Trasformazione, editing del genoma e fenotipizzazione dell'azoto-fixing Tropical Cannabaceae Tree Parasponia andersonii
In This Article
Summary
Parasponia andersonii è un albero tropicale in rapida crescita che appartiene alla famiglia Cannabis (Cannabaceae) e può formare noduli di radice che fissano l'azoto in associazione con il rizobium. Qui, descriviamo un protocollo dettagliato per le analisi genetiche inverse in P. andersonii basato su Agrobacterium tumefaciens- mediato trasformazione stabile e modifica del genoma CRISPR/Cas9.
Abstract
Parasponia andersonii è un albero tropicale in rapida crescita che appartiene alla famiglia Cannabis (Cannabaceae). Insieme a 4 specie aggiuntive, forma l'unico lignaggio non legume conosciuto in grado di stabilire una simbiosi nodule che fissa l'azoto con il rizobio. Studi comparativi tra legumi e P. andersonii potrebbero fornire preziose informazioni sulle reti genetiche alla base della formazione dei noduli delle radici. Per facilitare gli studi comparativi, abbiamo recentemente sequenziato il genoma di P. andersonii e stabilito Agrobacterium tumefaciens- mediato trasformazione stabile e editing del genoma basato su CRISPR/Cas9. Qui, forniamo una descrizione dettagliata delle procedure di trasformazione e di editing del genoma sviluppate per P. andersonii. Inoltre, descriviamo le procedure per la germinazione dei semi e la caratterizzazione dei fenotipi simbiotici. Utilizzando questo protocollo, possono essere generate linee mutanti transgeniche stabili in un periodo di 2-3 mesi. La propagazione vegetativa nella propagazione in vitro delle linee transgeniche T0 consente di avviare esperimenti di fenotipizzazione a 4 mesi dopo la co-coltivazione di A. tumefaciens. Pertanto, questo protocollo richiede solo marginalmente più tempo del metodo transitorio di trasformazione della radice basato su Agrobacterium rhizogenesdisponibile per P. andersonii, anche se offre diversi vantaggi chiari. Insieme, le procedure qui descritte permettono a P. andersonii di essere utilizzato come modello di ricerca per studi volti a comprendere le associazioni simbiotiche e potenzialmente altri aspetti della biologia di questo albero tropicale.
Introduction
Parasponia andersonii è un albero tropicale appartenente alla famiglia Cannabis (Cannabaceae) ed è nativo di Papua Nuova Guinea e diverse isole del Pacifico1,2,3. Insieme ad altre 4 specie di Parasponia, rappresenta l'unico lignaggio non legume in grado di stabilire una simbiosi nodule che fissa l'azoto con la rizobia. Questa simbiosi è ben studiata nei modelli di legume (Fabaceae) Medicago truncatula e Lotus japonicus, che ha portato all'acquisizione di una conoscenza dettagliata della natura genetica molecolare della formazione dei noduli e del funzionamento4. Inoltre, è stato dimostrato che la simbiosi nodule radice nei legumi si basa sulla simbiosi micorrizica molto più antica e diffusa5 . I confronti filogenomici suggeriscono che le simbiosi noduli che fissano l'azoto dei legumi, Parasponia, così come le cosiddette specie di piante actinorhizal che ospitano batteri Frankia diazotrofica che ospitano, hanno un'origine evolutiva condivisa 6,7,8. Per determinare se i geni identificati per essere coinvolti nella formazione del nodulo legume sono parte di una base genetica conservata, gli studi sulle specie non legumi sono essenziali. A tal fine, proponiamo di utilizzare P. andersonii come modello di ricerca comparativa, insieme ai legumi, per identificare le reti genetiche fondamentali alla base della formazione e del funzionamento dei noduli delle radici.
P. andersonii è un pioniere che si può trovare sulle pendici delle colline vulcaniche. Può soddisfare velocità di crescita di 45 cm al mese e raggiungere lunghezze fino a 10 metri9. P. andersonii alberi sono impollinati dal vento, che è facilitato dalla formazione di fiori maschili e femminili separati3,10. Recentemente abbiamo sequenziato e annotato il genoma diploide (2n - 20; 560 Mb/1C) di P. andersonii, e assemblato sequenze di genoma di progetto di 2 specie aggiuntive di Parasponia; P. rigida e P. rugosa6. Questo ha rivelato 35.000 dollari modelli genici P. andersonii che possono essere raggruppati in >20,000 ortogruppi insieme ageni da M. truncatula, soybean (Glycine max), Arabidopsis thaliana, fragola bosco ( Fragaria vesca), Trema orientalis, pioppo di cotone nero (Populus trichocarpa) e eucalipto (Eucalyptus grandis)6. Inoltre, i confronti tratrascrittomi tra M. truncatula e P. andersonii hanno identificato una serie di 290 ortologhi putativi che mostrano un modello di espressione con valori nodulo in entrambe le specie6. Ciò costituisce un'eccellente risorsa per gli studi comparativi.
Per studiare la funzione genica nelle radici e nei noduli di P. andersonii, è stato stabilito un protocollo per la trasformazione della radice mediata Agrobacterium -mediata11. Utilizzando questo protocollo, le piante composte che portano radici transgeniche possono essere generate in un lasso di tempo relativamente breve. Questo metodo è, inoltre, ampiamente applicato nella ricerca legume-simbiosi12,13,14. Tuttavia, lo svantaggio di questo metodo è che solo le radici vengono trasformate e che ogni radice transgenica rappresenta un evento di trasformazione indipendente, con conseguente variazione sostanziale. Inoltre, la trasformazione è transitoria e le linee transgeniche non possono essere mantenute. Questo rende la trasformazione della radice basata su A. rhizogenesmeno adatta per l'editing genomico mediato da CRISPR/Cas9. Inoltre, A. rhizogenes trasferisce i suoi geni locus (rol) che inducevano la radice al genoma della pianta, che una volta esprimeva interferiscono con l'omeostasi dell'ormone15. Questo rende difficile studiare il ruolo degli ormoni vegetali nelle radici trasformate da A. rhizogenes. Per superare questi limiti, abbiamo recentemente sviluppato un protocollo per la trasformazione basata su Agrobacterium tumefaciense la mutagenesi mediata CRISPR/Cas9 di P. andersonii10.
Qui, forniamo una descrizione dettagliata della procedura di trasformazione a. tumefaciense della pipeline di genetica inversa sviluppata per P. andersonii. Inoltre, forniamo protocolli per la gestione a valle delle piastrine transgeniche, inclusi i saggi per studiare le interazioni simbiotiche. Utilizzando il protocollo qui descritto, possono essere generate più linee transgeniche in un periodo di 2-3 mesi. In combinazione con la mutagenesi mediata DA CRISPR/Cas9, questo permette una generazione efficiente di linee mutanti ad eliminazione diretta. Queste linee mutanti possono essere propagate in vitro10,16,17, il che consente di generare materiale sufficiente per iniziare la caratterizzazione fenotipica a 4 mesi dopo che la procedura di trasformazione è stata stato avviato10. Insieme, questa serie di procedure dovrebbe consentire a qualsiasi laboratorio di adottare P. andersonii come modello di ricerca per studi volti a comprendere le associazioni rizobiali e micorriche, nonché potenzialmente altri aspetti della biologia di questo albero tropicale.
Protocol
1. Coltivare P. andersonii Alberi nella serra
-
Germina P. andersonii WU1 semi18.
- Utilizzare bacche fresche di Parasponia o immergere le bacche secche in acqua per 2 h per reidratarsi. Schiacciare le bacche su un pezzo di carta velina o strofinare contro l'interno di un setaccio di tè per rimuovere i semi.
- Disinfettare i semi utilizzando la candeggina commerciale (4% di ipoclotosi) per 15-20 min e successivamente lavare i semi 6 volte con acqua sterilizzata.
- Trasferire i semi in sterili tubi PCR da 200 . Riempire i tubi con acqua sterilizzata, in modo che i semi siano completamente sommersi. Incubare i tubi per 10 giorni in un termociclore che esegue il seguente programma: 30 cicli (7 gradi centigradi per 4 h, 28 gradi centigradi per 4 h). Non usare un coperchio riscaldato, in quanto questo potrebbe uccidere i semi.
- Preparare le piastre SH-0 (vedere Tabella 1). Trasferire i semi in piatti SH-0 e incubare a 28 c, 16 h:8 h giorno:notte. Chiudere le piastre con 2 strati di pellicola di guarnizione elastica per evitare l'essiccazione durante l'incubazione a 28 gradi centigradi.
- Dopo che le piantine hanno sviluppato il loro primo set di foglie vere (3-4 settimane dopo l'incubazione a 28 gradi centigradi), trasferiscono le piantine in vasi pieni di terriccio commerciale e coprono le piantine con una tazza di plastica traslucida per evitare la disidratazione. Collocate i vasi in una stanza clima o in una serra di 28 gradi centigradi, l'85% DI RH, sotto un regime di 16 h:8 h giorno:notte.
- Dopo 1 settimana, rimuovere la tazza di plastica traslucida. Innaffiare regolarmente i vasi e quando gli alberi crescono più grande integratore con fertilizzante per sostenere la crescita.
2. Clonazione di costrutti per la Mutagenesi mediata CRISPR/Cas9 di P. andersonii
NOT: I vettori di trasformazione binaria standard possono essere utilizzati per la trasformazione stabile di P. andersonii. Qui, ad esempio, è una procedura per generare costrutti per la mutagenesi mediata CRISPR/Cas9 utilizzando la clonazione modulare (ad esempio, Golden Gate)19.
- Identificare le sequenze di target dell'RNA guida per i geni di interesse, utilizzando un software di bioinformatica dotato di uno strumento di progettazione CRISPR integrato. Scegli sequenze di RNA guida situate all'estremità 5'della sequenza di codifica del gene bersaglio per aumentare la probabilità di ottenere knockout completi. Assicurarsi di controllare gli effetti fuori target cercando contro il genoma di P. andersonii 6.
NOTA: Utilizzare 2 sgRNA per gene bersaglio, preferibilmente a 200-300 bp l'uno dall'altro. Questo può generare delezioni che possono essere identificate dalla PCR e successivamente dall'elettroforesi gel agarose. - Genera costrutti Golden Gate di livello 1 contenenti le sequenze sgRNA.
- Progettare i primer per amplificare ogni singolo sgRNA inserendo la sequenza di guida di 20 bp nella posizione di N(20) nella seguente sequenza di primer: 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAATAGCAAG-3'.
NOTA: Se la sequenza guida è uguale a GN(19),rimuovere la G alla fine 5' della sequenza guida prima di inserire nella sequenza primer. - PCR amplifica gli sgRNA da pICH86966::AtU6p::sgRNA_PDS20 utilizzando i primer in avanti progettati al passaggio 2.2.1 e l'inversione universale: 5'-TGTGCTCCAGCGTAATGCCAACTTGTAC-3'. Utilizzare una polimerasi del DNA ad alta fedeltà stabile e le seguenti condizioni di PCR: 98 gradi centigradi per 30 s; 30 cicli (98 gradi centigradi per 10 s; 53 gradi centigradi per 20 s; 72 gradi centigradi per 10 s); 72 s per 7 min. Le reazioni PCR riuscite producono un'ampiacone da 165 bp.
- Purificazione a colonna dell'amplificatore PCR utilizzando un kit di purificazione PCR commerciale. Successivamente, impostare le reazioni Golden Gate per clonare sgRNA dietro l'Arabidopsis thaliana AtU6p piccolo RNA promoter: 10 ng del sgRNA PCR amplificatore, 150 ng di pICSL01009::AtU6p20, 60 ng del vettore accettatore di livello 1 appropriato, 2 L di T4 buffer di ligase, 2 L 0,1% dell'albumina del siero bovino (BSA), 0,5 l di BsaI, 0,5 l di T4 ligase, riempire fino a 20 l con acqua ultra-pura. Assicurarsi che tutti gli sgRNA siano clonati nello stesso orientamento per evitare la formazione di forcina.
- Incubare le reazioni in un termociclista che esegue il seguente programma: 37 C per 20 s; 26 cicli (37 gradi centigradi per 3 min; 16 gradi centigradi per 4 min); 50 gradi centigradi per 5 min; 80 s per 5 min. Trasformare le reazioni Golden Gate a Escherichia coli e piastra su LB medio21 contenente ampicillina (50 mg/L), X-Gal (200 mg/L) e IPTG (1 mM).
NOTA: Preparare soluzioni stock di IPTG e X-Gal rispettivamente in acqua ultrapura e dimetthylformamide. Filtro sterilizzare le soluzioni di stock di ampicillina e IPTG e conservare tutte le scorte a -20 gradi centigradi. Indossare guanti durante la manipolazione di dimethylformamide. - Seleziona colonie bianche e isola i plasmidi utilizzando un kit di isolamento plasmiera commerciale. Verifica sequenza verificare plasmidi isolati prima di continuare con l'assemblaggio di livello 2 di Golden Gate.
- Progettare i primer per amplificare ogni singolo sgRNA inserendo la sequenza di guida di 20 bp nella posizione di N(20) nella seguente sequenza di primer: 5'-TGTGGTCTCAATTGN(20) GTTTTAGAGCTAGAATAGCAAG-3'.
- Assembla i costrutti golden Gate di livello 2 per la trasformazione stabile.
- Eseguire una reazione Golden Gate utilizzando il livello 1 AtU6p::sgRNA costruisce (generato sotto la sezione 2.2) così come pICH47802::NPTII, pICH47742::35Spro:: linker finale (vedere Engler et al.22). Eseguire le reazioni come segue: utilizzare 100 fmol di ciascun vettore donatore e 20 fmol del vettore dell'acceleratore e aggiungere 2 l of T4 ligase buffer, 2 - L di 0,1% BSA, 0,5 L di BpiI, 0,5 dollari di Ligase T4, riempire a 20 -L con acqua ultrapura.
NOTA: Il livello 1 plasmids pICH47802::NPTII, pICH47742::35Spro:: NLS-aCas9::35Ster devono essere clonati prima (vedere Supplemental File 1), come descritto per sgRNA ai punti 2.220,22 ,23. - Incubare le reazioni come al punto 2.2.4 e trasformarsi in E. coli. Piastra su supporto LB contenente kanamycin. Il giorno dopo, selezionare le colonie bianche e isolare i plasmidi. Determinare l'assemblaggio plasmid corretto mediante l'analisi della digestione delle restrizioni.
- Eseguire una reazione Golden Gate utilizzando il livello 1 AtU6p::sgRNA costruisce (generato sotto la sezione 2.2) così come pICH47802::NPTII, pICH47742::35Spro:: linker finale (vedere Engler et al.22). Eseguire le reazioni come segue: utilizzare 100 fmol di ciascun vettore donatore e 20 fmol del vettore dell'acceleratore e aggiungere 2 l of T4 ligase buffer, 2 - L di 0,1% BSA, 0,5 L di BpiI, 0,5 dollari di Ligase T4, riempire a 20 -L con acqua ultrapura.
- Trasformare i costrutti di livello 2 in ceppo AGL124di Agrobacterium tumefaciens .
3. Trasformazione stabile di P. andersonii
- Inoculare 2 lastre Di LB contenenti gli antibiotici appropriati con ceppo A. tumefaciens AGL1 trasformato con il costrutto di interesse. Incubare piastre a 28 gradi centigradi per 2 giorni.
- Raccogliere rami giovani da alberi coltivati a serre. Utilizzare circa 5 rami di 5-8 cm di lunghezza per ogni trasformazione. Assicurarsi di utilizzare solo rami sani non infetti. Togliere le foglie tagliandole in quanto tali che 1 cm2 di tessuto fogliare viene lasciato alla fine di ogni picciolo. Scartare le foglie.
- Disinfettare il tessuto per 15 min utilizzando una candeggina commerciale diluita 1:1 (2% di ipocloto si dopo la diluizione) contenente alcune gocce di polisorbate 20. Quindi, sciacquare il tessuto 6 volte con acqua autoclaved.
NOTA: Questo passaggio, così come, i seguenti passaggi devono essere condotti all'interno di un armadio laminare down-flow per mantenere il tessuto sterile. - Sospendere nuovamente le cellule A. tumefaciens da 1-2 piastre in 25 mL di mezzo di infiltrazione (cfr. tabella 1) contenente acetosyringone (20 mg/L) e un surfactant non ionico (0,001% v/v) per raggiungere una densità ottica (OD600) di 5 dollari.
NOTA: Preparare la soluzione stock acetosyringone nel 70% di etanolo e conservare a -20 gradi centigradi. Il surfactant non ionico deve essere sterilizzato filtrando prima di aggiungere al mezzo di infiltrazione. - Tagliare sia il gambo che il tessuto del picciolo in pezzi di 1 cm di lunghezza all'interno della sospensione A. tumefaciens, creando così ferite fresche su entrambi i lati. Lasciare i pezzi di tessuto nella sospensione A. tumefaciens per 10-30 min.
- Preparare il supporto di radicamento (vedere la tabella1) e aggiungere acetosyringone (20 mg/L) dopo l'autoclaving. Pezzi di tessuto secco su un pezzo sterile di carta da filtro e posizionarlo sul mezzo (10 espianti /piastra). Incubare piastre al buio a 21 gradi centigradi per 2 giorni.
NOTA: Lasciare raffreddare il mezzo fino a 60 gradi centigradi prima di aggiungere l'acetosyringone. - Dopo 2 giorni, ispezionare le piastre per la contaminazione batterica fungina o evidente (batteri diversi da A. tumefaciens). Le piastre contaminate devono essere scartate.
- Preparare il liquido SH-10 medio (vedere la tabella 1). Dopo l'autoclaving, aggiungere il polisrizo 20 (0,01%, v/v). Trasferire i pezzi di tessuto a 10 mL di SH-10 contenenti polisorbate 20. Durante un periodo di almeno 10 min, agitare delicatamente ogni 2-3 min per lavare il tessuto.
- Lavare altre due volte con SH-10 fresco contenente polis ricoperto 20. Questi tempi, un tempo di incubazione 2-3 min per passo di lavaggio è sufficiente.
- Preparare il supporto di rooting (vedere la tabella 1). Dopo l'autoclaving, aggiungere cefotaxime (300 mg/L) e la kanamycin (50 mg/L) e versare le piastre. Per le trasformazioni secondarie (trasformazioni di linee transgeniche resistenti alla kanamicicina), applicare la selezione dell'igromiccina (15 mg/L).
- Pezzi di tessuto secco su pezzi sterili di carta da filtro. Successivamente, trasferire i pezzi di tessuto alle piastre preparate al passaggio 3.9.
- Incubare piastre per 7 giorni a 28 gradi centigradi, 16 h:8 h giorno:notte. Ogni 2 giorni controllare piastre per la contaminazione fungina o batterica e la crescita eccessiva di A. tumefaciens. In caso di contaminazione, trasferire i pezzi non infetti in una piastra fresca.
- Dopo 7 giorni, trasferire i pezzi di tessuto a medio di propagazione (cfr. tabella 1) contenenti cefotaxime (300 mg/L) e kanamycin (50 mg/L). Incubare piastre a 28 gradi centigradi, 16 h:8 h giorno:notte. Rinfrescare le piastre una volta alla settimana fino a quando non si sviluppano germogli transgenici. Assicurarsi di trasferire solo pezzi di tessuto non infetti su piastre fresche. Scartare i pezzi che sono ricoperti da A. tumefaciens.
- Una volta che i germogli putativamente transgenici sono di 1 cm di lunghezza, germogli tagliati e colture in modo indipendente nel mezzo di propagazione contenente cefotaxime (300 mg/L) e kanamicina (50 mg/L). Per garantire che i germogli rappresentino trasformazioni indipendenti, scatta un solo tiro da ogni lato di un espianto.
- I germogli vegetativi si propagano putagatamente transgenici come descritto al punto 5.2.
4. Genotipizzazione di germogli putativi-transgenici
- Primer di progettazione che coprono i siti di riconoscimento sgRNA. Per consentire il sequenziamento dell'amplificatore PCR, scegliere primer a 150-250 bp di distanza dai siti di riconoscimento sgRNA.
- Tagliare una punta di foglia (5 mm) da ogni germoglio transgenico per essere genotipato. Inoltre, raccogliere un campione di controllo di tipo selvatico.
- Eseguire 50 reazioni PCR da 50 luna utilizzando i primer progettati al passaggio 4.1 e un kit commerciale per amplificare direttamente il DNA da campioni di piante. In alternativa, le reazioni PCR possono essere eseguite su DNA purificato utilizzando una polimerasi ad alta fedeltà.
- Separare gli amplificatori PCR su un gel di agarose 1.5-2%.
- Analizzare i risultati dell'elettroforesi gel. Controllare i campioni che producono più bande (più di 1 allele) e amplificatori PCR con dimensioni diverse dal tipo selvaggio, che indica la presenza di indel di medie dimensioni.
- Amplificatori PCR di sequenza per identificare le mutazioni esatte. Per i campioni che producono un singolo amplificatore PCR, i prodotti PCR possono essere sequenziati direttamente. I campioni che producono più di 1 banda dopo l'elettroforesi del gel o che sembrano essere eterozici dopo il sequenziamento diretto dell'amplificatore PCR, devono prima essere clonati in un vettore di clonazione contundente.end. Successivamente, sequenziare più cloni per ogni campione per identificare tutti i possibili alleli presenti nel campione.
- Allineare i risultati del sequenziamento al gene di interesse e ispezionare l'allineamento per verificare la presenza di mutazioni vicino ai siti di destinazione dello sgRNA. Successivamente, controlla se queste mutazioni creano frameshift. Elimina le linee con > 2 alleli e le linee contenenti mutazioni in-frame.
- Selezionare più righe per un'ulteriore analisi.
- Propagare le righe selezionate come descritto al punto 5.2.
- Quando le linee hanno sviluppato diversi nuovi germogli, preleva nuovi campioni dalle punte delle foglie da 3 dollari e ripeti i passaggi da 4,3 a 4,7. Determinare se le mutazioni presenti in ciascuno dei campioni provenienti dalla stessa linea e dal campione PCR originale sono identiche. Le linee che producono le stesse mutazioni in tutti i campioni sono omogeneamente mutate e possono essere utilizzate per ulteriori sperimentazioni. Eliminare le linee che non producono gli stessi risultati di queste linee sono chiateriche.
5. Preparazione delle piantine Radicate P. andersonii per la sperimentazione
-
Avviare una nuova linea di coltura dei tessuti di P. andersonii.
- Raccogliere gemme ascellari, giovani germogli avventurosi o tessuti fogliati da alberi sani. In alternativa, le piantine possono essere utilizzate come materiale di partenza.
- Disinfettare il tessuto utilizzando una candeggina commerciale diluita 1:1 (2% di ipocloresi dopo la diluizione) contenente alcune gocce di polisorbate 20 per 15 min. In seguito, risciacquare il tessuto 6 volte usando acqua autoclatura.
NOTA: Questo passaggio, così come, i seguenti passaggi devono essere condotti all'interno di un deflusso laminare o armadietto a flusso incrociato laminare per mantenere il tessuto sterile. - Trasferire il tessuto al mezzo di propagazione (cfr. tabella 1). Chiudere le piastre con 2 strati di pellicola elastica di sigillamento e incubare piastre a 28 , 16 h:8 h giorno:notte.
- Ispezionare le piastre ogni pochi giorni durante le prime 2 settimane per garantire che il tessuto sia privo di contaminazione fungina o batterica.
- Propagare il tessuto mettendo 10 germogli su una piastra fresca di mezzo di propagazione e chiudere la piastra con 2 strati di pellicola di sigillazione elastica. Incubare piastre a 28 gradi centigradi, 16 h:8 h giorno:notte. Ripetere questo passaggio ogni 4 settimane.
- Quando i germogli sono >1 cm di lunghezza, tagliare germogli alla loro base e metterli sul mezzo di radicamento (vedere Tabella 1). Circa 10 germogli possono essere posizionati su una singola piastra di radicamento. La posizione scatta in posizione verticale inserendo la punta basale del tiro nel mezzo. Le radici appaiono a 10-14 giorni dopo l'incubazione delle piastre a 28 gradi centigradi, 16 h:8 h giorno:notte.
NOTA: Non radicare tutti i germogli, ma mantenere parte per la propagazione della coltura dei tessuti (vedere il passaggio 5.2).
6. Nodulation delle piantine P. andersonii in vasi
-
Preparare il rizobium inoculum.
- Inoculare 10 mL di liquido YEM medio (vedi tabella 2) da una singola colonia di Mesorhizobium plurifarium BOR26 e incubare a 28 gradi centigradi per 2 giorni.
NOTA: M. plurifarium BOR2 è preferito in quanto nodula in modo efficiente P. andersonii. Tuttavia, altri ceppi di rizobio possono essere utilizzati anche per annuire P. andersonii (ad esempio Bradyrhizobium elkanii WUR325, Rhizobium tropic CIAT89926,27 o Bradyrhizobium sp. Kelud2A4). - Utilizzare la coltura 10 mL per inoculare un volume maggiore di supporto liquido YEM. Il volume di questa coltura dipende dal numero di vasi che devono essere inoculati.
- Preparare il mezzo Liquido EKM (vedere tabelle 3, tabella 4). Centrifugare la coltura batterica per 10 min a 3.500 x g per raccogliere le cellule. Successivamente, sospendere nuovamente il pellet batterico in EKM liquido (utilizzare approssimativamente lo stesso volume della coltura originale yEM) e determinare la densità ottica (OD600).
- Inoculare 10 mL di liquido YEM medio (vedi tabella 2) da una singola colonia di Mesorhizobium plurifarium BOR26 e incubare a 28 gradi centigradi per 2 giorni.
- Per i vasi da 20 dollari, preparare 3 L di liquido EKM medio e inoculato con la sospensione rinozobiale preparata al gradirio 6.1.3. per raggiungere l'OD600 e 0,025.
- Mescolare 3 L di EKM contenente rizobia con 1.250 g di perlite. Successivamente, aggiungere 210 g di questa miscela a sterili vasi di polipropilene traslucido. In alternativa, invece di perlite, utilizzare la sabbia come substrato per i saggi di annuizione.
- Piantare 1-3 P. andersonii piantagioni in ogni vaso. Inoltre, preparare diversi vasi contenenti piastrine P. andersonii trasformate con il costrutto di controllo CRISPR (vedere tabella supplementare 1). Pesare diversi vasi per essere in grado di determinare la perdita d'acqua durante l'esperimento. Coprire il fondo di ogni vaso per proteggere le radici dall'esposizione alla luce.
- Incuba i vasi in una stanza di crescita climatizzata (28 gradi centigradi, 16 h:8 h giorno:notte) per 4-6 settimane. Una volta alla settimana, pesare diverse pentole per determinare la perdita d'acqua. Se la perdita d'acqua supera i 10 mL, integrare con acqua ultra-pura per compensare la perdita.
- Dopo 4-6 settimane, pulire le radici dalla perlite e determinare i numeri di nodulo utilizzando un binocolo per esaminare l'efficienza nodulazione.
7. Nodulation delle piastre P. andersonii sulle piastre
-
Preparare le membrane di cellophane 28.
- Tagliare la membrana del cellophane per adattarla a un quadrato di 12 cm x 12 cm piatto Petri. Tagliare le membrane un po 'più corte nella parte superiore per consentire lo spazio per i germogli a crescere.
- Per aumentare la permeabilità delle membrane di cellophane, far bollire le membrane in soluzione EDTA (1 g/L) per 20 min. Successivamente, risciacquare almeno 6x con acqua demineralizzata per rimuovere l'EDTA.
NOTA: Poiché la membrana secca tende a rughe quando a contatto con l'acqua, immergere le membrane secche una ad una nella soluzione. - Disporre le membrane orizzontalmente in un sottile strato d'acqua in una lastra di vetro rotonda. Sterilizzare le membrane autoclaving due volte.
- Posizionare 1 membrana di cellophane autoclaved su un quadrato 12 x 12 cm piatto Petri contenente agar-solidificato EKM medio (vedere tabella 3, tabella 4). Posizionare due piantine P. andersonii radicate di 3 settimane (vedi sezione 5) o piantine vecchie di 4 settimane (vedi sezione 1.1) sulla parte superiore della membrana. Assicurarsi di raccogliere solo piantine o piantine con radici che hanno punte di radice bianca, indicando che queste radici sono ancora in crescita.
- Coprire delicatamente le radici con una seconda membrana di cellophane, creando uno strato di sandwich. Sigillare la piastra con 3 strati di pellicola di sigillazione elastica. Avvolgere la metà inferiore delle piastre con un foglio di alluminio, per coprire le radici dall'esposizione alla luce.
- Incubare le piastre in una sala di crescita climatizzata (28 gradi centigradi, 16 h:8 h giorno:notte) per 3-4 settimane. Contrassegnare la posizione dei suggerimenti radice per seguire la crescita della radice nel tempo.
- Se le piastre EKM iniziano ad asciugarsi a causa di un'incubazione prolungata, trasferire le piante in piastre EKM fresche qualche giorno prima dell'inoculazione batterica.
- Preparare l'inoculum batterico come descritto al punto 6.1.
- Rimuovere la membrana superiore del cellophane e applicare 1 mL di coltura di rizobium (OD600 - 0,025) alle radici. Successivamente, posizionare una nuova membrana di cellophane sulle radici inocurate. Avvolgere l'esterno della piastra utilizzando un foglio di alluminio per coprire le radici dall'esposizione alla luce.
- Dopo 4 settimane, esaminare i numeri dei noduli utilizzando un binocolare per determinare l'efficienza del nodulation.
8. Nodulation of P. andersonii Seedlings in Pouches
- Semi di Germinate P. andersonii come descritto nella sezione 1.1. Dopo che i cotiledoni sono emersi (12 giorni su piastre SH-0 a 28 gradi centigradi), trasferiscono le piantine in sacchetti.
- Per preparare le sacche, strappare la sezione piegata dello stoppino di carta e aggiungere 7 mL di mezzo EKM modificato (vedere Tabella 3, Tabella 4).
- Inserire 1 o 2 piantine mettendo le radici tra entrambi i fogli di carta che formano lo stoppino di carta e il foglio di plastica anteriore del sacchetto.
- Proteggere le radici dall'esposizione alla luce, piegando lamina di alluminio intorno alla sacca. Sospendere le sacche in una scatola di plastica ricoperta da un coperchio traslucido per mantenere alta l'umidità. Posizionare la scatola in una stanza di crescita climatizzata (28 gradi centigradi, 16 h:8 h giorno:notte).
- Compensare l'evaporazione dell'acqua aggiungendo acqua sterile ultra-pura, in quanto lo stoppino di carta rimane umido (evitare l'acqua in piedi sul fondo della sacca). Dopo la prima settimana, questo richiede generalmente l'aggiunta di 2-3 ml ogni 4 giorni.
- Preparare l'inoculum batterico come descritto al punto 6.1.
- Dopo che le piantine sono state coltivate per 10-12 giorni in sacchetti, inoculare l'impianto radicale con 500 l di coltura di rizobium (OD600 - 0,025).
- Seguire la formazione di noduli nel tempo. Quattro settimane dopo l'inoculazione, i noduli possono essere contati e raccolti per determinare l'efficienza di nodulazione.
9. Nodule Cytoarchitecture Analisi
- Raccogliere 10-15 noduli in un tubo da 2 mL contenente fissativo (5% glutaraldeide in buffer fosfato da 0,1 M, pH 7.2). Applicare l'aspirapolvere per 1/2-1 h e incubare durante la notte a 4 gradi centigradi. Durante questo periodo, i campioni affondano sul fondo del tubo.
NOTA: La soluzione fissativa può essere conservata a 4 gradi centigradi per 2-4 settimane prima dell'utilizzo. Assicurarsi di indossare guanti quando si lavora con il fissativo dei tessuti. - Lavare i noduli 2x con buffer fosfato da 0,1 M, pH 7.2. Applicare intervalli di 10 min tra ogni passo di lavaggio.
- Disidratare i campioni incubando successivamente nel 30%, 50%, 70% e 100% etanolo. Per assicurarsi che tutta l'acqua venga rimossa dai campioni, ripetere il passaggio 3x di etanolo al 100%. Applicare intervalli di 10 min tra ogni fase di disidratazione.
- Preparare la miscela di polimerizzazione I (PM-I) aggiungendo 1 confezione di Hardener I a 2,5 mL di PEG400 mescolato con 100 mL di HEMA (2-hydroxyethyl methacrylate)-soluzione di resina. Mescolare la soluzione per 15 min per sciogliere completamente l'Hardener I. Successivamente, conservare PM-I a -20 gradi centigradi.
- Rimuovere l'etanolo dal punto 9.3. e infiltrarsi nei campioni nel seguente ordine: PM-I:100% etanolo (1:3, v/v), PM-I:100% etanolo (1:1, v/v) e PM-I:100% etanolo (3:1, v/v). Incubare i campioni in ogni soluzione a RT per 1/2-1 h o fino a quando i campioni affondano sul fondo.
- Incubare i campioni durante la notte a 4 gradi centigradi nella soluzione PM-I al 100%.
- Preparare la miscela di polimerizzazione II mescolando PM-I e Hardener II in un rapporto 15:1 (v/v). Riempire lo stampo di plastica con la soluzione di polimerizzazione, orientare i campioni orizzontalmente nella parte inferiore dello stampo e coprire con un pezzo di pellicola di sigillazione elastica. Evitare la formazione di bolle d'aria.
NOTA: Quando la soluzione inizia a polimerizzare dopo l'esposizione a RT, provare a orientare i campioni il più rapidamente possibile nel supporto di plastica. La polimerizzazione viene completata dopo un'incubazione notturna a RT, o 1 h a 37 gradi centigradi. - Rimuovere il coperchio elastico della pellicola di guaritura dal passaggio 9.7 e posizionare un supporto per i campioni polimerizzati. Per montare il supporto nei campioni, sciogliere 10 mL di polvere di resina a base di metilla metila in 5 mL di soluzione di resina a base di metilico. Aggiungere rapidamente la soluzione al foro nella parte superiore del supporto.
NOTA: Eseguire la fase di polimerizzazione nella cofafuta del fume (30 min a RT). - Campioni di sezione microtome per uno spessore di 4-5 m. Posizionare un vetrino del microscopio su una piastra calda di 58 gradi centigradi e aggiungere una grande goccia d'acqua ad ogni scivolo. Posizionare le sezioni sulla parte superiore dell'acqua. Una volta che l'acqua è evaporata, le sezioni aderiscono allo scivolo.
- Stain scivola immergendo in 0.05 % (w/v) toluidine blu per 2 min. Successivamente, risciacquare gli scivoli 3x con acqua ultra-pura. I vetrini possono essere osservati utilizzando un microscopio a campo luminoso.
10. Mycorrhization di P. andersonii Plantlets
-
Preparare l'inoculo delle spore di Rhizophagus irregularis
- Preparare una pila di filtri in tessuto in poliestere con le seguenti dimensioni (dall'alto verso il basso): 210 m, 120 m e 36 m di dimensioni mesh.
- Pipette la quantità necessaria di una sospensione spore commerciale sulla pila di filtri in poliestere. Risciacquare i filtri 3x con 100 mL di acqua demineralizzata autoclaved. Le spore vengono mantenute sulla superficie del filtro da 36 m.
NOTA: Preparare la sospensione delle spore nell'armadio del flusso incrociato laminare per evitare contaminazioni. - Smontare la pila di poliestere e mantenere solo il filtro da 36 m. Ripetere il passaggio di lavaggio con acqua demineralizzata autoclaved per almeno 6x.
- Posizionare il filtro su un piatto Petri e ri-sospendere le spore in acqua demineralizzata autoclaved. Utilizzare un volume d'acqua uguale al volume della sospensione spore utilizzata al punto 10.1.2. Trasferire la sospensione spore in un tubo sterile pipettando.
- Posizionare 5 gocce di 20 l della sospensione delle spore su un vetrino di vetro e contare il numero di spore utilizzando un microscopio a campo luminoso. Convertire i conteggi delle spore in un rapporto di spore/mL e diluire la sospensione delle spore fino a raggiungere 250 spore/mL. Conservare la sospensione spore a 4 gradi centigradi.
- Eseguire il saggio di micorrhizzazione. A tal fine, aggiungere 800 g di sabbia autoclaved integrata con 70 mL di 1/2-Hoagland medio a sterili vasi di polipropilene traslucido (vedi tabelle 5-6). Mescolare sabbia e mezzo direttamente nella pentola agitando vigorosamente.
- Posizionare una pianta p. andersonii in ogni vaso e una pipetta 1 mL della sospensione delle spore direttamente sulla radice della pianta p. andersonii. Assicurarsi di includere diversi vasi contenenti piastrine P. andersonii trasformate con un costrutto di controllo CRISPR (vedere tabella supplementare 1).
- Incubai vasi in una stanza di crescita climatizzata (28 gradi centigradi, 16 h:8 h giorno:notte) per 6 settimane.
- Estrarre le piante dai vasi e lavare le radici con acqua corrente per rimuovere quanta più sabbia possibile.
- Tagliare le radici a pezzi lunghi 1 cm e far bollire i pezzi di radice in 10% KOH (w/v) per 20 min a 90 gradi centigradi. Successivamente, posizionare le radici bollite su un colino cellulare con una dimensione della maglia di 100 m e risciacquare 3x con 50 mL di acqua.
- Radici di macchie con 0,05% (w/v) trypan blu in lattoglycerol (300 mL di acido lattico; 300 mL di glicerolo; e 400 mL di acqua demineralizzata) per 5 min a 90 gradi centigradi in un bagno d'acqua o un blocco di riscaldamento. Successivamente, trasferire le radici al 30% glicerolo. I campioni radice possono essere archiviati in RT.
- Posizionare 15-25 frammenti di radice su un singolo vetrino al microscopio. Aggiungere il 30% di glicerolo e coprire con un vetro di copertura e premere fino a quando i pezzi di radice diventano piatti. Osservare i frammenti di radice utilizzando un microscopio a campo luminoso e segnare la colonizzazione micorriza.
NOTA: Un metodo per segnare la micorra è descritto secondo Trouvelot etal. Questo metodo utilizza diverse classi (%F, %M e %A), che consente una rapida stima del livello di colonizzazione micorriza di ogni frammento radice e dell'abbondanza di arbuscules.
Representative Results
P. andersonii trees può essere coltivato in una serra condizionata a 28 e 85 % di umidità relativa (Figura 1A). In queste condizioni, gli alberi iniziano a fiorire a 6-9 mesi dopo la semina. I fiori p. andersonii femminili producono bacche che contengono ciascuno un singolo seme. Durante la maturazione, le bacche cambiano colore; prima dal verde al bianco e successivamente dal bianco al marrone (Figura 1B). I semi estratti dalle bacche marroni maturate germinano bene dopo un ciclo di temperatura di 10 giorni e un'incubazione di 7 giorni su piastre SH-0 (Figura 1C). Semi germinati continuano a svilupparsi in giovani piantine che possono essere utilizzati per la sperimentazione dopo 4 settimane (Figura 1D).
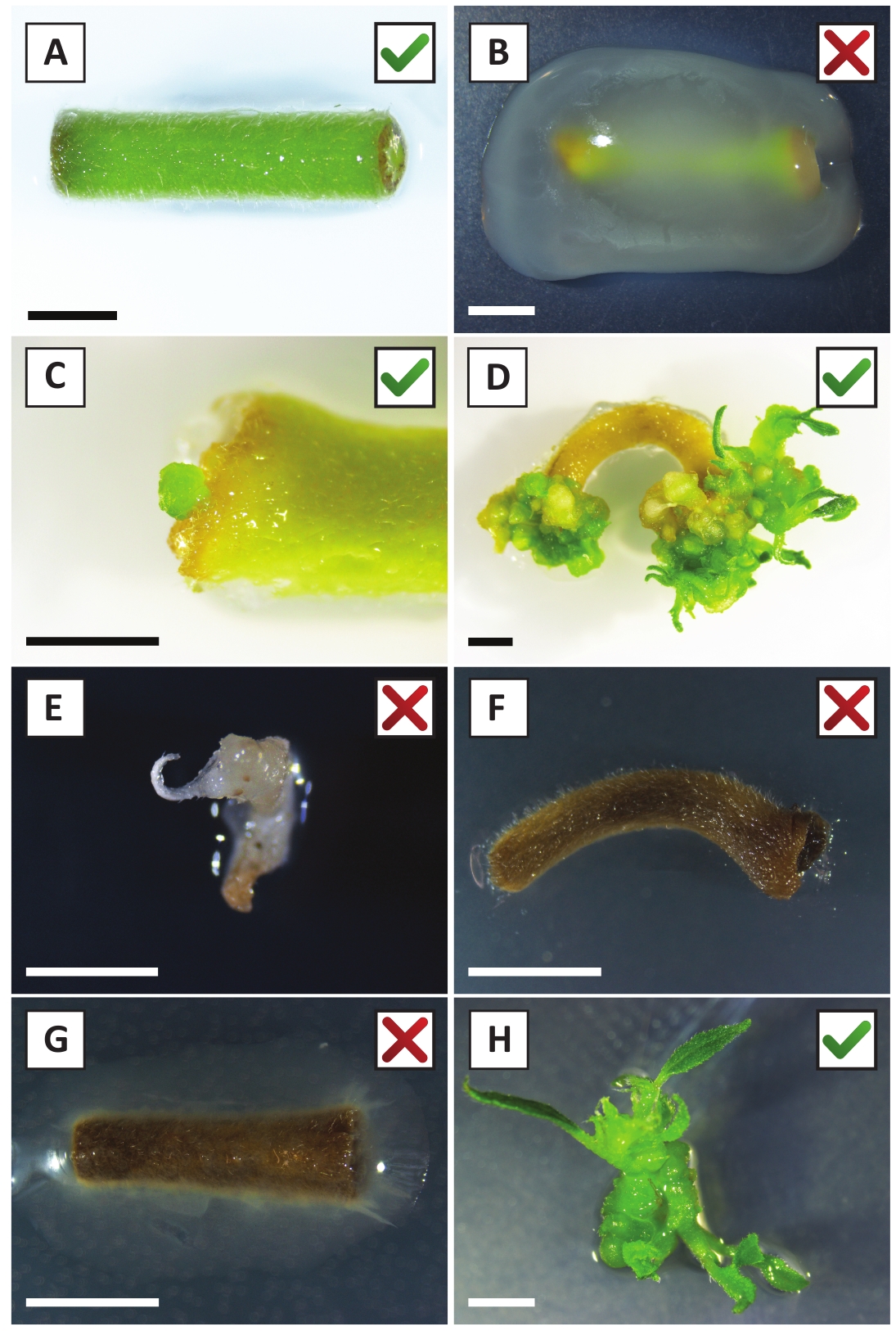
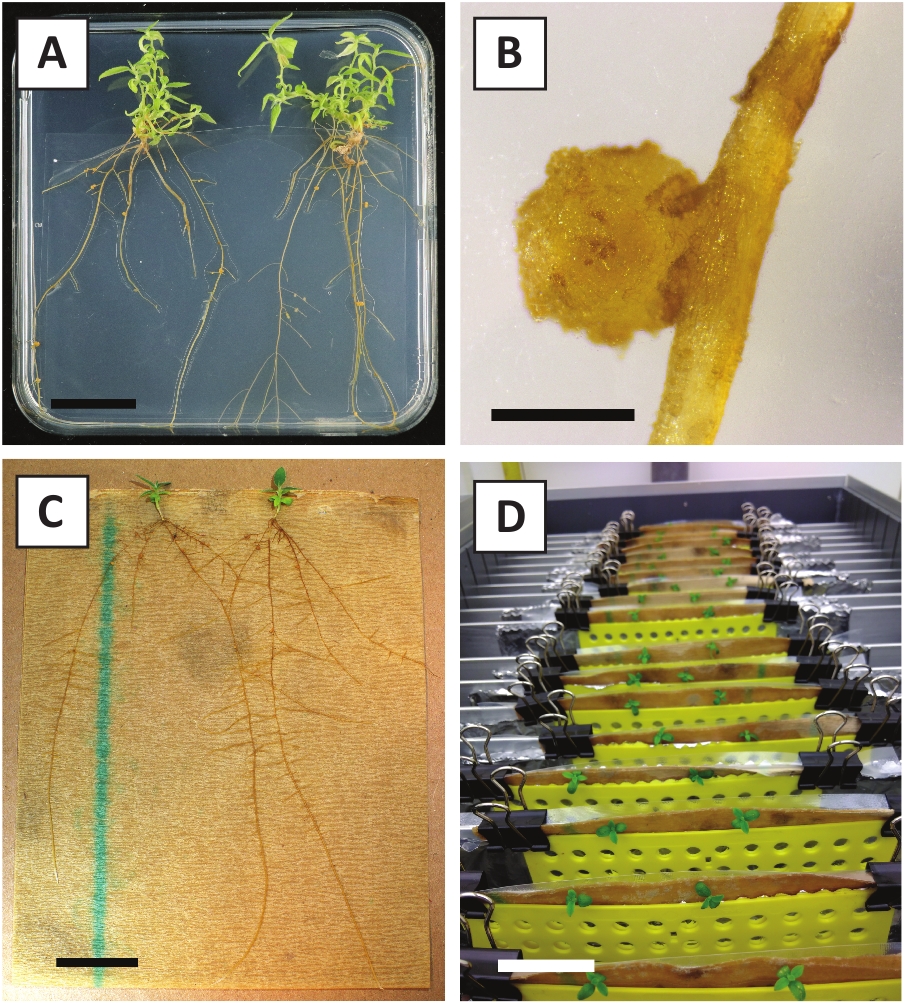
Abbiamo dimostrato in precedenza che i piccioli e i segmenti dei giovani steli P. andersonii possono essere trasformati in modo efficiente utilizzando il ceppo A. tumefaciens AGL110. All'inizio della procedura di trasformazione, gli esplants tissutali sono co-coltivati con A. tumefaciens per 2 giorni a 21 gradi centigradi (Figura 2A). La co-coltivazione prolungata comporta l'eccessiva colonizzazione degli espianti tissutali da parte di A. tumefaciens e dovrebbe quindi essere prevenuta (Figura 2B). Dopo il periodo di co-coltivazione, gli espianti dei tessuti vengono trasferiti a supporti selettivi, il che favorisce la crescita del tessuto trasformato. Due o tre settimane dopo, piccoli microcalli verdi sono generalmente osservati lungo la superficie originale della ferita (Figura 2C). Questi calli dovrebbero continuare a crescere e sviluppare 1 o più germogli trasformati in modo putativo a 6-8 settimane dopo l'avvio della procedura di trasformazione (Figura 2D). In questa fase, l'efficienza della trasformazione varia in genere dal 10 al 30% per le trasformazioni avviate con espespie tissutali prelevati da rami maturi e in parte legnosi (tabella 7). Se si avviano trasformazioni con espianti tratti dalle punte giovanili e in rapida crescita di rami che non portano ancora fiori, si possono ottenere efficienze di trasformazione del 65-75% (tabella7). Occasionalmente, i calli biancascali si formano sul lato di un espianto che non è in contatto con il mezzo e, pertanto, non sperimentano la selezione della kanamicina. Questi calli spesso non sono transgenici e qualsiasi germoglio formato da questi calli sarà generalmente candeggina e morire dopo il contatto diretto con la kanamicicina contenente mezzo (Figura 2E). Nel caso in cui il tasso di trasformazione sia basso e/o che il materiale iniziale non fosse ottimale, i pezzi di tessuto potrebbero diventare marroni (Figura 2F) e soffrire di eccessiva proliferazione da parte di A. tumefaciens (Figura 2G). Per evitare che A. tumefaciens si diffondano e crescano eccessivamente crescono gli espiante vicine, è necessario un rinfresco regolare del mezzo e gli espianti gravemente infetti devono essere rimossi. Una volta che i singoli germogli transgenici sono collocati nel mezzo di propagazione, la sovraproliferazione da parte di A. tumefaciens non si verifica più (Figura 2H). I germogli transgenici possono essere moltiplicati attraverso la propagazione in vitro, che darà origine a decine di germogli in un periodo di un mese (Figura 3A-B). Questi germogli possono essere posizionati su un mezzo di radicamento, che dovrebbe indurre la formazione della radice dopo 2 settimane (Figura3C-D). Le piastrine radicate possono essere successivamente utilizzate per la sperimentazione.
Per creare linee mutanti ad eliminazione diretta, ci facciamo uso della mutagenesi mediata CRISPR/Cas9. A tal fine, utilizziamo un vettore binario contenente il gene di resistenza della kanamicina NPTII, una sequenza di codifica Cas9 guidata dal promotore CaMV35S e 2 sgRNA per gene bersaglio che sono espressi dal piccolo promotore di RNA AtU6p20. Una rappresentazione grafica del costrutto utilizzato per la mutagenesi mediata CRISPR/Cas9 di P. andersonii è fornita nella Figura 4A. Utilizzando questo metodo, l'editing del genoma è osservato nel 40% dei germogli con trasformazione putativa10. Per identificare le linee mutanti, i germogli trasformati in modo putativo sono genotipizzati per le mutazioni nei siti bersaglio dello sgRNA utilizzando primer che attraversano la regione bersaglio. Un esempio dei risultati attesi è dato in Figura 4. Come si può vedere dalla foto scattata dopo l'elettroforesi gel, diversi campioni producono un amplificatore PCR con dimensioni simili al tipo selvaggio (Figura 4B). Queste piante possono contenere piccoli indel che non possono essere visualizzati dall'elettroforesi gel agarose o rimanere inediti dall'enzima Cas9. Inoltre, diversi campioni producono bande di dimensioni diverse dal tipo selvaggio (ad esempio, linee 2, 4, 7 e 8 nella figura 4B). In queste righe, 1 (linee 4, 7 e 8) o entrambi (linea 2) alleli contengono indels più grandi che possono essere facilmente visualizzati. L'esatta natura delle mutazioni nei siti di destinazione viene rivelata dopo il sequenziamento dell'amplio PCR. Come si può vedere dalla figura 4C, sia piccoli indels di 1-4 bp, così come, le eliminazioni più grandi possono essere ottenute dopo la mutagenesi CRISPR/Cas9. Nella figura 4C, la sequenza della riga 1 è identica a quella del tipo wild, a indicare che questa riga è stata sottoposta a escape nella modifica e, pertanto, deve essere eliminata. Tra le linee che contengono mutazioni, possono essere identificati mutanti etetozigosi, omozigi e biallelici (Figura 4C). Tuttavia, i mutanti eterozie sono generalmente rari10. I mutanti omozici o filtranti biallelici possono essere propagati vegetativamente per ottenere materiale sufficiente per l'analisi fenotipica. Poiché l'analisi fenotipica viene eseguita nella generazione T 0, è importante verificare se le linee mutanti potrebbero essere chimeriche. A tal fine, la genotipizzazione deve essere ripetuta su almeno 3 campioni diversi prelevati da ogni linea mutante. Se i risultati della genotipizzazione sono identici tra loro e il campione di genotipizzazione originale (ad esempio, la riga 8 nella Figura 4D), la riga viene mutata omogeneamente e può essere utilizzata per ulteriori analisi. Tuttavia, se i risultati della genotipizzazione differiscono tra campioni indipendenti (ad esempio, la linea 4 nella figura 4D), la linea mutante è chimerica e deve essere scartata.
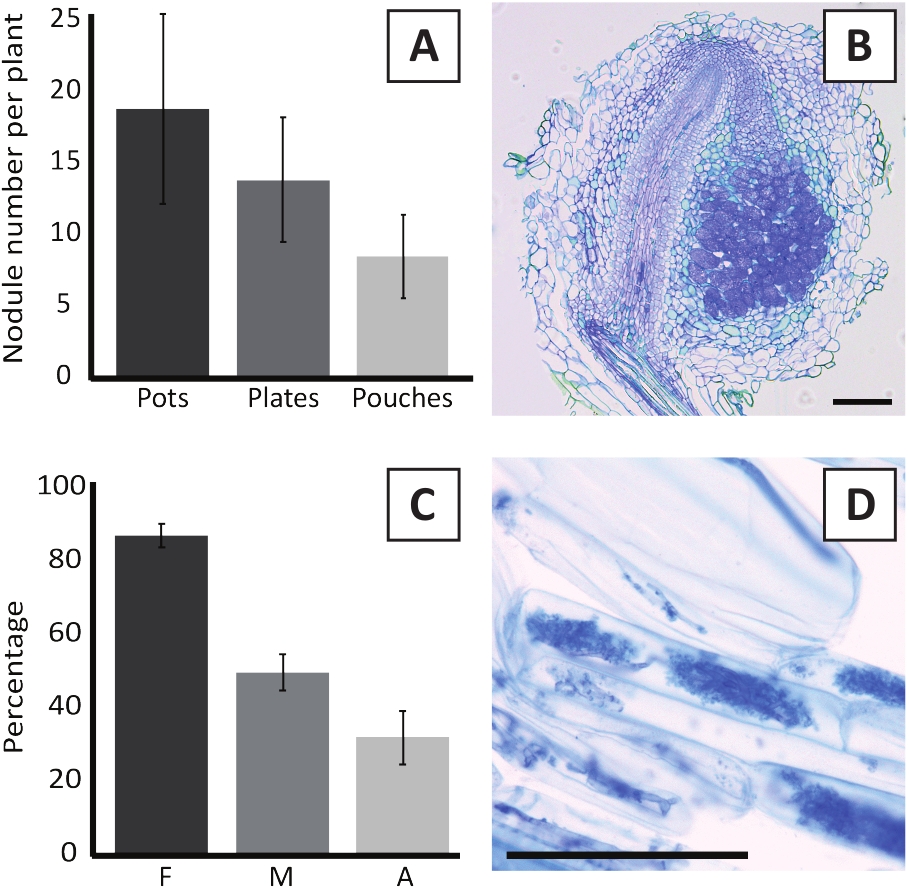
L'inoculazione di P. andersonii con M. plurifarium BOR2 determina la formazione di noduli di radice (Figura 5). Come si può vedere in Figura 5A, questi noduli sono distribuiti lungo il sistema radice. I noduli di P. andersonii sono di colore marrone chiaro, ma possono essere facilmente discriminati dal tessuto radicale in base alla loro forma (Figura 5B). Esperimenti di inoculazione in vasi e crescita successiva per 4-6 settimane in genere provocare la formazione di 10-30 noduli di dollari (Figura 6A). Un numero simile di noduli è formato dopo l'inoculazione delle piantagioni P. andersonii coltivate a piastra EKM a 4 settimane dopo l'inoculazione (Figura 6A). Nelle sacche, le piantine P. andersonii formano in genere noduli 5-15 a 5 settimane dopo l'inoculazione (Figura 5C-D, 6A). Per analizzare la citoarchitettura a nodulo, i noduli possono essere sezionato e osservato utilizzando la microscopia a campo luminoso. Figura 6B mostra un esempio di una sezione longitudinale attraverso il centro di un nodulo P. andersonii. Questa sezione mostra il fascio vascolare centrale di un nodulo P. andersonii, che è affiancato da lobi noduli contenenti cellule infette (Figura 6B).
Anche le piantine P. andersonii possono essere micorrhizzate. Dopo 6 settimane di inoculazione con R. irregolaris, frequenza di colonizzazione micorrizaina in genere raggiunge > 80% (Figura 6C). A questo punto, generalmente il 30% delle cellule contiene arbuscules (Figura 6C). Un'immagine rappresentativa di un segmento radice P. andersonii contenente arbuscles è illustrata nella Figura 6D.

Figura 1: Immagini rappresentative di un P. andersonii albero, semi e piantine. (A) Albero P. andersonii di sei mesi coltivato in terriccio in una serra condizionata a 28 gradi centigradi. (B) Immagine rappresentativa raffigurante bacche di P. andersonii in varie fasi della maturazione. Le bacche giovani di P. andersonii (non mature) cambieranno colore dal verde al bianco e infine al marrone (maturo) alla maturazione. (C) Semi di P. andersonii incubati su mezzo SH-0 per 1 settimana. Un cerchio nero indica una piantità germinata. (D) Le piantine P. andersonii di quattro settimane coltivate in mezzo SH-0. Le barre della scala sono pari a 25 cm in (A) e 1 cm in (B-D). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Immagini rappresentative di espianti nelle diverse fasi della procedura di trasformazione stabile. (A) Explant co-coltivato con A. tumefaciens. (B) Explant overgrown di A. tumefaciens durante le prime 2 settimane dopo la trasformazione. (C) Microcallo transgenico formatosi vicino al sito della ferita di un espianto a 2,5 settimane dopo la co-coltivazione. (D) Immagine rappresentativa di un'espianto a 6 settimane dopo la co-coltivazione che mostra l'emergere di germogli da calli (transgenici). (E) Immagine rappresentativa di un servizio fotografico che diventa biancastro e alla fine muore quando è in contatto diretto con il mezzo contenente kanamicina. Questo scatto è molto probabilmente non transgenico ed evaso nella selezione della kanamicicina quando è collegato all'espianto. (F) Immagine rappresentativa di un espianto trasformato senza successo. (G) Immagine rappresentativa di un espianto trasformato senza successo ricoperto da A. tumefaciens. (H) Singolo germoglio transgenico coltivato su mezzo di propagazione a 8 settimane dopo la co-coltivazione con A. tumefaciens. Le barre della scala sono uguali a 2,5 mm. Le caselle contenenti segni di spunta verdi o croci rosse indicano rispettivamente la trasformazione riuscita o non riuscita dei despighi. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Immagini rappresentative di in vitro propagazione. (A) Riprese coltivate su mezzo di propagazione. L'immagine è stata scattata 1 settimana dopo che le piastre sono state aggiornate. (B) Spara coltivato su mezzo di propagazione. L'immagine è stata scattata 4 settimane dopo che le piastre sono state aggiornate. (C) I germogli appena tagliati posizionati sul mezzo di radicamento. (D) Spara incubato su mezzo di radicamento per 2 settimane. Si noti la presenza di radici. Barre della scala sono uguali a 2,5 cm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Risultati rappresentativi dopo la genotipizzazione delle linee mutanti transgeniche CRISPR/Cas9 di P. andersonii T0. (A) Mappa rappresentativa di un vettore binario utilizzato per la mutagenesi mediata CRISPR/Cas9 di P. andersonii. (B) Risultato rappresentativo dopo la genotipizzazione basata su PCR di potenziali linee mutanti CRISPR/Cas9 utilizzando primer che coprono il sito target dello sgRNA. Mostrato è un'immagine dopo l'elettroforesi gel agarose di amplificatori. I campioni prelevati da singole linee transgeniche sono indicati da numeri. Il tipo selvaggio (WT) e nessun controllo modello (NTC) indicano rispettivamente corsie contenenti controlli positivi e negativi. (C) Rappresentazione schematica degli alleli mutanti ottenuta dopo l'editing genico mediato da CRISPR/Cas9. In colori blu e rosso sono evidenziati rispettivamente nei siti di destinazione del blu e del rosso i siti di destinazione dello sgRNA e le sequenze PAM. (D) Risultato rappresentativo dopo lo screening basato su PCR per potenziali linee mutanti chimeriche. Mostrato è un'immagine dopo l'elettroforesi gel agarose di 3 singoli campioni prelevati dalle linee mutanti 4 e 8. Si noti che la linea 4 mutante transgenica è chimerica. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Immagini rappresentative dei saggi di nodulation in piatti e sacchetti. (A) Nodulation su piastre contenenti mezzo EKM solidificato e inoculato con M. plurifarium BOR2 per 4 settimane. (B) Immagine rappresentativa di un nodulo di radice P. andersonii. L'immagine è stata scattata a 4 settimane dopo l'inoculazione con M. plurifarium BOR2. (C) Nodulation in sacchetti contenenti mezzo liquido EKM. Le piantine sono state inoculate con Bradyrhizobium sp. Kelud2A4 per 5 settimane. (D) Immagine rappresentativa di una configurazione completa utilizzata per il nodulation in sacchetti. Le barre di scala sono pari a 2,5 cm in (A,C), 1 mm in (B) e 5 cm in (D). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Risultati rappresentativi dei saggi di annuizione e micorraria. (A) Grafico a barre rappresentative che mostra il numero di noduli formati per pianta a 4 settimane dopo l'inoculazione con M. plurifarium BOR2 in vasi o su piatti e a 5 settimane dopo l'inoculazione con Bradyrhizobium sp. Kelud2A4 in sacchetti. I dati rappresentano la media : SD (n - 10). (B) Immagine rappresentativa di una sezione longitudinale attraverso un nodulo formato a 4 settimane dopo l'inoculazione con M. plurifarium BOR2. La sezione è macchiata di blu toluidine. (C) Grafico a barre rappresentative che mostra la quantificazione della micorra. Le variabili quantificate secondo Trouvelot et al.29 sono F, la frequenza dei frammenti di radice analizzati che vengono micorrizzati; M, l'intensità dell'infezione; A, l'abbondanza di arbuscules maturi nel sistema radicale totale. La micorrariizzazione è stata quantificata a 6 settimane dopo l'inoculazione con R. irregularis (ceppo DAOM197198). I dati rappresentano la media : SD (n - 10). (D) Immagine rappresentativa degli arbuscules maturi presenti nelle cellule corticali della radice di P. andersonii a 6 settimane dopo l'inoculazione con R. irregolaris. Barre di scala uguali a 75 m. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
| composto | SH-0 | SH-10 | Mezzo di propagazione | Mezzo di radicamento | Mezzo di infiltrazione |
| Mezzo sale SH-basal | 3,2 g | 3,2 g | 3,2 g | 3,2 g | 3,2 g |
| Miscela SH-vitamina | 1 g | 1 g | 1 g | 1 g | 1 g |
| saccarosio | - | 10 g | 20 g | 10 g | 10 g |
| BAP (1 mg/mL) | - | - | 1 mL (4,44 m) | - | - |
| IBA (1 mg/mL) | - | - | 100 l (0,49 m) | 1 mL (4,92 m) | - |
| NAA (1 mg/mL) | - | - | - | 100 l (0,54 M) | - |
| 1 M MES pH 5,8 | 3 mL | 3 mL | 3 mL | 3 mL | 3 mL |
| 1 M KOH | Regolare pH a 5,8 | Regolare pH a 5,8 | Regolare pH a 5,8 | Regolare pH a 5,8 | Regolare pH a 5,8 |
| Daishin agar | 8 g | - | 8 g | 8 g | - |
Tabella 1: Composizione di30 supporti basati su Schenk-Hildebrandt utilizzati per la crescita delle piantine di P. andersonii, la trasformazione stabile e la propagazione in vitro. Sciogliere i composti solidi in 750 mL di acqua ultra-pura prima di aggiungere scorte liquide. Successivamente, riempire il supporto completo a 1 L. Preparare BAP, IBA, NAA scorte in 0.1 M KOH e memorizzare a -20 oC.
| Prima dell'autoclaving: | ||
| composto | Importo per litro | Concentrazione finale |
| Mannitolo | 5 g | 27,45 mM |
| Na-Gluconate | 5 g | 22,92 mM |
| Estratto di lievito | 0,5 g | - |
| MgSO4X7O2O | 0,2 g | 0,81 mM |
| Nacl | 0,1 g | 1,71 mM |
| K2HPO4 | 0,5 g | 2,87 mM |
| Dopo l'autoclaving: | ||
| composto | Importo per litro | Concentrazione finale |
| 1,5 M CaCl2 | 1 mL | 1,5 mM |
Tabella 2: Composizione del mezzo Yeast-Mannitol (YEM) utilizzato per la coltivazione del rizobio. Regolare il pH a 7,0 e riempire con acqua ultra-pura a 1 L. Per preparare il supporto YEM agar-solidificato, aggiungere 15 g di microagar prima di autoclaving.
| Prima dell'autoclaving: | |||
| composto | Concentrazione di scorte | Importo per litro medio | Concentrazione finale |
| KH2PO4 | 0,44 M | Aggiungi 2 mL | 0,88 mM |
| K2HPO4 | 1,03 M | Aggiungi 2 mL | 2,07 mM |
| Soluzione di stock di microelementi 500x | - | Aggiungi 2 mL | - |
| MES pH 6,6 | 1 M | Aggiungi 3 mL | 3 mM |
| Hcl | 1 M | Regolare pH a 6,6 | - |
| Acqua ultra-pura | - | Riempire a 990 mL | - |
| Dopo l'autoclaving: | |||
| composto | Concentrazione di scorte | Importo per litro medio | Concentrazione finale |
| MgSO4X7O2O | 1,04 M | 2 mL | 2,08 mM |
| Na2SO4 | 0,35 M | 2 mL | 0,70 mM |
| NH4NO3 | 0,18 M | 2 mL | 0,36 mM |
| CaCl2o 2H2O | 0,75 M | 2 mL | 1,5 mM |
| Fe(III)-citrato | 27 mM | 2 mL | 54 M |
Tabella 3: Composizione di 1 L modificato EKM medio31 utilizzato per P. andersonii nodulation assay. La composizione della soluzione stock 500x micro-elementi è elencata nella tabella 4. Per preparare il mezzo EKM solidificato 2%, aggiungere 20 g di Daishin agar prima di autoclaving. Autoclave le scorte di citrate MgSO47H2O, Na2SO4, CaCl2x 2H2O e Fe(III) da sterilizzare. Filtro sterilizzare NH4NO3 soluzione stock per sterilizzare.
| composto | Importo per litro | Concentrazione di scorte |
| MnSO4 | 500 mg | 3,31 mM |
| NSO4x 7H2O | 125 mg | 0,43 mM |
| CuSO4x 5H2O | 125 mg | 0,83 mM |
| H3BO3 | 125 mg | 2,02 mM |
| Na2MoO4o 2H2O | 50 mg | 0,21 mM |
Tabella 4: Composizione della soluzione stock 500x micro-elementi utilizzata per la preparazione del supporto EKM modificato. Conservare la soluzione di stock di microelementi a 4 gradi centigradi.
| Composti | Concentrazione di scorte | Importo per litro medio | Concentrazione finale |
| K2HPO4 | 20 mM | 1 mL | 0,2 mM |
| NH4NO3 | 0,28 M | 10 mL | 2,8 mM |
| MGSO4 | 40 mM | 10 mL | 0,4 mM |
| K2SO4 | 40 mM | 10 mL | 0,4 mM |
| Fe(II)-EDTA | 9 mM | 10 mL | 0,9 mM |
| CaCl2 | 80mm | 10 mL | 0,8 mM |
| Soluzione di stock di microelementi 50x | - | 10 mL | - |
Tabella 5: Composizione del mezzo 1/2-Hoagland32 utilizzato per i saggi di micorraria. La composizione della soluzione stock 50x microelementi è riportata nella tabella 6. Preparare la soluzione Fe(II)-EDTA combinando FeSO4-7H2O (9 mM) e Na2 EDTA (9 mM) in 1 soluzione di stock, e conservare a 4 gradi centigradi. Regolare il pH del mezzo a 6,1 utilizzando 1 M KOH e riempire con acqua ultra pura a 1 L.
| Composti | Importo per litro | Concentrazione di scorte |
| H3BO3 | 71,1 mg | 1,15 mM |
| MnCl2s4H2O | 44,5 mg | 0,22 mM |
| CuSO4x 5H2O | 3,7 mg | 23.18 . |
| NCl2 (NCl) | 10,2 mg | 74,84 M |
| Na2MoO4o2H2O | 1,2 mg | 4,96 M |
Tabella 6: Composizione della soluzione di stock 50x micro-elementi utilizzata per la preparazione di un mezzo 1/2-Hoagland.
| Età degli espianti | Efficienza della trasformazione |
| prole di qualcosa | 69,4 x 6,2% (n - 2) |
| Maturo | 18,3 x 10,2% (n - 15) |
Tabella 7: Efficienza di trasformazione di P. andersonii. Qui, l'efficienza della trasformazione è definita come la percentuale di espianti che formano almeno 1 callo transgenico o sparare. L'efficienza di trasformazione è stata valutata in 6 settimane dopo la trasformazione ed è rappresentata come media: SD. n indica il numero di esperimenti di trasformazione da cui è stata determinata l'efficienza della trasformazione.
File supplementare 1: Panoramica dei costrutti di livello 1 e livello 2 utilizzati per la mutagenesi CRISPR/Cas9. Fare clic qui per scaricare questo file.
Discussion
I legumi e il genere Cannabaceae Parasponia rappresentano gli unici due cladi di specie vegetali in grado di stabilire una relazione endosimbiotica con rizobia che fissa l'azoto e formare noduli di radice. Gli studi comparativi tra specie di entrambi i cladi sono altamente rilevanti per fornire informazioni sulle reti genetiche di base che consentono questa simbiosi. Attualmente, gli studi genetici sono fatti principalmente nei legumi; specialmente le due specie modello M. truncatula e L. japonicus. Per fornire una piattaforma sperimentale aggiuntiva e facilitare gli studi comparativi con un nodulante non legume, descriviamo qui un protocollo dettagliato per la trasformazione stabile e le analisi genetiche inverse in P. andersonii. Il protocollo presentato utilizza la propagazione in vitro delle linee T0 transgeniche P. andersonii, consentendo l'avvio dell'analisi fenotipica entro 4 mesi dalla co-coltivazione A. tumefaciens. Questo è sostanzialmente più veloce rispetto ai protocolli attuali che sono stati stabiliti per la trasformazione stabile dei legumi33. Questo rende P. andersonii un modello di ricerca attraente.
Il protocollo descritto di seguito contiene diversi passaggi critici. Il primo dei quali riguarda la germinazione dei semi. Per preparare i semi di P. andersonii per la germinazione, i semi devono essere isolati dalle bacche. Questo viene fatto strofinando le bacche su un pezzo di carta velina o contro l'interno di un setaccio di tè. Questa procedura deve essere eseguita delicatamente per evitare danni al cappotto di semi. Se il mantello del seme viene danneggiato, la candeggina potrebbe entrare nel seme durante la sterilizzazione, riducendo la vitalità del seme. Per rompere la dormancy dei semi, i semi sono sottoposti a un ciclo di temperatura di 10 giorni. Tuttavia, nonostante questo trattamento, la germinazione non è completamente sincronizzata. Generalmente, i primi semi mostrano l'emergere del radicolo dopo 7 giorni, ma altri potrebbero richiedere diversi giorni in più per germinare.
I punti critici della procedura di trasformazione riguardano la scelta del materiale di partenza e la durata della fase di co-coltivazione. Per raggiungere una trasformazione efficiente, è meglio utilizzare steli sani e giovani o piccioli di piante coltivate in serra non sterili come materiale di partenza. Al fine di indurre la crescita di rami giovani, si consiglia di tagliare gli alberi Parasponia ogni 2-3 mesi e rinfrescare gli alberi una volta all'anno. Inoltre, la fase di co-coltivazione deve essere eseguita solo per 2 giorni. La co-coltivazione prolungata promuove l'eccessiva colonizzazione degli espianti tissutali da parte di A. tumefaciens e generalmente riduce l'efficienza di trasformazione. Per evitare l'eccessiva colonizzazione di A. tumefaciens è anche importante aggiornare regolarmente le piastre su cui vengono coltivati gli espianti. Nel caso in cui si verifichi una colonizzazione eccessiva, gli espianti tissutali potrebbero essere lavati (vedi Sezione 3.8) per rimuovere le cellule A. tumefaciens. Si consiglia di aggiungere candeggina alla soluzione SH-10 utilizzata per il lavaggio (concentrazione finale: ipocloreto del 2%). È importante notare che questa fase di lavaggio aggiuntiva potrebbe non funzionare su espianti fortemente infetti (Figura 2B). Nel caso in cui una trasformazione con un costrutto CRISPR/Cas9 produca solo un numero limitato di germogli con trasformazione putativa o se si prevede che la mutagenesi di un particolare gene causi problemi di rigenerazione, si consiglia di includere un costrutto di controllo vettoriale vuoto come il controllo positivo. Infine, è importante assicurarsi che tutte le linee transgeniche selezionate siano risultanti da eventi indipendenti di integrazione T-DNA. Pertanto, istruiamo a prendere solo un singolo germoglio putativamente transgenico da ogni lato di un eximpianto. Tuttavia, ci rendiamo conto che questo riduce il numero potenziale di linee indipendenti. Se sono necessarie molte linee, i ricercatori potrebbero decidere di separare calli formati in modo putativo dagli espianti originali quando questi calli hanno dimensioni e coltura di 2 mm in modo indipendente. In questo modo, più linee potrebbero essere isolate da ogni espianto, il che aumenta il numero di potenziali linee transgeniche.
Nel protocollo attuale, le linee transgeniche di P. andersonii vengono propagate vegetativamente attraverso la propagazione in vitro. Il vantaggio di questo è che molte pitole transgeniche possono essere generate in un periodo di tempo relativamente breve. Tuttavia, questo metodo ha anche diverse limitazioni. In primo luogo, il mantenimento delle linee transgeniche T0 attraverso la propagazione in vitro è laborioso e potrebbe provocare alterazioni genetiche o epigenetiche indesiderate34,35. In secondo luogo, le linee T0 contengono ancora una copia del T-DNA, compresa la cassetta di resistenza agli antibiotici. Questo limita il numero di possibili ritrasformazioni, poiché sono necessari marcatori di selezione diversi per ogni ritrasformazione. Attualmente, abbiamo testato la trasformazione solo utilizzando la kanamycin o la selezione dell'igromicina (dati non mostrati). Inoltre, la presenza della sequenza di codifica Cas9 e degli sgRNA nelle linee transgeniche T0 complica gli studi di complemento. I saggi di complemento sono possibili, ma richiedono che i siti di destinazione dello sgRNA siano mutati in quanto tale che sia impedito l'editing genico del costrutto di complemento. In terzo luogo, uno svantaggio di lavorare con le linee T0 è che i mutanti CRISPR/Cas9 potrebbero essere chimerici. Per prevenire l'analisi fenotipica delle linee mutanti chimetriche, si consiglia di ripetere l'analisi della genotipizzazione dopo la propagazione in vitro su almeno 3 diversi germogli. Anche se, il numero di mutanti chimerici ottenuti utilizzando il protocollo qui descritto è limitato, sono occasionalmente osservati10. Per superare i limiti di lavoro con le linee T 0, le linee mutanti P. andersonii potrebbero essere propagate generativamente. P. andersonii alberi sono dioecious e vento impollinato2. Ciò significa che ogni linea transgenica deve essere manipolata in modo tale che i fiori maschili e femminili siano prodotti su un singolo individuo e successivamente coltivati come tali che l'impollinazione incrociata non si verifichi. Poiché P. andersonii è un albero in rapida crescita, richiede una notevole quantità di spazio in una serra tropicale (28 , 85% di umidità relativa). Pertanto, anche se tecnicamente possibile, la propagazione generativa delle linee transgeniche P. andersonii è logisticamente impegnativa.
Nella sezione protocollo, abbiamo descritto 3 metodi per annuificare P. andersonii. Il vantaggio dei sistemi a piastre e sacca è che le radici sono facilmente accessibili, il che può consentire l'inoculazione spot dei batteri e seguire la formazione di noduli nel tempo. Tuttavia, il sistema di piastre è piuttosto laborioso, il che lo rende meno adatto per esperimenti di nodulation su larga scala. Uno svantaggio del sistema della sacca è che è difficile prevenire la contaminazione fungina. I sacchetti non sono sterili, e quindi la crescita fungina è spesso osservata nella metà superiore della sacca. Tuttavia, questo non influisce sulla crescita di P. andersonii, e quindi non interferisce con i saggi di annuizione. Inoltre, il sistema della sacca è adatto solo per piantine. Nonostante diversi tentativi, non siamo stati in grado di coltivare le piantine ottenute attraverso la propagazione in vitro nelle sacche.
La pipeline di genetica inversa P. andersonii descritta qui offre un miglioramento sostanzialerispetto al metodo di trasformazione delle radici basato su A. rhizogenes11 . Utilizzando le procedure descritte, le linee transgeniche stabili possono essere generate in modo efficiente e possono essere mantenute tramite la propagazione in vitro. Al contrario, la trasformazione di A. rhizogenes è transitoria e si traduce solo nella formazione di radici transgeniche. Poiché ogni radice transgenica deriva da una trasformazione indipendente, i saggi basati sulla trasformazione di A. rhizogenes soffrono di una sostanziale variazione fenotipica. Questa variazione è molto meno in caso di linee stabili, anche se la propagazione in vitro crea anche un certo livello di variazione. A causa di questa variazione ridotta e del fatto che più piastrine potrebbero essere fenotitizzate per ogni linea stabile, le linee stabili sono più adatte per i saggi quantitativi rispetto alle radici trasformate da A. rhizogenes. Inoltre, la trasformazione stabile non dipende dall'introduzione del locus che induce la radice di A. rhizogenes (rol) che colpisce l'equilibrio ormonale endogeno15. Pertanto, linee stabili sono più adatte per l'analisi genetica inversa dei geni coinvolti nell'omeostasi ormonale rispetto alle radici trasformate da A. rhizogenes. Un vantaggio più generale di P. andersonii come modello di ricerca è che non ha sperimentato una recente duplicazione dell'intero genoma (WGD). La sottofamiglia legume Papilionobiasi, che comprende il modello legumi M. truncatula e L. japonicus, così come il Salicaceae (ordine Malpighiales) che comprende l'albero modello Populus trichocarpa sperimentato WGDs 65 milioni di anni fa36,37. Molte copie geniche paralose risultanti da questi WGD sono conservate nei genomi di M. truncatula, L. japonicus e P. trichocarpa37,38,39, che crea ridondanza che potrebbe complicare le analisi genetiche inverse. Poiché P. andersonii non ha sperimentato un recente WGD, le analisi genetiche inverse su P. andersonii potrebbero essere meno influenzate dal funzionamento ridondante delle copie del gene paraloso.
Nel loro insieme, forniamo un protocollo dettagliato per l'analisi genetica inversa in P. andersonii. Utilizzando questo protocollo, singole linee mutanti possono essere generate in modo efficiente in un lasso di tempo di 2-3 mesi10. Questo protocollo può essere esteso per creare mutanti di ordine superiore attraverso il multiplexing di sgRNA che prendono di mira diversi geni contemporaneamente, come mostrato per altre specie vegetali40,41,42. Inoltre, la procedura di trasformazione stabile qui descritta non si limita al targeting genico CRISPR/Cas9, ma potrebbe anche essere utilizzata per introdurre altri tipi di costrutti (ad esempio, per i saggi promotore-reporter, espressione ectopica o trans- complementari). Abbiamo stabilito P. andersonii come un modello di ricerca comparativa per studiare simbiosi mutualistici con rizobia che fissa l'azoto o funghi endomycorrizal. Tuttavia, i protocolli qui descritti forniscono anche strumenti per studiare altri aspetti della biologia di questo albero tropicale, come la formazione del legno, lo sviluppo di fiori bisessuali o la biosintesi dei metaboliti secondari specifici di Cannabaceae.
Acknowledgements
Gli autori amano riconoscere Mark Youles, Sophien Kamoun e Sylvestre Marillonnet per aver reso disponibili le parti di clonazione di Golden Gate attraverso il database Addgene. Inoltre, vorremmo ringraziare E. James, P. Hadobas e T. J. Higgens per i semi di P. andersonii. Questo lavoro è stato sostenuto dall'Organizzazione olandese per la ricerca scientifica (NWO-VICI grant 865.13.001; NWO-Open Competition grant 819.01.007) e il Ministero della Ricerca, tecnologia e istruzione superiore della Repubblica d'Indonesia (riSET-PRO grant 8245-ID).
Materials
| Name | Company | Catalog Number | Comments |
| Sigma-Aldrich | N0640 | NAA | |
| Duchefa Biochemie | M1503.0250 | MES | |
| Sigma-Aldrich | D134406 | Acetosyringone | |
| Duchefa Biochemie | X1402.1000 | X-Gal | |
| Merck | 101236 | For nucleic acid electrophoresis gel | |
| - | - | Pouches box material, hangers | |
| Merck | 101188 | NH4NO3 | |
| Sigma-Aldrich | B3408-1G | BAP | |
| Merck | 100156 | H3BO3 | |
| Thermo-Fisher | ER1011 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Thermo-Fisher | 15561020 | Used in Golden Gate cloning assembly | |
| Merck | 137101 | CaCl2·2H2O | |
| Duchefa Biochemie | C0111.0025 | C16H16N5O7S2Na | |
| Thermo-Fisher | K1231 | Used for cloning the blunt-ended PCR amplicons in genotyping procedure | |
| Agronutrition | AP2011 | Containing Rhizophagus irregularis DAOM 197198 (1,000 spores/mL), used for mychorrization assay | |
| Merck | 102790 | CuSO4·5H2O | |
| Duchefa Biochemie | D1004.1000 | Used for plant tissue culture agar-based medium | |
| Merck | 105101 | K2HPO4 | |
| VWR Chemicals | 20302.293 | Na2·EDTA | |
| Duchefa Biochemie | M0803.1000 | C6H14O6 | |
| Thermo-Fisher | ER0291 | Used as restriction enzyme in Golden Gate cloning assembly | |
| Merck | 100983 | C2H5OH | |
| VWR Chemicals | BDH9232-500G | EDTA | |
| Sigma-Aldrich | Z377600-1PAK | Cellophane membrane | |
| Biomatters, Ltd. | R9 or higher | Bioinformatics software for in silico cloning and designing of sgRNAs | |
| Mega International | - | Technical information at https://mega-international.com/tech-info/ | |
| Sigma-Aldrich | 65882 | Used for fixating nodule tissues | |
| VWR Chemicals | 24385.295 | - | |
| Vink | 219341 | Pouches box material, bottom part | |
| Leica Biosystems | 14702218311 | Used as a template for plastic embedding | |
| Merck | 100317 | HCl | |
| Sigma-Aldrich | I5386-1G | IBA | |
| Merck | 103862 | C6H5FeO7 | |
| Merck | 103965 | FeSO4O·7H2O | |
| Duchefa Biochemie | I1401.0005 | IPTG | |
| Duchefa Biochemie | K0126.0010 | ||
| Sigma-Aldrich | L2000 | ||
| Merck | 105886 | MgSO4O·7H2O | |
| Merck | 105934 | MnCl2·4H2O | |
| Merck | 102786 | MnSO4O | |
| Duchefa Biochemie | M1002.1000 | Used for bacterial culture agar-based medium | |
| Manutan | 92007687 | Pouches material | |
| Paraxisdienst | 130774 | Elastic sealing foil | |
| Pull Rhenen | Agra-Perlite No.3 | Used as growing substrate in pots for nodulation assay | |
| VWR Chemicals | 391-0581 | Used as container for cellophane membranes | |
| Thermo-Fisher | F130WH | For genotyping transgenic lines | |
| Addgene | 50337 | Level 0 terminator, 3'UTR, 35s (Cauliflower Mosaic Virus) | |
| Addgene | 48017 | End-link 2 for assembling 2 level one part into a level 2 acceptor | |
| Addgene | 48018 | End-link 3 for assembling 3 level one part into a level 2 acceptor | |
| Addgene | 48001 | Level 1 acceptor. Position 5. Forward orientation | |
| Addgene | 48007 | Level 1 Acceptor. Position 1. Reverse orientation | |
| Addgene | 50268 | Level 0 promoter (0.4 kb), 35s (Cauliflower Mosaic Virus) + 5'UTR, Ω (Tobacco Mosaic Virus) | |
| Addgene | 46966 | Used for designing CRISPR/Cas9 module | |
| Addgene | 46968 | Used for designing CRISPR/Cas9 module | |
| Addgene | 50334 | Level 0 Kanamycin/Neomycin/Paromomycin resistance cassette | |
| Topzeven | - | Used as filters for washing spore suspension | |
| Sigma-Aldrich | 8.17003 | PEG400 | |
| Duchefa Biochemie | E1674.0001 | Pots to grow Parasponia plantlets/seedlings | |
| Merck | 104871 | KH2PO4 | |
| Merck | 105033 | KOH | |
| Merck | 105153 | K2SO4O | |
| Van Leusden b.v. | - | Used as growing substrate for mychorrhization assay | |
| Duchefa Biochemie | S0225.0050 | SH-basal salt medium | |
| Duchefa Biochemie | S0411.0250 | SH-vitamin mixture | |
| Lehle Seeds | VIS-02 | Used as non-ionic surfactant in the washing step of stable transformation | |
| Merck | 137017 | NaCl | |
| VWR Chemicals | 89230-072 | C6H11NaO7 | |
| Merck | 106521 | Na2MoO4·2H2O | |
| Merck | 106574 | Na2HPO4·7H2O | |
| Merck | 567549 | NaH2PO4·H2O | |
| Sigma-Aldrich | 239313 | Na2SO4O | |
| Duchefa Biochemie | S0809.5000 | C12H22O11 | |
| Thermo-Fisher | B69 | Used in Golden Gate cloning assembly | |
| Thermo-Fisher | EL0013 | Used in Golden Gate cloning assembly | |
| Kulzer-Mitsui Chemicals Group | 64708806 | Methyl methacrylate-based resin powder | |
| Kulzer-Mitsui Chemicals Group | 64709003 | HEMA (2-hydroxyethyl methacrylate)-based resin solution | |
| Kulzer-Mitsui Chemicals Group | 66022678 | Methyl methacrylate-based resin solution | |
| Merck | 1159300025 | ||
| Acros | 189350250 | ||
| VWR Chemicals | 663684B | Polysorbate 20 | |
| Stout Perspex | - | pouches box material, lid | |
| Duchefa Biochemie | Y1333.1000 | ||
| Merck | 108816 | ZnCl2 | |
| Alfa Aesar | 33399 | ZnSO4O·7H2O |
References
- Clason, E. W. THE VEGETATION OF THE UPPER-BADAK REGION OF MOUNT KELUT (EAST JAVA). Bulletin du Jardin Botanique de Buitenzorg. Serie III, 509-518 (1936).
- Soepadmo, E. Ulmaceae. Flora Malesiana-Series 1, Spermatophyta. 8, 31-76 (1974).
- Becking, J. H. The Rhizobium symbiosis of the nonlegume Parasponia. Biological Nitrogen Fixation. , 497-559 (1992).
- Oldroyd, G. E. D. Speak, friend, and enter: signalling systems that promote beneficial symbiotic associations in plants. Nature Reviews Microbiology. 11, 252-263 (2013).
- Gutjahr, C., Parniske, M. Cell and developmental biology of arbuscular mycorrhiza symbiosis. Annual Review of Cell and Developmental Biology. 29, 593-617 (2013).
- van Velzen, R., et al. Comparative genomics of the nonlegume Parasponia reveals insights into evolution of nitrogen-fixing rhizobium symbioses. Proceedings of the National Academy of Sciences of the United States of America. 115, E4700-E4709 (2018).
- van Velzen, R., Doyle, J. J., Geurts, R. A Resurrected Scenario: Single Gain and Massive Loss of Nitrogen-Fixing Nodulation. Trends in Plant Science. 24, 49-57 (2019).
- Griesmann, M., et al. Phylogenomics reveals multiple losses of the nitrogen-fixing root nodule symbiosis. Science. 1743, eaat1743 (2018).
- Becking, J. H. Root-Nodule Symbiosis Between Rhizobium And Parasponia (Ulmaceae). Plant and Soil. 51, 289-296 (1979).
- van Zeijl, A., et al. CRISPR/Cas9-Mediated Mutagenesis of Four Putative Symbiosis Genes of the Tropical Tree Parasponia andersonii Reveals Novel Phenotypes. Frontiers in Plant Science. 9, 284 (2018).
- Cao, Q., et al. Efficiency of Agrobacterium rhizogenes–mediated root transformation of Parasponia and Trema is temperature dependent. Plant Growth Regulation. 68, 459-465 (2012).
- Limpens, E., et al. RNA interference in Agrobacterium rhizogenes-transformed roots of Arabidopsis and Medicago truncatula. Journal of Experimental Botany. 55, 983-992 (2004).
- Boisson-Dernier, A., et al. Agrobacterium rhizogenes-Transformed Roots of Medicago truncatula for the Study of Nitrogen-Fixing and Endomycorrhizal Symbiotic Associations. Molecular Plant-Microbe Interactions. 14, 695-700 (2001).
- Kumagai, H., Kouchi, H. Gene Silencing by Expression of Hairpin RNA in Lotus japonicus Roots and Root Nodules. Molecular Plant-Microbe Interactions. 16, 663-668 (2003).
- Nilsson, O., Olsson, O. Getting to the root: The role of the Agrobacterium rhizogenes rol genes in the formation of hairy roots. Physiologia Plantarum. 100, 463-473 (1997).
- Davey, M. R., et al. Effective Nodulation of Micro-Propagated Shoots of the Non-Legume Parasponia andersonii by Bradyrhizobium. Journal of Experimental Botany. 44, 863-867 (1993).
- Webster, G., Poulton, P. R., Cocking, E. C., Davey, M. R. The nodulation of micro-propagated plants of Parasponia andersonii by tropical legume rhizobia. Journal of Experimental Botany. 46, 1131-1137 (1995).
- Op den Camp, R., et al. LysM-type mycorrhizal receptor recruited for rhizobium symbiosis in nonlegume Parasponia. Science. 331, 909-912 (2011).
- Weber, E., Engler, C., Gruetzner, R., Werner, S., Marillonnet, S. A Modular Cloning System for Standardized Assembly of Multigene Constructs. PLOS ONE. 6, e16765 (2011).
- Nekrasov, V., Staskawicz, B. J., Weigel, D., Jones, J. D. G., Kamoun, S. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease. Nature Biotechnology. 31, 691-693 (2013).
- Bertani, G. Studies On Lysogenesis. I. The Mode Of Phage Liberation By Lysogenic Escherichia Coli. Journal of Bacteriology. 62, 293-300 (1951).
- Engler, C., et al. A Golden Gate modular cloning toolbox for plants. ACS Synthetic Biology. 3, 839-843 (2014).
- Fauser, F., Schiml, S., Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. The Plant Journal. 79, 348-359 (2014).
- Lazo, G. R., Stein, P. A., Ludwig, R. A. A DNA Transformation-Competent Arabidopsis Genomic Library in Agrobacterium. Biotechnology. 9, 963-967 (1991).
- Op den Camp, R. H. M., et al. N Nonlegume Parasponia andersonii Deploys A Broad Rhizobium Host Range Strategy Resulting in Largely Variable Symbiotic Effectivenes. Molecular Plant-Microbe Interactions. 25, 954-963 (2012).
- Graham, P. H., Viteri, S. E., Mackie, F., Vargas, A. T., Palacios, A. Variation in acid soil tolerance among strains of Rhizobium phaseoli. Field Crops Research. 5, 121-128 (1982).
- Martinez-Romero, E., et al. Rhizobium tropici, A Novel Species Nodulating Phaseolus vulgaris L. Beans and Leucaena sp. Trees. International Journal of Systematic and Evolutionary Microbiology. 41, 417-421 (1991).
- Felten, J., et al. The Ectomycorrhizal Fungus Laccaria bicolor Stimulates Lateral Root Formation in Poplar and Arabidopsis through Auxin Transport and Signaling. Plant Physiology. 151, 1991-2005 (1991).
- Trouvelot, A., Kough, J. L., Gianinazzi-Pearson, V. Mesure du taux de mycorhization VA d’un systeme radiculaire. Recherche de methods d’estimation ayant une signification fonctionnelle. Aspects Physiologiques et Genetiques des Mycorhizes. , 217-221 (1986).
- Schenk, R. U., Hildebrandt, A. C. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Canadian Journal of Botany. 50, 199-204 (1972).
- Becking, J. H. The Parasponia parviflora - Rhizobium symbiosis. Host specificity, growth and nitrogen fixation under various conditions. Plant and Soil. 75, 309-342 (1983).
- Hoagland, D. R., Arnon Revised, D. I., Arnon, D. I. The Water-Culture Method for Growing Plants without Soil. Circular California Agricultural Experiment Station. 347, 1-32 (1950).
- Wang, K. . Methods in Molecular Biology: Agrobacterium Protocols. 1, (2015).
- Smulders, M. J. M., de Klerk, G. J. Epigenetics in plant tissue culture. Plant Growth Regulation. 63, 137-146 (2011).
- Larkin, P. J., Scowcroft, W. R. Somaclonal variation — a novel source of variability from cell cultures for plant improvement. Theoretical and Applied Genetics. 60, 197-214 (1981).
- Cannon, S. B., et al. Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proceedings of the National Academy of Sciences of the United States of America. 103, 14959-14964 (2006).
- Tuskan, G. A., et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science. 313, 1596-1604 (2006).
- Young, N. D., et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 480, 520 (2011).
- Sato, S., et al. Genome Structure of the Legume, Lotus japonicus. DNA Research. 15, 227-239 (2008).
- Xing, H. L., et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biology. 14, 327 (2014).
- Lowder, L. G., et al. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiology. 169, 971 (2015).
- van Zeijl, A. . Dissecting Hormonal Pathways in Nitrogen-Fixing Rhizobium Symbioses. , (2017).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved