Transforming, Genome Editing and Phenoty the Nitrogen-fixing Tropical Cannabaceae Tree Parasponia andersonii (en plus de la transformation, de l'édition du génome et du phénotypage de l'arbre tropical de Cannabaceae parasponia andersonii)
Dans cet article
Résumé
Parasponia andersonii est un arbre tropical à croissance rapide qui appartient à la famille du cannabis (Cannabaceae) et peut former des nodules de racine fixant l'azote en association avec le rhizobium. Ici, nous décrivons un protocole détaillé pour les analyses génétiques inversées dans P. andersonii basé sur la transformation stable d'Agrobacterium-négociée et l'édition de génome CRISPR/Cas9-basée.
Résumé
Parasponia andersonii est un arbre tropical à croissance rapide qui appartient à la famille cannabis (Cannabaceae). Avec 4 espèces supplémentaires, il forme la seule lignée connue de non-légumineuses capable d'établir une symbiose nodule fixatrice d'azote avec le rhizobium. Des études comparatives entre les légumineuses et P. andersonii pourraient fournir un aperçu précieux des réseaux génétiques sous-jacents à la formation de nodules racinaires. Pour faciliter les études comparatives, nous avons récemment séquencé le génome de P. andersonii et établi Agrobacterium tumefaciens-mediated transformation stable et CRISPR/Cas9-based genome editing. Ici, nous fournissons une description détaillée des procédures de transformation et d'édition du génome développées pour P. andersonii. En outre, nous décrivons des procédures pour la germination et la caractérisation de graine des phénotypes symbiotiques. Grâce à ce protocole, des lignées mutantes transgéniques stables peuvent être générées en 2-3 mois. La propagation in vitro végétative des lignées transgéniques T0 permet d'initier des expériences de phénotypage 4 mois après la coculture de A. tumefaciens. Par conséquent, ce protocole ne prend que légèrement plus de temps que la méthode transitoire de transformation des racines agrobacterium rhizogenesdisponible pour P. andersonii, mais offre plusieurs avantages évidents. Ensemble, les procédures décrites ici permettent à P. andersonii d'être utilisé comme modèle de recherche pour des études visant à comprendre les associations symbiotiques ainsi que potentiellement d'autres aspects de la biologie de cet arbre tropical.
Introduction
Parasponia andersonii est un arbre tropical appartenant à la famille Cannabis (Cannabaceae) et est originaire de Papouasie-Nouvelle-Guinée et de plusieurs îles du Pacifique1,2,3. Avec 4 espèces supplémentaires de Parasponia, il représente la seule lignée non légumineuse qui peut établir une symbiose nodule fixatrice d'azote avec le rhizobia. Cette symbiose est bien étudiée dans les modèles de légumineuses (Fabaceae) Medicago truncatula et Lotus japonicus, ce qui a abouti à l'acquisition de connaissances détaillées sur la nature génétique moléculaire de la formation de nodule et le fonctionnement4. En outre, il a été démontré que la symbiose nodule racine dans les légumineuses est fondée sur la symbiose mycorhizique arbusculaire beaucoup plus ancienne et répandue5. Les comparaisons phylogenomiques suggèrent que les symbioes nodules fixant l'azote des légumineuses, Parasponia,ainsi que, les espèces dites de plante actinorhizal qui hébergent les bactéries diazotrophiques de Frankia, ont une origine évolutive partagée 6,7,8. Pour déterminer si les gènes identifiés pour être impliqués dans la formation de nodule de légumineuses font partie d'une base génétique conservée, des études sur des espèces non légumineuses sont essentielles. À cette fin, nous proposons d'utiliser P. andersonii comme modèle de recherche comparative, aux côtés des légumineuses, pour identifier les réseaux génétiques de base sous-jacents à la formation et au fonctionnement des nodules racinaires.
P. andersonii est un pionnier que l'on peut trouver sur les pentes des collines volcaniques. Il peut atteindre des vitesses de croissance de 45 cm par mois et atteindre des longueurs allant jusqu'à 10 mètres9. Les arbres P. andersonii sont pollinisés par le vent, ce qui est facilité par la formation de fleurs mâles et femelles séparées3,10. Nous avons récemment séquencé et annoté le génome diploïde (2n - 20; 560 Mb/1C) de P. andersonii, et assemblé des séquences de génome de projet de 2 autres espèces de Parasponia; P. rigida et P. rugosa6. Cela a révélé 35 000 modèles de gènes P. andersonii qui peuvent être regroupés en 20 000 orthèses avec des gènes de M. truncatula, soja (Glycine max), Arabidopsis thaliana, fraise des bois ( Fragaria vesca), Trema orientalis, peuplier de coton noir (Populus trichocarpa) et eucalyptus (Eucalyptus grandis)6. De plus, les comparaisons de transcriptome entre M. truncatula et P. andersonii ont permis d'identifier un ensemble de 290 orthologues putatifs qui présentent un modèle d'expression amélioré par la nodule chez les deux espèces6. Cela constitue une excellente ressource pour les études comparatives.
Pour étudier la fonction génique dans les racines et les nodules de P. andersonii, un protocole pour la transformation des racines médiées par Agrobacterium rhizogenesa été établi11. À l'aide de ce protocole, les plantes composées portant des racines transgéniques peuvent être générées dans un laps de temps relativement court. Cette méthode est, aussi, largement appliquée dans la recherche de légumineuse-symbiose12,13,14. Cependant, l'inconvénient de cette méthode est que seules les racines sont transformées et que chaque racine transgénique représente un événement de transformation indépendant, entraînant une variation substantielle. En outre, la transformation est transitoire et les lignes transgéniques ne peuvent pas être maintenues. Cela rend la transformation des racines basée sur A. rhizogenesmoins adaptée à l'édition du génome à médiation CRISPR/Cas9. En outre, A. rhizogenes transfère ses gènes induisant la racine locus (rol) au génome de la plante, qui une fois exprimé interférer avec l'homéostasie hormonale15. Cela rend difficile l'étude du rôle des hormones végétales dans A. rhizogenes- racines transformées. Pour surmonter ces limitations, nous avons récemment développé un protocole pour la transformation basée sur Agrobacterium tumefacienset la mutagenèse CRISPR/Cas9-négociée de P. andersonii10.
Ici, nous fournissons une description détaillée de la procédure de transformation a. tumefacienset du pipeline de génétique inversée développé pour P. andersonii. En outre, nous fournissons des protocoles pour la manipulation en aval des plantules transgéniques, y compris des essais pour étudier les interactions symbiotiques. En utilisant le protocole décrit ici, plusieurs lignes transgéniques peuvent être générées dans une période de 2-3 mois. En combinaison avec la mutagénèse CRISPR/Cas9- négociée, cela permet une génération efficace de lignes mutantes knock-out. Ces lignées mutantes peuvent être végétativement propagées in vitro10,16,17, ce qui permet de générer suffisamment de matériel pour commencer la caractérisation phénotypique à 4 mois après la procédure de transformation a été initié10. Ensemble, cet ensemble de procédures devrait permettre à n'importe quel laboratoire d'adopter P. andersonii comme modèle de recherche pour des études visant à comprendre les associations rhizobial et mycorhiziennes, ainsi que potentiellement d'autres aspects de la biologie de cet arbre tropical.
Protocole
1. Cultivez des arbres P. andersonii dans la serre
-
Germer P. andersonii WU1 graines18.
- Utilisez des baies fraîches de Parasponia ou trempez les baies séchées dans l'eau pendant 2 h pour se réhydrater. Squash baies sur un morceau de papier de soie ou frotter contre l'intérieur d'un tamis de thé pour enlever les graines.
- Désinfecter les graines à l'aide d'eau de Javel commerciale (hypochlorite de 4 %) pendant 15 à 20 min et ensuite laver les graines 6 fois à l'aide d'eau stérilisée.
- Transférer les graines dans des tubes stériles de 200 l PCR. Remplissez les tubes d'eau stérilisée, de sorte que les graines sont complètement submergées. Incuber les tubes pendant 10 jours dans un thermocycleur exécutant le programme suivant : 30 cycles (7 oC pour 4 h, 28 oC pour 4 h). N'utilisez pas de couvercle chauffé, car cela pourrait tuer les graines.
- Préparer les plaques SH-0 (voir tableau 1). Transférer les graines dans les assiettes SH-0 et incuber à 28 oC, 16 h :8 h jour:nuit. Fermer les plaques avec 2 couches de papier d'étanchéité élastique pour éviter le séchage pendant l'incubation à 28 oC.
- Une fois que les semis ont mis au point leur premier ensemble de vraies feuilles (3-4 semaines après l'incubation à 28 oC), transférez les semis dans des pots remplis de terre à pot commercial et recouvrez les semis d'une tasse en plastique translucide pour prévenir la dessiccation. Placer les pots dans une salle climatique ou une serre de 28 oC, 85 % de RH, sous un régime de 16 h : 8 h jour : nuit.
- Après 1 semaine, retirer la tasse en plastique translucide. Arrosez les pots régulièrement et lorsque les arbres poussent plus grand supplément avec de l'engrais pour soutenir la croissance.
2. Clonage des constructions pour CRISPR/Cas9-mediated Mutagenesis of P. andersonii
REMARQUE: Les vecteurs de transformation binairestandard peuvent être utilisés pour la transformation stable de P. andersonii. Voici, par exemple, une procédure pour générer des constructions pour CRISPR/Cas9-mediated mutagenesis utilisant le clonage modulaire (par exemple, Golden Gate)19.
- Identifiez les séquences cibles d'ARN de guidage pour le gène (s) d'intérêt, en utilisant un logiciel bioinformatique comportant un outil de conception CRISPR intégré. Choisissez des séquences d'ARN de guidage situées à la 5'-extrémité de la séquence de codage du gène cible pour augmenter les chances d'obtenir des KO complets. Assurez-vous de vérifier les effets hors cible en recherchant contre le génome P. andersonii 6.
REMARQUE: Utilisez 2 sgARN par gène cible, de préférence 200-300 bp à part. Cela peut générer des suppressions qui peuvent être identifiées par PCR et par la suite par l'électrophorèse de gel d'agarose. - Générer des constructions golden Gate de niveau 1 contenant les séquences sgRNA.
- Concevoir des amorces pour amplifier chaque sgRNA individuel en insérant la séquence de guide de 20 pb à la position de N(20) dans la séquence d'amorce suivante : 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAATAGCAAG-3'.
REMARQUE : Si la séquence du guide est égale à GN(19), retirez le G à la fin de 5' de la séquence de guide avant d'insérer dans la séquence d'amorce. - PCR amplifier sgRNAs de pICH86966::AtU6p::sgRNA-PDS20 en utilisant les amorces avant conçus à l'étape 2.2.1 et l'amorce inverse universelle: 5'-TGTGGTCCaAGCGTAATAGACTTTGTAC-3'. Utilisez une polymérase d'ADN stable à haute fidélité et les conditions DE PCR suivantes : 98 oC pour 30 s; 30 cycles (98 oC pour 10 s; 53 oC pour 20 s; 72 oC pour 10 s); 72 oC pendant 7 min. Les réactions réussies de PCR donnent un amplicon de 165 bp.
- La colonne purifie l'amplicon PCR à l'aide d'un kit commercial de purification PCR. Par la suite, mettre en place des réactions Golden Gate à clone sgRNAs derrière l'Arabidopsis thaliana AtU6p petit promoteur d'ARN: 10 ng de l'amplicon sgRNA PCR, 150 ng de pICSL01009::AtU6p20, 60 ng du niveau approprié 1 vecteur d'accepteur, 2 'L de T4 tampon de ligase, 2 l 0,1 % de l'albumine de sérum bovin (BSA), 0,5 l de BsaI, 0,5 l de ligase T4, remplissage à 20 l avec de l'eau ultra-pure. Assurez-vous que tous les sgRNAs sont clonés dans la même orientation pour empêcher la formation d'épingle scapillée.
- Incuber les réactions dans un thermocycleur exécutant le programme suivant : 37 oC pour 20 s; 26 cycles (37 oC pour 3 min; 16 oC pour 4 min); 50 oC pendant 5 min; 80 oC pendant 5 min. Transformer les réactions du Golden Gate à Escherichia coli et à la plaque sur LB moyen21 contenant de l'ampicilline (50 mg/L), x-Gal (200 mg/L) et IPTG (1 mM).
REMARQUE : Préparez des solutions de stock d'IPTG et de X-Gal dans de l'eau ultra-pure et du diméthylformamide, respectivement. Filtrer stériliser les solutions d'ampicilline et de stock IPTG et stocker tous les stocks à -20 oC. Portez des gants lorsque vous manipulez du diméthylformamide. - Sélectionnez des colonies blanches et isolez les plasmides à l'aide d'une trousse commerciale d'isolement plasmide. La séquence vérifie les plasmides isolés avant de continuer avec l'assemblage de niveau 2 du Golden Gate.
- Concevoir des amorces pour amplifier chaque sgRNA individuel en insérant la séquence de guide de 20 pb à la position de N(20) dans la séquence d'amorce suivante : 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAATAGCAAG-3'.
- Assembler le niveau 2 Golden Gate construit pour la transformation stable.
- Effectuer une réaction Golden Gate en utilisant le niveau 1 AtU6p::sgRNA construit (généré sous la section 2.2) ainsi que pICH47802::NPTII, pICH47742:::35S pro::NLS-aCas9::35Ster, le niveau 2 accepteur pICSL4723 et le approprié end-linker (voir Engler et coll.22). Effectuer des réactions comme suit: utiliser 100 fmol de chaque vecteur de donneur et 20 fmol du vecteur d'accepteur et ajouter 2 L de tampon de ligase T4, 2 L de 0,1% BSA, 0,5 'L de BpiI, 0,5 'L de T4 ligase, remplir à 20 'L avec de l'eau ultra-pure.
REMARQUE: Le niveau 1 plasmids pICH47802::NPTII, pICH47742::35S pro::NLS-aCas9::35Ster besoin d'être cloné en premier (voir Fichier supplémentaire 1), comme il est décrit pour les sgRNAs en vertu de l'article 2.220,22 ,23. - Incuber les réactions sous l'étape 2.2.4 et se transformer en E. coli. Plaque sur le milieu LB contenant de la kanamycine. Le lendemain, sélectionnez les colonies blanches et isolez les plasmides. Déterminez l'assemblage plasmide correct par l'analyse de restriction-digestion.
- Effectuer une réaction Golden Gate en utilisant le niveau 1 AtU6p::sgRNA construit (généré sous la section 2.2) ainsi que pICH47802::NPTII, pICH47742:::35S pro::NLS-aCas9::35Ster, le niveau 2 accepteur pICSL4723 et le approprié end-linker (voir Engler et coll.22). Effectuer des réactions comme suit: utiliser 100 fmol de chaque vecteur de donneur et 20 fmol du vecteur d'accepteur et ajouter 2 L de tampon de ligase T4, 2 L de 0,1% BSA, 0,5 'L de BpiI, 0,5 'L de T4 ligase, remplir à 20 'L avec de l'eau ultra-pure.
- Transformez les constructions de niveau 2 à la souche AGL124d'Agrobacterium tumefaciens.
3. Transformation stable de P. andersonii
- Inoculer 2 plaques LB contenant les antibiotiques appropriés avec A. tumefaciens souche AGL1 transformé avec la construction de l'intérêt. Incuber les assiettes à 28 oC pendant 2 jours.
- Récoltez de jeunes branches à partir d'arbres cultivés en serre. Utilisez environ 5 branches de 5-8 cm de longueur pour chaque transformation. Assurez-vous d'utiliser uniquement des branches saines non infectées. Enlevez les feuilles en les coupant en tant que telles que 1 cm2 de tissu foliaire est laissé à l'extrémité de chaque pétiole. Jeter les feuilles.
- Désinfecter le tissu pendant 15 min à l'aide de l'eau de Javel commerciale diluée de 1:1 (hypochlorite après dilution) contenant quelques gouttes de polysorbate 20. Ensuite, rincer le tissu 6 fois avec de l'eau autoclaved.
REMARQUE: Cette étape, ainsi que les étapes suivantes doivent être effectuées à l'intérieur d'une armoire laminaire à flux descendant pour garder les tissus stériles. - Suspendre à nouveau les cellules A. tumefaciens de 1 à 2 plaques dans 25 ml de milieu d'infiltration (voir tableau 1) contenant de l'acétosyringone (20 mg/L) et un surfactant non ionique (0,001 % v/v) pour atteindre une densité optique (OD600) de 5.
REMARQUE: Préparer la solution de stock d'acétosyringone dans 70 % d'éthanol et le stocker à -20 oC. Le surfactant non ionique doit être stérilisé par filtre avant d'ajouter au milieu d'infiltration. - Couper à la fois la tige et le tissu pétiole en morceaux de 1 cm de longueur à l'intérieur de la suspension A. tumefaciens, créant ainsi de nouvelles blessures des deux côtés. Laissez des morceaux de tissu dans la suspension A. tumefaciens pendant 10-30 min.
- Préparer le milieu d'enracinement (voir tableau 1) et ajouter l'acetosyringone (20 mg/L) après l'autoclacnage. Sécher les morceaux de tissu sur un morceau stérile de papier filtre et le placer sur le milieu (10 explants/plaque). Incuber les assiettes dans l'obscurité à 21 oC pendant 2 jours.
REMARQUE: Laisser le milieu refroidir jusqu'à 60 oC avant d'ajouter de l'acetosyringone. - Après 2 jours, inspecter les plaques pour la contamination bactérienne fongique ou évidente (bactéries autres que A. tumefaciens). Les plaques contaminées doivent être jetées.
- Préparer le milieu sH-10 liquide (voir tableau 1). Après l'autoclacation, ajouter polysorbate 20 (0,01%, v/v). Transférer les morceaux de tissu à 10 ml de SH-10 contenant du polysorbate 20. Pendant une période d'au moins 10 min, agiter doucement toutes les 2-3 min pour laver le tissu.
- Laver deux fois de plus avec le SH-10 frais contenant le polysorbate 20. Ces temps,un temps d'incubation de 2-3 min par étape de lavage est suffisant.
- Préparer le milieu d'enracinement (voir tableau 1). Après l'autoclacage, ajouter le cefotaxime (300 mg/L) et la kanamycine (50 mg/L) et verser les assiettes. Pour les transformations secondaires (transformations de lignées transgéniques résistantes à la kanamycine), appliquer la sélection de l'hygromycine (15 mg/L).
- Sécher les morceaux de tissu sur des morceaux stériles de papier filtre. Ensuite, transférer les morceaux de tissu dans les plaques préparées à l'étape 3.9.
- Incuber les assiettes pendant 7 jours à 28 oC, 16 h :8 h jour:nuit. Tous les 2 jours vérifier les plaques pour la contamination fongique ou bactérienne et la croissance excessive de A. tumefaciens. En cas de contamination, transférer les morceaux non infectés dans une assiette fraîche.
- Après 7 jours, transférer les morceaux de tissu au milieu de propagation (voir tableau 1) contenant du cefotaxime (300 mg/L) et de la kanamycine (50 mg/L). Incuber les assiettes à 28 oC, 16 h:8 h jour:nuit. Rafraîchir les plaques une fois par semaine jusqu'à ce que les pousses transgéniques se développent. Assurez-vous de ne transférer que des morceaux de tissus non infectés dans des assiettes fraîches. Jetez les morceaux qui sont envahis par A. tumefaciens.
- Une fois que les pousses putativement transgéniques ont une longueur de 1 cm, les pousses coupées et les cultures indépendamment dans le milieu de propagation contenant du céfotaxime (300 mg/L) et de la kanamycine (50 mg/L). Pour s'assurer que les pousses représentent des transformateurs indépendants, ne prenez qu'une seule pousse de chaque côté d'une explantation.
- Propager végétativement les pousses putativement transgéniques telles que décrites sous l'étape 5.2.
4. Génotypage des pousses putativement transgéniques
- Concevoir des amorces couvrant le site de reconnaissance sgRNA(s). Pour permettre le séquençage de l'amplicon PCR, choisissez des amorces de 150 à 250 pb du site de reconnaissance sgRNA.
- Couper une pointe de feuille (5 mm) de chaque pousse transgénique pour être génotype. De plus, récoltez un échantillon de contrôle de type sauvage.
- Effectuer des réactions PCR de 50 L à l'aide des amorces conçues à l'étape 4.1 et d'un kit commercial pour amplifier directement l'ADN des échantillons de plantes. Alternativement, les réactions de PCR peuvent être exécutées sur l'ADN purifié utilisant une polymérase de haute fidélité.
- Amplicons PCR séparés sur un gel agarose de 1,5 à 2 %.
- Analyser les résultats de l'électrophoresis gel. Vérifier les échantillons produisant plusieurs bandes (plus de 1 allèle) et des amplicônes PCR avec des tailles différentes du type sauvage, ce qui indique la présence d'indels de taille moyenne.
- Séquence amplicons PCR pour identifier les mutations exactes. Pour les échantillons produisant un seul amplicon PCR, les produits PCR peuvent être séquencés directement. Les échantillons qui produisent plus d'une bande après l'électrophoresis de gel ou qui semblent être hétérozygotes après le séquençage direct de l'amplicon de PCR, doivent être clonés dans un vecteur de clonage émoussé d'abord. Par la suite, séquencez plusieurs clones pour chaque échantillon afin d'identifier tous les allèles possibles présents dans l'échantillon.
- Aligner les résultats de séquençage sur le gène d'intérêt et inspecter l'alignement pour vérifier les mutations près du site cible de l'ARSA. Par la suite, vérifiez si ces mutations créent des changements de trame. Jetez les lignes avec 2 alleles et des lignes contenant des mutations dans le cadre.
- Sélectionnez plusieurs lignes pour une analyse plus approfondie.
- Prosaguer des lignes sélectionnées telles que décrites sous l'étape 5.2.
- Lorsque les lignées ont mis au point plusieurs nouvelles pousses, prélever de nouveaux échantillons à partir de 3 pointes de feuilles et répéter les étapes 4,3-4,7. Déterminer si les mutations présentes dans chacun des échantillons provenant de la même lignée ainsi que l'échantillon PCR original sont identiques. Les lignées qui produisent les mêmes mutations dans tous les échantillons sont mutées de façon homogène et peuvent être utilisées pour d'autres expérimentations. Jeter les lignes qui ne donnent pas les mêmes résultats que ces lignes sont chimériques.
5. Préparation de planteulets P. andersonii enracinés pour l'expérimentation
-
Lancer une nouvelle ligne de culture tissulaire de P. andersonii.
- Récoltez les bourgeons axillaires, les jeunes pousses aventureuses ou les tissus foliaires des arbres sains. Alternativement, les semis peuvent être utilisés comme matériau de départ.
- Désinfecter les tissus à l'aide de l'eau de Javel commerciale diluée de 1:1 (hypochlorite après dilution) contenant quelques gouttes de polysorbate 20 pendant 15 min. Ensuite, rincer les tissus 6 fois à l'aide d'eau autoclave.
REMARQUE: Cette étape, ainsi que les étapes suivantes doivent être effectuées à l'intérieur d'un downflow laminaire ou d'un coffret à débit croisé laminaire pour garder les tissus stériles. - Transférer les tissus au milieu de propagation (voir tableau 1). Fermer les assiettes avec 2 couches de papier d'étanchéité élastique et couver les plaques à 28 oC, 16 h :8 h jour:nuit.
- Inspectez les plaques tous les quelques jours pendant les 2 premières semaines pour vous assurer que les tissus sont exempts de contamination fongique ou bactérienne.
- Proxaguer le tissu en plaçant 10 pousses sur une plaque fraîche de milieu de propagation et fermer la plaque avec 2 couches de papier d'étanchéité élastique. Incuber les assiettes à 28 oC, 16 h:8 h jour:nuit. Répétez cette étape toutes les 4 semaines.
- Lorsque les pousses sont de longueur de 1 cm, couper les pousses à leur base et les placer sur le milieu d'enracinement (voir tableau 1). Environ 10 pousses peuvent être placées sur une seule plaque d'enracinement. Position pousses à la verticale en insérant la pointe basale de la pousse dans le milieu. Les racines apparaissent à 10-14 jours après l'incubation des plaques à 28 oC, 16 h :8 h jour:nuit.
REMARQUE : N'enracinez pas toutes les pousses, mais gardez une partie de la propagation de la culture tissulaire (voir l'étape 5.2).
6. Nodulation des plantlets P. andersonii à Pots
-
Préparer le rhizobium inoculum.
- Inoculer 10 ml de milieu YEM liquide (voir tableau 2) d'une seule colonie de Mesorhizobium plurifarium BOR26 et incuber à 28 oC pendant 2 jours.
REMARQUE: M. plurifarium BOR2 est préféré car il nodule efficacement P. andersonii. Cependant, d'autres souches de rhizobium peuvent également être utilisées pour la nodulation de P. andersonii (par exemple Bradyrhizobium elkanii WUR325, Rhizobium tropici CIAT89926,27 ou Bradyrhizobium sp. Kelud2A4). - Utilisez la culture de 10 ml pour inoculer un plus grand volume de milieu YEM liquide. Le volume de cette culture dépend du nombre de pots qui doivent être inoculés.
- Préparer le milieu EKM liquide (voir les tableaux 3, tableau 4). Centrifuger la culture bactérienne pendant 10 min à 3500 x g pour récolter les cellules. Par la suite, suspendre à nouveau la pastille bactérienne dans eKM liquide (utiliser à peu près le même volume que la culture YEM d'origine) et déterminer la densité optique (OD600).
- Inoculer 10 ml de milieu YEM liquide (voir tableau 2) d'une seule colonie de Mesorhizobium plurifarium BOR26 et incuber à 28 oC pendant 2 jours.
- Pour 20 pots, préparer 3 L de liquide EKM moyen et inoculer avec la suspension rhizobial préparée à l'étape 6.1.3. pour atteindre600 OD 0,025.
- Mélanger 3 L d'EKM contenant du rhizobia avec 1 250 g de perlite. Par la suite, ajouter 210 g de ce mélange dans des pots stériles de polypropylène translucide. Alternativement, au lieu de perlite, utiliser le sable comme substrat pour les essais de nodulation.
- Plantez 1-3 p. andersonii plantlets dans chaque pot. Aussi, préparer plusieurs pots contenant des plantlets P. andersonii transformés avec la construction CRISPR-contrôle (voir tableau supplémentaire 1). Peser plusieurs pots pour pouvoir déterminer la perte d'eau pendant l'expérience. Couvrez le fond de chaque pot pour protéger les racines de l'exposition à la lumière.
- Incuber les pots dans une salle de croissance climatisée (28 oC, 16 h :8 h jour:nuit) pendant 4-6 semaines. Une fois par semaine, peser plusieurs pots pour déterminer la perte d'eau. Si la perte d'eau dépasse 10 ml, supplémentavec de l'eau ultra-pure pour compenser la perte.
- Après 4-6 semaines, nettoyez les racines de la perlite et déterminez les nombres de nodules à l'aide d'une jumelle pour examiner l'efficacité de la nodulation.
7. Nodulation des plantlets P. andersonii sur les plaques
-
Préparer les membranes de cellophane 28.
- Couper la membrane de cellophane pour s'adapter dans un plat carré de 12 cm x 12 cm Petri. Couper les membranes un peu plus courtes au sommet pour laisser de l'espace pour que les pousses se développent.
- Pour augmenter la perméabilité des membranes de cellophane, faire bouillir les membranes de la solution EDTA (1 g/L) pendant 20 min. Ensuite, rincer au moins 6x avec de l'eau déminéralisée pour enlever l'EDTA.
REMARQUE: Comme la membrane sèche a tendance à se froisser lorsqu'elle est en contact avec l'eau, immerger les membranes sèches une par une dans la solution. - Disposer les membranes horizontalement dans une fine couche d'eau dans une plaque de verre ronde. Stériliser les membranes en autoclant deux fois.
- Placer 1 membrane de cellophane autoclaved sur un plat carré De 12 x 12 cm contenant un milieu EKM solidifié en agar (voir tableau 3, tableau 4). Placez deux plantuées P. andersonii de 3 semaines (voir la section 5) ou des semis de 4 semaines (voir la section 1.1) sur le dessus de la membrane. Assurez-vous de ne cueillir que des plantlets ou des semis avec des racines qui ont des pointes de racines blanches, ce qui indique que ces racines sont encore en croissance.
- Couvrir délicatement les racines d'une deuxième membrane de cellophane, créant ainsi une couche sandwich. Sceller la plaque avec 3 couches de papier d'étanchéité élastique. Envelopper la moitié inférieure des assiettes avec du papier d'aluminium, pour couvrir les racines de l'exposition à la lumière.
- Incuber les plaques dans une salle de croissance climatisée (28 oC, 16 h:8 h jour:nuit) pendant 3-4 semaines. Marquez la position des pointes de racine pour suivre la croissance de racine au fil du temps.
- Si les plaques EKM commencent à sécher en raison d'une incubation prolongée, transférez les plantes dans des plaques EKM fraîches quelques jours avant l'inoculation bactérienne.
- Préparer l'inoculum bactérien tel que décrit à l'étape 6.1.
- Retirer la membrane supérieure de cellophane et appliquer 1 ml de culture de rhizobium (OD600 à 0,025) aux racines. Par la suite, placez une nouvelle membrane de cellophane sur les racines inoculées. Enveloppez l'extérieur de la plaque à l'aide d'une feuille d'aluminium pour couvrir les racines de l'exposition à la lumière.
- Après 4 semaines, examinez les nombres de nodules à l'aide d'une jumelle pour déterminer l'efficacité de la nodulation.
8. Nodulation des semis P. andersonii dans Pouches
- Germer les graines de P. andersonii telles que décrites dans la section 1.1. Une fois que les cotylédons ont complètement émergé (12 jours sur les plaques SH-0 à 28 oC), transférez les semis en sachets.
- Pour préparer les sachets, déchirer la section pliée de la mèche de papier et ajouter 7 ml de milieu EKM modifié (voir tableau 3, tableau 4).
- Insérez 1 ou 2 semis en plaçant les racines entre les deux feuilles de papier qui forment la mèche de papier et la feuille de plastique avant de la poche.
- Protégez les racines de l'exposition à la lumière, en pliant le papier d'aluminium autour de la poche. Suspendre les sachets dans une boîte en plastique recouverte d'un couvercle translucide pour maintenir une humidité élevée. Placez la boîte dans une salle de croissance climatisée (28 oC, 16 h : 8 h jour:nuit).
- Compenser l'évaporation de l'eau en ajoutant de l'eau stérile ultra-pure, en tant que telle que la mèche de papier reste humide (éviter l'eau stagnante au fond de la poche). Après la première semaine, cela nécessite généralement l'ajout de 2-3 ml tous les 4 jours.
- Préparer l'inoculum bactérien tel que décrit à l'étape 6.1.
- Après que les semis ont été cultivés pendant 10-12 jours dans des sachets, inoculer le système racinaire avec 500 L de culture de rhizobium (OD600 - 0,025).
- Suivez la formation de nodule à travers le temps. Quatre semaines après l'inoculation, les nodules peuvent être comptés et récoltés pour déterminer l'efficacité de nodulation.
9. Analyse De Cytoarchitecture Nodule
- Recueillir 10-15 nodules dans un tube de 2 ml contenant du fixatif (5 % de glutaraldéhyde dans un tampon de phosphate de 0,1 M, pH 7,2). Appliquer le vide pendant 1/2-1 h et couver toute la nuit à 4 oC. Pendant cette période, les échantillons coulent au fond du tube.
REMARQUE: La solution fixative peut être stockée à 4 oC pendant une utilisation de 2 à 4 semaines avant. Assurez-vous de porter des gants lorsque vous travaillez avec un fixatif tissulaire. - Laver les nodules 2x avec 0,1 M tampon de phosphate, pH 7,2. Appliquer 10 min d'intervalles entre chaque étape de lavage.
- Déshydrater les échantillons en couve par la suite dans 30%, 50%, 70%, et 100% d'éthanol. Pour vous assurer que toute l'eau est retirée des échantillons, répétez l'étape 100% éthanol 3x. Appliquer 10 min d'intervalles entre chaque étape de déshydratation.
- Préparer le mélange de polymérisation I (PM-I) en ajoutant 1 paquet de Hardener I à 2,5 ml de PEG400 mélangé avec 100 ml de HEMA (2-hydroxyethyl methacrylate) solution de résine à base. Remuer la solution pendant 15 min pour dissoudre complètement le Durcir I. Par la suite, entreposez le PM-I à -20 oC.
- Retirer l'éthanol de l'étape 9.3. et infiltrer les échantillons dans l'ordre suivant : PM-I:100% éthanol (1:3, v/v), PM-I:100% éthanol (1:1, v/v), et PM-I:100% éthanol (3:1, v/v). Incuber les échantillons dans chaque solution à RT pendant 1/2-1 h ou jusqu'à ce que les échantillons coulent au fond.
- Incuber des échantillons pendant la nuit à 4 oC dans une solution 100% PM-I.
- Préparer le mélange de polymérisation II en mélangeant PM-I et Hardener II dans un rapport 15:1 (v/v). Remplissez le moule en plastique avec la solution de polymérisation, orientez les échantillons horizontalement au fond du moule, et recouvrez-le d'un morceau de papier d'étanchéité élastique. Évitez la formation de bulles d'air.
REMARQUE: Lorsque la solution commence à polymériser lors de l'exposition à RT, essayez d'orienter les échantillons le plus rapidement possible dans le support en plastique. La polymérisation est terminée après l'incubation de nuit à RT, ou 1 h à 37 oC. - Retirez le couvercle élastique du papier d'étanchéité de l'étape 9.7 et placez un support sur les échantillons polymérisés. Pour monter le support aux échantillons, dissoudre 10 mL de poudre de résine méthylique à base de méthyle dans 5 ml de résine méthylique à base de résine. Ajouter rapidement la solution au trou dans le haut du support.
REMARQUE: Effectuer l'étape de polymérisation dans le capot de fumée (30 min à RT). - Échantillons de section de microtome jusqu'à une épaisseur de 4 à 5 m. Placez une lame de microscope sur une plaque chauffante de 58 oC et ajoutez une grande goutte d'eau à chaque lame. Placer les sections sur le dessus de l'eau. Une fois que l'eau s'est évaporée, les sections adhéreront à la glissière.
- Glissez en plongeant dans 0,05 % (w/v) bleu toluidine pendant 2 min. Par la suite, rincer les diapositives 3x avec de l'eau ultra-pure. Des diapositives peuvent être observées à l'aide d'un microscope à champ lumineux.
10. Mycorhisation des plantons P. andersonii
-
Préparer l'inoculum des spores de Rhizophagus irregularis
- Préparer une pile de filtres tissés en polyester avec les tailles suivantes (de haut en bas) : 210 m, 120 m et 36 m de taille en maille.
- Pipette la quantité requise d'une suspension de spores commerciales sur la pile de filtres en polyester. Rincer les filtres 3x avec 100 ml d'eau déminéralisée autoclave. Les spores sont conservées à la surface du filtre de 36 m.
REMARQUE: Préparer la suspension des spores dans l'armoire à débit croisé laminaire pour éviter la contamination. - Démontez la pile de polyester et conservez le filtre de 36 m seulement. Répétez l'étape de lavage avec de l'eau déminéralisée autoclave d'au moins 6 x.
- Placer le filtre sur un plat Petri et suspendre à nouveau les spores dans de l'eau déminéralisée autoclave. Utilisez un volume d'eau égal au volume de la suspension des spores utilisée à l'étape 10.1.2. Transférer la suspension des spores dans un tube stérile par pipetting.
- Placez 5 gouttes de 20 l de la suspension des spores sur une lame de verre et comptez le nombre de spores à l'aide d'un microscope à champ lumineux. Convertir le nombre de spores en un rapport spores/mL et diluer la suspension des spores jusqu'à ce qu'elle atteigne 250 spores/mL. Conserver la suspension des spores à 4 oC.
- Effectuer l'exemple de mycorhisation. À cette fin, ajouter 800 g de sable autoclaved complété de 70 ml de 1/2-Hoagland moyen à stérile s'accumulent dans des pots de polypropylène translucide (voir tableaux 5-6). Mélanger le sable et le milieu directement dans le pot en secouant vigoureusement.
- Placer un plantlet P. andersonii dans chaque pot, et la pipette 1 ml de la suspension des spores directement sur la racine du plantlet P. andersonii. Assurez-vous d'inclure plusieurs pots contenant des plantlets P. andersonii transformés avec une construction CRISPR-contrôle (voir tableau supplémentaire 1).
- Incuber les pots dans une salle de croissance climatisée (28 oC, 16 h :8 h jour:nuit) pendant 6 semaines.
- Sortez les plantes des pots et lavez les racines avec de l'eau courante pour enlever autant de sable que possible.
- Couper les racines en morceaux de 1 cm de long et faire bouillir les morceaux de racine en 10 % DE KOH (w/v) pendant 20 min à 90 oC. Par la suite, placez les racines bouillies sur une passoire cellulaire avec une taille de maille de 100 m et rincez 3x avec 50 ml d'eau.
- Racine s'intérioriser avec 0,05 % (w/v) trypan bleu dans le lactoglycérol (300 ml d'acide lactique; 300 ml de glycérol; et 400 ml d'eau déminéralisée) pendant 5 min à 90 oC dans un bain d'eau ou un bloc de chauffage. Par la suite, transférer les racines à 30 % de glycérol. Les échantillons de racines peuvent être stockés à RT.
- Placer 15 à 25 fragments de racines sur une seule lame de microscope. Ajouter 30% de glycérol et couvrir d'un verre de couverture et presser jusqu'à ce que les morceaux de racine deviennent plats. Observez les fragments de racines à l'aide d'un microscope à champ lumineux et marquez la colonisation mycorhizienne.
REMARQUE: Une méthode pour marquer la mycorhisation est décrite selon Trouvelot et coll.29. Cette méthode utilise plusieurs classes (%F, %M, et %A), ce qui permet d'estimer rapidement le niveau de colonisation mycorhilien de chaque fragment de racine et l'abondance des arbuscules.
Résultats
P. andersonii trees peut être cultivé dans une serre conditionnée à 28 oC et à 85 % d'humidité relative (figure 1A). Dans ces conditions, les arbres commencent à fleurir à 6-9 mois après la plantation. Les fleurs femelles de P. andersonii produisent des baies qui contiennent chacune une seule graine. Pendant la maturation, les baies changent de couleur; d'abord du vert au blanc, puis du blanc au brun (Figure 1B). Graines extraites des baies brunes mûries, germent bien après un cycle de température de 10 jours et une incubation de 7 jours sur des plaques SH-0 (figure 1C). Les graines germées continuent de se développer en jeunes semis qui peuvent être utilisés pour l'expérimentation après 4 semaines (figure1D).
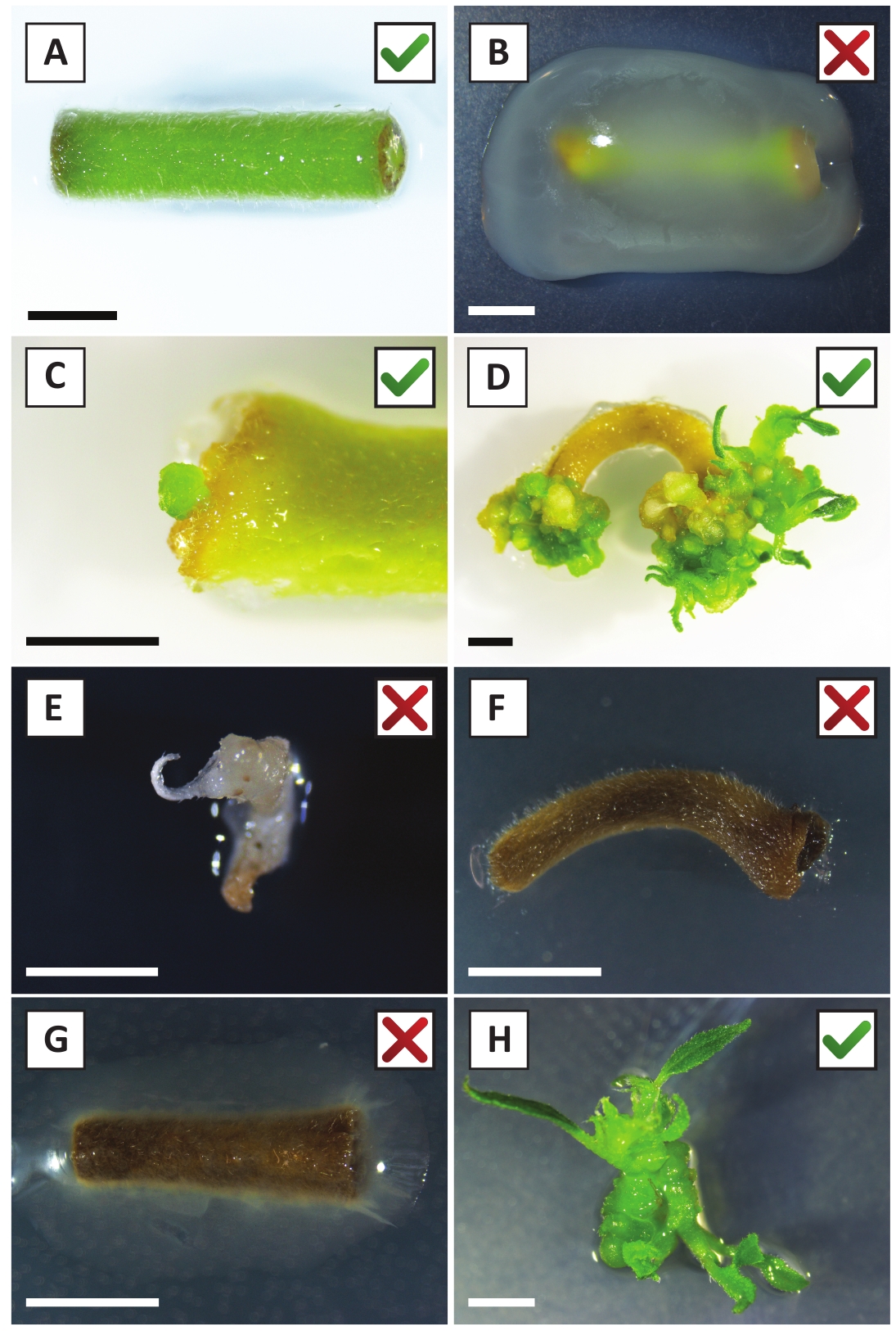
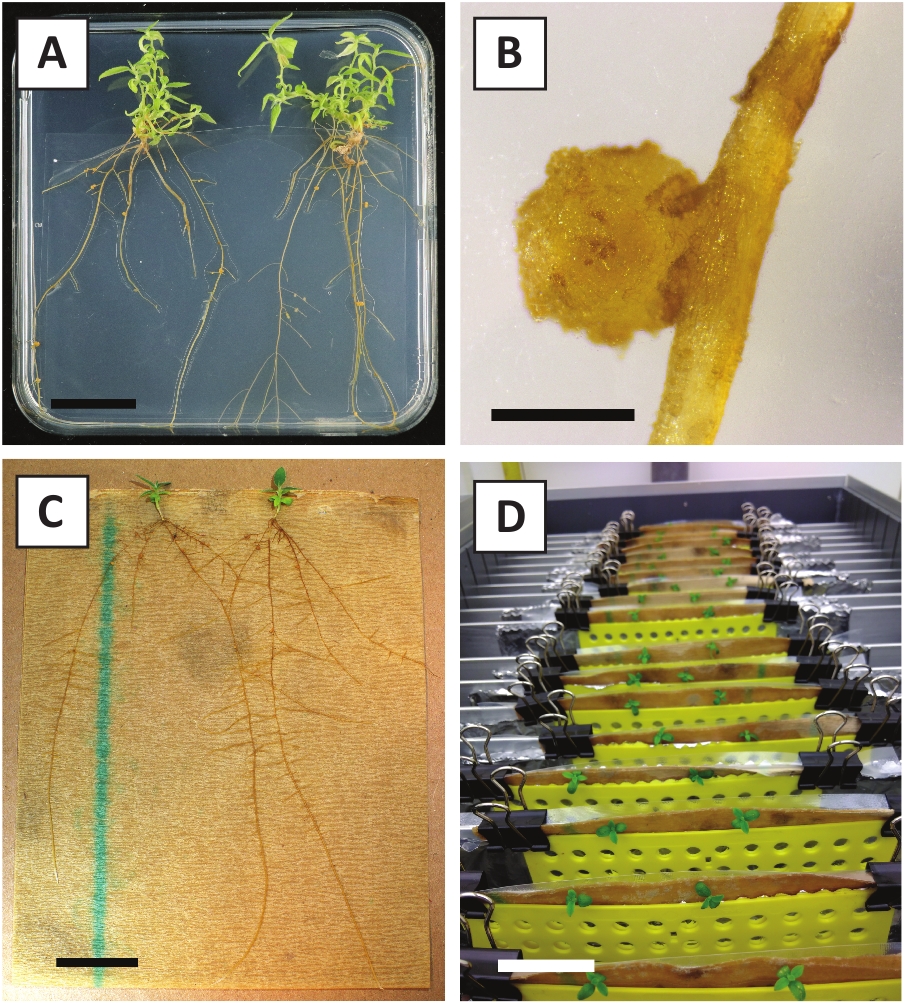
Nous avons déjà montré que les pétioles et les segments de jeunes tiges de P. andersonii peuvent être transformés efficacement à l'aide de la souche A. tumefaciens AGL110. Au début de la procédure de transformation, les explants tissulaires sont co-cultivés avec A. tumefaciens pendant 2 jours à 21 oC (figure2A). La co-culture prolongée entraîne une surcolonisation des explants tissulaires par A. tumefaciens et devrait donc être évitée (figure 2B). Après la période de co-culture, les explants tissulaires sont transférés dans des médias sélectifs, ce qui favorise la croissance des tissus transformés. Deux à trois semaines plus tard, de petits micro-calli verts sont généralement observés le long de la surface de la plaie d'origine (figure 2C). Ces calli devraient continuer à se développer et à développer 1 ou plus de pousses transformées de façon présumée à 6-8 semaines après le début de la procédure de transformation (Figure 2D). À ce stade, l'efficacité de la transformation varie généralement de 10 à 30 % pour les transformations amorcées avec des explants de tissus prélevés à partir de branches matures et partiellement ligneuses (tableau 7). Si des transformations sont amorcées avec des explants prélevés sur les pointes jeunes et en croissance rapide des branches qui ne portent pas encore de fleurs, des gains d'efficacité de transformation de 65 à 75 % peuvent être atteints (tableau 7). De temps en temps, les calli blanchâtres sont formés sur le côté d'une explante qui n'est pas en contact avec le milieu et, par conséquent, ne connaissent pas la sélection de kanamycine. Ces calli ne sont souvent pas transgéniques et les pousses formées à partir de ces calli blanchiront généralement et mourront après un contact direct avec le milieu contenant de la kanamycine (Figure 2E). Dans le cas où le taux de transformation est faible et/ou que le matériau de départ était sous-optimal, les morceaux de tissu pourraient brunir (Figure 2F) et souffrir d'une surprolifération de la part de A. tumefaciens (Figure 2G). Pour empêcher A. tumefaciens de se propager et de surcroître à proximité des explants, un rafraîchissement régulier du milieu est nécessaire, et les explants gravement infectés doivent être enlevés. Une fois que les pousses transgéniques individuelles sont placées dans le milieu de propagation, la sur-prolifération par A. tumefaciens ne se produit généralement plus (Figure 2H). Les pousses transgéniques peuvent être multipliées par la propagation in vitro, ce qui donnera lieu à des dizaines de pousses sur une période d'un mois (figure3A-B). Ces pousses peuvent être placées sur le milieu d'enracinement, ce qui devrait induire la formation des racines après 2 semaines (figure3C-D). Les plantons enracinés peuvent ensuite être utilisés pour l'expérimentation.
Pour créer des lignes mutantes knock-out, nous utilisons la mutagénèse CRISPR/Cas9-négociée. À cette fin, nous utilisons un vecteur binaire contenant le gène de résistance à la kanamycine NPTII, une séquence d'encodage Cas9 entraînée par le promoteur CaMV35S et 2 sgRNAR par gène cible qui sont exprimés à partir du promoteur atU6p petit ARN20. Une représentation graphique de la construction utilisée pour la mutagénèse de P. andersonii à médiation CRISPR/Cas9 est fournie à la figure 4A. À l'aide de cette méthode, l'édition du génome est observée dans 40 % des pousses transformées de façon putative10. Pour identifier les lignées mutantes, les pousses transformées de façon putative sont génotypes pour les mutations sur le site cible de l'ARSG à l'aide d'amorces couvrant la région ciblée. Un exemple des résultats attendus est donné à la figure 4. Comme on peut le voir sur la photo prise après l'électrophoresis de gel, plusieurs échantillons produisent un amplicon de PCR avec la taille semblable au type sauvage (figure 4B). Ces plantes peuvent contenir de petits indels qui ne peuvent pas être visualisés par électrophoresis de gel d'agarose ou rester non édités par l'enzyme Cas9. De plus, plusieurs échantillons produisent des bandes de taille différente de celle du type sauvage (p. ex., les lignes 2, 4, 7 et 8 de la figure 4B). Dans ces lignes, 1 (lignes 4, 7 et 8) ou les deux (ligne 2) allélées contiennent de plus grandes indels qui peuvent être facilement visualisés. La nature exacte des mutations au site cible (s) est indiquée après le séquençage d'amplicon de PCR. Comme on peut le voir à partir de la figure 4C, les deux petites indels de 1-4 bp, ainsi que, de plus grandes suppressions peuvent être obtenues après CRISPR / Cas9 mutagénèse. Dans la figure 4C, la séquence de la ligne 1 est identique à celle du type sauvage, ce qui indique que cette ligne a échappé à l'édition et, par conséquent, doit être écartée. Parmi les lignées qui contiennent des mutations, les mutants hétérozygotes, homozygotes et bi-alléliques peuvent être identifiés (Figure 4C). Cependant, les mutants hétérozygotes sont généralement rares10. Les mutants knock-out homozygotes ou bi-alléliques peuvent être propagés végétativement pour obtenir suffisamment de matériel pour l'analyse phénotypique. Comme l'analyse phénotypique est effectuée dans la génération T 0, il est important de vérifier si les lignées mutantes pourraient être chimériques. À cette fin, le génotypage doit être répété sur au moins 3 échantillons différents prélevés sur chaque lignée mutante. Si les résultats du génotypage sont identiques les uns aux autres et que l'échantillon de génotypage d'origine (p. ex., ligne 8 de la figure 4D),la ligne est mutée de façon homogène et peut être utilisée pour une analyse plus approfondie. Toutefois, si les résultats du génotypage diffèrent d'échantillons indépendants (p. ex., ligne 4 dans la figure 4D),la lignée mutante est chimérique et doit être jetée.
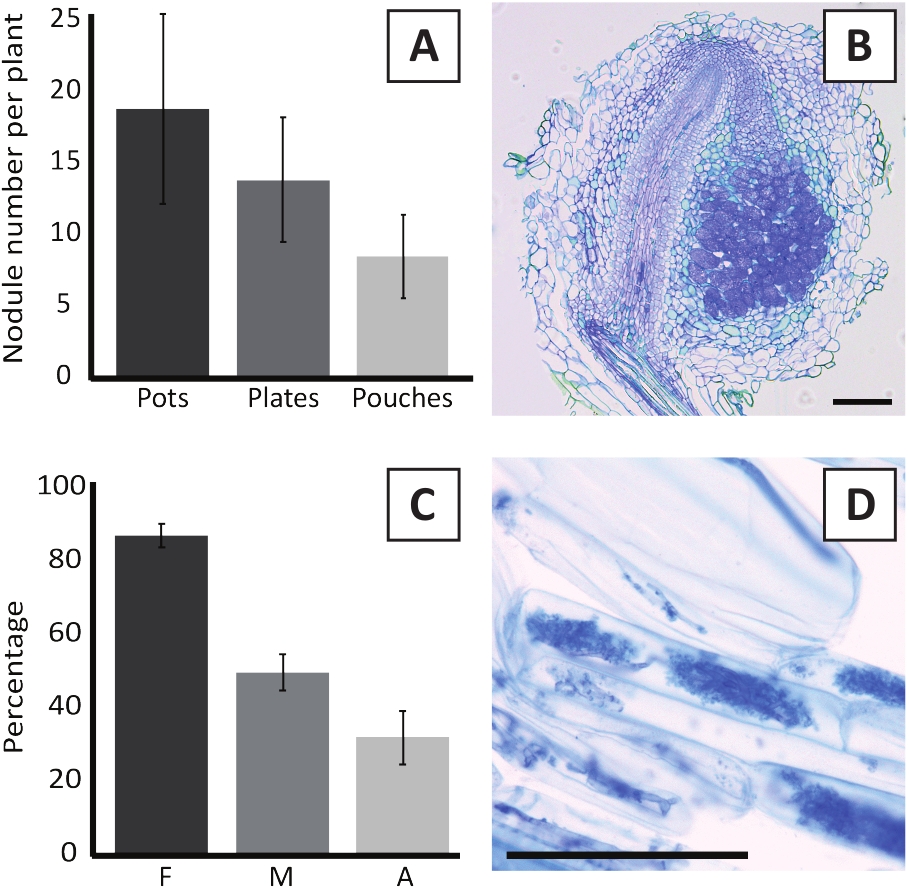
L'inoculation de P. andersonii avec M. plurifarium BOR2 entraîne la formation de nodules racinaires (figure 5). Comme on peut le voir dans la figure 5A, ces nodules sont répartis le long du système racinaire. Les nodules de P. andersonii sont de couleur brun clair, mais peuvent être facilement discriminés du tissu racinaire en fonction de leur forme (Figure 5B). Les expériences d'inoculation dans les pots et la croissance subséquente pendant 4-6 semaines se traduisent généralement par la formation de nodules de 10 à 30 euros (figure 6A). Un nombre similaire de nodules se forme après l'inoculation des plantuelles P. andersonii cultivées dans la plaque EKM à 4 semaines après l'inoculation (Figure 6A). Dans les sachets, les semis P. andersonii forment généralement des nodules de 5 à 15 semaines après l'inoculation (Figure 5C-D, 6A). Pour analyser la cytoarchitecture nodule, les nodules peuvent être sectionnés et observés à l'aide de la microscopie de champ lumineux. La figure 6B montre un exemple d'une section longitudinale au milieu d'un nodule P. andersonii. Cette section montre le faisceau vasculaire central d'un nodule De P. andersonii, qui est flanqué de lobes de nodule contenant des cellules infectées (figure 6B).
Les plantules P. andersonii peuvent également être mycorhisées. Après 6 semaines d'inoculation avec R. irregularis, la fréquence de colonisation mycorhizienne atteint généralement 'gt; 80% (Figure 6C). À l'heure actuelle, généralement 30 % des cellules contiennent des arbuscules (figure6C). Une image représentative d'un segment racinaire P. andersonii contenant des arbuscles est montrée dans la figure 6D.

Figure 1 : Images représentatives d'un P. andersonii arbre, graines et semis. (A) Arbre P. andersonii de six mois cultivé dans le sol de mise en pot dans une serre conditionnée à 28 oC. (B) Image représentative représentant les baies de P. andersonii à divers stades de maturation. Les jeunes baies de P. andersonii (non mûres) passeront de la couleur du vert au blanc et enfin au brun (mûr) à la maturation. (C) Graines de P. andersonii incubées sur le milieu SH-0 pendant 1 semaine. Un cercle noir indique un semis germé. (D) Quatre semaines p. andersonii semis cultivés dans sH-0 milieu. Les barres d'échelle sont égales à 25 cm dedans (A) et de 1 cm dans (B-D). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : Images représentatives d'explantations à différents stades de la procédure de transformation stable. (A) Explant co-cultivé avec A. tumefaciens. (B) Explant envahi par A. tumefaciens au cours des 2 premières semaines après la transformation. (C) Micro-callus transgénique formé près du site de la plaie d'une explante à 2,5 semaines après la co-culture. (D) Image représentative d'une explantation à 6 semaines après la co-culture montrant l'émergence de pousses de calli (transgéniques). (E) Image représentative d'une pousse qui devient blanchâtre et finit par mourir en contact direct avec le milieu contenant de la kanamycine. Cette pousse est très probablement non transgénique et a échappé à la sélection de kanamycine lorsqu'elle est attachée à l'explantation. (F) Image représentative d'une explantation transformée sans succès. (G) Image représentative d'une explantation transformée sans succès envahie par A. tumefaciens. (H) Pousse transgénique unique cultivée sur le milieu de propagation à 8 semaines après la co-culture avec A. tumefaciens. Les barres d'échelle égalent 2,5 mm. Les boîtes contenant des marques de contrôle vertes ou des croix rouges indiquent une transformation réussie ou infructueuse des explants, respectivement. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Images représentatives de in vitro propagation. (A) Pousses cultivées sur le milieu de propagation. L'image a été prise 1 semaine après que les plaques ont été rafraîchies. (B) Pousses cultivées sur le milieu de propagation. L'image a été prise 4 semaines après que les plaques ont été rafraîchies. (C) Pousses fraîchement coupées placées sur le milieu d'enracinement. (D) Pousses incubées sur le milieu d'enracinement pendant 2 semaines. Notez la présence de racines. Les barres d'échelle sont égales à 2,5 cm. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Résultats représentatifs après génotypage des lignées mutantes transgéniques CRISPR/Cas9 de P. andersonii T0. (A) Carte représentative d'un vecteur binaire utilisé pour la mutagénèse CRISPR/Cas9-négociée de P. andersonii. (B) Résultat représentatif après le génotypage basé sur PCR des lignes mutantes potentielles DE CRISPR/Cas9 utilisant des amorces couvrant le site cible de sgRNA(s). On voit une image après l'électrophoresis de gel d'agarose des amplicons. Les échantillons prélevés sur des lignées transgéniques individuelles sont indiqués par des nombres. Le type sauvage (WT) et l'absence de contrôle de modèle (NTC) indiquent des voies contenant des contrôles positifs et négatifs, respectivement. (C) Représentation schématique des allèles mutants obtenue après l'édition génétique CRISPR/Cas9-négociée. Les sites cibles sgRNA et les séquences PAM, respectivement, sont mis en évidence dans les couleurs bleues et rouges. (D) Résultat représentatif après le criblage PCR-basé pour les lignes mutantes chimériques potentielles. On voit une image après l'électrophorèse de gel d'agarose de 3 échantillons individuels prélevés sur les lignes mutantes 4 et 8. Notez que la ligne mutante transgénique 4 est chimérique. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Images représentatives des essais de nodulation dans les assiettes et les sachets. (A) Nodulation sur les plaques contenant un milieu EKM solidifié en agar et inoculé avec M. plurifarium BOR2 pendant 4 semaines. (B) Image représentative d'une nodule racine de P. andersonii. L'image a été prise à 4 semaines après l'inoculation avec M. plurifarium BOR2. (C) Nodulation dans des sachets contenant un milieu EKM liquide. Les semis ont été inoculés avec Le Bradyrhizobium sp. Kelud2A4 pendant 5 semaines. (D) Image représentative d'une configuration complète utilisée pour la nodulation dans les sachets. Les barres d'échelle sont égales à 2,5 cm(A,C),1 mm dedans (B), et 5 cm dans (D). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6 : Résultats représentatifs des essais de nodulation et de mycorhisation. (A) Graphique à barres représentatives montrant le nombre de nodules formés par plante à 4 semaines après l'inoculation avec M. plurifarium BOR2 dans des pots ou sur des assiettes et à 5 semaines après l'inoculation avec Bradyrhizobium sp. Kelud2A4 dans des sachets. Les données représentent la moyenne de SD (n ' 10). (B) Image représentative d'une section longitudinale à travers une nodule formée à 4 semaines après l'inoculation avec M. plurifarium BOR2. La section est tachée de bleu toluidine. (C) Graphique à barres représentatives montrant la quantification de la mycorhisation. Les variables quantifiées selon Trouvelot et coll.29 sont F, la fréquence des fragments de racines analysés qui sont mycorhisés; M, l'intensité de l'infection; A, l'abondance d'arbuscules matures dans le système racinaire total. La mycorhisation a été quantifiée à 6 semaines après l'inoculation avec R. irregularis (souche DAOM197198). Les données représentent la moyenne de SD (n ' 10). (D) Image représentative des arbuscules matures présents dans les cellules corticales de racine de P. andersonii à 6 semaines après l'inoculation avec R. irregularis. Les barres d'échelle sont égales à 75 m. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
| enceinte | SH-0 (SH-0) | SH-10 (en) | Moyen de propagation | Milieu d'enracinement | Milieu d'infiltration |
| Milieu salé sH-basal | 3,2 g | 3,2 g | 3,2 g | 3,2 g | 3,2 g |
| Mélange SH-vitamine | 1 g | 1 g | 1 g | 1 g | 1 g |
| Saccharose | - | 10 g | 20 g | 10 g | 10 g |
| BAP (1 mg/mL) | - | - | 1 ml (4,44 M) | - | - |
| IBA (1 mg/ml) | - | - | 100 l (0,49 M) | 1 ml (4,92 M) | - |
| NAA (1 mg/mL) | - | - | - | 100 l (0,54 M) | - |
| 1 M MES pH 5,8 | 3 mL | 3 mL | 3 mL | 3 mL | 3 mL |
| 1 M KOH | Ajuster le pH à 5,8 | Ajuster le pH à 5,8 | Ajuster le pH à 5,8 | Ajuster le pH à 5,8 | Ajuster le pH à 5,8 |
| Agar Daishin | 8 g | - | 8 g | 8 g | - |
Tableau 1 : Composition des30 supports à base de Schenk-Hildebrandt utilisés pour la culture des semis de P. andersonii, la transformation stable et la propagation in vitro. Dissoudre les composés solides dans 750 ml d'eau ultra-pure avant d'ajouter des stocks liquides. Ensuite, remplissez le support complet à 1 L. Préparer BAP, IBA, NAA stocks en 0,1 M KOH et stocker à -20 oC.
| Avant l'autoclacage : | ||
| enceinte | Montant par litre | Concentration finale |
| Mannitol | 5 g | 27,45 mM |
| Na-Gluconate | 5 g | 22,92 mM |
| extrait | 0,5 g | - |
| MgSO47H2O | 0,2 g | 0,81 mM |
| Nacl | 0,1 g | 1,71 mM |
| K2HPO4 | 0,5 g | 2,87 mM |
| Après l'autoclacage : | ||
| enceinte | Montant par litre | Concentration finale |
| 1,5 M CaCl2 Annonces | 1 mL | 1,5 mm |
Tableau 2 : Composition du milieu de levure-mannitol (YEM) utilisé pour la culture du rhizobium. Ajuster le pH à 7,0 et remplir d'eau ultra-pure à 1 L. Pour préparer le milieu YEM solidifié par l'agar, ajouter 15 g de microagar avant d'autoclage.
| Avant l'autoclacage : | |||
| enceinte | Concentration des stocks | Montant par litre moyen | Concentration finale |
| KH2PO4 | 0,44 M | Ajouter 2 mL | 0,88 mM |
| K2HPO4 | 1,03 M | Ajouter 2 mL | 2,07 mM |
| 500x micro-éléments solution de stock | - | Ajouter 2 mL | - |
| MES pH 6,6 | 1 M | Ajouter 3 mL | 3 mM |
| Hcl | 1 M | Ajuster le pH à 6,6 | - |
| Eau ultra-pure | - | Remplir à 990 ml | - |
| Après l'autoclacage : | |||
| enceinte | Concentration des stocks | Montant par litre moyen | Concentration finale |
| MgSO47H2O | 1,04 M | 2 mL | 2,08 mM |
| Na2SO4 Annonces | 0,35 M | 2 mL | 0,70 mm |
| NH4NO3 | 0,18 M | 2 mL | 0,36 mm |
| CaCl2h 2H2O | 0,75 M | 2 mL | 1,5 mm |
| Fe(III)-citrate | 27 mM | 2 mL | 54 M |
Tableau 3 : Composition de 1 L modifié EKM medium31 utilisé pour l'assay de nodulation P. andersonii. La composition de la solution de stock de micro-éléments 500x est indiquée au tableau 4. Pour préparer 2% de moyenne EKM solidifiée d'agar, ajouter 20 g d'agar daishin avant d'autoclaciner. Autoclave les mgSO4x 7H2O, Na2SO4, CaCl2x 2H2O, et Fe(III)-citrate stocks à stériliser. Filtrer stériliser NH4NO3 solution de stock pour stériliser.
| enceinte | Montant par litre | Concentration des stocks |
| MnSO4 | 500 mg | 3,31 mM |
| ZnSO47H2O | 125 mg | 0,43 mM |
| CuSO45H2O | 125 mg | 0,83 mM |
| H3BO3 | 125 mg | 2,02 mM |
| Na2MoO42H2O | 50 mg | 0,21 mM |
Tableau 4 : Composition de la solution de stock de micro-éléments 500x utilisée pour la préparation du support EKM modifié. Entreposer la solution de stock de micro-éléments à 4 oC.
| Composés | Concentration des stocks | Montant par litre moyen | Concentration finale |
| K2HPO4 | 20 mM | 1 mL | 0,2 mm |
| NH4NO3 | 0,28 M | 10 ml | 2,8 mM |
| MgSO4 | 40 mM | 10 ml | 0,4 mm |
| K2SO4 | 40 mM | 10 ml | 0,4 mm |
| Fe(II)-EDTA | 9 mM | 10 ml | 0,9 mm |
| CaCl2 (en) | 80 mM | 10 ml | 0,8 mm |
| 50x micro-éléments solution de stock | - | 10 ml | - |
Tableau 5 : Composition d'un milieu de 1/2-Hoagland32 utilisé pour les essais de mycorhisation. La composition de la solution de stock de micro-éléments 50x est indiquée dans le tableau 6. Préparer la solution Fe(II)-EDTA en combinant FeSO4'7H2O (9 mM) et Na2' EDTA (9 mM) en 1 solution de stock, et stocker à 4 oC. Ajuster le pH du milieu à 6,1 à l'aide de 1 M KOH et remplir d'eau ultra-pure à 1 L.
| Composés | Montant par litre | Concentration des stocks |
| H3BO3 | 71,1 mg | 1,15 mm |
| MnCl24H2O | 44,5 mg | 0,22 mM |
| CuSO45H2O | 3,7 mg | 23,18 M |
| ZnCl2 | 10,2 mg | 74,84 M |
| Na2MoO42H2O | 1,2 mg | 4,96 M |
Tableau 6 : Composition de la solution de stock de micro-éléments 50x utilisée pour la préparation du milieu de 1/2-Hoagland.
| Age des explantations | Efficacité de transformation |
| jeunes | 69,4 à 6,2 % (n et 2) |
| Mature | 18,3 à 10,2 % (n 15) |
Tableau 7 : Efficacité de transformation de P. andersonii. Ici, l'efficacité de transformation est définie comme le pourcentage d'explants qui forment au moins 1 callus transgénique ou tirer. L'efficacité de transformation a été notée à 6 semaines après la transformation et est décrite comme moyenne - SD. n indique le nombre d'expériences de transformation à partir desquelles l'efficacité de transformation a été déterminée.
Fichier supplémentaire 1 : Aperçu des constructions de niveau 1 et de niveau 2 utilisées pour la mutagénèse CRISPR/Cas9. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Les légumineuses et le genre Cannabaceae Parasponia, très apparenté, représentent les deux seuls clades d'espèces végétales capables d'établir une relation endosymbiotique avec les rhizobia fixant l'azote et de former des nodules racinaires. Les études comparatives entre les espèces des deux clades sont très pertinentes pour fournir un aperçu des réseaux génétiques de base permettant cette symbiose. Actuellement, les études génétiques sont principalement effectuées chez les légumineuses; en particulier les deux espèces modèles M. truncatula et L. japonicus. Pour fournir une plate-forme expérimentale supplémentaire et faciliter des études comparatives avec un non-légumineuse nodulating, nous décrivons ici un protocole détaillé pour la transformation stable et les analyses génétiques inversées dans P. andersonii. Le protocole présenté utilise la propagation in vitro des lignées Transgéniques De 0 P. andersonii, ce qui permet d'initier une analyse phénotypique dans les 4 mois suivant la coculture d'A. tumefaciens. C'est beaucoup plus rapide que les protocoles actuels qui ont été établis pour une transformation stable des légumineuses33. Cela fait de P. andersonii un modèle de recherche attrayant.
Le protocole décrit ici contient plusieurs étapes critiques. Le premier concerne la germination des graines. Pour préparer les graines de P. andersonii pour la germination, les graines doivent être isolées des baies. Cela se fait en frottant les baies sur un morceau de papier de soie ou contre l'intérieur d'un tamis à thé. Cette procédure doit être effectuée en douceur afin d'éviter des dommages à la couche de graines. Si la couche de graines est endommagée, l'eau de Javel pourrait pénétrer dans la graine pendant la stérilisation, ce qui réduit la viabilité des semences. Pour briser la dormance des graines, les graines sont soumises à un cycle de température de 10 jours. Cependant, en dépit de ce traitement, la germination n'est pas entièrement synchronisée. En général, les premières graines montrent l'émergence du radicle après 7 jours, mais d'autres peuvent prendre plusieurs jours de plus pour germer.
Des points critiques dans la procédure de transformation concernent le choix du matériel de départ et la durée de l'étape de co-culture. Pour parvenir à une transformation efficace, il est préférable d'utiliser des tiges saines et jeunes ou des pétioles de plantes non stériles cultivées en serre comme matériau de départ. Afin d'induire la croissance des jeunes branches, il est conseillé de couper les arbres Parasponia tous les 2-3 mois et rafraîchir les arbres une fois par an. En outre, l'étape de co-culture doit être effectuée pendant 2 jours seulement. La co-culture prolongée favorise la surcolonisation des explants de tissus par A. tumefaciens et réduit généralement l'efficacité de la transformation. Pour éviter la surcolonisation par A. tumefaciens, il est également important de rafraîchir régulièrement les assiettes sur lesquelles les explants sont cultivés. En cas de surcolonisation, les explants tissulaires pourraient être lavés (voir la section 3.8) pour enlever les cellules A. tumefaciens. Nous conseillons d'ajouter de l'eau de Javel à la solution SH-10 utilisée pour le lavage (concentration finale : hypochlorite de 2 %). Il est important de noter que cette étape de lavage supplémentaire pourrait ne pas fonctionner sur les explants fortement infectés (figure 2B). Dans le cas où une transformation avec une construction CRISPR/Cas9 ne donne qu'un nombre limité de pousses transformées de façon putative ou si la mutagénèse d'un gène particulier est censée causer des problèmes de régénération, il est conseillé d'inclure une construction de contrôle vectorielle vide comme le contrôle positif. Enfin, il est important de s'assurer que toutes les lignes transgéniques sélectionnées résultent d'événements indépendants d'intégration de l'ADN T. Par conséquent, nous instruisons de ne prendre qu'une seule pousse putativement transgénique de chaque côté d'une explante. Cependant, nous nous rendons compte que cela réduit le nombre potentiel de lignes indépendantes. Si de nombreuses lignes sont nécessaires, les chercheurs pourraient décider de séparer les calli transformés de façon putative des explants d'origine lorsque ces calli sont de 2 mm de taille et de culture de ces calli indépendamment. De cette façon, plusieurs lignes pourraient être isolées de chaque explante, ce qui augmente le nombre de lignes transgéniques potentielles.
Dans le protocole actuel, les lignées transgéniques de P. andersonii se propagent végétativement par la propagation in vitro. L'avantage de ceci est que beaucoup de plantlets transgéniques peuvent être produits dans une période relativement courte de temps. Cependant, cette méthode a également plusieurs limitations. Tout d'abord, le maintien des lignées transgéniques T0 par la propagation in vitro est laborieux et pourrait entraîner des altérations génétiques ou épigénétiques indésirables34,35. Deuxièmement, les lignes T0 contiennent encore une copie de l'ADN T, y compris la cassette de résistance aux antibiotiques. Cela limite le nombre de retransformations possibles, car différents marqueurs de sélection sont nécessaires pour chaque transformation. Actuellement, nous n'avons testé la transformation qu'à l'aide de la sélection de kanamycine ou d'hygromycine (données non affichées). De plus, la présence de la séquence et des sgARN de Cas9 dans les lignes transgéniques T0 complique les études de complémentarité. Des essais de complémentation sont possibles, mais exigent que le site cible sgRNA (s) soit muté en tant que tel que l'édition génétique de la construction de complémentation est empêchée. Troisièmement, un inconvénient de travailler avec des lignes T0 est que les mutants CRISPR/Cas9 pourraient être chimériques. Pour empêcher l'analyse phénotypique des lignes mutantes chimériques, nous recommandons de répéter l'analyse de génotypage après la propagation in vitro sur au moins 3 pousses différentes. Bien que, le nombre de mutants chimériques obtenus à l'aide du protocole décrit ici est limité, ils sont parfois observés10. Pour surmonter les limites du travail avec des lignes T 0, les lignées mutantes de P. andersonii pourraient être propagées de façon générative. Les arbres P. andersonii sont dioïques et pollinisés par le vent2. Cela signifie que chaque lignée transgénique doit être manipulée en tant que telle que les fleurs mâles et femelles sont produites sur un seul individu, et ensuite cultivées en tant que telle que la pollinisation croisée ne se produit pas. Comme P. andersonii est un arbre à croissance rapide, il nécessite une quantité substantielle d'espace dans une serre tropicale (28 oC, 85 % d'humidité relative). Par conséquent, bien que techniquement possible, la propagation générative des lignées transgéniques de P. andersonii est difficile sur le plan logistique.
Dans la section de protocole, nous avons décrit 3 méthodes pour nodulation de P. andersonii. L'avantage des systèmes de plaque et de poche est que les racines sont facilement accessibles, ce qui peut permettre l'inoculation ponctuelle des bactéries et la formation de nodule suivant au fil du temps. Cependant, le système de plaque est assez laborieux, ce qui le rend moins adapté aux expériences de nodulation à grande échelle. Un inconvénient du système de poche est qu'il est difficile de prévenir la contamination fongique. Les sachets ne sont pas stériles, et donc la croissance fongique est souvent observée sur la moitié supérieure de la poche. Cependant, cela n'affecte pas la croissance de P. andersonii, et n'interfère donc pas avec les essais de nodulation. En outre, le système de poche est seulement approprié pour les semis. Malgré plusieurs tentatives, nous avons été incapables de cultiver des plantons obtenus par la propagation in vitro dans des sachets.
Le pipeline de génétique inversée P. andersonii décrit ici offre une amélioration substantielle parrapport à la méthode de transformation des racines A. rhizogenesexistante 11 . En utilisant les procédures décrites, des lignées transgéniques stables peuvent être générées efficacement et peuvent être maintenues par propagation in vitro. En revanche, la transformation de A. rhizogenes est transitoire et n'entraîne que la formation de racines transgéniques. Puisque chaque racine transgénique résulte d'une transformation indépendante, les essais basés sur la transformation de A. rhizogenes souffrent d'une variation phénotypique importante. Cette variation est beaucoup moins en cas de lignées stables, bien que la propagation in vitro crée également un certain niveau de variation. En raison de cette variation réduite et du fait que plusieurs planctueux pourraient être phénotypés pour chaque ligne stable, les lignes stables sont plus adaptées aux essais quantitatifs que les racines transformées de A. rhizogenes. En outre, la transformation stable ne dépend pas de l'introduction de la racine A. rhizogenes induisant locus (rol) qui affecte l'équilibre hormonal endogène15. Par conséquent, les lignées stables sont mieux adaptées pour l'analyse génétique inversée des gènes impliqués dans l'homéostasie hormonale par rapport à A. rhizogenes-racines transformées. Un avantage plus général de P. andersonii comme modèle de recherche est qu'il n'a pas connu une duplication récente du génome entier (WGD). La sous-famille de légumineuses Papilionoideae, qui comprend les légumineuses modèles M. truncatula et L. japonicus, ainsi que les Salicaceae (ordre Malpighiales) qui comprend le modèle d'arbre Populus trichocarpa expérimenté WGDs 65 millions d'années il ya36,37. De nombreuses copies de gènes paralogues résultant de ces WGD sont conservées dans les génomes de M. truncatula, L. japonicus et P. trichocarpa37,38,39, qui crée redondance qui pourrait compliquer les analyses génétiques inversées. Comme P. andersonii n'a pas connu un WGD récent, les analyses génétiques inversées sur P. andersonii pourraient être moins affectées par le fonctionnement redondant des copies de gène paralogue.
Pris ensemble, nous fournissons un protocole détaillé pour l'analyse génétique inversée dans P. andersonii. En utilisant ce protocole, les lignes mutantes simples peuvent être générées efficacement dans un délai de 2-3 mois10. Ce protocole peut être étendu pour créer des mutants d'ordre supérieur par le multiplexage des sgRNAs ciblant différents gènes simultanément, comme indiqué pour d'autres espèces végétales40,41,42. En outre, la procédure de transformation stable décrite ici ne se limite pas au ciblage génétique CRISPR/Cas9, mais pourrait également être utilisée pour introduire d'autres types de constructions (p. ex., pour les essais de promoteurs-journalistes, l'expression extra-utérine ou trans- complémentation). Nous avons établi P. andersonii comme modèle de recherche comparatif pour étudier les symbioses mutualistes avec des rhizobia fixant l'azote ou des champignons endomycorhiziens. Cependant, les protocoles décrits ici fournissent également des outils pour étudier d'autres aspects de la biologie de cet arbre tropical, tels que la formation de bois, le développement de fleurs bi-sexuelles ou la biosynthèse des métabolites secondaires spécifiques aux Cannabacées.
Déclarations de divulgation
Les auteurs n'ont rien à révéler.
Remerciements
Les auteurs aiment remercier Mark Youles, Sophien Kamoun et Sylvestre Marillonnet d'avoir rendu disponibles les pièces de clonage Golden Gate via la base de données Addgene. De plus, nous tenons à remercier E. James, P. Hadobas et T. J. Higgens pour les graines de P. andersonii. Ces travaux ont été soutenus par l'Organisation néerlandaise pour la recherche scientifique (subvention NWO-VICI 865.13.001; NWO-Open Competition grant 819.01.007) and The Ministry of Research, Technology and Higher Education of the Republic of Indonesia (RISET-PRO grant 8245-ID).
matériels
| Name | Company | Catalog Number | Comments |
| Sigma-Aldrich | N0640 | NAA | |
| Duchefa Biochemie | M1503.0250 | MES | |
| Sigma-Aldrich | D134406 | Acetosyringone | |
| Duchefa Biochemie | X1402.1000 | X-Gal | |
| Merck | 101236 | For nucleic acid electrophoresis gel | |
| - | - | Pouches box material, hangers | |
| Merck | 101188 | NH4NO3 | |
| Sigma-Aldrich | B3408-1G | BAP | |
| Merck | 100156 | H3BO3 | |
| Thermo-Fisher | ER1011 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Thermo-Fisher | 15561020 | Used in Golden Gate cloning assembly | |
| Merck | 137101 | CaCl2·2H2O | |
| Duchefa Biochemie | C0111.0025 | C16H16N5O7S2Na | |
| Thermo-Fisher | K1231 | Used for cloning the blunt-ended PCR amplicons in genotyping procedure | |
| Agronutrition | AP2011 | Containing Rhizophagus irregularis DAOM 197198 (1,000 spores/mL), used for mychorrization assay | |
| Merck | 102790 | CuSO4·5H2O | |
| Duchefa Biochemie | D1004.1000 | Used for plant tissue culture agar-based medium | |
| Merck | 105101 | K2HPO4 | |
| VWR Chemicals | 20302.293 | Na2·EDTA | |
| Duchefa Biochemie | M0803.1000 | C6H14O6 | |
| Thermo-Fisher | ER0291 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Merck | 100983 | C2H5OH | |
| VWR Chemicals | BDH9232-500G | EDTA | |
| Sigma-Aldrich | Z377600-1PAK | Cellophane membrane | |
| Biomatters, Ltd. | R9 or higher | Bioinformatics software for in silico cloning and designing of sgRNAs | |
| Mega International | - | Technical information at https://mega-international.com/tech-info/ | |
| Sigma-Aldrich | 65882 | Used for fixating nodule tissues | |
| VWR Chemicals | 24385.295 | - | |
| Vink | 219341 | Pouches box material, bottom part | |
| Leica Biosystems | 14702218311 | Used as a template for plastic embedding | |
| Merck | 100317 | HCl | |
| Sigma-Aldrich | I5386-1G | IBA | |
| Merck | 103862 | C6H5FeO7 | |
| Merck | 103965 | FeSO4O·7H2O | |
| Duchefa Biochemie | I1401.0005 | IPTG | |
| Duchefa Biochemie | K0126.0010 | ||
| Sigma-Aldrich | L2000 | ||
| Merck | 105886 | MgSO4O·7H2O | |
| Merck | 105934 | MnCl2·4H2O | |
| Merck | 102786 | MnSO4O | |
| Duchefa Biochemie | M1002.1000 | Used for bacterial culture agar-based medium | |
| Manutan | 92007687 | Pouches material | |
| Paraxisdienst | 130774 | Elastic sealing foil | |
| Pull Rhenen | Agra-Perlite No.3 | Used as growing substrate in pots for nodulation assay | |
| VWR Chemicals | 391-0581 | Used as container for cellophane membranes | |
| Thermo-Fisher | F130WH | For genotyping transgenic lines | |
| Addgene | 50337 | Level 0 terminator, 3'UTR, 35s (Cauliflower Mosaic Virus) | |
| Addgene | 48017 | End-link 2 for assembling 2 level one part into a level 2 acceptor | |
| Addgene | 48018 | End-link 3 for assembling 3 level one part into a level 2 acceptor | |
| Addgene | 48001 | Level 1 acceptor. Position 5. Forward orientation | |
| Addgene | 48007 | Level 1 Acceptor. Position 1. Reverse orientation | |
| Addgene | 50268 | Level 0 promoter (0.4 kb), 35s (Cauliflower Mosaic Virus) + 5'UTR, Ω (Tobacco Mosaic Virus) | |
| Addgene | 46966 | Used for designing CRISPR/Cas9 module | |
| Addgene | 46968 | Used for designing CRISPR/Cas9 module | |
| Addgene | 50334 | Level 0 Kanamycin/Neomycin/Paromomycin resistance cassette | |
| Topzeven | - | Used as filters for washing spore suspension | |
| Sigma-Aldrich | 8.17003 | PEG400 | |
| Duchefa Biochemie | E1674.0001 | Pots to grow Parasponia plantlets/seedlings | |
| Merck | 104871 | KH2PO4 | |
| Merck | 105033 | KOH | |
| Merck | 105153 | K2SO4O | |
| Van Leusden b.v. | - | Used as growing substrate for mychorrhization assay | |
| Duchefa Biochemie | S0225.0050 | SH-basal salt medium | |
| Duchefa Biochemie | S0411.0250 | SH-vitamin mixture | |
| Lehle Seeds | VIS-02 | Used as non-ionic surfactant in the washing step of stable transformation | |
| Merck | 137017 | NaCl | |
| VWR Chemicals | 89230-072 | C6H11NaO7 | |
| Merck | 106521 | Na2MoO4·2H2O | |
| Merck | 106574 | Na2HPO4·7H2O | |
| Merck | 567549 | NaH2PO4·H2O | |
| Sigma-Aldrich | 239313 | Na2SO4O | |
| Duchefa Biochemie | S0809.5000 | C12H22O11 | |
| Thermo-Fisher | B69 | Used in Golden Gate cloning assembly | |
| Thermo-Fisher | EL0013 | Used in Golden Gate cloning assembly | |
| Kulzer-Mitsui Chemicals Group | 64708806 | Methyl methacrylate-based resin powder | |
| Kulzer-Mitsui Chemicals Group | 64709003 | HEMA (2-hydroxyethyl methacrylate)-based resin solution | |
| Kulzer-Mitsui Chemicals Group | 66022678 | Methyl methacrylate-based resin solution | |
| Merck | 1159300025 | ||
| Acros | 189350250 | ||
| VWR Chemicals | 663684B | Polysorbate 20 | |
| Stout Perspex | - | pouches box material, lid | |
| Duchefa Biochemie | Y1333.1000 | ||
| Merck | 108816 | ZnCl2 | |
| Alfa Aesar | 33399 | ZnSO4O·7H2O |
Références
- Clason, E. W. THE VEGETATION OF THE UPPER-BADAK REGION OF MOUNT KELUT (EAST JAVA). Bulletin du Jardin Botanique de Buitenzorg. Serie III, 509-518 (1936).
- Soepadmo, E. Ulmaceae. Flora Malesiana-Series 1, Spermatophyta. 8, 31-76 (1974).
- Becking, J. H. The Rhizobium symbiosis of the nonlegume Parasponia. Biological Nitrogen Fixation. , 497-559 (1992).
- Oldroyd, G. E. D. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nature Reviews Microbiology. 11, 252-263 (2013).
- Gutjahr, C., Parniske, M. Cell and developmental biology of arbuscular mycorrhiza symbiosis. Annual Review of Cell and Developmental Biology. 29, 593-617 (2013).
- van Velzen, R., et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proceedings of the National Academy of Sciences of the United States of America. 115, E4700-E4709 (2018).
- van Velzen, R., Doyle, J. J., Geurts, R. A Resurrected Scenario: Single Gain and Massive Loss of Nitrogen-Fixing Nodulation. Trends in Plant Science. 24, 49-57 (2019).
- Griesmann, M., et al. Phylogenomics reveals multiple losses of the nitrogen-fixing root nodule symbiosis. Science. 1743, eaat1743 (2018).
- Becking, J. H. Root-Nodule Symbiosis Between Rhizobium And Parasponia (Ulmaceae). Plant and Soil. 51, 289-296 (1979).
- van Zeijl, A., et al. CRISPR/Cas9-Mediated Mutagenesis of Four Putative Symbiosis Genes of the Tropical Tree Parasponia andersonii Reveals Novel Phenotypes. Frontiers in Plant Science. 9, 284 (2018).
- Cao, Q., et al. Efficiency of Agrobacterium rhizogenes–mediated root transformation of Parasponia and Trema is temperature dependent. Plant Growth Regulation. 68, 459-465 (2012).
- Limpens, E., et al. RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicago truncatula. Journal of Experimental Botany. 55, 983-992 (2004).
- Boisson-Dernier, A., et al. Agrobacterium rhizogenes-Transformed Roots of Medicago truncatula for the Study of Nitrogen-Fixing and Endomycorrhizal Symbiotic Associations. Molecular Plant-Microbe Interactions. 14, 695-700 (2001).
- Kumagai, H., Kouchi, H. Gene Silencing by Expression of Hairpin RNA in Lotus japonicus Roots and Root Nodules. Molecular Plant-Microbe Interactions. 16, 663-668 (2003).
- Nilsson, O., Olsson, O. Getting to the root: The role of the Agrobacterium rhizogenes rol genes in the formation of hairy roots. Physiologia Plantarum. 100, 463-473 (1997).
- Davey, M. R., et al. Effective Nodulation of Micro-Propagated Shoots of the Non-Legume Parasponia andersonii by Bradyrhizobium. Journal of Experimental Botany. 44, 863-867 (1993).
- Webster, G., Poulton, P. R., Cocking, E. C., Davey, M. R. The nodulation of micro-propagated plants of Parasponia andersonii by tropical legume rhizobia. Journal of Experimental Botany. 46, 1131-1137 (1995).
- Op den Camp, R., et al. LysM-type mycorrhizal receptor recruited for rhizobium symbiosis in nonlegume Parasponia. Science. 331, 909-912 (2011).
- Weber, E., Engler, C., Gruetzner, R., Werner, S., Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLOS ONE. 6, e16765 (2011).
- Nekrasov, V., Staskawicz, B. J., Weigel, D., Jones, J. D. G., Kamoun, S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nature Biotechnology. 31, 691-693 (2013).
- Bertani, G. Studies On Lysogenesis. I. The Mode Of Phage Liberation By Lysogenic Escherichia Coli. Journal of Bacteriology. 62, 293-300 (1951).
- Engler, C., et al. A Golden Gate modular cloning toolbox for plants. ACS Synthetic Biology. 3, 839-843 (2014).
- Fauser, F., Schiml, S., Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. The Plant Journal. 79, 348-359 (2014).
- Lazo, G. R., Stein, P. A., Ludwig, R. A. A DNA Transformation-Competent Arabidopsis Genomic Library in Agrobacterium. Biotechnology. 9, 963-967 (1991).
- Op den Camp, R. H. M., et al. N Nonlegume Parasponia andersonii Deploys A Broad Rhizobium Host Range Strategy Resulting in Largely Variable Symbiotic Effectivenes. Molecular Plant-Microbe Interactions. 25, 954-963 (2012).
- Graham, P. H., Viteri, S. E., Mackie, F., Vargas, A. T., Palacios, A. Variation in acid soil tolerance among strains of Rhizobium phaseoli. Field Crops Research. 5, 121-128 (1982).
- Martinez-Romero, E., et al. Rhizobium tropici, A Novel Species Nodulating Phaseolus vulgaris L. Beans and Leucaena sp. Trees. International Journal of Systematic and Evolutionary Microbiology. 41, 417-421 (1991).
- Felten, J., et al. The Ectomycorrhizal Fungus Laccaria bicolor Stimulates Lateral Root Formation in Poplar and Arabidopsis through Auxin Transport and Signaling. Plant Physiology. 151, 1991-2005 (1991).
- Trouvelot, A., Kough, J. L., Gianinazzi-Pearson, V. Mesure du taux de mycorhization VA d’un systeme radiculaire. Recherche de methods d’estimation ayant une signification fonctionnelle. Aspects Physiologiques et Genetiques des Mycorhizes. , 217-221 (1986).
- Schenk, R. U., Hildebrandt, A. C. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Canadian Journal of Botany. 50, 199-204 (1972).
- Becking, J. H. The Parasponia parviflora - Rhizobium symbiosis. Host specificity, growth and nitrogen fixation under various conditions. Plant and Soil. 75, 309-342 (1983).
- Hoagland, D. R., Arnon Revised, D. I., Arnon, D. I. The Water-Culture Method for Growing Plants without Soil. Circular California Agricultural Experiment Station. 347, 1-32 (1950).
- Wang, K. . Methods in Molecular Biology: Agrobacterium Protocols. 1, (2015).
- Smulders, M. J. M., de Klerk, G. J. Epigenetics in plant tissue culture. Plant Growth Regulation. 63, 137-146 (2011).
- Larkin, P. J., Scowcroft, W. R. Somaclonal variation — a novel source of variability from cell cultures for plant improvement. Theoretical and Applied Genetics. 60, 197-214 (1981).
- Cannon, S. B., et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proceedings of the National Academy of Sciences of the United States of America. 103, 14959-14964 (2006).
- Tuskan, G. A., et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313, 1596-1604 (2006).
- Young, N. D., et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 480, 520 (2011).
- Sato, S., et al. Genome Structure of the Legume, Lotus japonicus. DNA Research. 15, 227-239 (2008).
- Xing, H. L., et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biology. 14, 327 (2014).
- Lowder, L. G., et al. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiology. 169, 971 (2015).
- van Zeijl, A. . Dissecting Hormonal Pathways in Nitrogen-Fixing Rhizobium Symbioses. , (2017).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.