Transforming, GenomEditing und Phenotypisierung der Stickstoff-fixierenden tropischen Cannabaceae Baum Parasponia andersonii
In This Article
Summary
Parasponia andersonii ist ein schnell wachsender tropischer Baum, der zur Cannabis-Familie (Cannabaceae) gehört und in Verbindung mit dem Rhizobium stickstofffixende Wurzelknollen bilden kann. Hier beschreiben wir ein detailliertes Protokoll für reverse genetische Analysen in P. andersonii basierend auf Agrobacterium tumefaciens-vermittelte stabile Transformation und CRISPR/Cas9-basierte Genombearbeitung.
Abstract
Parasponia andersonii ist ein tropischer Baum, der zur Familie der Cannabis (Cannabaceae) gehört. Zusammen mit 4 weiteren Arten bildet es die einzige bekannte nicht-legume Linie, die in der Lage ist, eine stickstofffixende Knötchensymbiose mit Rhizobium herzustellen. Vergleichende Studien zwischen Hülsenfrüchten und P. andersonii könnten wertvolle Einblicke in die genetischen Netzwerke liefern, die der Wurzelknötbildung zugrunde liegen. Um vergleichende Studien zu erleichtern, haben wir vor kurzem das Genom von P. andersonii sequenziert und Agrobacterium tumefaciens- vermittelte stabile Transformation und CRISPR/Cas9-basierte Genombearbeitung etabliert. Hier finden Sie eine detaillierte Beschreibung der Transformations- und Genombearbeitungsverfahren, die für P. andersoniientwickelt wurden. Darüber hinaus beschreiben wir Verfahren zur Keimung und Charakterisierung symbiotischer Phänotypen. Mit diesem Protokoll können stabile transgene mutierte Leitungen in einem Zeitraum von 2-3 Monaten erzeugt werden. Die vegetative In-vitro-Vermehrung von Transgenlinien T0 ermöglicht die Einleitung von Phänotypisierungsexperimenten 4 Monate nach der Kokultivierung von A. tumefaciens. Daher dauert dieses Protokoll nur geringfügig länger als die transiente Agrobacterium rhizogenes-basierteWurzeltransformationsmethode, die für P. andersoniiverfügbar ist, obwohl es mehrere klare Vorteile bietet. Zusammen erlauben die hier beschriebenen Verfahren P. andersonii als Forschungsmodell für Studien zum Verständnis symbiotischer Assoziationen sowie potenziell anderer Aspekte der Biologie dieses tropischen Baumes zu verwenden.
Introduction
Parasponia andersonii ist ein tropischer Baum aus der Familie der Cannabis (Cannabaceae) und stammt aus Papua-Neuguinea und mehreren Pazifischen Inseln1,2,3. Zusammen mit 4 zusätzlichen Parasponia-Arten stellt sie die einzige nicht-legume-Linie dar, die eine stickstofffixende Knötchensymbiose mit Rhizobia herstellen kann. Diese Symbiose ist gut in den Hülsenfrüchte (Fabaceae) Modellen Medicago truncatula und Lotus japonicusuntersucht, was zu detaillierten Kenntnissen über die molekulargenetische Natur der Knötselbildung undFunktionsweise4 geführt hat. Darüber hinaus wurde gezeigt, dass die Wurzelknötsymbiose in Hülsenfrüchten auf der viel älteren und weit verbreiteten arbuskulären Mykorrhizasymbiose5beruht. Phylogenomische Vergleiche deuten darauf hin, dass die stickstofffixierenden Knötchensymbiosen von Hülsenfrüchte, Parasponia, sowie die sogenannten actinorhizal Pflanzenarten, die diazotrophe Frankienbakterien beherbergen, einen gemeinsamen evolutionären Ursprung haben. 6,7,8. Um festzustellen, ob die Gene, die als an der Legume-Knötichbildung beteiligt identifiziert werden, Teil einer konservierten genetischen Grundlage sind, sind Studien an Nicht-Legume-Arten unerlässlich. Zu diesem Zweck schlagen wir vor, P. andersonii als vergleichendes Forschungsmodell neben Hülsenfrüchten zu verwenden, um die genetischen Kernnetzwerke zu identifizieren, die der Wurzelknötbildung und -funktion zugrunde liegen.
P. andersonii ist ein Pionier, der an den Hängen vulkanischer Hügel zu finden ist. Es kann Wachstumsgeschwindigkeiten von 45 cm pro Monat erfüllen und Längen von bis zu 10 Metern9erreichen. P. andersonii Bäume sind windbefruchtet, was durch die Bildung von getrennten männlichen und weiblichen Blüten erleichtert wird3,10. Wir haben vor kurzem das diploide Genom (2n = 20; 560 Mb/1C) von P. andersoniisequenziert und mit Anmerkungen bezeichnet und Entwürfe von Genomsequenzen von 2 zusätzlichen Parasponia-Arten zusammengestellt; P. rigida und P. rugosa6. Dies ergab 35.000 P. andersonii Genmodelle, die in >20.000 Orthogruppen zusammen mit Genen aus M. truncatula, Sojabohnen (Glycine max), Arabidopsis thaliana, Wald Erdbeere ( gruppiert werden können Fragaria vesca), Trema orientalis, schwarze Baumwollpappel (Populus trichocarpa) und Eukalyptus (Eukalyptus grandis)6. Zusätzlich identifizierten Transkriptomvergleiche zwischen M. truncatula und P. andersonii eine Reihe von 290 vermeintlichen Orthologen, die ein knöchernverstärktes Expressionsmuster bei beiden Arten anzeigen6. Dies bietet eine ausgezeichnete Ressource für vergleichende Studien.
Um die Genfunktion in P. andersonii Wurzeln und Knötchen zu untersuchen, wurde ein Protokoll für Agrobacterium rhizogenes-vermittelte Wurzeltransformation 11 etabliert. Mit diesem Protokoll können Verbundpflanzen mit transgenen Wurzeln in relativ kurzer Zeit erzeugt werden. Diese Methode ist auch in der Hülsenfrüchte-Symbiose-Forschung weit verbreitet12,13,14. Der Nachteil dieser Methode besteht jedoch darin, dass nur Wurzeln transformiert werden und dass jede transgene Wurzel ein unabhängiges Transformationsereignis darstellt, was zu erheblichen Variationen führt. Außerdem ist die Transformation transient und transgene Linien können nicht aufrechterhalten werden. Dadurch ist die A. rhizogenes-basierteWurzeltransformation weniger geeignet für die CRISPR/Cas9-vermittelte Genombearbeitung. Zusätzlich überträgt A. rhizogenes seine Wurzel induzierenden Lokus -rol) Gene auf das Pflanzengenom, das einmal mit dem Hormon Homöostase15stören. Dies macht das Studium der Rolle von Pflanzenhormonen in A. rhizogenes-transformierten Wurzeln herausfordernd. Um diese Einschränkungen zu überwinden, haben wir vor kurzem ein Protokoll für Agrobacterium tumefaciens-basierteTransformation und CRISPR/Cas9-vermittelte Mutagenese von P. andersonii10entwickelt.
Hier bieten wir eine detaillierte Beschreibung des A. tumefaciens-basiertenTransformationsverfahrens und der Reverse-Genetik-Pipeline, die für P. andersoniientwickelt wurde. Darüber hinaus bieten wir Protokolle für den nachgelagerten Umgang mit transgenen Pflanzen, einschließlich Assays zur Untersuchung symbiotischer Wechselwirkungen. Mit dem hier beschriebenen Protokoll können mehrere transgene Leitungen in einem Zeitraum von 2-3 Monaten erzeugt werden. In Kombination mit CRISPR/Cas9-vermittelter Mutagenese ermöglicht dies eine effiziente Erzeugung von Knockout-Mutantenlinien. Diese mutierten Linien können in vitro 10,16,17vegetativ vermehrt werden, wodurch genügend Material erzeugt werden kann, um 4 Monate nach dem Transformationsverfahren mit der phänotien Charakterisierung zu beginnen. initiiert wurde10. Zusammen sollte diese Reihe von Verfahren jedem Labor erlauben, P. andersonii als Forschungsmodell für Studien zu übernehmen, die auf das Verständnis von rhizobialen und mykorrhizalen Assoziationen sowie möglicherweise anderen Aspekten der Biologie dieses tropischen Baumes abzielen.
Protocol
1. Wachsen P. andersonii Bäume im Gewächshaus
-
Germinate P. andersonii WU1 Samen18.
- Verwenden Sie frische Parasponia-Beeren oder tauchen Sie getrocknete Beeren in Wasser für 2 h ein, um zu rehydrieren. Squash Beeren auf einem Stück Tissuepapier oder reiben Sie an der Innenseite eines Teesiebs, um die Samen zu entfernen.
- Desinfizieren Sie Samen mit kommerzieller Bleichmittel (ca. 4% Hypochlorit) für 15-20 min und waschen Sie die Samen anschließend 6 Mal mit sterilisiertem Wasser.
- Übertragen Sie die Samen in sterile 200 L PCR-Röhren. Füllen Sie die Schläuche mit sterilisiertem Wasser, so dass die Samen vollständig untergetaucht sind. Inkubieren Sie die Rohre für 10 Tage in einem Thermocycler mit folgendem Programm: 30 Zyklen (7 °C für 4 h, 28 °C für 4 h). Verwenden Sie keinen beheizten Deckel, da dies die Samen töten könnte.
- SH-0-Platten vorbereiten (siehe Tabelle 1). Die Samen auf SH-0 Teller geben und bei 28 °C, 16 h:8 h Tag:Nacht inkubieren. Platten mit 2 Lagen elastischer Dichtfolie schließen, um eine Trocknung während der Inkubation bei 28 °C zu verhindern.
- Nachdem die Sämlinge ihren ersten Satz echter Blätter entwickelt haben (ca. 3-4 Wochen nach der Inkubation bei 28 °C), die Sämlinge in Töpfe mit kommerziellen Blumenerde übertragen und die Sämlinge mit einem durchscheinenden Plastikbecher bedecken, um eine Austrocknung zu verhindern. Legen Sie Töpfe in einem 28 °C Klimaraum oder Gewächshaus, 85% RH, unter einem 16 h:8 h Tag:Nacht-Regime.
- Nach 1 Woche den lichtdurchlässigen Kunststoffbecher entfernen. Gießen Sie die Töpfe regelmäßig und wenn Bäume wachsen größere Ergänzung mit Dünger, um das Wachstum zu erhalten.
2. Klonen von Konstrukten für CRISPR/Cas9-vermittelte Mutagenese von P. andersonii
HINWEIS: Standard-Binärtransformationsvektoren können für die stabile Transformation von P. andersoniiverwendet werden. Hier ist z.B. ein Verfahren zum Generieren von Konstrukten für CRISPR/Cas9-vermittelte Mutagenese mittels modularem Klonen (z.B. Golden Gate)19.
- Identifizieren Sie Die folgenden RNA-Zielsequenzen für die von Interesse sind, indem Sie die Bioinformatik-Software mit einem integrierten CRISPR-Design-Tool verwenden. Wähle Guide-RNA-Sequenzen am 5'-Ende der Kodierungssequenz des Zielgens, um die Chance zu erhöhen, volle Knockouts zu erhalten. Achten Sie darauf, nach Off-Target-Effekten zu suchen, indem Sie nach dem P. andersonii Genom6suchen.
HINWEIS: Verwenden Sie 2 sgRNAs pro Zielgen, vorzugsweise 200-300 bp voneinander entfernt. Dies kann Zulessen erzeugen, die durch PCR und anschließend durch Agarose-Gel-Elektrophorese identifiziert werden können. - Generieren Sie Golden Gate-Konstrukte der Stufe 1, die die sgRNA-Sequenzen enthalten.
- Design-Primer zur Verstärkung jeder einzelnen sgRNA durch Einfügen der 20 bp-Führungssequenz an der Position N(20) in der folgenden Primersequenz: 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAAATAGCAAG-3'.
HINWEIS: Wenn die Führungssequenz GN(19)entspricht, entfernen Sie das G am 5' Ende der Führungssequenz, bevor Sie es in die Primersequenz einfügen. - PCR verstärken sgRNAs von pICH86966::AtU6p::sgRNA_PDS20 mit den Vorwärtsgrundierungen, die in Schritt 2.2.1 entworfen wurden, und der universellen Reverse Primer: 5'-TGTGGTCTCAAGCGTAATGCCAACTTTGTAC-3'. Verwenden Sie eine hochgradig hitzestabile DNA-Polymerase und die folgenden PCR-Bedingungen: 98 °C für 30 s; 30 Zyklen (98 °C für 10 s; 53 °C für 20 s; 72 °C für 10 s); 72 °C für 7 min. Erfolgreiche PCR-Reaktionen ergeben ein 165 bp Amplikon.
- Säulenreinigen Sie das PCR-Amplikon mit einem kommerziellen PCR-Reinigungskit. Anschließend, setzen Golden Gate Reaktionen auf Klon sgRNAs hinter der Arabidopsis thaliana AtU6p kleinen RNA-Promotor: 10 ng der sgRNA PCR Amplicon, 150 ng von pICSL01009::AtU6p20, 60 ng der entsprechenden Level 1 Akzeptor vektor, 2 l von T4 Ligase-Puffer, 2 l 0,1% Rinderserumalbumin (BSA), 0,5 l BsaI, 0,5 l T4-Ligase, füllung bis 20 l mit reinem Reinstwasser. Stellen Sie sicher, dass alle sgRNAs in der gleichen Ausrichtung geklont werden, um haarserhaltende Bildung zu verhindern.
- Inkubationsreaktionen in einem Thermocycler mit folgendem Programm: 37 °C für 20 s; 26 Zyklen (37 °C für 3 min; 16 °C für 4 min); 50 °C für 5 min; 80 °C für 5 min. Transform Golden Gate Reaktionen auf Escherichia coli und Platte auf LB Medium21 mit Ampicillin (50 mg/L), X-Gal (200 mg/L) und IPTG (1 mM).
HINWEIS: Bereiten Sie Lagerlösungen von IPTG und X-Gal in reinem Wasser bzw. Dimethylformamid vor. Filter sterilisieren die Ampicillin- und IPTG-Lagerlösungen und lagern alle Bestände bei -20 °C. Tragen Sie Handschuhe beim Umgang mit Dimethylformamid. - Wählen Sie weiße Kolonien und isolieren Sie Plasmide mit einem kommerziellen Plasmid-Isolationskit. Sequenz verifizieren isolierte Plasmide, bevor Sie mit der Golden Gate Level 2-Baugruppe fortfahren.
- Design-Primer zur Verstärkung jeder einzelnen sgRNA durch Einfügen der 20 bp-Führungssequenz an der Position N(20) in der folgenden Primersequenz: 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAAATAGCAAG-3'.
- Montieren Sie Level 2 Golden Gate-Konstrukte für die stabile Transformation.
- Führen Sie eine Golden Gate-Reaktion mit den AtU6p-Konstrukten der Stufe 1::sgRNA (generiert unter Abschnitt 2.2) sowie pICH47802::NPTII, pICH47742::35Spro::'NLS-aCas9::35Ster, der Level 2-Akzeptor pICSL4723 und der entsprechende Endlinker (siehe Engler et al.22). Führen Sie Reaktionen wie folgt durch: Verwenden Sie 100 fmol pro Spendervektor und 20 fmol des Akzeptorvektors und fügen Sie 2 l T4-Ligase-Puffer, 2 l 0,1% BSA, 0,5 l BpiI, 0,5 l T4-Ligase, füllung auf 20 l mit ultrareinem Wasser.
HINWEIS: Die Plasmide der Stufe 1 pICH47802::NPTII, pICH47742::35Spro::'NLS-aCas9::35Ster müssen zuerst geklont werden (siehe Ergänzungsdatei 1), wie für sgRNAs unter Abschnitt 2.220,22 beschrieben ,23. - Inkubieren Sie Reaktionen unter Schritt 2.2.4 und wandeln Sie sich in E. colium. Platte auf LB-Medium, das Kanamycin enthält. Am nächsten Tag wählen Sie weiße Kolonien aus und isolieren Sie Plasmide. Bestimmen Sie die richtige Plasmid-Baugruppe durch Restriktions-Verdauungsanalyse.
- Führen Sie eine Golden Gate-Reaktion mit den AtU6p-Konstrukten der Stufe 1::sgRNA (generiert unter Abschnitt 2.2) sowie pICH47802::NPTII, pICH47742::35Spro::'NLS-aCas9::35Ster, der Level 2-Akzeptor pICSL4723 und der entsprechende Endlinker (siehe Engler et al.22). Führen Sie Reaktionen wie folgt durch: Verwenden Sie 100 fmol pro Spendervektor und 20 fmol des Akzeptorvektors und fügen Sie 2 l T4-Ligase-Puffer, 2 l 0,1% BSA, 0,5 l BpiI, 0,5 l T4-Ligase, füllung auf 20 l mit ultrareinem Wasser.
- Transformlevel 2 konstruktiert in Agrobacterium tumefaciens Stamm AGL124.
3. Stabile Transformation von P. andersonii
- Impfen Sie 2 LB-Platten, die die entsprechenden Antibiotika mit A. tumefaciens-Stamm AGL1 enthalten, der mit dem Konstrukt des Interesses transformiert wurde. Platten bei 28 °C für 2 Tage inkubieren.
- Ernten Sie junge Zweige von Gewächshausbäumen. Verwenden Sie ca. 5 Zweige von 5-8 cm Länge für jede Transformation. Stellen Sie sicher, dass Sie nur gesunde, nicht infizierte Zweige verwenden. Entfernen Sie die Blätter, indem Sie sie als solche schneiden, dass 1 cm2 Blattgewebe am Ende jeder Blattbals hinterlassen wird. Entsorgen Sie die Blätter.
- Desinfektiongewebe für 15 min mit 1:1-verdünnter kommerzieller Bleichmittel (ca. 2% Hypochlorit nach Verdünnung), die ein paar Tropfen Polysorbat 20 enthalten. Dann spülen Sie das Gewebe 6 Mal mit autoklavierten Wasser.
HINWEIS: Dieser Schritt sowie die folgenden Schritte müssen in einem laminaren Down-Flow-Schrank durchgeführt werden, um Gewebe steril zu halten. - Unterbrechen Sie die A. tumefaciens-Zellen von 1-2 Platten in 25 ml Infiltrationsmedium (siehe Tabelle 1), die Acetosyringon (20 mg/L) und ein nichtionisches Tensid (0,001% v/v) enthalten, um eine optische Dichte (OD600) von 5 zu erreichen.
HINWEIS: Bereiten Sie die Acetosyringon-Lagerlösung in 70% Ethanol vor und lagern Sie sie bei -20 °C. Das nichtionische Tensid muss vor dem Infiltrationsmedium filtersterilisiert werden. - Schneiden Sie sowohl den Stamm als auch das Blattgewebe in Stücke von 1 cm Länge innerhalb der Suspension von A. tumefaciens, wodurch beidseitig frische Wunden entstehen. Gewebeteile in der A. tumefaciens Suspension für 10-30 min lassen.
- Bereiten Sie das Verwurzelungsmedium vor (siehe Tabelle 1) und fügen Sie Acetosyringon (20 mg/L) nach dem Autoklavieren hinzu. Trocknen Sie Gewebestücke auf ein steriles Stück Filterpapier und legen Sie es auf das Medium (ca. 10 Explanten/Platte). Platten bei Dunkelheit bei 21 °C für 2 Tage inkubieren.
ANMERKUNG: Lassen Sie das Medium auf 60 °C abkühlen, bevor Acetosyringon hinzugefügt wird. - Nach 2 Tagen, überprüfen Sie Platten auf Pilz oder offensichtliche bakterielle Kontamination (Bakterien außer A. tumefaciens). Kontaminierte Platten müssen entsorgt werden.
- Bereiten Sie flüssiges SH-10 Medium vor (siehe Tabelle 1). Fügen Sie nach dem Autoklavieren Polysorbat 20 (0,01 %, v/v) hinzu. Gewebeteile auf 10 ml SH-10 mit Polysorbat 20 übertragen. Während eines Zeitraums von mindestens 10 min, sanft alle 2-3 min rühren, um das Gewebe zu waschen.
- Waschen Sie zwei zusätzliche Male mit frischem SH-10 enthaltendem Polysorbat 20. Diese Zeiten reicht eine Inkubationszeit von 2-3 min pro Waschschritt aus.
- Bereiten Sie das Verwurzelungsmedium vor (siehe Tabelle 1). Nach dem Autoklavieren Cefotaxim (300 mg/L) und Kanamycin (50 mg/L) hinzufügen und Platten gießen. Für die sekundären Transformationen (Transformationen transgener Kanamycin-resistenter Linien) wenden Sie Hygromycin (15 mg/L) Selektion an.
- Trockene Gewebestücke auf sterilen Stücken Filterpapier. Anschließend Gewebeteile auf die in Schritt 3.9 vorbereiteten Platten übertragen.
- Inkubieren Teller für 7 Tage bei 28 °C, 16 h:8 h Tag:Nacht. Alle 2 Tage überprüfen Platten auf Pilz- oder Bakterienkontamination und übermäßiges Wachstum von A. tumefaciens. Im Falle einer Kontamination nicht infizierte Stücke auf eine frische Platte übertragen.
- Nach 7 Tagen Gewebestücke auf Vermehrungsmedium (siehe Tabelle 1) übertragen, das Cefotaxim (300 mg/L) und Kanamycin (50 mg/L) enthält. Inkubieren Teller bei 28 °C, 16 h:8 h Tag:Nacht. Erfrischen Sie die Platten einmal pro Woche, bis sich transgene Triebe entwickeln. Stellen Sie sicher, dass Sie nur nicht infizierte Gewebestücke auf frische Platten übertragen. Entsorgen Sie die Stücke, die von A. tumefaciensüberwuchert sind.
- Wenn vermeintlich-transgene Triebe 1 cm lang sind, steilieren und sie unabhängig voneinander im Vermehrungsmedium mit Cefotaxim (300 mg/L) und Kanamycin (50 mg/L) kultivieren. Um sicherzustellen, dass Triebe unabhängige Transformationsmittel darstellen, nehmen Sie nur einen einzigen Trieb von jeder Seite einer Explantation.
- Vegetativ propagieren vermeintlich-transgene Triebe, wie unter Schritt 5.2 beschrieben.
4. Genotypisierung von vermeintlich-transgenen Trieben
- Design-Primer, die die sgRNA-Erkennungsstelle(n) umfassen. Um die PCR-Amplicon-Sequenzierung zu ermöglichen, wählen Sie Primer 150-250 bp von der sgRNA-Erkennungsstelle(n entfernt).
- Schneiden Sie eine Blattspitze (ca. 5 mm) von jedem transgenen Trieb, um genotypisiert zu werden. Ernten Sie auch eine Wildtyp-Kontrollprobe.
- Führen Sie 50-L-PCR-Reaktionen mit den in Schritt 4.1 entworfenen Primern und einem kommerziellen Kit durch, um die DNA aus Pflanzenproben direkt zu verstärken. Alternativ können PCR-Reaktionen auf gereinigte DNA mit einer Hochtreue-Polymerase durchgeführt werden.
- Separate PCR-Amplicons auf einem 1,5-2% Agarose-Gel.
- Analysieren Sie die Ergebnisse der Gelelektrophorese. Überprüfen Sie, ob Samples mehrere Bänder (mehr als 1 Allel) und PCR-Amplicons mit größen von wildtyp produzieren, was auf das Vorhandensein von mittelgroßen Indels hinweist.
- Sequenz PCR-Amplicons, um die genauen Mutationen zu identifizieren. Für Proben, die ein einzelnes PCR-Amplikon erzeugen, können PCR-Produkte direkt sequenziert werden. Proben, die nach der Gelelektrophorese mehr als 1 Band produzieren oder nach direkter Sequenzierung des PCR-Amplicons heterozygot zu sein scheinen, müssen zuerst in einen stumpfen Klonvektor geklont werden. Anschließend sequenzieren Sie mehrere Klone für jede Probe, um alle möglichen Allele in der Probe zu identifizieren.
- Richten Sie die Sequenzierungsergebnisse an das gen von Interesse aus, und überprüfen Sie die Ausrichtung, um Mutationen in der Nähe der sgRNA-Zielstelle(n) zu überprüfen. Anschließend überprüfen Sie, ob diese Mutationen Frameshifts erzeugen. Verwerfen Sie Linien mit > 2 Allelen und Linien, die Inframe-Mutationen enthalten.
- Wählen Sie mehrere Zeilen für die weitere Analyse aus.
- Vermehren Sie ausgewählte Linien, wie unter Schritt 5.2 beschrieben.
- Wenn Linien mehrere neue Triebe entwickelt haben, nehmen Sie neue Proben von 3 Blattspitzen und wiederholen Sie die Schritte 4.3-4.7. Bestimmen Sie, ob die Mutationen in jeder der Proben, die aus derselben Zeile stammen, sowie die ursprüngliche PCR-Probe identisch sind. Linien, die in allen Proben die gleichen Mutationen ergeben, werden homogen mutiert und können für weitere Experimente verwendet werden. Verwerfen Sie Linien, die nicht die gleichen Ergebnisse liefern wie diese Linien chimeric sind.
5. Herstellung von Verwurzelten P. andersonii Plantlets für Experimente
-
Initiieren Sie eine neue Gewebekulturlinie von P. andersonii.
- Ernten Sie axilläre Knospen, junge zufällige Triebe oder Blattgewebe von gesunden Bäumen. Alternativ können Sämlinge als Ausgangsmaterial verwendet werden.
- Desinfizieren Sie Gewebe mit 1:1-verdünnter kommerzieller Bleichmittel (ca. 2% Hypochlorit nach Verdünnung), die 15 min ein paar Tropfen Polysorbat 20 enthalten. Anschließend geweben Sie 6 Mal mit autoklaviertem Wasser.
HINWEIS: Dieser Schritt sowie die folgenden Schritte müssen in einem laminaren Abfluss- oder laminaren Crossflow-Schrank durchgeführt werden, um Gewebe steril zu halten. - Gewebe auf Vermehrungsmedium übertragen (siehe Tabelle 1). Platten mit 2 Lagen elastischer Dichtfolie schließen und Platten bei 28 °C, 16 h:8 h Tag:Nacht inkubieren.
- Überprüfen Sie die Platten alle paar Tage während der ersten 2 Wochen, um sicherzustellen, dass Gewebe frei von Pilz- oder Bakterienkontamination ist.
- Vermehren Sie Gewebe, indem Sie 10 Triebe auf eine frische Platte aus Vermehrungsmedium legen und die Platte mit 2 Schichten elastischer Dichtfolie schließen. Inkubieren Teller bei 28 °C, 16 h:8 h Tag:Nacht. Wiederholen Sie diesen Schritt alle 4 Wochen.
- Wenn die Triebe >1 cm lang sind, schneiden Sie Triebe an ihrer Basis und legen Sie sie auf das Wurzelmedium (siehe Tabelle 1). Etwa 10 Triebe können auf eine einzelne Wurzelplatte gelegt werden. Position schießt aufrecht, indem die Basalspitze des Triebes in das Medium eingesetzt wird. Die Wurzeln erscheinen 10-14 Tage nach der Inkubation der Platten bei 28 °C, 16 h:8 h Tag:Nacht.
HINWEIS: Nicht alle Triebe wurzeln, sondern Einen Teil für die Gewebekulturvermehrung halten (siehe Schritt 5.2).
6. Nodulation von P. andersonii Plantlets in Töpfen
-
Bereiten Sie Rhizobium inoculum vor.
- 10 ml flüssiges YEM-Medium (siehe Tabelle 2) aus einer einzigen Kolonie von Mesorhizobium plurifarium BOR26 impfen und 2 Tage lang bei 28 °C inkubieren.
ANMERKUNG: M. plurifarium BOR2 wird bevorzugt, da es P. andersoniieffizient noduiert. Andere Rhizobium-Stämme können jedoch auch zur Nodulation von P. andersonii (z.B. Bradyrhizobium elkanii WUR325, Rhizobium tropici CIAT89926,27 oder Bradyrhizobium sp. Kelud2A4). - Verwenden Sie die 10 ml-Kultur, um ein größeres Volumen an flüssigem YEM-Medium zu impfen. Das Volumen dieser Kultur hängt von der Anzahl der Töpfe ab, die geimpft werden müssen.
- Bereiten Sie flüssiges EKM-Medium vor (siehe Tabellen 3, Tabelle 4). Zentrifugieren Sie die Bakterienkultur für 10 min bei 3.500 x g, um die Zellen zu ernten. Anschließend das bakterielle Pellet in flüssigem EKM wieder aufhängen (etwa das gleiche Volumen wie die ursprüngliche YEM-Kultur verwenden) und die optische Dichte bestimmen (OD600).
- 10 ml flüssiges YEM-Medium (siehe Tabelle 2) aus einer einzigen Kolonie von Mesorhizobium plurifarium BOR26 impfen und 2 Tage lang bei 28 °C inkubieren.
- Für 20 Töpfe 3 L flüssiges EKM-Medium zubereiten und mit der in Schritt 6.1.3 zubereiteten Rhizobialsuspension impfen. OD600 = 0,025 zu erreichen.
- Mischen Sie 3 L EKM mit Rhizobia mit 1.250 g Perlit. Anschließend 210 g dieser Mischung in sterile transluzente Polypropylentöpfe geben. Alternativ, anstelle von Perlit, verwenden Sie Sand als Substrat für Nodulation sbeidenk.
- 1-3 P. andersonii Pflanzen in jeden Topf pflanzen. Bereiten Sie außerdem mehrere Töpfe mit P. andersonii-Pflanzenzupfen vor, die mit dem CRISPR-Kontrollkonstrukt umgewandelt wurden (siehe Ergänzende Tabelle 1). Wiegen Sie mehrere Töpfe, um den Wasserverlust während des Experiments zu bestimmen. Bedecken Sie den Boden jedes Topfes, um die Wurzeln vor Lichteinwirkung zu schützen.
- Inkubieren Sie Töpfe in einem klimatisierten Wachstumsraum (28 °C, 16 h:8 h Tag:Nacht) für 4-6 Wochen. Einmal pro Woche mehrere Töpfe wiegen, um Wasserverlust zu bestimmen. Wenn der Wasserverlust 10 ml übersteigt, mit reinem reinem Wasser ergänzen, um den Verlust auszugleichen.
- Nach 4-6 Wochen reinigen Sie die Wurzeln von Perlit und bestimmen Sie Diele-Zahlen mit einem Binokular, um die Nodulationseffizienz zu untersuchen.
7. Nodulation von P. andersonii Plantlets auf Platten
-
Vorbereiten von Zellophanmembranen 28.
- Schneiden Sie die Zellophanmembran, um in eine quadratische 12 cm x 12 cm Petrischale zu passen. Schneiden Sie die Membranen an der Spitze etwas kürzer, damit die Triebe wachsen können.
- Um die Durchlässigkeit von Zellophanmembranen zu erhöhen, kochen Sie die Membranen in EDTA-Lösung (1 g/L) für 20 min. Danach spülen Sie mindestens 6x mit demineralisiertem Wasser, um die EDTA zu entfernen.
HINWEIS: Da die trockene Membran dazu neigt, bei Kontakt mit Wasser zu falten, tauchen Sie die trockenen Membranen nacheinander in die Lösung ein. - Ordnen Sie die Membranen horizontal in einer dünnen Wasserschicht in einer runden Glasplatte an. Sterilisieren Sie die Membranen durch Zweimal-Autoklavieren.
- 1 autoklavierte Zellophanmembran auf eine quadratische 12 x 12 cm Petrischale mit agar-erstarrtem EKM-Medium legen (siehe Tabelle 3, Tabelle 4). Legen Sie zwei drei Wochen alte verwurzelte P. andersonii Pflanzen (siehe Abschnitt 5) oder 4 Wochen alte Sämlinge (siehe Abschnitt 1.1) auf die Oberseite der Membran. Achten Sie darauf, nur Pflanzchen oder Sämlinge mit Wurzeln mit weißen Wurzelspitzen zu pflücken, was darauf hinweist, dass diese Wurzeln noch wachsen.
- Bedecken Sie die Wurzeln vorsichtig mit einer zweiten Zellophanmembran und bilden Sie so eine Sandwichschicht. Versiegeln Sie die Platte mit 3 Schichten elastischer Dichtfolie. Wickeln Sie die untere Hälfte der Platten mit Aluminiumfolie, um die Wurzeln vor Lichtbelichtung zu bedecken.
- Inkubieren Sie die Platten in einem klimatisierten Wachstumsraum (28 °C, 16 h:8 h Tag:Nacht) für 3-4 Wochen. Markieren Sie die Position der Wurzelspitzen, um dem Wurzelwachstum im Laufe der Zeit zu folgen.
- Wenn die EKM-Platten aufgrund einer längeren Inkubation auszutrocknen beginnen, übertragen Sie die Pflanzen einige Tage vor der bakteriellen Impfung auf frische EKM-Platten.
- Bereiten Sie das bakterielle Inokulum wie in Schritt 6.1 beschrieben vor.
- Entfernen Sie die obere Zellophanmembran und tragen Sie 1 ml Rhizobiumkultur (OD600 = 0,025) auf die Wurzeln auf. Anschließend legen Sie eine neue Zellophanmembran auf die geimpften Wurzeln. Wickeln Sie die Außenseite der Platte mit Aluminiumfolie, um die Wurzeln vor Lichtbelichtung zu bedecken.
- Nach 4 Wochen, untersuchen Knötnurnzahlen mit einem Binokular, um die Nodulationseffizienz zu bestimmen.
8. Nodulation von P. andersonii Sämlingen in Beuteln
- Keimen Sie P. andersonii Samen, wie in Abschnitt 1.1 beschrieben. Nachdem die Kotyledonen vollständig entstanden sind (ca. 12 Tage auf SH-0-Platten bei 28 °C), die Sämlinge in Beutel geben.
- Zur Vorbereitung der Beutel den gefalteten Teil des Papierdochts zerreißen und 7 ml modifiziertes EKM-Medium hinzufügen (siehe Tabelle 3, Tabelle 4).
- Legen Sie 1 oder 2 Sämlinge ein, indem Sie die Wurzeln zwischen beiden Blatt Papier, die den Papierdocht bilden, und die vordere Kunststofffolie des Beutels platzieren.
- Schützen Sie die Wurzeln vor Lichtbelichtung, indem Sie Aluminiumfolie um den Beutel falten. Hängen Sie die Beutel in einer Kunststoffbox auf, die mit einem durchscheinenden Deckel bedeckt ist, um eine hohe Luftfeuchtigkeit zu erhalten. Legen Sie die Box in einen klimatisierten Wachstumsraum (28 °C, 16 h:8 h Tag:Nacht).
- Kompensieren Sie die Wasserverdunstung durch Zugabe von sterilem reinem Wasser, so dass der Papierdocht feucht bleibt (vermeiden Sie stehendes Wasser am Boden des Beutels). Nach der ersten Woche, Dies erfordert in der Regel 2-3 ml alle 4 Tage.
- Bereiten Sie das bakterielle Inokulum wie in Schritt 6.1 beschrieben vor.
- Nachdem Sämlinge 10-12 Tage lang in Beuteln angebaut wurden, impfen Sie das Wurzelsystem mit 500 l Rhizobiumkultur (OD600 = 0,025).
- Folgen Sie der Knötaberbildung im Laufe der Zeit. Vier Wochen nach der Impfung können Knötchen gezählt und geerntet werden, um die Nodulationseffizienz zu bestimmen.
9. Nodule Cytoarchitecture Analyse
- Sammeln Sie 10-15 Knötchen in einem 2 ml-Rohr, das fixative (5% Glutaraldehyd in 0,1 M Phosphatpuffer, pH 7,2) enthält. Vakuum für 1/2-1 h auftragen und über Nacht bei 4 °C inkubieren. Während dieser Zeit sinken die Proben auf den Boden des Rohres.
HINWEIS: Die Fixierlösung kann bei 4 °C für 2-4 Wochen vor der Verwendung gelagert werden. Achten Sie darauf, Handschuhe zu tragen, wenn Sie mit Gewebefixierung arbeiten. - Waschen Sie die Knötchen 2x mit 0,1 M Phosphatpuffer, pH 7,2. Tragen Sie 10 min Intervalle zwischen jedem Waschschritt auf.
- Dehydrieren Sie die Proben, indem Sie anschließend in 30%, 50%, 70% und 100% Ethanol inkubieren. Um sicherzustellen, dass das gesamte Wasser aus den Proben entfernt wird, wiederholen Sie den 100% EthanolSchritt 3x. Tragen Sie 10 min Intervalle zwischen jedem Austrocknungsschritt auf.
- Bereiten Sie polymerisationsgemisch I (PM-I) vor, indem Sie 1 Packung Härter I zu 2,5 ml PEG400 gemischt mit 100 ml HEMA (2-Hydroxyethylmethacrylat)-basierten Harzlösung hinzufügen. Rühren Sie die Lösung für 15 min, um den Härter I vollständig aufzulösen. Anschließend PM-I bei -20 °C lagern.
- Entfernen Sie das Ethanol aus Schritt 9.3. und infiltrieren Sie die Proben in der folgenden Reihenfolge: PM-I:100% Ethanol (1:3, v/v), PM-I:100% Ethanol (1:1, v/v) und PM-I:100% Ethanol (3:1, v/v). Inkubieren Sie die Proben in jeder Lösung bei RT für 1/2-1 h oder bis die Proben auf den Boden sinken.
- Proben über Nacht bei 4 °C in 100% PM-I-Lösung inkubieren.
- Bereiten Sie polymerisationsgemisch II durch Mischen von PM-I und Härter II im Verhältnis 15:1 (v/v) vor. Füllen Sie die Kunststoffform mit der Polymerisationslösung, richten Sie die Proben horizontal an der Unterseite der Form aus und bedecken Sie sie mit einem Stück elastischer Dichtfolie. Vermeiden Sie die Bildung von Luftblasen.
HINWEIS: Wenn die Lösung bei der Exposition gegenüber RT zu polymerisieren beginnt, versuchen Sie, die Proben so schnell wie möglich im Kunststoffhalter auszurichten. Die Polymerisation wird nach einer nächtlichen Inkubation bei RT oder 1 h bei 37 °C abgeschlossen. - Entfernen Sie die elastische Dichtfolienabdeckung aus Schritt 9.7 und legen Sie einen Halter an die polymerisierten Proben. Um den Halter an den Proben zu montieren, lösen Sie 10 ml Methylmethacrylat-basiertes Harzpulver in 5 ml Methylmethacrylat-basierten Harzlösung auf. Fügen Sie die Lösung schnell zum Loch in der Oberseite des Halters hinzu.
HINWEIS: Führen Sie den Polymerisationsschritt in der Dunstabzugshaube (ca. 30 min bei RT) durch. - Mikrotome-Sektionsproben bis zu einer Dicke von 4-5 m. Legen Sie ein Mikroskopschlitten auf eine 58 °C-Heißplatte und fügen Sie jedem Dia einen großen Tropfen Wasser hinzu. Platzieren Sie die Abschnitte auf der Oberseite des Wassers. Sobald das Wasser verdunstet ist, haften die Abschnitte an der Rutsche.
- Stain gleitet durch Eintauchen in 0,05 % (w/v) Toluidinblau für 2 min. Anschließend rutschen die Dias 3x mit reinem Wasser. Dias können mit einem Hellfeldmikroskop beobachtet werden.
10. Mykorrhiisation von P. andersonii Plantlets
-
Bereiten Sie Rhizophagus irregularis spores' inoculum vor
- Bereiten Sie einen Stapel von Polyester-Gewebefiltern mit den folgenden Größen (von oben nach unten) vor: 210 m, 120 m und 36 m Maschenöffnung.
- Pipette die erforderliche Menge einer kommerziellen Sporensuspension auf den Stapel von Polyesterfiltern. Spülen Sie die Filter 3x mit 100 ml autoklaviertem demineralisiertem Wasser. Die Sporen werden auf der Oberfläche des 36-mm-Filters zurückgehalten.
ANMERKUNG: Bereiten Sie die Sporensuspension im laminaren Querlaufschrank vor, um eine Kontamination zu verhindern. - Zerlegen Sie den Polyester-Stack und halten Sie den 36-mm-Filter nur. Wiederholen Sie den Waschschritt mit autoklaviertem demineralisiertem Wasser mindestens 6x.
- Legen Sie den Filter auf eine Petrischale und setzen Sie die Sporen in autoklaviertem demineralisiertem Wasser wieder auf. Verwenden Sie ein Wasservolumen, das dem Volumen der sporensuspension entspricht, die in Schritt 10.1.2 verwendet wird. Übertragen Sie die Sporensuspension durch Pipettieren auf ein steriles Rohr.
- Legen Sie 5 Tropfen von 20 l der Sporensuspension auf ein Glasschlitten und zählen Sie die Anzahl der Sporen mit einem Hellfeldmikroskop. Konvertieren Sie Sporenzahlen in ein Verhältnis von Sporen/ml und verdünnen Sie die Sporensuspension, bis sie 250 Sporen/ml erreicht. Die Sporenaufhängung bei 4 °C aufbewahren.
- Führen Sie Mykorrtherisierungstest durch. Dazu 800 g autoklavierter Sand hinzufügen, ergänzt mit 70 ml 1/2-Hoagland-Medium bis sterilen transluzenten Polypropylentöpfen (siehe Tabellen 5-6). Sand und Medium direkt im Topf mischen, indem Sie kräftig schütteln.
- Legen Sie eine P. andersonii Pflanze in jedem Topf und Pipette 1 ml der Sporensuspension direkt auf die Wurzel des P. andersonii Plantlets. Stellen Sie sicher, dass mehrere Töpfe mit P. andersonii-Pflanzenmitpflanzen enthalten, die mit einem CRISPR-Kontrollkonstrukt umgewandelt wurden (siehe Ergänzende Tabelle 1).
- Inkubieren Sie Töpfe in einem klimatisierten Wachstumsraum (28 °C, 16 h:8 h Tag:Nacht) für 6 Wochen.
- Nehmen Sie die Pflanzen aus den Töpfen und waschen Sie die Wurzeln mit fließendem Wasser, um so viel Sand wie möglich zu entfernen.
- Wurzeln in 1 cm lange Stücke schneiden und die Wurzelstücke in 10% KOH (w/v) für 20 min bei 90 °C kochen. Anschließend die gekochten Wurzeln auf ein Zellsieb mit einer Maschenöffnung von 100 m legen und 3x mit 50 ml Wasser abspülen.
- Fleckenwurzeln mit 0,05% (w/v) Trypan blau in Lactoglycerol (300 ml Milchsäure; 300 ml Glycerin; und 400 ml demineralisiertes Wasser) für 5 min bei 90 °C in einem Wasserbad oder Heizblock. Anschließend Wurzeln auf 30% Glycerin übertragen. Die Stammproben können bei RT gespeichert werden.
- Platzieren Sie 15-25 Wurzelfragmente auf einem einzigen Mikroskopschlitten. 30% Glycerin hinzufügen und mit einem Deckglas abdecken und drücken, bis die Wurzelstücke flach werden. Beobachten Sie die Wurzelfragmente mit einem Hellfeldmikroskop und bewerten Sie die Mykorrhizabebe.
ANMERKUNG: Eine Methode zur Bewertung der Mykorrhiisation wird gemäß Trouvelot et al.29beschrieben. Diese Methode verwendet mehrere Klassen (%F, %M und %A), was eine schnelle Schätzung des Niveaus der mykorrhizalen Kolonisierung jedes Wurzelfragments und der Fülle von Arbuskeln ermöglicht.
Representative Results
P. andersonii trees kann in einem klimatisierten Gewächshaus bei 28 °C und einer relativen Luftfeuchtigkeit von 85 % angebaut werden (Abbildung 1A). Unter diesen Bedingungen beginnen die Bäume 6-9 Monate nach der Pflanzung zu blühen. Weibliche P. andersonii Blüten produzieren Beeren, die jeweils einen einzigen Samen enthalten. Während der Reifung ändern die Beeren ihre Farbe; zuerst von grün nach weiß und anschließend von weiß nach braun (Abbildung 1B). Samen, die aus den gereiften braunen Beeren gewonnen werden, keimen gut nach einem 10-tägigen Temperaturzyklus und einer 7-tägigen Inkubation auf SH-0-Platten (Abbildung 1C). Gekeimte Samen entwickeln sich weiterhin zu jungen Sämlingen, die nach 4 Wochen für Experimente verwendet werden können (Abbildung 1D).
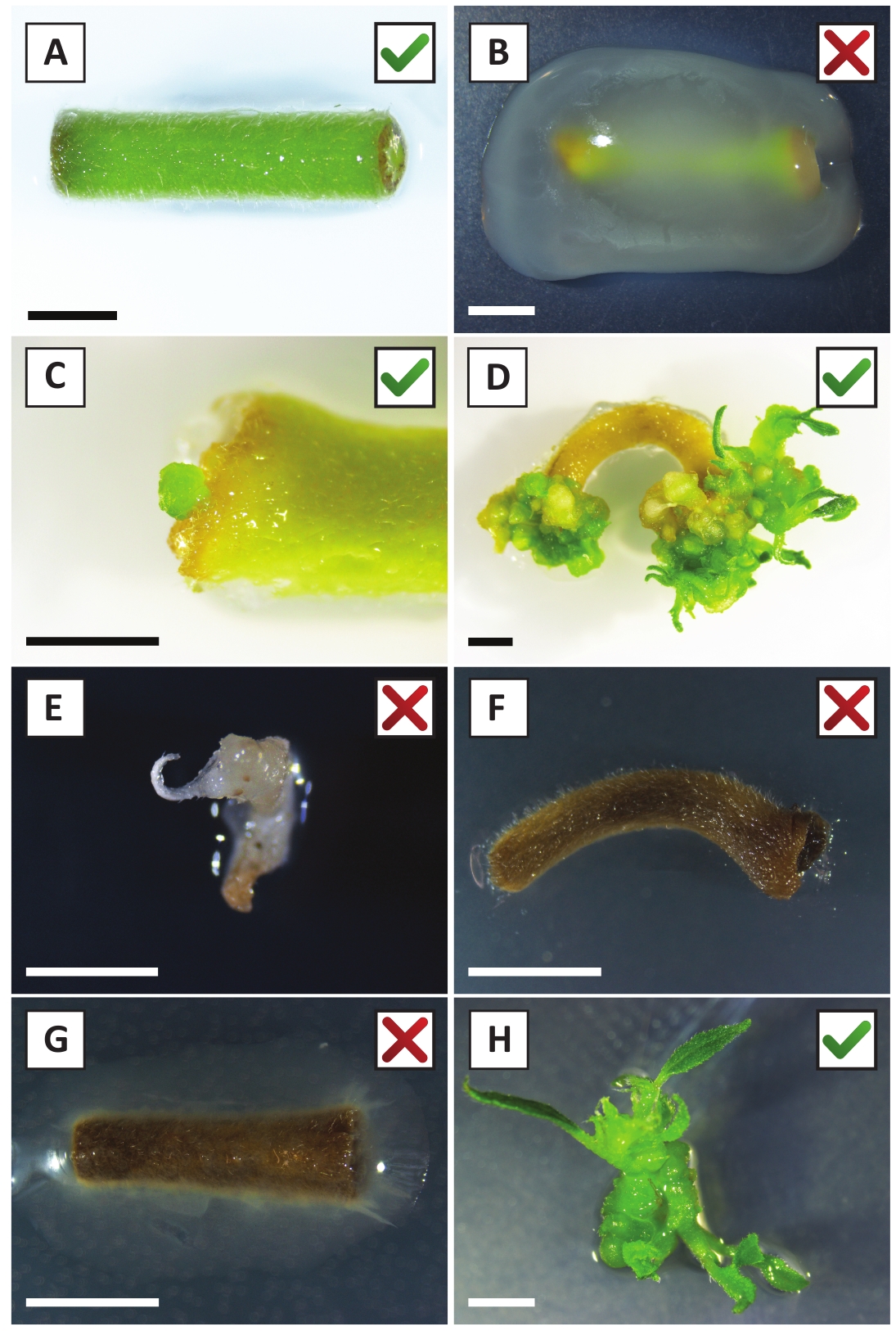
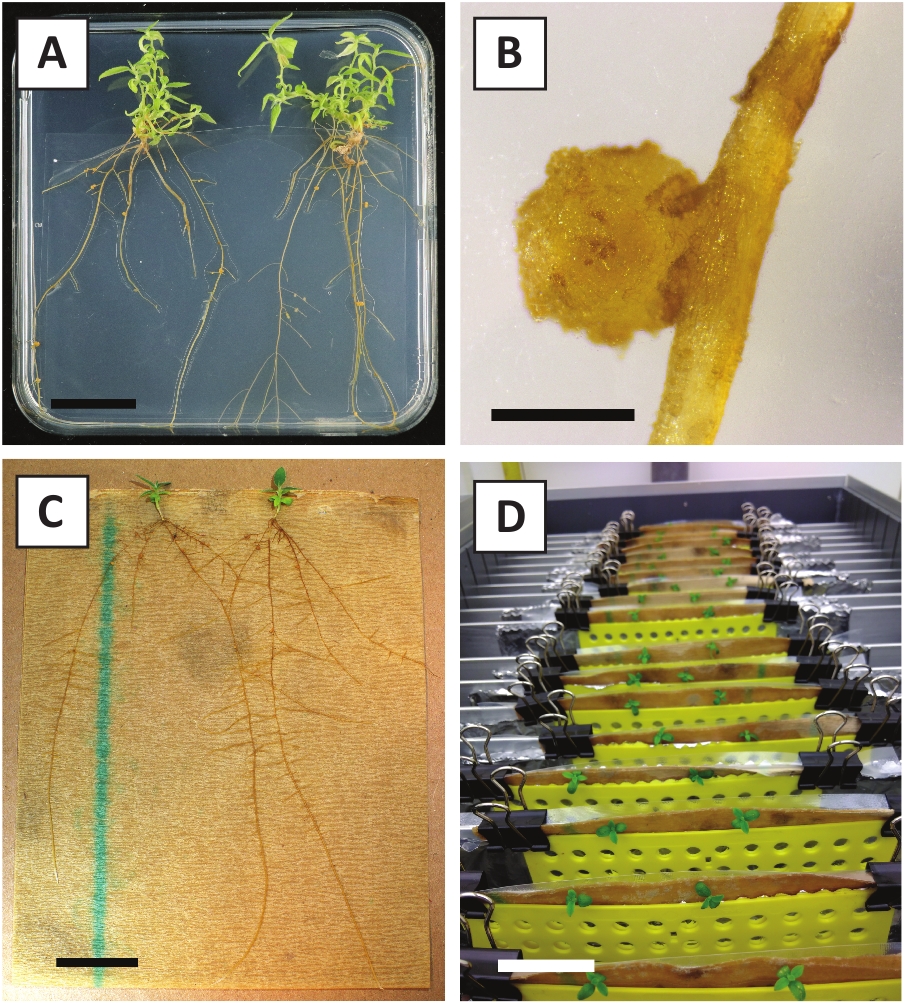
Wir haben bereits gezeigt, dass Petioles und Segmente junger P. andersonii-Stämme mit A. tumefaciens-Stamm AGL110effizient transformiert werden können. Zu Beginn des Transformationsverfahrens werden die Gewebeexplanten 2 Tage lang bei 21 °C mit A. tumefaciens kokultiviert (Abbildung 2A). Eine längere Kokultivierung führt zur Überkolonialisierung der Gewebeexplantationen durch A. tumefaciens und sollte daher verhindert werden (Abbildung 2B). Nach der Kokultivierungwerden werden Gewebeexpflanzen auf selektive Medien übertragen, was das Auswachsen von transformiertem Gewebe fördert. Zwei bis drei Wochen später werden kleine grüne Mikro-Calli in der Regel entlang der ursprünglichen Wundoberfläche beobachtet (Abbildung 2C). Diese Calli sollten weiter wachsen und 1 oder mehr vermeintlich transformierte Triebe nach 6-8 Wochen nach Einleitung des Transformationsverfahrens entwickeln (Abbildung 2D). In diesem Stadium reichen die Transformationseffizienzen in der Regel zwischen 10 und 30 % für Transformationen, die mit Gewebeexpflanzen aus reifen und teilweise holzigen Zweigen eingeleitet wurden (Tabelle 7). Wenn Transformationen mit Expflanzen eingeleitet werden, die den jungen und schnell wachsenden Spitzen von Zweigen entnommen werden, die noch keine Blüten tragen, können Transformationseffizienzen von 65-75% erreicht werden (Tabelle 7). Gelegentlich bilden sich auf der Seite einer Explantation weißliche Kali, die nicht in Kontakt mit dem Medium ist und daher keine Kanamycin-Auswahl erlebt. Diese Calli sind oft nicht transgen und alle Triebe, die aus diesen Calli gebildet werden, bleichen und sterben in der Regel nach direktem Kontakt mit Kanamycin-haltigem Medium (Abbildung 2E). Falls die Transformationsrate niedrig ist und/oder das Ausgangsmaterial suboptimal war, könnten Gewebeteile braun werden (Abbildung 2F) und an einer Überproliferation durch A. tumefaciens leiden (Abbildung 2G). Um zu verhindern, dass sich A. tumefaciens in der Nähe ausbreitet und überwächst, ist eine regelmäßige Erfrischung des Mediums erforderlich, und schwer infizierte Expflanzen müssen entfernt werden. Sobald einzelne transgene Triebe in das Vermehrungsmedium gelegt werden, tritt eine Überproliferation durch A. tumefaciens in der Regel nicht mehr auf (Abbildung 2H). Transgene Triebe können durch In-vitro-Vermehrung multipliziert werden, was zu Dutzenden von Trieben in einem Zeitraum von einem Monat führen wird (Abbildung 3A-B). Diese Triebe können auf das Wurzelmedium gelegt werden, was die Wurzelbildung nach 2 Wochen induzieren sollte (Abbildung 3C-D). Verwurzelte Pflanzen können anschließend für Experimente verwendet werden.
Um Knockout-Mutationslinien zu erstellen, nutzen wir CRISPR/Cas9-vermittelte Mutagenese. Zu diesem Zweck verwenden wir einen binären Vektor, der das Kanamycin-Resistenzgen NPTIIenthält, eine Cas9-kodienierende Sequenz, die vom CaMV35S-Promotor angetrieben wird, und 2 sgRNAs pro Zielgen, die aus dem AtU6p-Kleinen RNA-Promotor20exprimiert werden. Eine grafische Darstellung des Konstrukts, das für CRISPR/Cas9-vermittelte Mutagenese von P. andersonii verwendet wird, ist in Abbildung 4Adargestellt. Mit dieser Methode wird die Genombearbeitung bei 40 % der vermeintlich transformierten Triebe10beobachtet. Um mutierte Linien zu identifizieren, werden vermeintlich transformierte Triebe für Mutationen an der sgRNA-Zielstelle(n) genotypisiert, indem Primer verwendet werden, die die Zielregion überspannen. Ein Beispiel für die erwarteten Ergebnisse finden Sie in Abbildung 4. Wie aus dem Foto nach der Gelelektrophorese zu sehen ist, erzeugen mehrere Proben ein PCR-Amplikon mit ähnlicher Größe wie der Wildtyp (Abbildung 4B). Diese Pflanzen können kleine Indels enthalten, die nicht durch Agarose-Gel-Elektrophorese visualisiert werden können oder durch das Cas9-Enzym unbearbeitet bleiben. Darüber hinaus ergeben mehrere Samples Bänder, die sich in der Größe vom Wildtyp unterscheiden (z. B. Zeilen 2, 4, 7 und 8 in Abbildung 4B). In diesen Zeilen enthalten 1 (Linien 4, 7 und 8) oder beide (Linie 2) Allele größere Indels, die leicht visualisiert werden können. Die genaue Art der Mutationen am Zielstandort(en) wird nach der PCR-Ampliconsequenzierung aufgedeckt. Wie aus Abbildung 4Cersehen kann, können sowohl kleine Indels von 1-4 bp als auch größere Deletionen nach CRISPR/Cas9-Mutagenese erhalten werden. In Abbildung 4Cist die Reihenfolge von Zeile 1 identisch mit der des Wild-Typs, was darauf hinweist, dass diese Zeile der Bearbeitung entkommen ist und daher verworfen werden sollte. Unter den Linien, die Mutationen enthalten, können heterozygote, homozygote und bi-allelische Mutanten identifiziert werden (Abbildung 4C). Heterozygote Mutanten sind jedoch in der Regel selten10. Homozygote oder bi-allel Knockout-Mutanten können vegetativ vermehrt werden, um genügend Material für die phänotypische Analyse zu erhalten. Da die phänotische Analyse in der T 0-Generation durchgeführt wird, ist es wichtig zu prüfen, ob mutierte Linien chimerisch sein könnten. Zu diesem Zweck muss die Genotypisierung an mindestens 3 verschiedenen Proben wiederholt werden, die aus jeder mutierten Linie entnommen wurden. Wenn die Genotypisierungsergebnisse identisch sind und die ursprüngliche Genotypisierungsprobe (z. B. Zeile 8 in Abbildung 4D), ist die Linie homogen mutiert und kann für weitere Analysen verwendet werden. Wenn sich die Genotypisierungsergebnisse jedoch zwischen unabhängigen Proben unterscheiden (z. B. Zeile 4 in Abbildung 4D), ist die mutierte Linie chimerisch und muss verworfen werden.
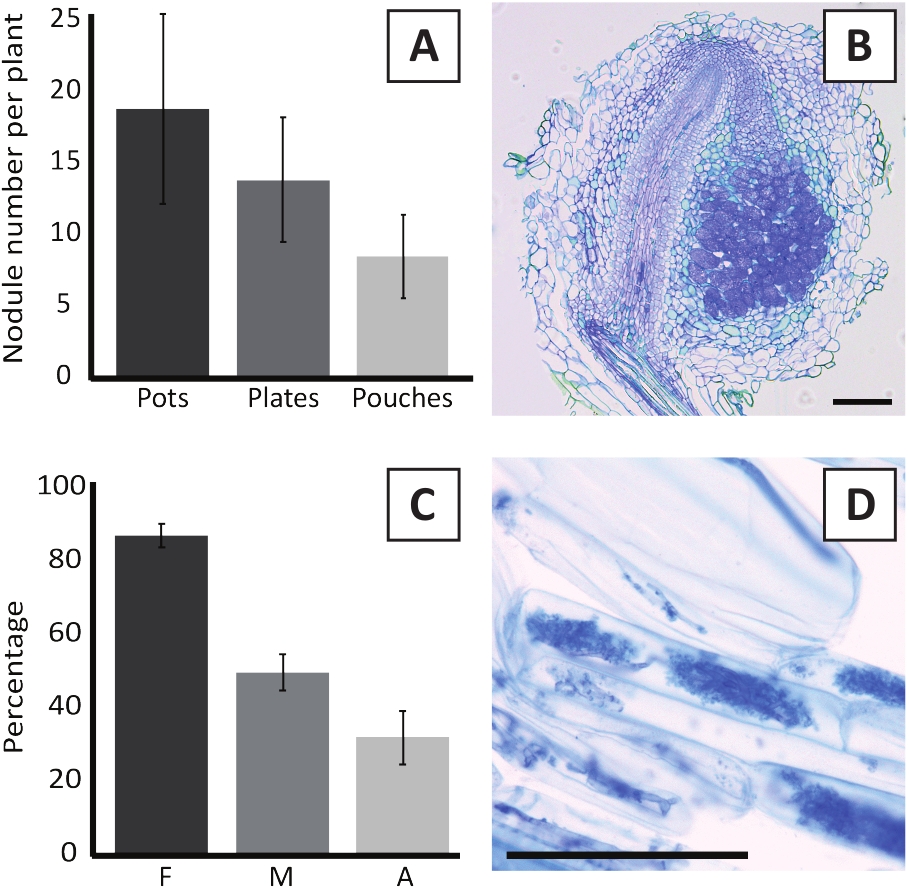
Die Impfung von P. andersonii mit M. plurifarium BOR2 führt zur Bildung von Wurzelknollen (Abbildung 5). Wie in Abbildung 5Azu sehen ist, werden diese Knötchen entlang des Wurzelsystems verteilt. Knötchen von P. andersonii sind hellbraun in der Farbe, können aber leicht vom Wurzelgewebe aufgrund ihrer Form diskriminiert werden (Abbildung 5B). Impfexperimente in Töpfen und anschließendes Wachstum für 4-6 Wochen führen in der Regel zur Bildung von 10-30 Knötchen (Abbildung 6A). Eine ähnliche Anzahl von Knötchen wird nach der Impfung von EKM-Platten-Anbau P. andersonii Pflanzen nach 4 Wochen nach der Impfung gebildet (Abbildung 6A). In Beuteln bilden P. andersonii Sämlinge typischerweise 5-15 Knötchen nach 5 Wochen nach der Impfung (Abbildung 5C-D, 6A). Zur Analyse der Knötchenzytoarchitektur können Knötchen mit Derinemik geschnitten und beobachtet werden. Abbildung 6B zeigt ein Beispiel für einen Längsschnitt durch die Mitte eines P. andersonii Knötlees. Dieser Abschnitt zeigt das zentrale Gefäßbündel eines P. andersonii-Knotens, der von Knötchenlappen flankiert wird, die infizierte Zellen enthalten (Abbildung 6B).
P. andersonii Plantlets können auch mykorreliert werden. Nach 6 Wochen Impfung mit R. irregulariserreicht die Mykorrhiza-Kolonisationshäufigkeit in der Regel > 80% (Abbildung 6C). Zu diesem Zeitpunkt enthalten in der Regel 30 % der Zellen Arbuscules (Abbildung 6C). Ein repräsentatives Bild eines P. andersonii Wurzelsegments, das Arbuscles enthält, ist in Abbildung 6Ddargestellt.

Abbildung 1: Repräsentative Bilder eines P. andersonii Baum, Samen und Sämlinge. (A) Sechs Monate alter P. andersonii-Baum, der in Blumenerde in einem Gewächshaus angebaut wird, das bei 28 °C konditioniert ist. (B) Repräsentatives Bild, das P. andersonii Beeren in verschiedenen Reifestadien darstellt. Junge P. andersonii Beeren (unreif) wird Farbe von grün zu weiß und schließlich braun (reif) nach der Reifung ändern. (C) P. andersonii Samen auf SH-0 Medium für 1 Woche inkubiert. Ein schwarzer Kreis zeigt einen gekeimten Sämling an. (D) Vier Wochen alte P. andersonii Sämlinge, die in SH-0 medium angebaut werden. Die Skalenbalken sind gleich 25 cm in (A) und 1 cm in (B-D). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Repräsentative Bilder von Expflanzen in verschiedenen Stadien des stabilen Transformationsverfahrens. (A) Explant ko-kultiviert mit A. tumefaciens. (B) Explant von A. tumefaciens während der ersten 2 Wochen nach der Transformation überwuchert. (C) Transgener Mikro-Callus bildete sich in der Nähe der Wundstelle einer Explantation nach 2,5 Wochen nach der Kokultivierung. (D) Repräsentatives Bild einer Explantation nach 6 Wochen nach der Kokultivierung, das die Entstehung von Trieben von (transgenen) Kali zeigt. (E) Repräsentatives Bild eines Triebes, der weißlich wird und schließlich stirbt, wenn er in direktem Kontakt mit kanamycinhaltigem Medium steht. Dieser Trieb ist höchstwahrscheinlich nicht transgen und entwichen Kanamycin-Auswahl, wenn an der Explantation befestigt. (F) Repräsentatives Bild einer erfolglos transformierten Explantation. (G) Repräsentatives Bild einer erfolglos transformierten Explantation, die von A. tumefaciensüberwuchert wurde. (H) Einzelner transgener Trieb, der auf Vermehrungsmedium nach 8 Wochen nach der Kokultivierung mit A. tumefaciensangebaut wird. Skalenbalken entsprechen 2,5 mm. Boxen mit grünen Häkchen oder roten Kreuzen weisen auf eine erfolgreiche bzw. erfolglose Umwandlung von Expflanzen hin. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Repräsentative Bilder von in vitro Vermehrung. (A) Triebe, die auf dem Vermehrungsmedium angebaut werden. Das Bild wurde 1 Woche nach dem Auffrischen der Platten aufgenommen. (B) Triebe, die auf dem Vermehrungsmedium angebaut werden. Das Bild wurde 4 Wochen nach der Aktualisierung der Platten aufgenommen. (C) Frisch geschnittene Triebe auf Wurzelmedium gelegt. (D) Triebe, die 2 Wochen lang auf dem Wurzelmedium inkubiert werden. Beachten Sie das Vorhandensein von Wurzeln. Die Maßstabsleisten sind gleich 2,5 cm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Repräsentative Ergebnisse nach Genotypisierung von P. andersonii T0 transgenen CRISPR/Cas9-Mutationslinien. (A) Repräsentative Karte eines binären Vektors, der für CRISPR/Cas9-vermittelte Mutagenese von P. andersoniiverwendet wird. (B) Repräsentatives Ergebnis nach PCR-basierter Genotypisierung potenzieller CRISPR/Cas9-Mutantenlinien mit Primern, die die sgRNA-Zielsite(n) überspannen. Gezeigt wird ein Bild nach der Agarose-Gel-Elektrophorese von Amplicons. Die Proben aus einzelnen transgenen Linien sind durch Zahlen gekennzeichnet. Wild-Typ (WT) und kein Vorlagensteuerelement (NTC) zeigen Bahnen an, die positive bzw. negative Steuerelemente enthalten. (C) Schematische Darstellung von mutierten Allelen, die nach CRISPR/Cas9-vermittelter Genbearbeitung erhalten wurden. Hervorgehoben in blauen und roten Farben sind die sgRNA-Zielsites bzw. PAM-Sequenzen. (D) Repräsentatives Ergebnis nach PCR-basiertem Screening auf potenzielle chimäre Mutantenlinien. Gezeigt wird ein Bild nach der Agarose-Gel-Elektrophorese von 3 Einzelproben aus den mutierten Linien 4 und 8. Beachten Sie, dass die transgene Mutantlinie 4 chimer ist. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Repräsentative Bilder von Nodulationstests in Platten und Beuteln. (A) Nodulation auf Platten, die agar-verfestigtes EKM-Medium enthalten und 4 Wochen lang mit M. plurifarium BOR2 geimpft werden. (B) Repräsentatives Bild eines P. andersonii Wurzelknotens. Das Bild wurde nach 4 Wochen nach der Impfung mit M. plurifarium BOR2 aufgenommen. (C) Nodulation in Beuteln, die flüssiges EKM-Medium enthalten. Die Samen wurden 5 Wochen lang mit Bradyrhizobium sp. Kelud2A4 geimpft. (D) Repräsentatives Bild eines vollständigen Setups, das für das Nicken in Beuteln verwendet wird. Die Skalenstäbe sind gleich 2,5 cm in (A,C), 1 mm in (B), und 5 cm in (D). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Repräsentative Ergebnisse der Nodulations- und Mykorrhiisierungstests. (A) Repräsentatives Balkendiagramm, das die Anzahl der knötper pflanzenweise bei 4 Wochen nach der Impfung mit M. plurifarium BOR2 in Töpfen oder auf Tellern und bei 5 Wochen nach der Impfung mit Bradyrhizobium sp. Kelud2A4 in Beuteln zeigt. Die Daten stellen den Mittelwert sD (n = 10) dar. (B) Repräsentatives Bild eines Längsschnitts durch einen Knötohne, der bei 4 Wochen nach der Impfung mit M. plurifarium BOR2 gebildet wurde. Der Abschnitt ist mit Toluidinblau befleckt. (C) Repräsentatives Balkendiagramm mit Quantifizierung der Mykorrhiisation. Variablen, die nach Trouvelot et al.29 quantifiziert werden, sind F, die Häufigkeit der analysierten Wurzelfragmente, die mykorrelisiert sind; M, die Intensität der Infektion; A, die Fülle der reifen Arbuscules im gesamten Wurzelsystem. Die Mykorrhiisation wurde nach 6 Wochen nach der Impfung mit R. irregularis (Stamm DAOM197198) quantifiziert. Die Daten stellen den Mittelwert sD (n = 10) dar. (D) Repräsentatives Bild von reifen Arbuskeln, die in P. andersonii Wurzelkortikalzellen bei 6 Wochen nach der Impfung mit R. irregularisvorhanden sind. Skalenbalken entsprechen 75 'm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| umzäuntes grundstück | SH-0 | SH-10 | Ausbreitungsmedium | Verwurzelungsmedium | Infiltrationsmedium |
| SH-Basalsalzmedium | 3,2 g | 3,2 g | 3,2 g | 3,2 g | 3,2 g |
| SH-Vitamin-Mischung | 1 g | 1 g | 1 g | 1 g | 1 g |
| sacharose | - | 10 g | 20 g | 10 g | 10 g |
| BAP (1 mg/ml) | - | - | 1 ml (4,44 x M) | - | - |
| IBA (1 mg/ml) | - | - | 100 l (0,49 x M) | 1 ml (4,92 m) | - |
| NAA (1 mg/ml) | - | - | - | 100 l (0,54 m) | - |
| 1 M MES pH=5,8 | 3 mL | 3 mL | 3 mL | 3 mL | 3 mL |
| 1 M KOH | pH auf 5,8 einstellen | pH auf 5,8 einstellen | pH auf 5,8 einstellen | pH auf 5,8 einstellen | pH auf 5,8 einstellen |
| Daishin-Agar | 8 g | - | 8 g | 8 g | - |
Tabelle 1: Zusammensetzung von30 Medien auf Schenk-Hildebrandt-Basis für den Anbau von P. andersonii Sämlingen, stabile Transformation und In-vitro-Vermehrung. Lösen Sie feste Verbindungen in 750 ml reines Wasser, bevor Sie flüssige Vorräte hinzufügen. Danach füllen Sie das komplette Medium bis 1 L. Bereiten Sie BAP, IBA, NAA-Bestände in 0,1 M KOH vor und lagern Sie bei -20 oC.
| Vor dem Autoklavieren: | ||
| umzäuntes grundstück | Betrag pro Liter | Endgültige Konzentration |
| Mannitol | 5 g | 27,45 mM |
| Na-Gluconat | 5 g | 22,92 mM |
| Hefe-Extrakt | 0,5 g | - |
| MgSO4bei 7H2O | 0,2 g | 0,81 mM |
| Nacl | 0,1 g | 1,71 mM |
| K2HPO4 | 0,5 g | 2,87 mM |
| Nach dem Autoklavieren: | ||
| umzäuntes grundstück | Betrag pro Liter | Endgültige Konzentration |
| 1,5 M CaCl2 | 1 ml | 1,5 mM |
Tabelle 2: Zusammensetzung des Hefe-Mannitol-Mediums (YEM), das für den Anbau von Rhizobium verwendet wird. Stellen Sie den pH-Wert auf 7,0 ein und füllen Sie ihn mit reinem Randwasser auf 1 L. Um das agar-verfestigte YEM-Medium vorzubereiten, fügen Sie 15 g Mikroagar vor dem Autoklavieren hinzu.
| Vor dem Autoklavieren: | |||
| umzäuntes grundstück | Lagerkonzentration | Menge pro Liter medium | Endgültige Konzentration |
| KH2PO4 | 0,44 M | 2 ml hinzufügen | 0,88 mM |
| K2HPO4 | 1,03 M | 2 ml hinzufügen | 2,07 mM |
| 500x Mikro-Elemente-Lagerlösung | - | 2 ml hinzufügen | - |
| MES pH=6,6 | 1 M | Hinzufügen von 3 ml | 3 mM |
| Hcl | 1 M | pH auf 6,6 einstellen | - |
| Ultrareines Wasser | - | Füllung auf 990 ml | - |
| Nach dem Autoklavieren: | |||
| umzäuntes grundstück | Lagerkonzentration | Menge pro Liter medium | Endgültige Konzentration |
| MgSO4bei 7H2O | 1,04 M | 2 mL | 2,08 mM |
| Na2SO4 | 0,35 M | 2 mL | 0,70 mM |
| NH4NR.3 | 0,18 M | 2 mL | 0,36 mM |
| CaCl2bei 2H2O | 0,75 M | 2 mL | 1,5 mM |
| Fe(III)-Citrat | 27 mM | 2 mL | 54 m |
Tabelle 3: Zusammensetzung von 1 L modifiziertem EKM Medium31, das für Denp. andersonii Nodulationstest verwendet wird. Die Zusammensetzung der 500x Mikroelement-Stammlösung ist in Tabelle 4 aufgeführt. Um 2% agar-verfestigtes EKM-Medium vorzubereiten, 20 g Daishin-Agar vor dem Autoklavieren hinzufügen. Autoklaven Siedie MgSO 4-7H2O-, Na 2-SO-4-, CaCl-2-H2-O- und Fe(III)-Citrat-Bestände, um sie zu sterilisieren. Filter sterilisieren NH4NO3 Lagerlösung zu sterilisieren.
| umzäuntes grundstück | Betrag pro Liter | Lagerkonzentration |
| MnSO4 | 500 mg | 3,31 mM |
| ZnSO4bei 7H2O | 125 mg | 0,43 mM |
| CuSO4bei 5H2O | 125 mg | 0,83 mM |
| H3BO3 | 125 mg | 2,02 mM |
| Na2MoO4bei 2H2O | 50 mg | 0,21 mM |
Tabelle 4: Zusammensetzung der 500x Mikroelement-Stammlösung zur Herstellung modifizierter EKM-Mittel. Bewahren Sie die Mikroelement-Lagerlösung bei 4 °C auf.
| Verbindungen | Lagerkonzentration | Menge pro Liter medium | Endgültige Konzentration |
| K2HPO4 | 20 mM | 1 ml | 0,2 mM |
| NH4NR.3 | 0,28 M | 10 ml | 2,8 mM |
| MgSO4 | 40 mM | 10 ml | 0,4 mM |
| K2SO4 | 40 mM | 10 ml | 0,4 mM |
| Fe(II)-EDTA | 9 mM | 10 ml | 0,9 mM |
| CaCl2 | 80 mM | 10 ml | 0,8 mM |
| 50x Mikro-Elemente-Lagerlösung | - | 10 ml | - |
Tabelle 5: Zusammensetzung des 1/2-Hoagland32 Mediums, das für Mykorrhiisationstests verwendet wird. Die Zusammensetzung der 50-fachen Mikroelement-Stammlösung ist in Tabelle 6 aufgeführt. Bereiten Sie die Fe(II)-EDTA-Lösung vor, indem Sie FeSO47H2O (9 mM) und Na2 EDTA (9 mM) in 1 Lagerlösung einlagern und bei 4 °C lagern. Stellen Sie den pH-Wert des Mediums mit 1 M KOH auf 6,1 ein und füllen Sie ihn mit reinem reinem Wasser auf 1 L.
| Verbindungen | Betrag pro Liter | Lagerkonzentration |
| H3BO3 | 71,1 mg | 1,15 mM |
| MnCl24H2O | 44,5 mg | 0,22 mM |
| CuSO4bei 5H2O | 3,7 mg | 23,18 M |
| ZnCl2 | 10,2 mg | 74,84 M |
| Na2MoO4'2H2O | 1,2 mg | 4,96 Mio. m |
Tabelle 6: Zusammensetzung der 50x Mikroelement-Stammlösung zur Herstellung des 1/2-Hoagland-Mediums.
| Alter der Expflanzen | Transformationseffizienz |
| Jung | 69,4 bis 6,2 % (n = 2) |
| Reifen | 18,3 bis 10,2% (n = 15) |
Tabelle 7: Transformationseffizienz von P. andersonii. Hier wird die Transformationseffizienz definiert als der Prozentsatz der Expflanzen, die mindestens 1 transgenen Callus bilden oder schießen. Die Transformationseffizienz wurde nach 6 Wochen nach der Transformation bewertet und wird als Mittelwert dargestellt. n gibt die Anzahl der Transformationsexperimente an, aus denen die Transformationseffizienz bestimmt wurde.
Ergänzende Datei 1: Übersicht über Level-1- und Level 2-Konstrukte, die für CRISPR/Cas9-Mutagenese verwendet werden. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Discussion
Hülsenfrüchte und die entfernt verwandte Cannabaceae-Gattung Parasponia stellen die einzigen beiden Klades von Pflanzenarten dar, die in der Lage sind, eine endosymbiotische Beziehung zu stickstofffixierenden Rhizobia herzustellen und Wurzelknollen zu bilden. Vergleichende Studien zwischen Arten beider Klades sind hoch relevant, um Einblicke in die genetischen Kernnetzwerke zu geben, die diese Symbiose ermöglichen. Derzeit werden genetische Studien hauptsächlich in Hülsenfrüchte durchgeführt; insbesondere die beiden Modellarten M. truncatula und L. japonicus. Um eine zusätzliche experimentelle Plattform zu schaffen und vergleichende Studien mit einer nickenden Nicht-Legume zu ermöglichen, beschreiben wir hier ein detailliertes Protokoll für stabile Transformation und umgekehrte genetische Analysen in P. andersonii. Das vorgestellte Protokoll verwendet die In-vitro-Vermehrung von T0 transgenen P. andersonii-Linien, so dass die phänotypische Analyse innerhalb von 4 Monaten nach der Kokultivierung von A. tumefaciens eingeleitet werden kann. Dies ist wesentlich schneller als aktuelle Protokolle, die für eine stabile Transformation von Hülsenfrüchte33eingerichtet wurden. Das macht P. andersonii zu einem attraktiven Forschungsmodell.
Das hier beschriebene Protokoll enthält mehrere kritische Schritte. Die erste betrifft die Keimung von Samen. Um P. andersonii Samen für die Keimung vorzubereiten, müssen die Samen von den Beeren isoliert werden. Dies geschieht durch Reiben der Beeren auf einem Stück Tissuepapier oder gegen das Innere eines Teesiebes. Dieses Verfahren muss schonend durchgeführt werden, um Schäden an der Samenschicht zu vermeiden. Wenn die Samenschicht beschädigt wird, könnte Bleichmittel während der Sterilisation in das Saatgut gelangen, was die Samenlebensfähigkeit verringert. Um die Saatruhe zu durchbrechen, werden die Samen einem Temperaturzyklus von 10 Tagen unterzogen. Trotz dieser Behandlung ist die Keimung jedoch nicht vollständig synchronisiert. Im Allgemeinen zeigen die ersten Samen Radicle-Emergence nach 7 Tagen, aber andere können mehrere Tage länger dauern, um zu keimen.
Kritische Punkte im Transformationsverfahren betreffen die Wahl des Ausgangsmaterials und die Dauer des Kokultivierungsschritts. Um eine effiziente Transformation zu erreichen, ist es am besten, gesunde und junge Stiele oder Blütenvon nicht sterilen Gewächshauspflanzen als Ausgangsmaterial zu verwenden. Um das Wachstum junger Zweige zu induzieren, ist es ratsam, Parasponia-Bäume alle 2-3 Monate zu trimmen und bäume einmal im Jahr zu erfrischen. Zusätzlich muss der Kokulturschritt nur für 2 Tage durchgeführt werden. Die verlängerte Kokultivierung fördert die Überkolonialisierung von Gewebeexpflanzen durch A. tumefaciens und reduziert in der Regel die Transformationseffizienz. Um eine Überkolonialisierung durch A. tumefaciens zu verhindern, ist es auch wichtig, die Platten, auf denen die Expflanzen angebaut werden, regelmäßig aufzufrischen. Im Falle einer Überbesiedlung könnten Gewebeexplanten gewaschen werden (siehe Abschnitt 3.8), um A. tumefaciens-Zellen zu entfernen. Wir empfehlen, der zum Waschen verwendeten SH-10-Lösung Bleichmittel hinzuzufügen (Endkonzentration: 2 % Hypochlorit). Es ist wichtig zu beachten, dass dieser zusätzliche Waschschritt bei stark infizierten Explanten möglicherweise nicht funktioniert (Abbildung 2B). Ergibt eine Transformation mit einem CRISPR/Cas9-Konstrukt nur eine begrenzte Anzahl von vermeintlich transformierten Trieben oder wenn die Mutagenese eines bestimmten Gens Probleme bei der Regeneration verursachen dürfte, ist es ratsam, ein leeres Vektorkontrollkonstrukt als die positive Kontrolle. Schließlich ist es wichtig sicherzustellen, dass alle ausgewählten transgenen Linien aus unabhängigen T-DNA-Integrationsereignissen resultieren. Daher weisen wir an, nur einen einzigen vermeintlich transgenen Trieb von jeder Seite einer Explantation zu nehmen. Wir sind uns jedoch bewusst, dass dies die potenzielle Anzahl unabhängiger Linien reduziert. Wenn viele Leitungen benötigt werden, könnten die Forscher entscheiden, vermeintlich transformierte Calli von den ursprünglichen Explants zu trennen, wenn diese Calli 2 mm groß sind und diese Calli unabhängig voneinander bebauen. Auf diese Weise könnten mehrere Linien von jeder Explantation isoliert werden, was die Anzahl potenzieller transgener Linien erhöht.
Im aktuellen Protokoll werden transgene Linien von P. andersonii vegetativ durch In-vitro-Vermehrung vermehrt. Der Vorteil dabei ist, dass viele transgene Pflanzen in relativ kurzer Zeit erzeugt werden können. Diese Methode hat jedoch auch mehrere Einschränkungen. Erstens ist die Aufrechterhaltung von T0 transgenen Linien durch In-vitro-Vermehrung arbeitsintensiv und könnte zu unerwünschten genetischen oder epigenetischen Veränderungen 34,35führen. Zweitens enthalten T 0-Linien noch eine Kopie der T-DNA, einschließlich der Antibiotika-Resistenzkassette. Dadurch wird die Anzahl möglicher Umtransformationen begrenzt, da für jede Neutransformation unterschiedliche Auswahlmarkierungen erforderlich sind. Derzeit haben wir die Transformation nur mit Kanamycin- oder Hygromycin-Auswahl getestet (Daten werden nicht angezeigt). Darüber hinaus erschwert das Vorhandensein der Cas9-Kodierungssequenz und der sgRNAs in den transgenen T 0-Linien Komplementierungsstudien. Komplementierungs-Assays sind möglich, erfordern aber, dass die sgRNA-Ziel-Site(n) als solche mutiert wird, damit die Genbearbeitung des Komplementierungskonstrukts verhindert wird. Drittens ist ein Nachteil der Arbeit mit T 0-Linien, dass CRISPR/Cas9-Mutanten chimerisch sein könnten. Um eine phänotypische Analyse von chimermutierten Linien zu verhindern, empfehlen wir, die Genotypisierungsanalyse nach In-vitro-Vermehrung an mindestens 3 verschiedenen Trieben zu wiederholen. Obwohl die Anzahl der chimarischen Mutanten, die mit demhier beschriebenen Protokoll erhalten wurden, begrenzt ist, werden sie gelegentlich 10 beobachtet. Um die Grenzen der Arbeit mit T 0-Linien zu überwinden, könnten P. andersonii mutierte Linien generativ propagiert werden. P. andersonii Bäume sind dioecious und windbefruchtet2. Das bedeutet, dass jede transgene Linie als solche manipuliert werden muss, dass männliche und weibliche Blüten auf einer einzigen Person produziert werden und anschließend als solche angebaut werden, dass eine Kreuzbestäubung nicht auftritt. Da P. andersonii ein schnell wüchsender Baum ist, benötigt er viel Platz in einem tropischen Gewächshaus (28 °C, 85 % relative Luftfeuchtigkeit). Daher ist die generative Vermehrung von transgenen P. andersonii-Linien, obwohl technisch möglich, logistisch anspruchsvoll.
Im Protokollabschnitt haben wir 3 Methoden zur Nodulation von P. andersoniibeschrieben. Der Vorteil der Platten- und Beutelsysteme ist, dass die Wurzeln leicht zugänglich sind, was eine Spot-Impfung von Bakterien und die anschließende Knötulation im Laufe der Zeit ermöglichen kann. Das Plattensystem ist jedoch recht arbeitsintensiv, was es weniger geeignet für groß angelegte Nodulationsexperimente macht. Ein Nachteil des Beutelsystems ist, dass es schwierig ist, Pilzkontaminationen zu verhindern. Beutel sind nicht steril, und daher wird Pilzwachstum oft auf der oberen Hälfte des Beutels beobachtet. Dies wirkt sich jedoch nicht auf das Wachstum von P. andersonii aus und stört daher nicht die Nodulationstests. Zusätzlich ist das Beutelsystem nur für Sämlinge geeignet. Trotz mehrerer Versuche konnten wir keine Pflanzen, die durch In-vitro-Vermehrung in Beuteln gewonnen wurden, anbauen.
Die hier beschriebene P. andersonii Reverse Genetics Pipeline bietet eine wesentliche Verbesserung gegenüber der bestehenden A. rhizogenes-basiertenWurzeltransformationsmethode11. Mit den beschriebenen Verfahren können stabile transgene Leitungen effizient erzeugt und durch In-vitro-Vermehrung aufrechterhalten werden. Im Gegensatz dazu ist die Umwandlung von A. rhizogenes vorübergehend und führt nur zur Bildung transgener Wurzeln. Da jede transgene Wurzel aus einer unabhängigen Transformation resultiert, leiden A. rhizogenes transformationsbasierte Assays unter erheblichen phänotypischen Variationen. Bei stabilen Linien ist diese Streuung viel geringer, obwohl die In-vitro-Vermehrung auch eine gewisse Streuung erzeugt. Aufgrund dieser reduzierten Variation und der Tatsache, dass mehrere Pflanzenfürst für jede stabile Linie phänotypisiert werden konnten, eignen sich stabile Linien besser für quantitative Assays als a. rhizogenes-transformierteWurzeln. Darüber hinaus hängt die stabile Transformation nicht von der Einführung der A. rhizogenes Wurzel induzierenden Lokus (rol) ab, die den endogenen Hormonhaushalt15beeinflusst. Daher eignen sich stabile Linien besser für die umgekehrte genetische Analyse von Genen, die an der Hormonhomöostase beteiligt sind, im Vergleich zu A. rhizogenes-transformierten Wurzeln. Ein allgemeinerer Vorteil von P. andersonii als Forschungsmodell ist, dass es keine kürzliche ganze Genomduplizierung (WGD) erfahren hat. Die Legume Papilionoideae Unterfamilie, die das Modell Hülsenfrüchte M. truncatula und L. japonicusumfasst, sowie die Salicaceae (Ordnung Malpighiales), die den Modellbaum Populus trichocarpa erfahrene WGDs umfasst 65 vor 1million Jahren36,37. Viele paralogische Genkopien, die sich aus diesen WGDs ergeben, werden in den Genomen von M. truncatula, L. japonicus und P. trichocarpa37,38,39, beibehalten, was Redundanz, die umgekehrte genetische Analysen erschweren könnte. Da P. andersonii vor kurzem keine WGD erlebt hat, könnten umgekehrte genetische Analysen auf P. andersonii weniger durch redundante Funktion paralogusischer Genkopien beeinflusst werden.
Zusammen stellen wir ein detailliertes Protokoll für die umgekehrte genetische Analyse in P. andersoniizur Verfügung. Mit diesem Protokoll können einzelne mutierte Leitungen effizient in einem Zeitrahmen von 2-3 Monaten10erzeugt werden. Dieses Protokoll kann erweitert werden, um Mutanten höherer Ordnung durch Multiplexing von sgRNAs zu erzeugen, die auf verschiedene Gene gleichzeitig abzielen, wie für andere Pflanzenarten40,41,42gezeigt. Darüber hinaus beschränkt sich das hier beschriebene stabile Transformationsverfahren nicht auf CRISPR/Cas9-Gen-Targeting, sondern könnte auch zur Einführung anderer Arten von Konstrukten (z.B. für Promoter-Reporter-Assays, ektopische Expression oder Trans- Ergänzung). Wir haben P. andersonii als vergleichendes Forschungsmodell etabliert, um mutualistische Symbiosen mit stickstofffixierenden Rhizobia oder Endomykorrhizapilzen zu untersuchen. Die hier beschriebenen Protokolle bieten jedoch auch Werkzeuge, um andere Aspekte der Biologie dieses tropischen Baumes zu untersuchen, wie die Holzbildung, die Entwicklung von bisexuellen Blüten oder die Biosynthese von Cannabaceae-spezifischen sekundären Metaboliten.
Acknowledgements
Die Autoren würdigen gerne Mark Youles, Sophien Kamoun und Sylvestre Marillonnet dafür, dass sie Golden Gate-Klonteile über die Addgene-Datenbank verfügbar gemacht haben. Darüber hinaus möchten wir E. James, P. Hadobas und T. J. Higgens für P. andersonii Samen danken. Diese Arbeit wurde von der niederländischen Organisation für wissenschaftliche Forschung (NWO-VICI-Stipendium 865.13.001; NWO-Open Competition Grant 819.01.007) und das Ministerium für Forschung, Technologie und Hochschulbildung der Republik Indonesien (RISET-PRO Grant 8245-ID).
Materials
| Name | Company | Catalog Number | Comments |
| Sigma-Aldrich | N0640 | NAA | |
| Duchefa Biochemie | M1503.0250 | MES | |
| Sigma-Aldrich | D134406 | Acetosyringone | |
| Duchefa Biochemie | X1402.1000 | X-Gal | |
| Merck | 101236 | For nucleic acid electrophoresis gel | |
| - | - | Pouches box material, hangers | |
| Merck | 101188 | NH4NO3 | |
| Sigma-Aldrich | B3408-1G | BAP | |
| Merck | 100156 | H3BO3 | |
| Thermo-Fisher | ER1011 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Thermo-Fisher | 15561020 | Used in Golden Gate cloning assembly | |
| Merck | 137101 | CaCl2·2H2O | |
| Duchefa Biochemie | C0111.0025 | C16H16N5O7S2Na | |
| Thermo-Fisher | K1231 | Used for cloning the blunt-ended PCR amplicons in genotyping procedure | |
| Agronutrition | AP2011 | Containing Rhizophagus irregularis DAOM 197198 (1,000 spores/mL), used for mychorrization assay | |
| Merck | 102790 | CuSO4·5H2O | |
| Duchefa Biochemie | D1004.1000 | Used for plant tissue culture agar-based medium | |
| Merck | 105101 | K2HPO4 | |
| VWR Chemicals | 20302.293 | Na2·EDTA | |
| Duchefa Biochemie | M0803.1000 | C6H14O6 | |
| Thermo-Fisher | ER0291 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Merck | 100983 | C2H5OH | |
| VWR Chemicals | BDH9232-500G | EDTA | |
| Sigma-Aldrich | Z377600-1PAK | Cellophane membrane | |
| Biomatters, Ltd. | R9 or higher | Bioinformatics software for in silico cloning and designing of sgRNAs | |
| Mega International | - | Technical information at https://mega-international.com/tech-info/ | |
| Sigma-Aldrich | 65882 | Used for fixating nodule tissues | |
| VWR Chemicals | 24385.295 | - | |
| Vink | 219341 | Pouches box material, bottom part | |
| Leica Biosystems | 14702218311 | Used as a template for plastic embedding | |
| Merck | 100317 | HCl | |
| Sigma-Aldrich | I5386-1G | IBA | |
| Merck | 103862 | C6H5FeO7 | |
| Merck | 103965 | FeSO4O·7H2O | |
| Duchefa Biochemie | I1401.0005 | IPTG | |
| Duchefa Biochemie | K0126.0010 | ||
| Sigma-Aldrich | L2000 | ||
| Merck | 105886 | MgSO4O·7H2O | |
| Merck | 105934 | MnCl2·4H2O | |
| Merck | 102786 | MnSO4O | |
| Duchefa Biochemie | M1002.1000 | Used for bacterial culture agar-based medium | |
| Manutan | 92007687 | Pouches material | |
| Paraxisdienst | 130774 | Elastic sealing foil | |
| Pull Rhenen | Agra-Perlite No.3 | Used as growing substrate in pots for nodulation assay | |
| VWR Chemicals | 391-0581 | Used as container for cellophane membranes | |
| Thermo-Fisher | F130WH | For genotyping transgenic lines | |
| Addgene | 50337 | Level 0 terminator, 3'UTR, 35s (Cauliflower Mosaic Virus) | |
| Addgene | 48017 | End-link 2 for assembling 2 level one part into a level 2 acceptor | |
| Addgene | 48018 | End-link 3 for assembling 3 level one part into a level 2 acceptor | |
| Addgene | 48001 | Level 1 acceptor. Position 5. Forward orientation | |
| Addgene | 48007 | Level 1 Acceptor. Position 1. Reverse orientation | |
| Addgene | 50268 | Level 0 promoter (0.4 kb), 35s (Cauliflower Mosaic Virus) + 5'UTR, Ω (Tobacco Mosaic Virus) | |
| Addgene | 46966 | Used for designing CRISPR/Cas9 module | |
| Addgene | 46968 | Used for designing CRISPR/Cas9 module | |
| Addgene | 50334 | Level 0 Kanamycin/Neomycin/Paromomycin resistance cassette | |
| Topzeven | - | Used as filters for washing spore suspension | |
| Sigma-Aldrich | 8.17003 | PEG400 | |
| Duchefa Biochemie | E1674.0001 | Pots to grow Parasponia plantlets/seedlings | |
| Merck | 104871 | KH2PO4 | |
| Merck | 105033 | KOH | |
| Merck | 105153 | K2SO4O | |
| Van Leusden b.v. | - | Used as growing substrate for mychorrhization assay | |
| Duchefa Biochemie | S0225.0050 | SH-basal salt medium | |
| Duchefa Biochemie | S0411.0250 | SH-vitamin mixture | |
| Lehle Seeds | VIS-02 | Used as non-ionic surfactant in the washing step of stable transformation | |
| Merck | 137017 | NaCl | |
| VWR Chemicals | 89230-072 | C6H11NaO7 | |
| Merck | 106521 | Na2MoO4·2H2O | |
| Merck | 106574 | Na2HPO4·7H2O | |
| Merck | 567549 | NaH2PO4·H2O | |
| Sigma-Aldrich | 239313 | Na2SO4O | |
| Duchefa Biochemie | S0809.5000 | C12H22O11 | |
| Thermo-Fisher | B69 | Used in Golden Gate cloning assembly | |
| Thermo-Fisher | EL0013 | Used in Golden Gate cloning assembly | |
| Kulzer-Mitsui Chemicals Group | 64708806 | Methyl methacrylate-based resin powder | |
| Kulzer-Mitsui Chemicals Group | 64709003 | HEMA (2-hydroxyethyl methacrylate)-based resin solution | |
| Kulzer-Mitsui Chemicals Group | 66022678 | Methyl methacrylate-based resin solution | |
| Merck | 1159300025 | ||
| Acros | 189350250 | ||
| VWR Chemicals | 663684B | Polysorbate 20 | |
| Stout Perspex | - | pouches box material, lid | |
| Duchefa Biochemie | Y1333.1000 | ||
| Merck | 108816 | ZnCl2 | |
| Alfa Aesar | 33399 | ZnSO4O·7H2O |
References
- Clason, E. W. THE VEGETATION OF THE UPPER-BADAK REGION OF MOUNT KELUT (EAST JAVA). Bulletin du Jardin Botanique de Buitenzorg. Serie III, 509-518 (1936).
- Soepadmo, E. Ulmaceae. Flora Malesiana-Series 1, Spermatophyta. 8, 31-76 (1974).
- Becking, J. H. The Rhizobium symbiosis of the nonlegume Parasponia. Biological Nitrogen Fixation. , 497-559 (1992).
- Oldroyd, G. E. D. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nature Reviews Microbiology. 11, 252-263 (2013).
- Gutjahr, C., Parniske, M. Cell and developmental biology of arbuscular mycorrhiza symbiosis. Annual Review of Cell and Developmental Biology. 29, 593-617 (2013).
- van Velzen, R., et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proceedings of the National Academy of Sciences of the United States of America. 115, E4700-E4709 (2018).
- van Velzen, R., Doyle, J. J., Geurts, R. A Resurrected Scenario: Single Gain and Massive Loss of Nitrogen-Fixing Nodulation. Trends in Plant Science. 24, 49-57 (2019).
- Griesmann, M., et al. Phylogenomics reveals multiple losses of the nitrogen-fixing root nodule symbiosis. Science. 1743, eaat1743 (2018).
- Becking, J. H. Root-Nodule Symbiosis Between Rhizobium And Parasponia (Ulmaceae). Plant and Soil. 51, 289-296 (1979).
- van Zeijl, A., et al. CRISPR/Cas9-Mediated Mutagenesis of Four Putative Symbiosis Genes of the Tropical Tree Parasponia andersonii Reveals Novel Phenotypes. Frontiers in Plant Science. 9, 284 (2018).
- Cao, Q., et al. Efficiency of Agrobacterium rhizogenes–mediated root transformation of Parasponia and Trema is temperature dependent. Plant Growth Regulation. 68, 459-465 (2012).
- Limpens, E., et al. RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicago truncatula. Journal of Experimental Botany. 55, 983-992 (2004).
- Boisson-Dernier, A., et al. Agrobacterium rhizogenes-Transformed Roots of Medicago truncatula for the Study of Nitrogen-Fixing and Endomycorrhizal Symbiotic Associations. Molecular Plant-Microbe Interactions. 14, 695-700 (2001).
- Kumagai, H., Kouchi, H. Gene Silencing by Expression of Hairpin RNA in Lotus japonicus Roots and Root Nodules. Molecular Plant-Microbe Interactions. 16, 663-668 (2003).
- Nilsson, O., Olsson, O. Getting to the root: The role of the Agrobacterium rhizogenes rol genes in the formation of hairy roots. Physiologia Plantarum. 100, 463-473 (1997).
- Davey, M. R., et al. Effective Nodulation of Micro-Propagated Shoots of the Non-Legume Parasponia andersonii by Bradyrhizobium. Journal of Experimental Botany. 44, 863-867 (1993).
- Webster, G., Poulton, P. R., Cocking, E. C., Davey, M. R. The nodulation of micro-propagated plants of Parasponia andersonii by tropical legume rhizobia. Journal of Experimental Botany. 46, 1131-1137 (1995).
- Op den Camp, R., et al. LysM-type mycorrhizal receptor recruited for rhizobium symbiosis in nonlegume Parasponia. Science. 331, 909-912 (2011).
- Weber, E., Engler, C., Gruetzner, R., Werner, S., Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLOS ONE. 6, e16765 (2011).
- Nekrasov, V., Staskawicz, B. J., Weigel, D., Jones, J. D. G., Kamoun, S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nature Biotechnology. 31, 691-693 (2013).
- Bertani, G. Studies On Lysogenesis. I. The Mode Of Phage Liberation By Lysogenic Escherichia Coli. Journal of Bacteriology. 62, 293-300 (1951).
- Engler, C., et al. A Golden Gate modular cloning toolbox for plants. ACS Synthetic Biology. 3, 839-843 (2014).
- Fauser, F., Schiml, S., Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. The Plant Journal. 79, 348-359 (2014).
- Lazo, G. R., Stein, P. A., Ludwig, R. A. A DNA Transformation-Competent Arabidopsis Genomic Library in Agrobacterium. Biotechnology. 9, 963-967 (1991).
- Op den Camp, R. H. M., et al. N Nonlegume Parasponia andersonii Deploys A Broad Rhizobium Host Range Strategy Resulting in Largely Variable Symbiotic Effectivenes. Molecular Plant-Microbe Interactions. 25, 954-963 (2012).
- Graham, P. H., Viteri, S. E., Mackie, F., Vargas, A. T., Palacios, A. Variation in acid soil tolerance among strains of Rhizobium phaseoli. Field Crops Research. 5, 121-128 (1982).
- Martinez-Romero, E., et al. Rhizobium tropici, A Novel Species Nodulating Phaseolus vulgaris L. Beans and Leucaena sp. Trees. International Journal of Systematic and Evolutionary Microbiology. 41, 417-421 (1991).
- Felten, J., et al. The Ectomycorrhizal Fungus Laccaria bicolor Stimulates Lateral Root Formation in Poplar and Arabidopsis through Auxin Transport and Signaling. Plant Physiology. 151, 1991-2005 (1991).
- Trouvelot, A., Kough, J. L., Gianinazzi-Pearson, V. Mesure du taux de mycorhization VA d’un systeme radiculaire. Recherche de methods d’estimation ayant une signification fonctionnelle. Aspects Physiologiques et Genetiques des Mycorhizes. , 217-221 (1986).
- Schenk, R. U., Hildebrandt, A. C. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Canadian Journal of Botany. 50, 199-204 (1972).
- Becking, J. H. The Parasponia parviflora - Rhizobium symbiosis. Host specificity, growth and nitrogen fixation under various conditions. Plant and Soil. 75, 309-342 (1983).
- Hoagland, D. R., Arnon Revised, D. I., Arnon, D. I. The Water-Culture Method for Growing Plants without Soil. Circular California Agricultural Experiment Station. 347, 1-32 (1950).
- Wang, K. . Methods in Molecular Biology: Agrobacterium Protocols. 1, (2015).
- Smulders, M. J. M., de Klerk, G. J. Epigenetics in plant tissue culture. Plant Growth Regulation. 63, 137-146 (2011).
- Larkin, P. J., Scowcroft, W. R. Somaclonal variation — a novel source of variability from cell cultures for plant improvement. Theoretical and Applied Genetics. 60, 197-214 (1981).
- Cannon, S. B., et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proceedings of the National Academy of Sciences of the United States of America. 103, 14959-14964 (2006).
- Tuskan, G. A., et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313, 1596-1604 (2006).
- Young, N. D., et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 480, 520 (2011).
- Sato, S., et al. Genome Structure of the Legume, Lotus japonicus. DNA Research. 15, 227-239 (2008).
- Xing, H. L., et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biology. 14, 327 (2014).
- Lowder, L. G., et al. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiology. 169, 971 (2015).
- van Zeijl, A. . Dissecting Hormonal Pathways in Nitrogen-Fixing Rhizobium Symbioses. , (2017).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved