Isolation and Purification of Murine Cardiac Pericytes
In This Article
Summary
We have optimized a protocol to isolate and purify murine cardiac pericytes for basic research and investigation of their biology and therapeutic potential.
Abstract
Pericytes, perivascular cells of microvessels and capillaries, are known to play a part in angiogenesis, vessel stabilization, and endothelial barrier integrity. However, their tissue-specific functions in the heart are not well understood. Moreover, there is currently no protocol utilizing readily accessible materials to isolate and purify pericytes of cardiac origin. Our protocol focuses on using the widely used mammalian model, the mouse, as our source of cells. Using the enzymatic digestion and mechanical dissociation of heart tissue, we obtained a crude cell mixture that was further purified by fluorescence activating cell sorting (FACS) by a plethora of markers. Because there is no single unequivocal marker for pericytes, we gated for cells that were CD31-CD34-CD45-CD140b+NG2+CD146+. Following purification, these primary cells were cultured and passaged multiple times without any changes in morphology and marker expression. With the ability to regularly obtain primary murine cardiac pericytes using our protocol, we hope to further understand the role of pericytes in cardiovascular physiology and their therapeutic potential.
Introduction
Perivascular cells known as pericytes surround the microvessels and capillaries of the vascular tree1,2. Physiologically, they are known to promote and play a part in angiogenesis, increase barrier integrity due to their close relationship with endothelial cells as well as stabilize and mature vessels1,2. Moreover, the dysfunction and/or loss of these cells have been implicated in diseases such as Alzheimer's disease2,3 and various cardiovascular diseases4. These cells are found throughout the entire body, but the cell numbers are tissue-dependent. Pericytes have most notably been studied in the brain due to high vascularization of the blood-brain barrier1,2. However, in the heart, the biology of pericytes is understudied.
Recently, there are increased interests in the field for cardiac pericytes, but there is currently no streamlined protocol available for their isolation from one of the most used tools in biology — the mouse. There are protocols in the literature on isolating pericytes from the brain5, retina6, placenta7, and skeletal muscle8,9; however, few protocols are on isolating pericytes from the heart. There are several groups that have isolated cardiac pericytes. Nees et al. were able to isolate an abundant amount of cardiac pericytes from multiple species including the mouse; however, their methods used specific in-house built equipment which decreases reproducibility10. Avolio et al.11, Chen et al.12, and Baily et al.13 also successfully isolated cardiac pericytes from human heart tissue, but human tissues are not always available and hard to obtain for some investigators. Here, we have developed an isolation method to obtain cardiac pericytes from mouse models for investigators to further study their biology with readily available materials.
Using enzymatic digestion and fluorescence activated cell sorting (FACS) with known key pericyte markers14, our protocol allows us to isolate and purify a population of pericytes that are characterized by CD31-CD34-CD45-CD140b+NG2+CD146+. Our panel of markers contain both inclusion and exclusion markers. CD45 is used as a marker to exclude hematopoietic cells. CD31 is used as a marker to exclude endothelial cells. CD34 is used as a marker to exclude both hematopoietic and endothelial progenitor cells. CD146 is a marker for perivascular cells. Lastly, NG2 and CD140b (also known as platelet derived growth factor receptor beta — PDGFRβ) are both accepted markers for pericytes14.The primary culture obtained can be cultured and passaged multiple times with no changes in morphology or marker expression. Furthermore, these cells can be co-cultured with endothelial cells to study their interactions and crosstalk with each other. This cell isolation method will allow investigators to study the biology and pathophysiology of cardiac pericytes from wild type, disease, and genetically variant mouse models.
Protocol
All animals were housed and used in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) accredited facility and all animal work was conducted under appropriate veterinary oversight and under the Institutional Animal Care and Use Committee (IACUC) approved protocol of Amgen Inc.
1. Preparation of Tools and Culture Media
- Autoclave surgical 9 cm straight tip fine point scissors and 10 cm angled serrated forceps.
- Add 25 mL of 5% fetal bovine serum (FBS) and 5 mL of 1% penicillin streptomycin (P/S) into a 500 mL bottle of calcium magnesium free Dulbecco's phosphate-buffered saline (CMF-DPBS). Place solution in an ice bath to ensure it will be cold at the time of use. Aliquot 50 mL into a 50 mL conical tube for heart isolation. Add in 250 units/mL of heparin sodium solution into the 50 mL aliquot. This will be referred to as heparinized CMF-DPBS.
- Add 20% FBS (100 mL) and 1% P/S (5 mL) into a 500 mL bottle of high glucose Dulbecco's modified eagle's medium (DMEM). This will be referred to as the enzyme-free culture media. Aliquot 20 mL of DMEM + 20% FBS + 1% P/S and add in 500 µg/mL of collagenase B. This will be referred to as the enzyme solution. Keep both the enzyme-free culture media and enzyme solution warm at 37 °C in an incubator or water bath.
2. Preparation of Animal and Procurement of Cardiac Tissue
- Intraperitoneally inject a mouse with 250 units of heparin sodium solution with a 31 G needle syringe. Then wait 10−15 min while the mouse remains active in its home cage.
NOTE: Representative data in this study were obtained from a 4 month-old male C57BL/6 mouse. However, this protocol can be used on any mouse regardless of strain, age, gender, weight, etc. - Anesthetize the mouse with 5% isoflurane. Check the anesthesia depth of the mouse by pinch reflex.
- Place the anesthetized mouse in supine position and tape down its forelimbs. Carefully open the chest cavity and cannulate the descending aorta using a 25 G butterfly needle.
- Make a nick in the right atrium and perfuse the heart with at least 20 mL of 250 units/mL heparinized CMF-DPBS at 2 mL/min with a variable-flow peristaltic pump. When the PBS comes out of the right atria clean, perfusion is complete.
- Cut the heart out at the aorta and place it into the ice-cold CMF-DPBS.
3. Dissociation of Heart Tissue
- Transfer the heart into a 15 cm x 15 cm Petri dish. Cut the heart into tiny pieces (1 mm/piece) using spring scissors and fine point forceps with enough enzyme solution to cover the pieces (10−15 mL).
- Transfer the pieces and solution into a 50 mL conical tube, seal with paraffin plastic film, and incubate at 37 °C on an orbital shaker at 120 rpm for 75 min.
- After collagenase digestion with the enzyme solution, decant the liquid through a 100 µm cell strainer into a new 50 mL tube but leave enough solution to make sure the pieces do not dry out.
- Using fine point forceps, take out the tissue from the tube and place a few pieces on a microscope slide. Then grind the tissue between two microscope slides to break up the tissue.Rinse the slides with enzyme free culture media into a new 50 mL conical tube.
- Repeat step 3.4 until all tissue pieces are dissociated.
- Combine the solutions from steps 3.3−3.5 into one tube. Strain the resulting suspension through a 100 µm cell strainer into a new 50 mL conical tube.
- Centrifuge at 220 x g, 4 °C, for 5 min. Aspirate off previous solution and gently resuspend cell pellet in fresh enzyme-free culture media.
- Count cells and check viability using a cell counter. Dilute cells to 1 x 106/mL with cold FACS staining buffer containing 500 mL of DPBS and 10−25 mL of 2−5% bovine serum albumin (BSA). The cells are ready to be stained and sorted.
4. Purification of Pericytes from Crude Cell Mixture Using FACS
- Prepare and label 5 mL FACS tubes for all controls and cell samples. Aliquot out cells (1 mL of cells per tube) for an unstained sample, fluorescence minus one (FMO) controls, and isotype-matched controls. Use the remaining cells for the sort. All controls and samples can be prepared and stained at the same time.
NOTE: A total of 13 mL at 0.5 x 106 cells/mL were used for the representative sort from one heart. However, the volume depends on how many cells the investigator obtains from their isolation, how many hearts they use, and how well the heart tissue is digested; the size of each heart is also a variable that can alter the volume.- Use compensation beads (Table of Materials) to optimize fluorescence compensation controls. Prepare one compensation control for each fluorochrome in the experiment in a labelled 5 mL FACS tube. For this experiment, prepare a total of 9 compensation controls — 2 kinds of unstained beads plus 7 different fluorochromes from the marker panel including NG2-FITC, CD31-APC, CD140b-PE, CD146-BV605, CD34-BV421, CD45-PE-Cy7, and cell viability-APC-Cy7 (Table of Materials).
- Add one drop of compensation beads (~ 50 µL) from the squeeze vial to each tube. Then add 1 µL of antibody to the beads. Repeat for each antibody from the marker panel. Mix vigorously by pulse-vortexing. Incubate for 30 min at 4 °C protected from light except for the cell viability beads which can be left at room temperature protected from light.
- Next, add 3 mL of FACS staining buffer to each tube and centrifuge at 300 x g for 5 min at 4 °C. Aspirate off solution and resuspend each bead pellet in 400 µL of FACS staining buffer. The compensation controls are ready to be used. Keep on ice.
- Use FMO controls to optimize background staining due to spectral overlap.
- Prepare FMO controls by using 1 mL of cells that was aliquoted from section 4.1 in a 5 mL FACS tube and adding in all antibodies from the marker panel described in step 4.1.1 at a 1:100 dilution but excluding one antibody. For example, prepare an NG2-AF488 FMO by including antibodies for CD31-APC, CD140b-PE, CD146-BV605, CD34-BV421, CD45-PE-Cy7, cell viability dye but not the NG2-AF488 antibody. Mix gently by pulse-vortexing. Repeat for each antibody for a total of 7 controls. Incubate for 30 min at 4 °C protected from light.
- Next, add 3 mL of FACS staining buffer to each tube and centrifuge at 300 x g for 5 min at 4 °C. Aspirate off solution and resuspend each cell pellet in 400 µL of FACS staining buffer. The FMO controls are ready to be used. Keep on ice.
- Use isotype-matched control antibodies (Table of Materials) for nonspecific staining.
- Prepare isotype controls by adding the isotype-matched control antibody (Table of Materials) to 1 mL of cell sample prepared from section 4.1 at a 1:100 dilution each in a 5 mL FACS tube. Mix gently by pulse-vortexing. Incubate for 30 min at 4 °C protected from light.
- Next, add 3 mL of FACS staining buffer to each tube and centrifuge at 300 x g for 5 min at 4 °C. Aspirate off solution and resuspend each cell pellet in 400 µL of FACS staining buffer. The isotype controls are ready to be used. Keep on ice.
- Prepare cells to be sorted by adding antibody cocktail to freshly isolated cells.
- Prepare cell sample from section 4.1 by adding in an antibody cocktail containing anti-mouse NG2-AF488, CD31-APC, CD140b-PE, CD146-BV605, CD34-BV421, CD45-PE-Cy7 at 1:100 dilution each and cell viability dye at 1:1,000 dilution. Gently vortex to mix. Incubate samples at 4 °C for 30 min protected from light.
- After staining, wash cells with FACS staining buffer by centrifugation at 300 x g for 5 min 4 °C. Aspirate off solution and resuspend the cell pellet in FACS staining buffer to 0.5 x 106 cells/mL.
- Using new FACS tubes that have 35 µm filter tops, pipette stained cell samples onto the lids and gravity filtrate to obtain single cell suspensions. Keep on ice.
- Use compensation beads (Table of Materials) to optimize fluorescence compensation controls. Prepare one compensation control for each fluorochrome in the experiment in a labelled 5 mL FACS tube. For this experiment, prepare a total of 9 compensation controls — 2 kinds of unstained beads plus 7 different fluorochromes from the marker panel including NG2-FITC, CD31-APC, CD140b-PE, CD146-BV605, CD34-BV421, CD45-PE-Cy7, and cell viability-APC-Cy7 (Table of Materials).
- Use a cell sorter to purify cells.
- Run the unstained cells on the cell sorter to set voltages and correct for the background signal (for example, set voltages for forward scatter to 490−560 and for side scatter to 180−250).
- Run each single-color compensation bead sample one at a time to adjust voltages for each channel and adjust gates for the positive signal. Collect data. Use the software to calculate for spectral overlap by calculating the compensation matrix. All voltages are ready and set.
- Run each isotype control one at a time and this data can be used to adjust gates for nonspecific binding if there are any.
- Run each FMO sample one at time and adjust voltages for each channel to correct for spectral bleed through due to a multi-color panel.
- Run the stained cell samples in the cell sorter and collect cells in 10 mL enzyme-free culture media (DMEM + 20% FBS + 1% P/S) in a 15 mL conical collection tube. Use the following gating strategy: gate for single cells, gate for live cells, gate for CD45 negative cells, gate for CD34 and CD31 negative cells, gate for NG2 positive cells, and finally gate for CD146 and CD140b positive cells.
5. Culturing of Pericytes
- Coat a 24-well plate with 0.2% gelatin for 5 min and aspirate off gelatin solution. Seed freshly obtained cells from step 4.2.5 in DMEM + 20% FBS + 1% P/S up to 2 x 104 cells/cm2. Culture cells in a cell incubator set at 37 °C, 5% CO2 and 95% O2.
- Passaging of pericytes
- Once the cells are 95% confluent, wash cells with warm 1x DPBS, and lift cells with 200 µL of 0.1% trypsin in each well at room temperature for 3−5 min.
- Gently tap the plate to loosen the cells.
- Neutralize the trypsin with 3.5x the amount of culture media (700 µL DMEM + 20% FBS + 1% P/S) and seed passage two (P2) cells onto an uncoated 6-well plate at to 2 x 104 cells/cm2.
- Each well, when confluent, can be moved into a single T-75 flask as P3 cells which then can be split at a 1:6 ratio.
6. Characterization of Pericytes
- Flow cytometry analysis
- Use the same FACS staining protocol and gating strategy as previously described in section 4.
- Run controls and stained samples on the flow cytometer. Collect data and analyze data using the analysis software (Table of Materials).
- To collect brightfield images, grow cells in a flask in a cell incubator set at 37 °C, 5% CO2 and 95% O2. Capture images on a microscope after cells attach to the surface.
- Immunocytochemistry
- Grow cells in a 96-well plate until 90% confluent. Wash cells with warm 1x DPBS and fix with 4% paraformaldehyde for 30 min at room temperature.
- Wash cells 3x with 1x DPBS and permeabilize with 0.1% detergent for 10 min at room temperature.
- Incubate cells with blocking buffer for 1 h at room temperature. After blocking, add primary antibodies (one antibody per well) diluted 1:100 in blocking buffer and incubate at 4 °C overnight. Primary antibodies are: anti-NG2, anti-CD140b, anti-CD31, anti-vimentin, anti-desmin, and anti-alpha smooth muscle actin.
- Next day, wash cells 3x with wash buffer (Table of Materials). Add secondary antibody diluted 1:1,000 in blocking buffer and incubate for 2 h at room temperature in the dark. Secondary antibody is an anti-rabbit conjugated to FITC.
- Wash cells 3x with wash buffer. Add 300 µM nuclear stain diluted at 1:1,000 for 5 min at room temperature.
- Wash cells 3x with 1x DPBS and mount with mounting media.
- Image cells with a confocal microscope.
Representative Results
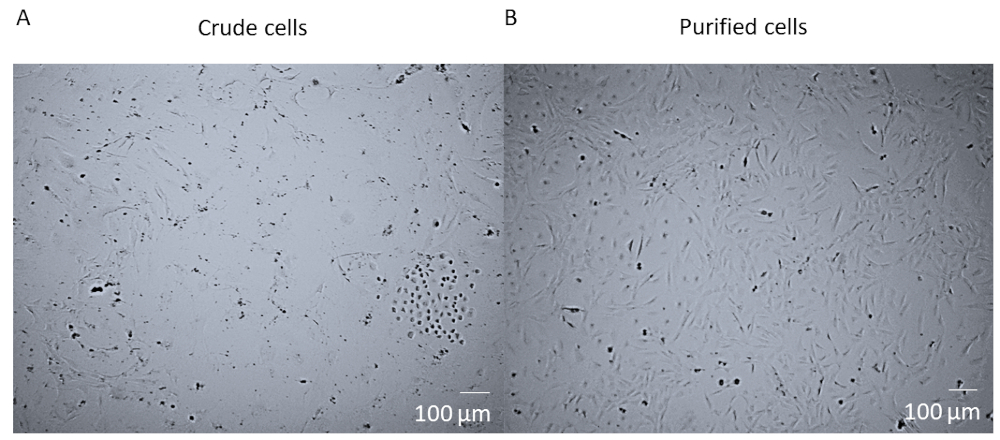
After enzymatic digestion and dissociation of the whole heart and before FACS purification of the cells, cells are a crude mixture that contains many different cell types from the heart (Figure 1A). After FACS purification and culturing, cells are homogenous. They are single nucleated, quite flat, and have the typical pericyte rhomboid morphology (Figure 1B).
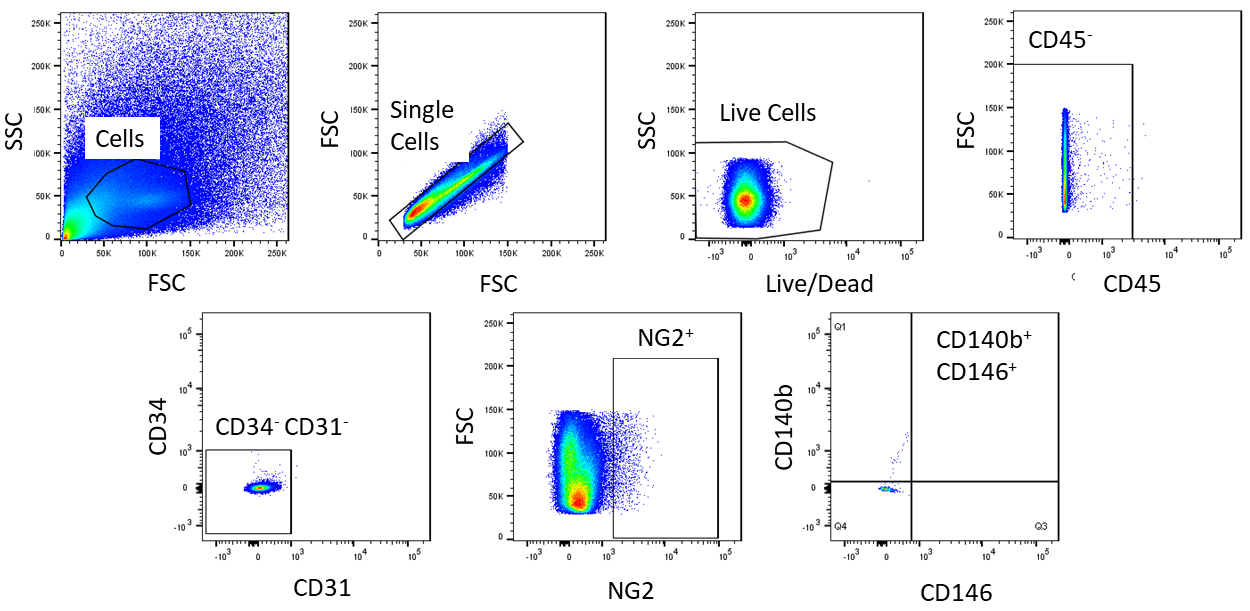
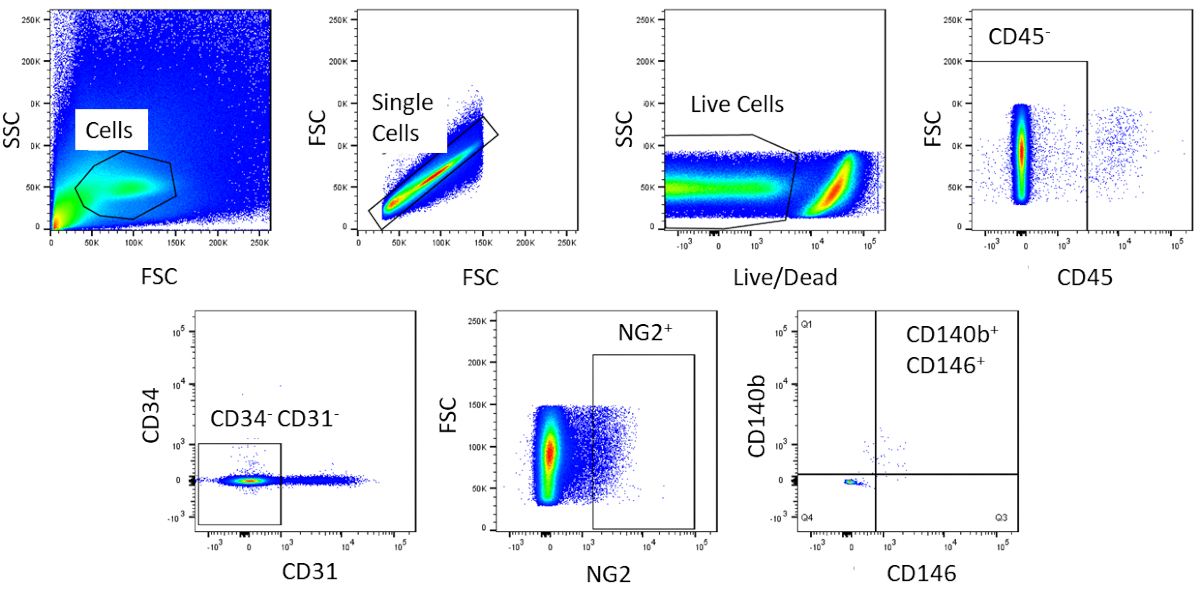
Using FACS, cells are purified to homogeny. The unstained control cell sample is used to show the gating strategy (Figure 2). First, debris and doublets were gated out based on forward and side scatter distributions. Then dead cells were gated out due to their amine reaction with the dye which produces a signal greater and more intense than live cells. Of the live cells, hematopoietic cells were gated out by being CD45+. To further remove hematopoietic and endothelial cells, CD34+ and CD31+ cells were gated out. Finally, NG2+ and CD140b+/CD146+ cells were selected for being perivascular cells with expression of typical pericyte markers (Figure 3). The marker panel was also tested on mouse coronary endothelial cells as a control (Supplemental Figure 1). Only about 1% of crude cell mixture consisted of pericytes after sorting.
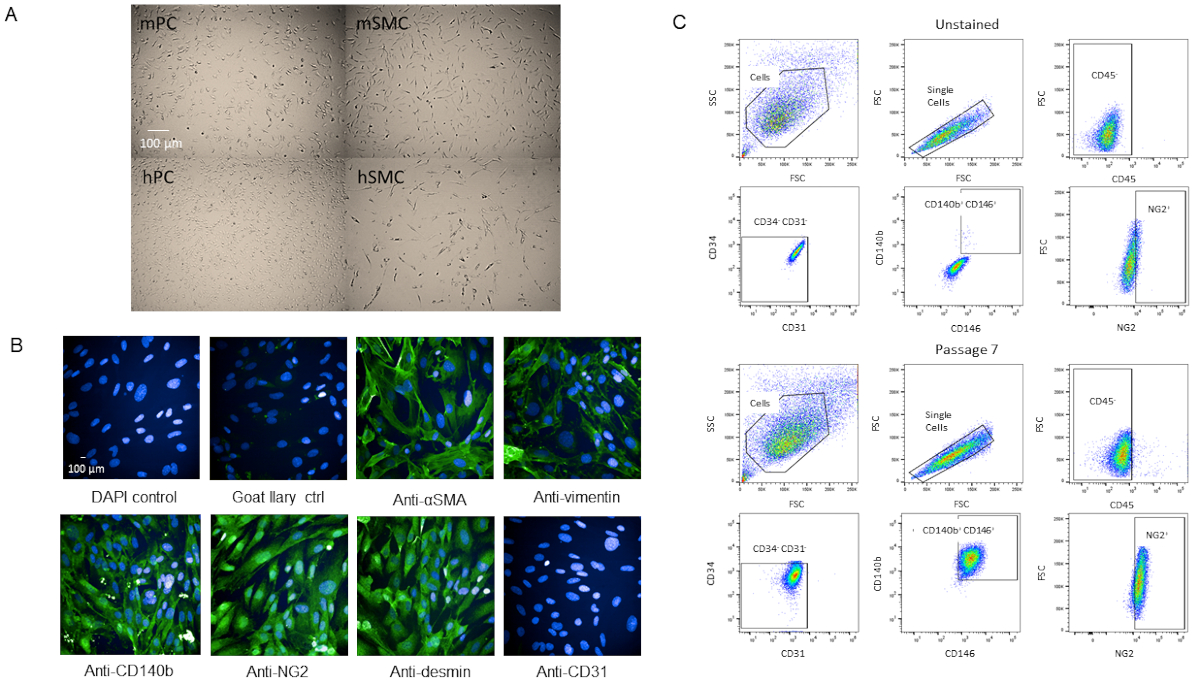
To validate that the cells were indeed pericytes, we passaged the cells for further characterization. Cells grew rapidly once they reached P3 in the T-75 flasks without changes in viability as they became older (Supplemental Figure 2). When compared with human brain pericytes, the cells had a similar morphology (Figure 4A). When compared with mouse and human smooth muscle cells, the cells had a different morphology (Figure 4A). There were also no observed changes in morphology or marker expression at P7 when immunostained or by flow cytometry analysis after passaging (Figure 4B,C).

Figure 1: Crude cells versus purified cells. (A) The brightfield image of crude cell mixture post whole heart enzymatic digestion and dissociation which has been cultured in a T25 flask for 14 days. (B) The brightfield image of a homogenous population of cardiac pericytes post-sorting and culturing after 14 days. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative images of FACS analysis of unstained cells. Schematic representation of the gating strategy used to purify crude cell mixture. Gate for cells that are single, live, CD45-, CD31-, CD34-, NG2+, CD146+, and CD140b+. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Representative images of FACS analysis of crude cells. Schematic representation of the sorting used to obtain a homogenous population of cardiac pericytes. Roughly 1% of crude cells are CD31-CD34-CD45-CD140b+NG2+CD146+. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Characterization of primary isolated cardiac pericytes (A) Brightfield images of cultured cells from human brain (hPC) and mouse hearts (mPC) show similar pericyte cell morphology but different morphology from human smooth muscle cells hSMC) and mouse smooth muscle cells (mSMC). Scale bar = 100 µm. (B) Phenotypic characterization of cells at P7 by immunocytochemistry for pericyte markers. Scale bar = 100 µm. (C) Analysis by flow cytometry of the pericytes at P7 where they were gated for negative markers CD31, CD34, CD45 and positive markers NG2, CD140b, and CD146. Population remains homogenous. Please click here to view a larger version of this figure.
{kind=link}
Supplemental Figure 1: Representative images of flow cytometry analysis of endothelial cells using marker panel. A mouse coronary endothelial cell line was used as control for the binding specificity for the markers. Using the same gating strategy that was used in the sort except for a positive gate for CD31 instead of a negative gate, the endothelial cells were negative for CD45, CD34, NG2, CD140b, and CD146 but positive for CD31 as expected. Please click here to download this figure.
Supplemental Figure 2: Representative images of flow cytometry analysis of different passages of mPC. Primary isolated cardiac pericytes were cultured and passaged up to passage 12. Cells were stained with propidium iodide and analyzed on a flow cytometer. Control population is a mixture of dead cells and live cells. There were no significant differences in number of viable cells between passages. Please click here to download this figure.
Discussion
As studies on cardiac pericytes are relatively new, the role of pericytes in cardiovascular physiology and pathophysiology have yet to be defined. In other organs, they have been shown to play key roles in vessel homeostasis and perfusion1,2. Compared to the literature of pericytes from other organs such as the brain, there are significantly fewer publications on cardiac pericytes. The isolation of cardiac pericytes is critical to the understanding of their functional characteristics and signaling mechanisms. Therefore, this protocol will provide investigators with an easier way to access cardiac pericytes from a more readily available tissue source and promote studies on their biology. It will help answer questions on how cardiac pericytes contribute to cardiac homeostasis and pathophysiology as well as investigate their therapeutic potential.
The pericyte population isolated from murine heart and characterized by CD31-CD34-CD45-CD140b+NG2+CD146+ has been passaged multiple times (up to P12 and was still going strong), which does not decrease in viability and propagates quickly (Supplementary Figure 2). The cells have also been cryofrozen and recovered with at least 95% viability. However, we prefer to use cells P7 or younger for our experiments. Comparing brightfield images of our pericytes with human brain pericytes, the two cell lines have comparable cell morphology (Figure 4A) while they differ in morphology from smooth muscle cells (Figure 4A). Our P7 cells were characterized by immunocytochemistry for pericyte markers, some from our FACS panel (NG2 and CD140b), and a few not in the panel (vimentin, desmin, αSMA) and we found that the cells expressed pericyte markers homogenously (Figure 4B). Additionally, our P7 cells were analyzed by flow cytometry again with the same marker panel to assess for changes in marker expression due to passaging and we found that there were no changes (Figure 4C). Therefore, both phenotypically and morphologically, our cells are pericytes.
The studies by Nees et al.10, Avolio et al.11, Chen et al.12, and Baily et al.13 have shown successful cardiac pericyte isolations. However, the use of an in-house custom built equipment to detach the pericytes from the microvessels by Nees et al.10 involved two chambers with pumps that perfused protease solution back and forth through a mesh net stack, which was hard to replicate as they did not provide a schematic and/or picture of the apparatus and how it was built. Although Nees et al.10 successfully isolated cardiac pericytes from many species, we were never able to reproduce their method. Our pericyte detachment step in our protocol simply uses an orbital shaker (to dissociate all cells) which is available in most, if not all laboratories, with the tissue and enzyme solution in a conical tube followed by a mechanical dissociation step. There is no custom apparatus required. Secondly, the remaining protocols involve the use of human tissues and thus the procurement of human tissue is limiting to investigators. Our protocol is a modification and optimization of current protocols9,12 using mouse models (wild type, genetically modified, diseased) and materials that are readily available to all investigators.
Because perivascular cells in general are sensitive, viability of the cells is critical to obtain a good yield. During procurement of cardiac tissue and staining of cells, the tissue/cells need to be kept ice cold. Secondly, the enzymatic digestion of the tissue may require optimization on an individual basis. Depending on the units of activity on one's vials of enzymes, concentration and digestion time may need to be optimized. Make sure that the enzymatic solution is prepared fresh each time otherwise yield will decrease. Thirdly, the crude mixture contains a lot of cells, some dead and/or dying, it is best to lower the concentration of FBS in the staining buffer from 5% to 2%. If you are having trouble with cells clogging the nozzle during sort, enrich the cells first by using a dead cell removal kit. You can also add EDTA/HEPES buffer or DNase treatment to the cell pre-sort to prevent cell clumping. Lastly, because our panel of antibodies is rather large and uses many fluorophores, be sure your FMO controls and compensation controls are done correctly.
One limitation to this method is the amount of cardiac pericytes that can be obtained per heart. In our case, only 1.1% of our crude mixture from one mouse heart were pericytes which is comparable to the percent in the human heart isolations, but the number of cells is significantly less due to the amount of heart tissue a mouse provides. Because the starting number of cells is so low after FACS, it would be better to isolate from multiple hearts at once. However, the problem with that is the sheer number of cells that you need to sort through in one day. If you have more than 30 million cells, it will be difficult to get through the sort without affecting the viability of the cells. If the investigator had multiple cell sorters, isolating from multiple hearts in a day would be doable. Another limitation is that because we do not know if there are subpopulations of pericytes in the heart like there is skeletal muscle15,16, we do not know if we are eliminating a subtype in our gating strategy. We are in the process of characterizing our cardiac pericytes and thus far in our unpublished data, they are functionally like other pericytes in the literature.
Our protocol will enable investigators to answer questions on cardiac pericyte properties, characteristics, functionality, and other aspects that will help define their contribution to cardiac homeostasis and hemodynamics. These cells could have therapeutic potential to cardiovascular disease once their biology is better understood.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank the Amgen Flow Cytometry Core for their help with fluorophore panel design, troubleshooting, and cell sorting.
Materials
| Name | Company | Catalog Number | Comments |
| 0.25% Trypsin-EDTA | Corning | 25-053-Cl | dilute with 1x DPBS to get 0.1% |
| 100 μM Cell strainer | FisherSci | 22363549 | |
| 15 mL Falcon conical tubes | BD | 352096 | |
| 24-well plate | Corning | CLS3527 | |
| 25 gauge butterfly needle | FisherSci | 22-253-146 | |
| 31 gauge needle syringe | FisherSci | B328446 | |
| 50 mL Falcon conical tubes | BD | 352098 | |
| 6-well plate | Corning | CLS3516 | |
| anti-alpha smooth muscle actin rabbit mAb | abcam | ab32575 | Antibody used in ICC 1:100 dilution |
| anti-CD140b rabbit mAb | Cell Signaling | 28E1 | Antibody used in ICC 1:100 dilution |
| anti-CD31 rabbit pAb | abcam | ab28364 | Antibody used in ICC 1:100 dilution |
| anti-desmin rabbit pAb | abcam | ab8592 | Antibody used in ICC 1:100 dilution |
| anti-NG2 conjugated to AF488 | Millipore | MAB5384A4 | Antibody used in ICC 1:100 dilution |
| anti-vimentin rabbit mAb | abcam | ab92547 | Antibody used in ICC 1:100 dilution |
| ArC Amine Reactive Compensation bead kit | Invitrogen | A10346 | compensation beads for Live/Dead Near IR dye |
| Brightfield Microscope | camera attached | ||
| CD140b-PE (clone APB5) | eBioscience | 12-1402-81 | Antibody used in FACS 1:100 dilution |
| CD146-BV605 (clone ME-9F1) | BD | 740434 | Antibody used in FACS 1:100 dilution |
| CD31-APC (clone MEC 13.3) | BD | 551262 | Antibody used in FACS 1:100 dilution |
| CD34-BV421 (clone RAM 34) | BD | 56268 | Antibody used in FACS 1:100 dilution |
| CD45-PE-Cy7 (clone 30-F11) | BD | 552848 | Antibody used in FACS 1:100 dilution |
| Centrifuge | eppendorf | ||
| Collagenase B | Roche | 11088815001 | 0.226 U/mg lyo. |
| Confocal Microscope | |||
| DAPI | ThermoFisher | D1306 | nuclear stain |
| DMEM with 4.5 g/L glucose, L-glutamine & sodium pyruvate | Corning | 10-013-CV | 500 mL |
| Dowell scissors | FST | 15040-11 | |
| Dulbecco's Phosphate-Buffered Saline (DPBS) | Corning | 21-030-CV | 500 mL |
| Dulbecco's Phosphate-Buffered Saline without Ca and Mg (CMF-DPBS) | Corning | 21-031-CV | 500 mL |
| Dumont #5 Fine Forceps | FST | 11254-20 | |
| FACSAria cell sorter | BD | Lasers: 405 nm 50 mW, 488 nm 100 mW, 561 nm 50mW, 633 nm 11 mW | |
| FACSAria software | BD | ||
| Falcon tube round-bottom polypropylene, 5 mL | BD | 38057 | |
| Falcon tube with cell strainer cap, 5 mL | BD | 08-771-23 | |
| Fetal Bovine Serum | Corning | 35-015-CV | 500 mL |
| Fine scissors | FST | 14060-09 | |
| FlowJo software | FlowJo LLC | ||
| Fortessa LSR flow cytometer | BD | Lasers: 405 nm 50 mW, 488 nm 100 mW, 561 nm 50mW, 633 nm 11 mW | |
| Gelatin-based coating | Cell Biologics | 6950 | |
| Goat anti-rabbit IgG (H+L) Cross-Absorbed Secondary antibody, Alexa Fluor 488 | Invitrogen | A-11008 | Antibody used in ICC 1:1000 dilution |
| Graefe Forceps | FST | 11049-10 | |
| Heparin sodium solution | Hospira | NDC 0409-2720-02 | 10,000 USP units/10 mL; from porcine intestines |
| Incubator | set at 37 °C, 5% CO2, 95% O2 | ||
| Live/Dead-Near IR | Life Technologies | L10119 | |
| Microscope slides | FisherSci | 12-550-343 | |
| NG2-FITC | Millipore | AB5320A4 | Antibody used in FACS 1:100 dilution |
| Oribital shaker | VWR | Inside 37 °C incubator or room | |
| Paraformaldehyde | FisherSci | 50-980-487 | dilute with 1x DPBS to get 4% |
| Penicillin-Streptomycin | Corning | 30-002-CI | |
| Petri dish | FisherSci | FB0875714 | |
| Pipette and tips | |||
| ProLong Diamond | ThermoFisher | P36965 | mounting media |
| Propidum Iodide | ThermoFisher | cell viability dye for supplemental figure 2 | |
| Rabbit IgG FITC | eBiosciences | 11-4614-80 | Isotype control antibody - FITC |
| Rat IgG2a APC | Biolegend | 400512 | Isotype control antibody - APC |
| Rat IgG2a BV421 | Biolegend | 400536 | Isotype control antibody - BV421 |
| Rat IgG2a BV605 | BD | 563144 | Isotype control antibody - BV605 |
| Rat IgG2a PE | Biolegend | 400308 | Isotype control antibody - PE |
| Rat IgG2b PE-Cy7 | Biolegend | 400617 | Isotype control antibody - PE-Cy7 |
| SuperBlock | ThermoFisher | 37515 | blocking buffer |
| T75 | ThermoFisher | 156499 | |
| Triton X-100 | Sigma | X100 | detergent, dilute with x DPBS to get 0.1% |
| UltraComp beads | Invitrogen | 01-2222-42 | compensation beads |
| Variable-Flow Peristaltic Pump | FisherSci | 13-876-1 | |
| ViCell Cell counter | Beckman | ||
| Wash buffer | 1:10 dilution of Superblock in 1x DPBS |
References
- Armulik, A., Abramsson, A., Betsholtz, C. Endothelial/pericyte interactions. Circulation Research. 97 (6), 512-523 (2005).
- Armulik, A., Genove, G., Betsholtz, C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell. 21 (2), 193-215 (2011).
- Sengillo, J. D., et al. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathology. 23 (3), 303-310 (2013).
- Avolio, E., Madeddu, P. Discovering cardiac pericyte biology: From physiopathological mechanisms to potential therapeutic applications in ischemic heart disease. Vascular Pharmacology. 86, 53-63 (2016).
- Dore-Duffy, P. Isolation and characterization of cerebral microvascular pericytes. Methods in Molecular Medicine. 89, 375-382 (2003).
- Bryan, B. A., D'Amore, P. A. Pericyte isolation and use in endothelial/pericyte coculture models. Methods in Enzymology. 443, 315-331 (2008).
- Maier, C. L., Shepherd, B. R., Yi, T., Pober, J. S. Explant outgrowth, propagation and characterization of human pericytes. Microcirculation. 17 (5), 367-380 (2010).
- Crisan, M., Corselli, M., Chen, W. C., Peault, B. Perivascular cells for regenerative medicine. Journal of Cellular Molecular Medicine. 16 (12), 2851-2860 (2012).
- Crisan, M., et al. Purification and long-term culture of multipotent progenitor cells affiliated with the walls of human blood vessels: myoendothelial cells and pericytes. Methods in Cellular Biology. 86, 295-309 (2008).
- Nees, S., et al. Isolation, bulk cultivation, and characterization of coronary microvascular pericytes: the second most frequent myocardial cell type in vitro. American Journal of Physiology Heart Circulatory Physiology. 302 (1), H69-H84 (2012).
- Avolio, E., et al. Expansion and characterization of neonatal cardiac pericytes provides a novel cellular option for tissue engineering in congenital heart disease. Journal of the American Heart Association. 4 (6), e002043 (2015).
- Chen, W. C., et al. Human myocardial pericytes: multipotent mesodermal precursors exhibiting cardiac specificity. Stem Cells. 33 (2), 557-573 (2015).
- Baily, J. E., et al. Isolation of Perivascular Multipotent Precursor Cell Populations from Human Cardiac Tissue. Journal of Visualized Experiments. (116), e54252 (2016).
- Murray, I. R., et al. Skeletal and cardiac muscle pericytes: Functions and therapeutic potential. Pharmacology & Therapeutics. 171, 65-74 (2017).
- Birbrair, A., et al. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem Cells Development. 22 (16), 2298-2314 (2013).
- Birbrair, A., et al. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Research. 10 (1), 67-84 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved