Using Human Induced Pluripotent Stem Cells for the Generation of Tumor Antigen-specific T Cells
* These authors contributed equally
In This Article
Summary
This article describes a method to generate functional tumor antigen-specific induced pluripotent stem cell-derived CD8αβ+ single positive T cells using OP9/DLL1 co-culture system.
Abstract
The generation and expansion of functional T cells in vitro can lead to a broad range of clinical applications. One such use is for the treatment of patients with advanced cancer. Adoptive T cell transfer (ACT) of highly enriched tumor antigen-specific T cells has been shown to cause durable regression of metastatic cancer in some patients. However, during expansion, these cells may become exhausted or senescent, limiting their effector function and persistence in vivo. Induced pluripotent stem cell (iPSC) technology may overcome these obstacles by leading to in vitro generation of large numbers of less differentiated tumor antigen-specific T cells. Human iPSC (hiPSC) have the capacity to differentiate into any type of somatic cell, including lymphocytes, which retain the original T cell receptor (TCR) genomic rearrangement when a T cell is used as a starting cell. Therefore, reprogramming of human tumor antigen-specific T cells to hiPSC followed by redifferentiation to T cell lineage has the potential to produce rejuvenated tumor antigen-specific T cells. Described here is a method for generating tumor antigen-specific CD8αβ+ single positive (SP) T cells from hiPSC using OP9/DLL1 co-culture system. This method is a powerful tool for in vitro T cell lineage generation and will facilitate the development of in vitro derived T cells for use in regenerative medicine and cell-based therapies.
Introduction
In addition to physiological advantages, T cells have many potential therapeutic applications. The generation and expansion of T cells in vitro can be used for disease modeling and therapeutic validation as well as a source of treatment for hereditary and acquired immunodeficiency states (i.e., viral immunodeficiencies and lymphodepletion secondary to chemotherapy or transplantation) and for the eradication of cancer. This latter quality has led to the development of adoptive T cell transfer (ACT) for the treatment of patients with advanced cancer1.
ACT consists of resecting a patient’s tumor, extracting tumor-infiltrating lymphocytes (TILs), expanding TILs ex vivo, then reinfusing the expanded cells into the patient2. It has been shown to be an effective treatment modality for some patients with metastatic cancer. Unfortunately, not all patients respond to this therapy. Previous reports have shown that the differentiation state of transferred cells3,4,5,6,7,8,9, use of large numbers of highly enriched cancer antigen-specific T cells10, and persistence of T cells after transfer11,12 are all correlated with more durable responses13,14. Therefore, when ACT fails to elicit an anti-tumor response, it may in part be due to a low yield of cancer antigen-specific T cells, inefficient ex vivo expansion leading to the exhaustion and loss of reactive clones, or lack of persistence after transfer4. It has been postulated that these obstacles may be overcome by the generation of large numbers of less differentiated cancer antigen-specific T cells in vitro15,16.
Hematopoietic stem/progenitor cells (HSPCs) are a conventional source for in vitro T cell generation, although this method is limited by the small number of cells able to be recovered from a single donor1. Embryonic stem cells (ESCs) have also been shown to produce T cells but with low yield17, making it inefficient for clinical applications. Furthermore, since T lineage cells experience stochastic genetic recombination of their T cell receptors (TCRs) in early developmental stages, it is not possible to use HSPCs or ESCs to generate a pure population of antigen-specific T cells without further genomic modifications like TCR gene transduction.
One approach to overcome these caveats is to reprogram TILs to human induced pluripotent stem cells (hiPSCs), which may provide a limitless source for in vitro T cell generation. It has been shown that cancer antigen-specific TILs can be reprogrammed into hiPSCs and re-differentiated to T cell lineage, which retains the same T cell receptor (TCR) gene rearrangement as the original T cell18,19. This detail is important for ACT because individual patient tumors have unique mutational profiles, and very few cancer antigens have been shown to be shared among patients20. Therefore, using cancer antigen-specific TILs as a source for in vitro generation of hiPSC-derived T cells may provide a new strategy for the personalized treatment of patients with metastatic cancer.
Presented here in detail is a protocol for differentiating hiPSC-derived T lineage cells into functional antigen-specific CD8αβ+ single positive (SP) T cells using OP9/DLL1 co-culture system. This method is a powerful tool for in vitro T cell differentiation of hiPSCs, hematopoietic progenitors, and embryonic stem cells, as well as their further applications in regenerative medicine and cell-based therapies.
Protocol
1. Culturing Human iPSCs (hiPSCs) on Mouse Embryonic Fibroblasts (MEF)
NOTE: Alternative methods for culturing hiPSCs can also be used, including but not limited to: seeding onto a 6 well plate pre-coated with gelatin, a gelatinous protein mixture, recombinant laminin 511, or any other extracellular matrix used in hiPSC expansion, and cultured using defined media specially formulated for human pluripotent stem cell culture.
- Culturing MEF
- Coat a 10 cm cell culture Petri dish with 4 mL of 0.1% gelatin and incubate for 30 min at 37 °C.
- Thaw a vial of 4 x 106 irradiated MEF quickly into 10 mL of 37 °C MEF media (DMEM + 10% FBS + 1x penicillin-streptomycin + 1x L-glutamine supplement). Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the supernatant and resuspend the cell pellet in 9 mL of MEF media.
- Remove the gelatin-coated dish from the incubator. Aspirate gelatin and add 7 mL of MEF media. Plate 3 mL of MEF suspension (from step 1.1.2) onto the gelatin-coated dish. Rock the dish side-to-side and front-to-back to ensure even distribution of MEF over the dish. Incubate at 37 °C for 8-36 h.
- Passaging hiPSC on MEF
NOTE: The data was generated using MART-1 iPSC derived from long-term cultured melanoma TIL, which specifically recognize MART-1 peptide in the context of HLA-A*02:01, as previously described18.- Passage hiPSCs when colonies are between 0.8 - 1.2 mm in diameter. Prior to passaging, check hiPSC colonies in a stereo-microscope and remove any areas of differentiation from the culture using the plastic edge of a 200 µL tip.
- Aspirate spent media and add 10 mL hiPSC media (human ES culture media [Table of Materials] + 10 ng/mL human basic fibroblast growth factor [hbFGF]) supplemented with 10 μM ROCK inhibitor.
- Hold the cell culture dish in one hand and roll a disposable cell passaging tool across the entire dish in one direction. Apply enough pressure so that the entire roller blade touches the culture dish and maintain a uniform pressure during rolling action.
- Rotate the culture dish 90° and repeat step 1.2.3. View the plate in the microscope to visually confirm proper cutting of the colonies, which should appear checkered. Detach cut colonies by gentle mechanical flushing using a 200 μL pipette.
NOTE: Detachment of cut colonies by mechanical flushing must be done immediately after cutting colonies with the roller, because after 3 min the cut colonies will start to reattach to the dish, and it will become difficult to detach colonies of homogeneous size by flushing. - Transfer 350 - 600 clumps of cut colonies onto a new 10 cm dish of MEF (plated 8 - 36 h prior to hiPSC passaging) with 10 mL of fresh hiPSC media supplemented with 10 μM ROCK inhibitor. Incubate at 37 °C.
NOTE: 600 clumps represents approximately 1.0 x 106 MART-1 iPSC and will yield 0.5-1.0 x 106 DP cells on day 35. However, expected numbers will vary depending on the potency of the starting cell line and culture conditions. - The following day, aspirate spent media and add 10 mL of fresh hiPSC media. Change hiPSC media every 1-2 days depending on hiPSC growth rate.
2. Preparation of OP9/DLL1 Cells for Co-culture with hiPSCs
- Culture OP9/DLL1 cells in OP9 media [α-minimum essential medium (α-MEM) + 20% fetal bovine serum (FBS) + 1x penicillin-streptomycin] at 37 °C. When OP9/DLL1 cells reach confluency, aspirate media and wash once with 5 mL of 1x magnesium, calcium, and phenol red-free phosphate buffered saline (PBS).
- Aspirate PBS and add 2 mL of 0.05% Trypsin-EDTA. Incubate for 5 min at 37 °C. Then, add 4 mL of OP9 media and mechanically dissociate the cell layer by pipetting to make a single-cell suspension.
- Transfer the cell suspension into a 50 mL conical tube through a 100 μm cell strainer to avoid cell clumps. Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the supernatant and resuspend in 12 mL of OP9 media.
- Add 8 mL of OP9 media to each of six new 10 cm cell culture Petri dishes. Plate 2 mL of OP9/DLL1 cell suspension from step 2.3 onto each new 10 cm dish. Rock the dish side-to-side then front-to-back to ensure even distribution of OP9/DLL1 over the dish.
- Incubate at 37 °C. Repeat passage every 2 - 3 days when cells reach confluency.
NOTE: It is important to make enough frozen stock of OP9/DLL1 cells and thaw a new stock every 4-6 weeks.
3. In Vitro Differentiation of hiPSCs into CD8αβ+ Single Positive (SP) T Cells
- Prepare gelatinized OP9/DLL1 dishes one week prior to co-culture with hiPSCs. To prepare 0.1% gelatin solution, add 5 mL of room temperature (RT) tissue-grade stock gelatin solution to 500 mL of PBS.
- Coat 3 new 10 cm cell culture Petri dishes by adding 4 mL per dish of 0.1% gelatin. Incubate 30 min at 37 °C.
- Aspirate gelatin and add 8 mL of OP9 media to each dish. Passage one confluent dish of OP9/DLL1 (as done in section 2 above) to three gelatin pre-coated dishes.
- After 4 days, add 10 mL of OP9 media to each 10 cm dish of OP9/DLL1 on gelatin, for a total of 20 mL of media per dish.
- After 7 - 8 days, begin the hiPSC co-culture on OP9/DLL1 confluent dishes (differentiation day 0).
- Aspirate spent media from confluent 10 cm dish of hiPSCs on MEF. Add 10 mL of OP9 media. Cut and detach hiPSC colonies using a disposable cell passaging tool as done in steps 1.2.3 and 1.2.4.
- Transfer 350 - 600 clumps of cut colonies onto a 10 cm pre-gelatinized OP9/DLL1 dish (step 3.1) with 10 mL of fresh OP9 media using a 200 μL pipette. Rock the culture dish side-to-side then front-to-back to ensure even distribution of colonies.
NOTE: Alternatively, pre-formed hiPSC embryoid bodies (EBs) or small clump suspension may be used. However, the use of a disposable cell passaging tool or EB formation system is preferred to produce hiPSC clumps of uniform size.
- On day 1, aspirate spent media and replace with 20 mL of fresh OP9 media. hiPSC clumps co-cultured on OP9/DLL1 for 1 day will appear as small round monolayer colonies (Figure 1).
- On day 5, aspirate 10 mL of spent media and add 10 mL of fresh OP9 media. hiPSC colonies will begin to differentiate into primitive mesoderm, which is characterized by a multilayered dark center.
- On day 9, aspirate 10 mL of spent media and add 10 mL of fresh OP9 media. At this point, multilayered center structures will evolve into dome-like shapes, and a peripheral network-like area will start to become evident.
- On day 13, harvest hematopoietic progenitor cells (HPCs) (Figure 1). hiPSC-derived structures on day 13 are characterized by a dark central organoid surrounded by a network of dome-like areas, representative of hematopoietic zones (HZs) previously reported to enclose human embryonic stem cell-derived hematopoietic progenitors21.
NOTE: The presence of the dome-like structures indicates a successful process even in the absence of dark centers. The inability to produce HPCs may be due to poor quality of OP9/DLL1, quality of FBS lot, confluency of iPSC clumps seeded onto OP9/DLL1 (350-600 clumps is optimal), and/or variations in potency of iPSC lines to produce hematopoietic precursors.- Aspirate spent media and wash 1x with 5 mL of 1x phenol red-free Hanks' balanced salt solution modified with calcium and magnesium (HBSS).
- Aspirate HBSS and add 250 μL of 5000 Units/mL collagenase IV in 10 mL of HBSS. Incubate at 37 °C for 45 min. Aspirate HBSS with collagenase IV and wash once with 5 mL of PBS.
- Aspirate PBS and add 5 mL of 0.25% Trypsin-EDTA. Incubate at 37 °C for 20 min. Then, add 4 mL of OP9 media and dissociate the cell layer by pipetting to make a single-cell suspension.
- Transfer the cell suspension into a 50 mL conical tube through a 100 μm cell strainer. Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the supernatant and resuspend in 10 mL of OP9 media.
- Plate cell suspension onto a new gelatinized 10 cm cell-culture Petri dish (see steps 3.1.1 and 3.1.2). Incubate at 37 °C for 45 min. Then, collect non-adherent cells by gentle pipetting.
- Transfer collected cell suspension into a 50 mL conical tube through a 100 μm cell strainer. Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the supernatant and resuspend in 10 mL of differentiation media [OP9 media with 5 ng/mL human stem cell factor (hSCF), 5 ng/mL human Flt3 ligand (hFLT3L), and 5 ng/mL human interleukin 7 (hIL-7)].
- Plate the cell suspension onto a new 10 cm OP9/DLL1 confluent dish.
- On day 16, passage the cells.
- Mechanically detach non-adherent cells by gentle pipetting and filter through a 100 μm cell strainer. Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the supernatant and resuspend in 10 mL differentiation media.
- Plate the cell suspension onto a new 10 cm OP9/DLL1 confluent dish.
- Continue passaging non-adherent cells every 5-7 days thereafter by repeating step 3.8.
- On day 35, enrich CD4+CD8+ double positive (DP) population and stimulate to produce CD8αβ+ SP T cells (Figure 2).
- Mechanically detach non-adherent cells by gentle pipetting and filter through a 100 μm cell strainer to remove cell clumps. Enrich CD4+ cell population by CD4 magnetic bead isolation according to the manufacturer’s protocol.
NOTE: The rationale for using CD4 magnetic beads is to remove CD4-CD8- DN cells from the culture, as these have been demonstrated to cause direct killing of CD4+CD8+ DP cells after stimulation22. - Count live CD4 enriched cells using a Neubauer hemocytometer and Trypan blue dye. Suspend in OP9 media at total concentration 0.5 x 106 cells/mL. Aliquot 1 mL of the cell suspension (0.5 x 106 cells) into each well of a tissue culture flat bottom 24 well plate of confluent OP9/DLL1.
- Add 100 IU human interleukin 2 (hIL-2), 5 ng/mL hIL-7, 500 ng/mL anti-human CD3 antibody, and 2 μg/mL anti-human CD28 antibody, then culture at 37 °C.
- On day 4 - 7 after stimulation, collect cells for molecular analysis (Figure 3) or co-culture with peptide-pulsed antigen presenting cells (APCs).
- Mechanically detach non-adherent cells by gentle pipetting and filter through a 100 μm cell strainer to remove cell clumps. Enrich CD4+ cell population by CD4 magnetic bead isolation according to the manufacturer’s protocol.
4. Measuring Antigen Specificity of hiPSC Derived CD8αβ+ SP T Cells
NOTE: The type of APCs to be used for this experiement is dependent on the MHC restriction of hiPSC-derived T cells. Here, T2 cell line is used, which is a hybrid of T and B lymphoblastoid cell lines. T2 cells express HLA-A*02:0123, which is recognized by JKF6 cells from which MART1-iPSC was derived18. This T2 cell line can be expanded in RPMI 1640 + 20% FBS + 1x penicillin-streptomycin and is passaged when cells reach a density of 5 x 105 cells/mL.
- Count live HLA-A*02:01+ T-B hybrid lymphoblastoid T2 cells using a Neubauer hemocytometer and Trypan blue dye. Incubate APCs in 24 well tissue culture plate with 1 μg/mL MART-1 peptide for 2 h at 37 °C.
NOTE: Optimal peptide concentration is variable, depending on cell line and antigen specificity. - Collect APCs and wash 2x with 10 mL of PBS to remove any extra peptide.
- Count APCs and suspend at 2 – 5 x 105 cells/mL in OP9 media with 100 IU IL-2 and 5 ng/mL IL-7. Aliquot 100 μL of cell suspension (2-5 x 104 cells) into each well of an ultra-low attachment U bottom 96 well plate or directly into a pre-coated ELISpot plate.
- Sort hiPSC-derived CD8αβ+ SP T cells (1 week after anti-human CD3/CD28 antibody stimulation) using a cell sorter and suspend at 1 x 106 cells/mL in OP9 media with 100 IU IL-2 and 5 ng/mL IL-7. Aliquot 100 μL of cell suspension (1 x 105 cells) into each well of APCs and culture for 16 - 20 h at 37 °C.
- After 16 - 20 h, analyze the cytokine secretion profile by ELISpot assay per the manufacturer's protocol (Figure 4).
Representative Results
After 13 days hiPSCs co-culturing with OP9/DLL1, CD34+CD43+ hematopoietic progenitor cells appeared (Figure 1). After an additional 22 days of culture on non-gelatinized OP9/DLL1 in the presence of hSCF, hFLT3L, and hIL-7, hematopoietic progenitors differentiated into CD3+CD7+CD4+CD8+ double-positive (DP) T lineage cells, the majority of which expressed TCR specific to the MART-1 epitope (tetramer) (Figure 2).
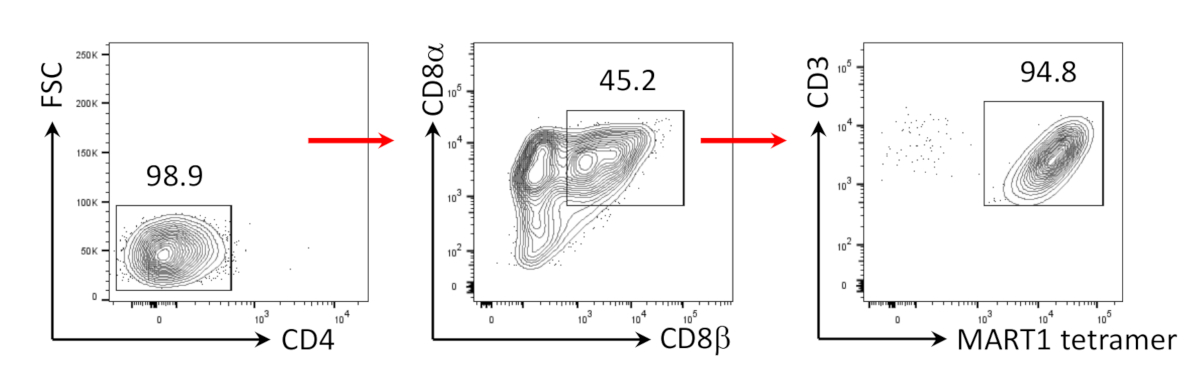
It has previously been shown that CD8+ SP T cells can be induced from CD4+CD8+ DP T cells by TCR signaling via agonist peptide or antibody-driven TCR stimulation24,25. Therefore, on day 35 of culture, hiPSC-derived CD4+CD8+ DP T cells were stimulated with anti-human CD3 and anti-human CD28 antibodies in the presence of hIL-7 and hIL-2. Four days after stimulation, the number of CD3+CD8αβ+ SP cells increased dramatically and remained specific for the MART-1 epitope, confirming the preservation of their inherited antigen-specificity (Figure 3).
To determine the functional properties of hiPSC-derived CD8αβ+ SP T cells, antigen-dependent activation and secretion of interferon gamma (IFN-γ) was analyzed. After stimulation with anti-human CD3 and anti-human CD28 antibodies for 1 week, hiPSC-derived CD8αβ+ SP T cells were isolated using a cell sorter and co-cultured with T2 cell line expressing HLA-A*02:01 with or without cognate MART1 peptide for 16 - 20 h. The ELISpot assay revealed that hiPSC-derived CD8αβ+ SP T cells secrete higher amounts of IFN-γ compared with CD8αα+ SP T cells, when cultured in the presence of MART-1 peptide. IFN-γ expression was null for T cells and APCs alone, demonstrating that human T-iPSC-derived T cells are antigen-specific and functional (Figure 4).

Figure 1: Generation of hiPSC-derived hematopoietic progenitor cells. (A) Schematic overview of the differentiation of hiPSCs to hematopoietic lineage using OP9/DLL1 co-culture. (B) Appearance of hiPSC-derived structures on days 1 (top left), 3 (top right), 7 (bottom left), and 13 (bottom right). Scale bars = 100 µm. (C) Flow cytometric analysis of hiPSC derived CD34+CD43+ hematopoietic progenitor cells on day 13. Data are representative of six independent experiments (n = 1 to 2). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: hiPSC differentiation into Mart1+ CD4+CD8αβ+ DP T cells. (A) Schematic overview of the differentiation of hiPSC-derived hematopoietic lineage to immature T cells using OP9/DLL1 co-culture. (B) Flow cytometric analysis of CD4 vs. CD8α, CD3 vs. CD8β, and MART-1 tetramer expression in hiPSC-derived T cells on day 35. Gated on lymphocytes, single cells, PI negative. Data are representative of three independent experiments (n = 3 - 8). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Induction of CD8αβ+ SP T cell phenotype. Flow cytometric analysis of CD4- hiPSC-derived T cells 4 days after human anti-CD3 and human anti-CD28 antibody-driven stimulation. Gated on lymphocytes, single cells, PI negative (n = 4). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Antigen specificity of hiPSC-derived CD8αβ+ SP T cells. IFN-γ secretion by ELISpot assay of hiPSC-derived CD8αβ+ SP, CD8αα+ SP, and bulk T cells after 20 h co-culture with or without T2 cells pulsed (or not) with MART-1 peptide. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The co-culture of OP9 murine stromal cells is a well-established system for in vitro generation of lymphocytes (i.e., NK, B, and T cells) from HSPCs and pluripotent stem cells. Notch signaling is required to induce T lineage commitment and can be accomplished by the ectopic expression of Notch ligand DLL1 or DLL4, which have comparable efficacy for T cell generation1. Therefore, the OP9/DLL1 co-culture system has become a widely used method for producing T cells in vitro. Furthermore, this method is applicable for use with several types and sources of human cells, including cord blood, bone marrow HSPCs and ESCs. However, the generation of T cells from these sources is limited either by insufficient retrieval of source cells or by inefficient differentiation to T cells1. Additionally, a T cell product with a single TCR recombination cannot be generated from these open repertoire sources. By using regenerative medicine techniques, namely induced pluripotent stem cell (iPSC) technology, it may be possible to produce massive numbers of antigen-specific T cells for use in cell-based therapeutics15.
hiPSCs are similar to pluripotent ESCs in their capacity for self-renewal, limitless expansion, and potential to differentiate to any type of somatic cell in the body; however, they lack the ethical concerns surrounding the use of products of embryonic origin for clinical applications. Moreover, hiPSCs can be produced from any somatic cell, allowing the development of cell products for personalized medicine. In previous reports, hiPSCs have been produced from human T cells using whole peripheral mononuclear cells, CD3+ cells, or isolated cytotoxic T lymphocytes (CTLs) as a source18,19,22,26. When hiPSCs are generated from a T cell source (T-iPSCs), the original TCR gene rearrangement is inherited. Therefore, patient T-iPSC derived T cells may provide a model for personalized ACT treatment by targeting a patient’s distinct cancer antigens.
The differentiation of human pluripotent stem cells into T lineage cells is divided into two steps: the generation of hematopoietic progenitor cells (HPCs)27 and their further differentiation into T lineage cells21. Both steps can be accomplished using the OP9/DLL1 co-culture system. Importantly, the quality of OP9/DLL1 feeder cells is critical to the success of T cell differentiation. Since OP9/DLL1 cells are not an immortalized homogenous cell line, the quality of the FBS and culture conditions are critical to maintaining their expansion without losing the ability to support hiPSC differentiation. Therefore, it is recommended to pre-evaluate the lot of FBS and passage consistently when cell-to-cell cytoplasmic contact begins to occur, in order to prevent cell differentiation and senescence. One point to take into consideration is that cell-to-cell contact can appear indistinguishable from the background depending on the phase contrast and magnification of the microscope. In our experience, most OP9/DLL1 dishes will appear to be 80% confluent when ready to passage.
It has been shown that the redifferentiated T lineage cells generated from T-iPSCs by OP9/DLL1 co-culture can produce CD8+ SP T cells upon stimulation18,19. However, regenerated CD8+ SP T cells acquire the innate-like CD8αα homodimer22,28, which is an ineffective co-receptor for TCR signaling29. Additionally, these regenerated CD8+ SP T cells have shown strong TCR-independent cytotoxicity, making these cells unfavorable for clinical use30. This protocol describes a recent method involving stimulation of purified CD4+CD8+ DP cells to generate CD8αβ+ SP T cells with a more conventional phenotype and improved antigen-specific cytotoxicity22. Although the loss of antigen specificity due to secondary TCRα allelic rearrangement occurs in the DP stage after prolonged long-term culture, this can be overcome by genome editing in T-iPSCs31. In our experience, hiPSC-derived DP cells start to appear on day 30 - 35 of culture, and these newly generated DP cells have not yet undergone secondary TCRα rearrangement. Therefore, most DP cells on day 35 retain antigen specificity and can be used to generate antigen-specific CD8αβ+ SP T cells.
Prior to human anti-CD3 and anti-CD28 stimulation on day 35, CD4-CD8- DN cells must be removed from the culture, as these have been demonstrated to cause direct killing of CD4+CD8+ DP cells after stimulation22. Using CD4 magnetic bead enrichment (step 3.10) will enrich for both DP and CD4+CD8- intermediate single positive (ISP) cells1, which we have been shown to have no negative effects22. Alternatively, fluorescence-activated cell sorting by flow cytometry can be performed to isolate DP cells. However, magnetic bead separation is preferred as it avoids the mechanical stress induced by flow cytometry.
The generation of CD8αβ+ SP T cells from human pluripotent stem cells without activation-mediated agonist selection has subsequently been demonstrated by the use of 3D murine stromal cell culture32. However, physiological positive selection is dependent on the interaction of TCR with self-peptide-MHC complexes, which are uniquely processed and presented by thymic cortical epithelial cells33. Furthermore, TCR affinity for selection peptides has been shown to determine the subsequent functional capabilities of mature CD8αβ+ SP T cells34. Currently, there is no evidence to suggest that a Notch stromal cell-based co-culture system can provide the defined selection peptide and MHC complex required for physiological positive selection.
It has been previously reported in a murine model that T lineage cells generated from tumor antigen-specific T cell-derived hiPSCs using OP9/DLL1 alone fail to experience conventional maturation. However, iPSC-derived immature T cells generated by the OP9/DLL1 system can mature into naïve-like T cells by further physiological thymic education in a 3D culture system 28,35. Therefore, the protocol presented here to produce iPSC-derived immature T cells generated by the OP9/DLL1 system is vital for further attempts to generate real human tumor antigen-specific post-thymic T cells capable of long-term persistence in vivo with efficiency to treat established vascularized tumors.
Acknowledgements
We thank Alan B. Hoofring and Erina H. He for graphical assistance. This research was supported by the Intramural Research Program of the National Cancer Institute (ZIA BC010763) and the Intramural NCI Cancer Moonshot Initiative for Cell-based Cancer Immunotherapy.
Materials
| Name | Company | Catalog Number | Comments |
| 10 cm dish | Corning, Inc. | 353003 | |
| Anti-CD3, human | BD Biosciences | Cat# 561812, RRID:AB_1089628 | |
| Anti-CD34, human | BD Biosciences | Cat# 348791, RRID:AB_400381 | |
| Anti-CD4, human | Biolegend | Cat# 344612, RRID:AB_2028479 | |
| Anti-CD43, human | BD Biosciences | Cat# 560198, RRID:AB_1645460 | |
| Anti-CD7, human | BD Biosciences | Cat# 555361, RRID:AB_395764 | |
| Anti-CD8a, human | BD Biosciences | Cat# 555369, RRID:AB_398595 | |
| Anti-CD8b, human | BD Biosciences | Cat# 641057, RRID:AB_1645747 | |
| Anti-TCRb, human | BD Biosciences | Cat# 555548, RRID:AB_395932 | |
| CD28 human monoclonal antibody (15E8), pure functional grade | Miltenyl Biotec | 130-093-375 | |
| CD3 human monoclonal antibody (OKT3), pure functional grade | Miltenyl Biotec | 130-093-387 | |
| CD4 Microbeads, human | Miltenyl Biotec | 130-045-101 | |
| Cell strainer 100 um | Fisher Scientific | 22-363-549 | |
| Fetal Bovine Serum (FBS) | Gemini | 100-500 | |
| Flt-3 ligand | R&D Systems | 427-FL | |
| Gelatin Solution 2% | SIGMA-Aldritch | G1393-100ML | |
| GlutaMAX (100X) | Thermo Fisher Scientific | 35050-061 | L-Glutamine supplement |
| HBSS Mg+Ca+ Phenol-Red Free | Gibco | 14025-092 | |
| Interleukin-2 | R&D Systems | 202-IL | |
| Interleukin-7 | R&D Systems | 407-ML | |
| iTAG MHC Tetramer HLA-A*0201 Mart1 Tetramer -ELAGIGILTV | MBL | Cat#TB-0009-2 | |
| Mart1-hiPSC | Vizcardo et al., Cell Stem Cell 2013 | RIKEN-IMS | |
| Melan-A, MART 1 (26-35) | InnoPep | 3146-0100 | |
| MEM Non-Essential Amino Acids Solution | Gibco | 11140050 | |
| αMEM powder | Gibco | 61100061 | |
| Mouse Embryonic Fibroblasts (MEF) | Thermo Fisher Scientific | C57BL/6 MEF MITC-TREATED 4M EACH; A34962 | |
| OP9/N-DLL1 | Riken Bioresource center | Cat# RCB2927; RRID:CVCL_B220 | OP9/DLL1 |
| Penicillin/streptomycin | Thermo Fisher Scientific | 15140-122 | |
| Phosphate buffered saline pH 7.4 (1x) | Thermo Fisher Scientific | 10010-023 | |
| Primate ES Cell Medium | Reprocell | RCHEMD001 | Human ESC Culture Media |
| Rhok inhibitor (Y-27632 dihydrochloride) | Tocris | 1254 | |
| RPMI 1640 | Gibco | 11875093 | |
| Stem Cell Factor (SCF) | R&D Systems | 455-MC | |
| StemPro | EZPassage | 23181-010 | |
| T2-tumor | ATCC | T2 (174 x CEM.T2) (ATCC® CRL-1992™) | |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher Scientific | 25300-062 | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | 25200-072 | |
| U Bottom 96 well plate | Corning, Inc. | 3799 |
References
- Brauer, P. M., Singh, J., Xhiku, S., Zuniga-Pflucker, J. C. T Cell Genesis: In Vitro Veritas Est. Trends in Immunology. 37 (12), 889-901 (2016).
- Rosenberg, S. A., Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 348 (6230), 62-68 (2015).
- Gattinoni, L., et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. Journal of Clinical Investigation. 115 (6), 1616-1626 (2005).
- Rosenberg, S. A., et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clinical Cancer Research. 17 (13), 4550-4557 (2011).
- Crompton, J. G., et al. Lineage relationship of CD8(+) T cell subsets is revealed by progressive changes in the epigenetic landscape. Cellular and Molecular Immunology. 13 (4), 502-513 (2016).
- Henning, A. N., Klebanoff, C. A., Restifo, N. P. Silencing stemness in T cell differentiation. Science. 359 (6372), 163-164 (2018).
- Henning, A. N., Roychoudhuri, R., Restifo, N. P. Epigenetic control of CD8(+) T cell differentiation. Nature Reviews Immunology. 18 (5), 340-356 (2018).
- Vodnala, S. K., et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 363 (6434), (2019).
- Restifo, N. P., Gattinoni, L. Lineage relationship of effector and memory T cells. Current Opinion in Immunology. 25 (5), 556-563 (2013).
- Tran, E., et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 344 (6184), 641-645 (2014).
- Gattinoni, L., et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nature Medicine. 15 (7), 808-813 (2009).
- Gautam, S., et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nature Immunology. 20 (3), 337-349 (2019).
- Klebanoff, C. A., et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clinical Cancer Research. 17 (16), 5343-5352 (2011).
- Klebanoff, C. A., Gattinoni, L., Restifo, N. P. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy. Journal of Immunotherapy. 35 (9), 651-660 (2012).
- Crompton, J. G., Clever, D., Vizcardo, R., Rao, M., Restifo, N. P. Reprogramming antitumor immunity. Trends in Immunology. 35 (4), 178-185 (2014).
- Crompton, J. G., Rao, M., Restifo, N. P. Memoirs of a reincarnated T cell. Cell Stem Cell. 12 (1), 6-8 (2013).
- Kennedy, M., et al. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Reports. 2 (6), 1722-1735 (2012).
- Vizcardo, R., et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 12 (1), 31-36 (2013).
- Nishimura, T., et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 12 (1), 114-126 (2013).
- Lo, W., et al. Immunologic recognition of a shared p53 mutated neoantigen in a patient with metastatic colorectal cancer. Cancer Immunology Research. , (2019).
- Timmermans, F., et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. Journal of Immunology. 182 (11), 6879-6888 (2009).
- Maeda, T., et al. Regeneration of CD8alphabeta T Cells from T-cell-Derived iPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Research. 76 (23), 6839-6850 (2016).
- Salter, R. D., Howell, D. N., Cresswell, P. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics. 21 (3), 235-246 (1985).
- Snauwaert, S., et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia. 28 (4), 830-841 (2014).
- Takahama, Y., Suzuki, H., Katz, K. S., Grusby, M. J., Singer, A. Positive selection of CD4+ T cells by TCR ligation without aggregation even in the absence of MHC. Nature. 371 (6492), 67-70 (1994).
- Seki, T., et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell. 7 (1), 11-14 (2010).
- Vodyanik, M. A., Slukvin, I. I. Hematoendothelial differentiation of human embryonic stem cells. Current Protocols in Cell Biology. , (2007).
- Vizcardo, R., et al. Generation of Tumor Antigen-Specific iPSC-Derived Thymic Emigrants Using a 3D Thymic Culture System. Cell Reports. 22 (12), 3175-3190 (2018).
- McNicol, A. M., et al. CD8alpha/alpha homodimers fail to function as co-receptor for a CD8-dependent TCR. European Journal of Immunology. 37 (6), 1634-1641 (2007).
- Themeli, M., Riviere, I., Sadelain, M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 16 (4), 357-366 (2015).
- Minagawa, A., et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell. 23 (6), 850-858 (2018).
- Montel-Hagen, A., et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell. 24 (3), 376-389 (2019).
- Takada, K., Kondo, K., Takahama, Y. Generation of Peptides That Promote Positive Selection in the Thymus. Journal of Immunology. 198 (6), 2215-2222 (2017).
- Takada, K., et al. TCR affinity for thymoproteasome-dependent positively selecting peptides conditions antigen responsiveness in CD8(+) T cells. Nature Immunology. 16 (10), 1069-1076 (2015).
- Vizcardo, R., et al. A Three-dimensional Thymic Culture System to Generate Murine Induced Pluripotent Stem Cell-derived Tumor Antigen-specific Thymic Emigrants. JoVE. , e58672 (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved