eluate ("CE") fractions. In the acid eluate ("AE") a second signal with a slightly larger MW is visible. This represents the proportion of TAP-tagged RpdA bound to the IgG resin that was not released by TEV cleavage. The difference in size corresponds to 16 kDa of the protein A repeat of uncleaved RpdA::TAP. As expected, no bands could be detected by the anti-CBP antibody in the wild-type control fractions (panel B, "wild type").
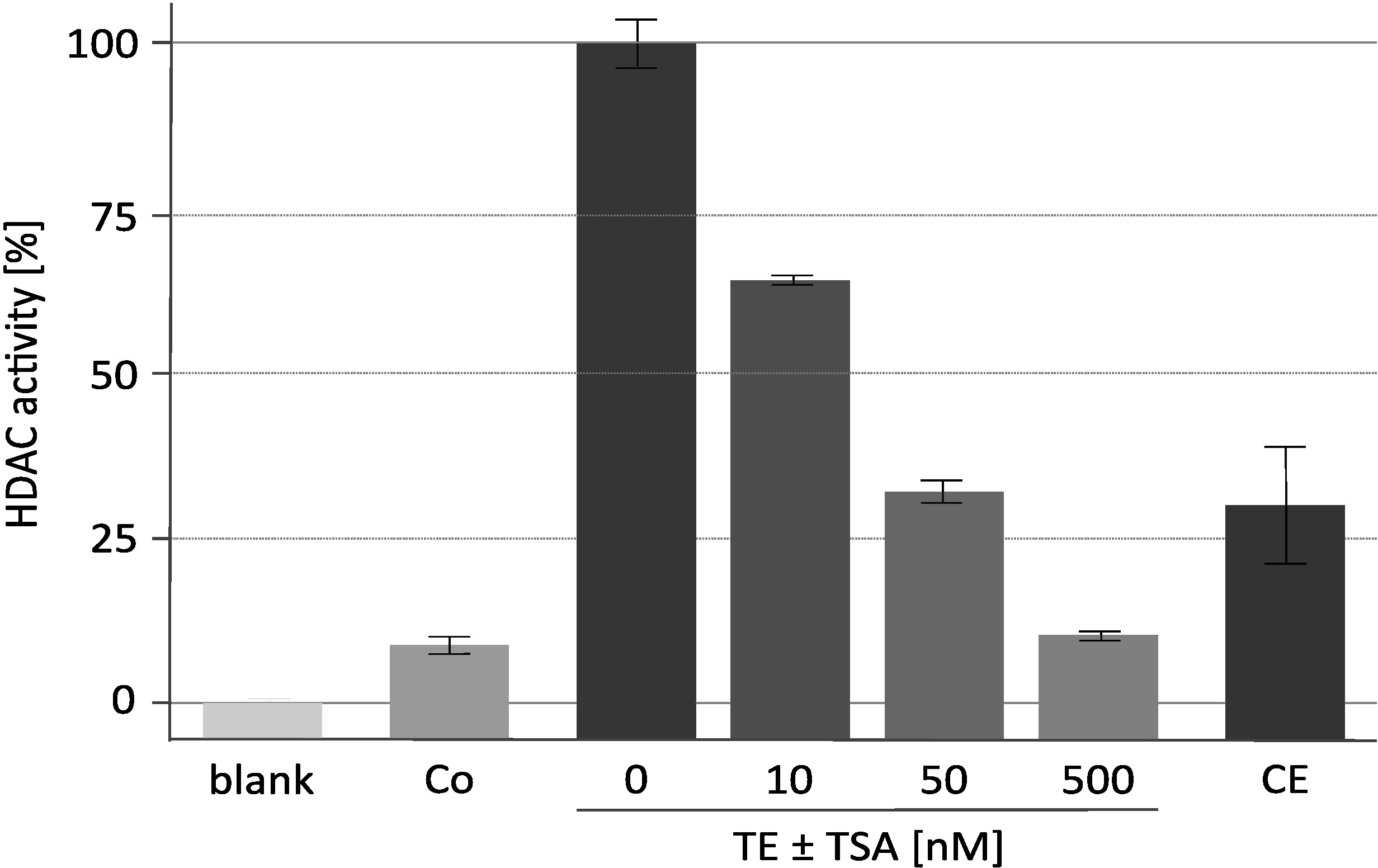
The results of a representative deacetylase activity assay with the specific HDAC inhibitor trichostatin A (TSA) are displayed in Figure 2. This assay was originally developed for plants18 and has also been used for inhibitor screening against mammalian deacetylases19,20. The histones used for the assay were labeled and prepared as described16. The effect of increasing concentrations of TSA on the catalytic activity of the enriched RpdA complex ("TE ± TSA") is shown. The sensitivity of the activity confirms that measured cpm values are indeed due to RpdA and not caused by unspecific protease activity. This is an important observation as it indicates that TEV, which is present at rather high concentration, does not interfere with the HDAC activity assay. In order to assign measured HDAC activity to RpdA, a wild-type strain was used as negative control ("Co"). As expected, only marginal HDAC activity (approx. 5-10% of RpdA-enriched fractions) was detected in the TEV eluate of the wild type. Interestingly, HDAC activity is significantly reduced after the second affinity purification step ("CE") when compared to the TEV eluate.

Figure 1. Tandem Affinity purificationof TAP-tagged RpdA. A silver-stained 10% SDS-polyacrylamide gel (A) and a western blot probed with the anti-CBP antibody (B) are displayed. Lane labeling and loaded volumes are as follows: "M": 2 µL of 1:10 diluted unstained protein marker (silver stain), 3.5 µL of prestained protein marker (western blot); "Ex": protein extract sample as prepared in step 2.2.8 (2 µL of 1:10 dilution, 5 µL); "FT": IgG resin flow-through sample as prepared in step 2.4.3 (2 µL of 1:10 dilution, 5 µL); "AE": acid eluate of step 2.4.14 (10 µL, 10 µL); "TE": TEV eluate of overnight elution by TEV cleavage, step 2.4.12 (10 µL, 10 µL); "CFT": calmodulin flow-through (20 µL, 20 µL); "CE": calmodulin eluate (10 µL, 10 µL). Size of selected marker proteins is indicated on the left side of the panels. The volumes given in parentheses correspond to sample loadings for silver stain and western blot, respectively. Asterisks in the silver-stained gel indicate the RpdA fusion protein. The immunoblot (B) was detected with alkaline phosphatase using the BCIP/NBT color development system. Please click here to view a larger version of this figure.
{kind=link}

Figure 2. HDAC activity assay under increasing concentrations of trichostatin A. Efficacy of RpdA inhibition was tested with 25 µL of affinity-purified recombinant RpdA ("TE") and 25 µL of 0, 10, 50, and 500 nM of the HDAC inhibitor TSA diluted in RPMI-1640 medium. RPMI was used to assess background activity ("blank"). Activities of the final eluate after the second calmodulin affinity step ("CE") and of an untagged strain after the first affinity purification step (negative control, "Co") are displayed. Activities are shown as percent of enriched RpdA without TSA (100 %, "TE"). Error bars indicate the standard deviation of three replicates. This figure has been modified from Bauer et al. 20162. Please click here to view a larger version of this figure.
{kind=link}
Discussion
This protocol describes a single-step enrichment of a TAP-tagged class 1 HDAC from the filamentous fungus A. nidulans for the assessment of in vitro deacetylase activity. The TAP tag was originally introduced in baker's yeast for the identification of protein-protein interaction partners of the tagged protein6. Subsequently, the tag was codon-optimized for its use in A. nidulans5. Here we provide a straight-forward step-by-step protocol for the application of the first affinity purification step of the TAP strategy for single-step enrichment of the class 1 HDAC RpdA. The second affinity purification step clearly increases the level of purification, which is particularly important for the identification of bait-interacting proteins. Nevertheless, just the first step is recommended for subsequent activity testing, since the lack of significant contamination after the first step was confirmed by a control experiment using a wild-type strain. Furthermore, eluted activity is considerably higher after single-step enrichment when compared to that of the full TAP. In addition to RpdA2, this protocol was also successfully used for purifying A. nidulans complexes of the second class 1 HDAC HosA21.
Due to our observations that fungal class 1 HDACs build up stable complexes4, we have succeeded to modify the protocol by Bayram et al. 201212 that represents the basis of this method. Nevertheless, some critical steps have to be mentioned. The preparation of highly concentrated protein extracts to ensure near-physiological conditions is critical for complex stability. Therefore, it is important to mind the given recommendations regarding the biomass/extraction buffer ratio. In this respect, it is also critical to use well ground fine mycelial powders to ensure proper extraction. Here, the use of a grinding machine is clearly advantageous. As mentioned in the protocol section, it is worth to try the TEV-cleavage step for 1-2 h at room temperature in order to speed up the purification. This was tested for RpdA without observing any deleterious effects on stability (unpublished observation, Bauer I, 2018). In addition, replacement of NP40 (used in the original protocol) with TX-100 might not be suitable for other protein complexes. When using this method for purification of other TAP-tagged proteins, one should also refer to the Bayram protocol, which contains a number of valuable hints that might be helpful for a sufficient purification of other protein complexes12.
Besides the here described TAP-method, other affinity tags and techniques are commonly used for single-step enrichment of protein of filamentous fungi, including His- and GFP-tags. However, as class 1 HDACs generally are functional as high-molecular-weight complexes, native elution conditions are a prerequisite for the enrichment of catalytically active HDACs. Importantly, many other affinity purifications are performed under unfavorable conditions. For instance, enrichment of HDACs via GFP-trap, which is based on antigen-antibody interaction, is not suitable due to the acidic elution conditions interfering with protein-protein interaction of HDAC complexes bound to resins. Moreover, attempts to purify His-tagged RpdA by metal chelate affinity chromatography22, resulted in a significant loss of catalytic activity during the purification procedure although imidazole instead of low-pH conditions was used for elution (unpublished data, Bauer, I, 2010).
One limitation of the described enzymatic assay protocol is the use of the radioactive substrate. However, assays on a fluorescent basis have been developed as well23,24 and are commercially available. These assays are performed in well-plates and thus are suitable also for high-throughput screening of HDAC inhibitors. In that case, an upscale of the presented procedure would be required.
Potential upcoming applications of this protocol include the enrichment of specific sub-complexes of class 1 HDACs to assess their specific physiological roles and/or differences in their susceptibility to HDAC inhibitors. When establishing the described method for other enzymes, it is strongly recommended to perform a negative control experiment with an untagged strain. This ensures specificity of measured enzyme activities and would reveal contamination by unspecifically bound enzymes.
Purification and activity assay described here can be performed within two days and the enriched enzymes are stable for at least several months, when stored in aliquots at -80 °C. In conclusion, this protocol provides a relatively simple and cost-effective way to achieve class 1 HDAC complexes for activity measurement and determination of inhibitor efficacy.
Acknowledgements
We would like to thank Petra Merschak, Division of Molecular Biology (Biocenter, Medical University of Innsbruck), for her help and support regarding this manuscript. Additionally, we would like to thank the reviewers for their valuable comments.
This work was funded by the Austrian Science Fund (P24803 to SG and P21087 to GB) and by intramural funding (MUI Start, ST201405031 to IB).
Materials
| Name | Company | Catalog Number | Comments |
| 10 x SDS-PAGE running buffer | Novex | ||
| 2-mercaptoethanol (EtSH) | Roth | 4227 | |
| 25 cm2 cell culture flasks with vent cap | Sarstedt | 833910002 | For spore production |
| 47 mm vacuum filtration unit | Roth | EYA7.1 | |
| AccuFLEX LSC-8000 | HITACHI | – | Scintillation counter |
| Acetic acid | Roth | 7332 | |
| Anti-Calmodulin Binding Protein Epitope Tag Antibody | Millipore | 07-482 | Used at 1:1333 dilution |
| Anti-Rabbit IgG (whole molecule)–Alkaline Phosphatase (AP) antibody | Sigma-Aldrich | A3687 | |
| Ball mill | Retsch | 207450001 | Mixer Mill MM 400 |
| BCIP/NBT | Promega | S3771 | Color development substrate for AP |
| Cell strainer | Greiner | 542040 | |
| Cheese cloth for harvesting mycelia | BioRen | H0028 | Topfentuch |
| Dimethylsulfoxid (DMSO) | Roth | 4720 | |
| EDTA | Prolabo | 20309.296 | |
| Ethyl acetate | Scharlau | Ac0155 | |
| Freeze Dryer | LABCONCO | 7400030 | FreeZone Triad |
| Glycerol | Roth | 3783 | |
| HCl | Roth | 4625 | |
| IgG resin | GE Healthcare | 17-0969-01 | IgG Sepharose 6 Fast Flow |
| Inoculation loops | VWR | 612-2498 | |
| KOH | Merck | 5033 | |
| Laminar flow cabinet | Thermo Scientific | – | Hera Safe KS |
| Mixed Cellulose Esters Membrane Filters | Millipore | GSWP04700 | |
| NaCl | Roth | 3957 | |
| NaOH | Roth | 6771 | |
| Neubauer counting chamber improved | Roth | T728 | |
| Novex gel system | Thermo Scientific | For SDS-PAGE | |
| Novex Tris-glycine SDS running buffer (10X) | Thermo Scientific | LC2675 | Running buffer for SDS-PAGE |
| peqGold protein-marker II | VWR | 27-2010P | Protein ladder used for silver stain |
| Peristaltic Pump P-1 | GE Healthcare | 18111091 | |
| Pipette controller | Brand | 26302 | accu-jet pro |
| Poly-Prep chromatography columns | Bio-Rad | 731-1550 | |
| ProSieve QuadColor protein marker | Biozym | 193837 | Prestained protein ladder used for western blot |
| Protease inhibitor cocktail tablets | Sigma-Aldrich | 11873580001 | cOmplete, EDTA-free |
| Rotary mixer | ELMI | – | Intelli-Mixer RM-2 S |
| Rotiszint eco plus | Roth | 0016 | |
| RPMI-1640 | Sigma-Aldrich | R6504 | |
| Scintillation vials | Greiner | 619080 | |
| Sorvall Lynx 4000 | Thermo Scientific | ||
| Thermomixer comfort | Eppendorf | ||
| TIB32.1 | A. nidulans rpdA::TAP strain. Genotype: alcA(p)::rpdA; veA1; argB2; yA2; pIB32::argB; ArgB+; PyrG+ | ||
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad | 1704271 | |
| Trans-Blot Turbo Transfer System | Bio-Rad | 1704150 | |
| Trichostatin A (TSA) | Sigma-Aldrich | T8552 | 5 mM stock in DMSO |
| Tris (free base) | Serva | 37190 | |
| Tris-HCl | Roth | 9090 | |
| Polysorbate 20 | Roth | 9127 | Tween 20 |
| Polysorbate 80 | Sigma-Aldrich | P1754 | Tween 80 |
| TX-100 | Acros Organics | 215682500 | Triton X-100, Octoxynol-9 detergent |
References
- Gregoretti, I. V., Lee, Y. -. M., Goodson, H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. Journal of Molecular Biology. 338 (1), 17-31 (2004).
- Bauer, I., et al. A Class 1 Histone Deacetylase with Potential as an Antifungal Target. mBio. 7 (6), (2016).
- Stefan, A., et al. Purification of active recombinant human histone deacetylase 1 (HDAC1) overexpressed in Escherichia coli. Biotechnology Letters. 40 (9-10), 1355-1363 (2018).
- Trojer, P., et al. Histone deacetylases in fungi: novel members, new facts. Nucleic Acids Research. 31 (14), 3971-3981 (2003).
- Bayram, &. #. 2. 1. 4. ;., et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 320 (5882), 1504-1506 (2008).
- Puig, O., et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 24 (3), 218-229 (2001).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
- Hill, T. W., Kafer, E. Improved protocols for Aspergillus minimal medium: trace element and minimal medium salt stock solutions. Fungal Genetics Reports. 48, (2001).
- Todd, R. B., Davis, M. A., Hynes, M. J. Genetic manipulation of Aspergillus nidulans: meiotic progeny for genetic analysis and strain construction. Nature Protocols. 2 (4), 811-821 (2007).
- Zadra, I., Abt, B., Parson, W., Haas, H. xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Applied and Environmental Microbiology. 66 (11), 4810-4816 (2000).
- Stolz, D. J., et al. Histological Quantification to Determine Lung Fungal Burden in Experimental Aspergillosis. Journal of Visualized Experiments. (133), (2018).
- Bayram, &. #. 2. 1. 4. ;., et al. Identification of protein complexes from filamentous fungi with tandem affinity purification. Methods in Molecular Biology. 944, 191-205 (2012).
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- Blum, H., Beier, H., Gross, H. J. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 8 (2), 93-99 (1987).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Biotechnology. 24, 145-149 (1992).
- Kölle, D., et al. Biochemical methods for analysis of histone deacetylases. Methods. 15 (4), 323-331 (1998).
- Tribus, M., et al. A novel motif in fungal class 1 histone deacetylases is essential for growth and development of Aspergillus. Molecular Biology of the Cell. 21 (2), 345-353 (2010).
- Sendra, R., Rodrigo, I., Salvador, M. L., Franco, L. Characterization of pea histone deacetylases. Plant Molecular Biology. 11 (6), 857-866 (1988).
- Valente, S., et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells. Journal of Medicinal Chemistry. 57 (14), 6259-6265 (2014).
- Mai, A., et al. Synthesis and biological properties of novel, uracil-containing histone deacetylase inhibitors. Journal of Medicinal Chemistry. 49 (20), 6046-6056 (2006).
- Pidroni, A., et al. A Class 1 Histone Deacetylase as Major Regulator of Secondary Metabolite Production in Aspergillus nidulans. Frontiers in Microbiology. 9, 2212 (2018).
- Porath, J., Carlsson, J., Olsson, I., Belfrage, G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 258 (5536), 598-599 (1975).
- Wegener, D., Wirsching, F., Riester, D., Schwienhorst, A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chemistry & Biology. 10 (1), 61-68 (2003).
- Peng, L., Yuan, Z., Seto, E. Histone deacetylase activity assay. Methods in Molecular Biology. 1288, 95-108 (2015).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved