Subcellular Fractionation of Primary Chronic Lymphocytic Leukemia Cells to Monitor Nuclear/Cytoplasmic Protein Trafficking
In This Article
Summary
This protocol enables the optimization and subsequent efficient generation of nuclear and cytoplasmic fractions from primary chronic lymphocytic leukemia cells. These samples are used to determine protein localization as well as changes in protein trafficking that take place between the nuclear and cytoplasmic compartments upon cell stimulation and drug treatment.
Abstract
Nuclear export of macromolecules is often deregulated in cancer cells. Tumor suppressor proteins, such as p53, can be rendered inactive due to aberrant cellular localization disrupting their mechanism of action. The survival of chronic lymphocytic leukaemia (CLL) cells, among other cancer cells, is assisted by the deregulation of nuclear to cytoplasmic shuttling, at least in part through deregulation of the transport receptor XPO1 and the constitutive activation of PI3K-mediated signaling pathways. It is essential to understand the role of individual proteins in the context of their intracellular location to gain a deeper understanding of the role of such proteins in the pathobiology of the disease. Furthermore, identifying processes that underlie cell stimulation and the mechanism of action of specific pharmacological inhibitors, in the context of subcellular protein trafficking, will provide a more comprehensive understanding of the mechanism of action. The protocol described here enables the optimization and subsequent efficient generation of nuclear and cytoplasmic fractions from primary chronic lymphocytic leukemia cells. These fractions can be used to determine changes in protein trafficking between the nuclear and cytoplasmic fractions upon cell stimulation and drug treatment. The data can be quantified and presented in parallel with immunofluorescent images, thus providing robust and quantifiable data.
Introduction
The transportation of macromolecules between the nucleus and cytoplasm has long been established to play a key role in normal cellular function and is often deregulated in cancer cells1,2. Such deregulation can result from overexpression/mutation of proteins that control nuclear export. One such protein Exportin-1 (XPO1), is a transport receptor that exports >200 nuclear export signal (NES)-containing proteins into the cytoplasm from the nucleus2. XPO1-cargos include p53, FOXO family members and IB, contributing to their inactivation by inhibiting their mechanism of action1,2,3. Further protein mislocalization can occur when microenvironmental signals impinge upon the cancer cells, leading to the activation of intracellular signaling pathways such as the phosphatidyl-inositol-3-kinase (PI3K)/Akt pathway, resulting in inactivation of FOXO family members and subsequent export from the nucleus4,5. Such mislocalization of tumor suppressor proteins has been implicated in the progression of a number of hematological and solid tumors1,2,6.
The development of small molecule inhibitors for clinical use in hematological malignancies (acute myeloid leukemia (AML)/CLL), which bind to and selectively inhibit XPO1 function, underlines the importance of developing appropriate techniques to address the impact of pharmacological agents on the shuttling of proteins between the nuclear and cytoplasmic compartments6,7,8. Imaging techniques have advanced significantly enabling the identification of proteins in subcellular compartments upon external stimulation of drug treatments, however, the importance of robust and supportive parallel techniques is critical to reliably inform a scientific audience of the validity of a result.
Resting lymphocytes and malignant CLL-B cells isolated from patient blood samples represent a challenge in the generation of nuclear and cytoplasmic fractions due to the high nuclear: cytoplasmic ratio. The optimization of experimental conditions to generate robust and reliable experimental data is of course critical in order to plan future experimental programs. The method described here enables quantification of proteins in the nuclear and cytoplasmic fractions and determines how these proteins can be impacted by cellular stimulation and/or drug treatment.
Protocol
The use of primary samples from CLL patients described here have been approved by the West of Scotland Research Ethics Service, NHS Greater Glasgow and Clyde (UK) and all work was carried out in accordance with the approved guidelines.
1. Isolation of CLL Cells from Patient Blood Samples
- Peripheral blood samples from previously consented CLL patients are received from the clinic in EDTA blood collection tubes, accompanied by the white cell count (WCC). Purify the peripheral blood CLL samples according to the WCC. For WCC < 40 x 106 cells/mL, proceed to step 1.1.1; for WCC ≥ 40 x 106 cells/mL, proceed to step 1.1.2.
- Pour the contents of all the EDTA blood tubes into a 50 mL conical centrifuge tube and add 50 µL of Human B Cell Enrichment Cocktail per 1 mL of blood. Incubate at room temperature (RT) for 20 min. Proceed to step 1.1.2.
- Dilute the sample at a ratio of 1:1 with RT CLL wash buffer (phosphate-buffered salin (PBS), 0.5% Fetal Bovine Serum (FBS) and 2 mM EDTA).
- Aliquot RT density gradient media into an appropriately sized conical centrifuge tube for the sample (10 mL into a 50 mL tube for 30 mL of sample or 4 mL into a 15 mL tube for 10 mL of sample).
- Carefully layer the sample on top of the density gradient media and centrifuge at 400 x g for 30 min at RT.
NOTE: Ensure that the centrifuge is at RT before the samples are placed in the centrifuge as a change in temperature will result in poor enrichment of mononuclear cells, and switch off the brake on the centrifuge, as sudden braking can disrupt the liquid interface. - Gently harvest the white layer of mononuclear cells that collect at the interface of the density gradient media and CLL wash buffer, into a fresh 50 mL conical centrifuge tube using a plastic Pasteur pipette.
- Add 40 mL of CLL wash buffer to the isolated monolayer to wash the cells and centrifuge at 300 x g for 10 min at RT.
- Discard the supernatant, resuspend the pellet by flicking the bottom of the tube, then repeat the wash step described in step 1.5.
- Discard the supernatant, resuspend the pellet as described in step 1.6, then resuspend the pellet in a set volume of CLL wash buffer (up to 40 mL, depending on the size of the cell pellet).
- Count the cells using trypan blue and a haemocytometer. Then proceed to flow cytometry to check the purity of CLL cells.
NOTE: At this stage the CLL cells can be cultured at a concentration of 10 x 106 cells/mL in media to be used in experiments, and/or cryopreserved in 10% dimethyl sulfoxide (DMSO)/FBS for future work at concentrations of up to 100 x 106 cells/vial.
2. Flow Cytometry of CLL Cells
- Label 12 mm x 75 mm round bottomed polystyrene tubes as described in Table 1.
- In tubes 2 – 5, put one drop of compensation beads and store on ice. Add 1 µL of the appropriate antibody (anti-CD5, CD19, CD23 or CD45, as indicated in Table 1) to tubes 2 – 5, and incubate on ice for 20 min, protected from light by placing tin foil over the ice bucket.
NOTE: These tubes serve as compensation controls for setting up the flow cytometry template. - Put up to 1 x 106 CLL cells into tubes 1, 6 and 7, add 2 mL of FACS buffer (PBS + 2% FBS) to each tube and centrifuge at 300 x g for 5 min at RT to wash the cells. Discard the supernatant and store the tubes containing cell pellets on ice.
- Resuspend the cell pellets and add the appropriate combination of antibodies to the cells in tube 7 as instructed in Table 1, in a final volume of 100 µL with FACS buffer. Antibodies are used at an appropriate concentration according to the manufacturer guidelines.
- Resuspend the cell pellets in tubes 2 and 6 in 100 µL of FACS buffer.
- Incubate the cells on ice, alongside the stained beads in tubes 2 – 5, protected from light for 20 min.
- After the incubation, add 2 mL of FACS buffer to all the tubes and centrifuge at 300 x g for 5 min at RT to wash the cells. Discard the supernatant and resuspend the bead/cell pellets by gently flicking the tubes.
- Resuspend tubes 1 - 5 in 100 µL of FACS buffer and place on ice until ready to analyze on the flow cytometer.
- Dilute the DAPI solution to 0.05-0.2 µg/mL in FACS buffer immediately prior to use. The optimal concentration may vary, and titration is recommended.
- Resuspend tubes 6 and 7 with 100 µL of diluted DAPI solution and incubate the tubes on ice for a minimum of 5 min to allow the cells to stain.
NOTE: No further washing is necessary as DAPI must be present in the buffer for dead cells to remain labelled. Once DAPI has been added, cells must be analyzed on the flow cytometer within 4 h. - Analyze cells using a flow cytometer.
3. Preparation of Subcellular Fractions from CLL Cells
NOTE: When planning the experimental set-up, include a well of unstimulated/untreated cells from which the whole cell extract can be generated.
- Perform the desired stimulation and/or drug treatment of the MEC1 CLL cell line or isolated primary CLL cells using 10 – 20 x 106 cells/condition. Cells will then be used for subcellular fractionation (steps 3.4 & 3.5) or to generate whole cell extract (step 3.6).
- Preparation of solutions/tubes: Prepare all solutions/buffers freshly on the day of the fractionation, before the cells are harvested. Store the solutions on ice until required and use within 4 h of preparation.
- PBS/phosphatase inhibitor solution: Prepare the phosphatase inhibitors in PBS by diluting the phosphatase inhibitors 1:20 in 1x PBS (i.e., 0.5 mL of phosphatase inhibitors in 9.5 mL of 1x PBS).
NOTE: Ensure the phosphatase inhibitors have not precipitated. If a precipitate is present, heat to 50 °C for 10 min. - Hypotonic buffer: Prepare 1x hypotonic Buffer by making a 1:10 dilution of 10x hypotonic buffer in distilled water (i.e., 50 μL of 10x hypotonic Buffer into 450 μ L of dH2O).
- 10 mM dithiothreitol (DTT): Prepare 10 mM DTT by making a 1:100 dilution of 1M DTT with distilled water (i.e., 10 μL of 1 M DTT in 990 μL of dH2O).
NOTE: DTT is highly labile so prepare this freshly each time. Avoid repeated freeze/thaw cycles. - Complete lysis buffer: Determine how much buffer is required for each experiment. Each sample requires 50 μL of complete lysis buffer, so add 5 μL of 10 mM DTT (step 3.2.3) to 44.5 μL of lysis buffer and then add 0.5 μL of protease inhibitor cocktail. This amount can be scaled up depending on the number of samples in the experiment.
- Label four sets of 1.5 mL microfuge tubes for each stimulation and/or drug treatment for the freshly stimulated cells (step 3.3), the freshly generated cytoplasmic fractions (step 3.4.3), the freshly generated nuclear fractions (step 3.5.3), and the whole cell lysates (step 3.6.3). Pre-chill these microfuge tubes on ice until required.
- PBS/phosphatase inhibitor solution: Prepare the phosphatase inhibitors in PBS by diluting the phosphatase inhibitors 1:20 in 1x PBS (i.e., 0.5 mL of phosphatase inhibitors in 9.5 mL of 1x PBS).
- Transfer the cells into individually labelled 1.5 mL microfuge tubes and pellet by centrifuging at 200 x g for 5 min at 4 °C. Remove the supernatant and resuspend the cells in 1 mL of ice-cold PBS/phosphatase inhibitors (step 3.2.1). Pellet the cells by centrifugation at 200 x g for 5 min at 4 °C. Remove the supernatant and keep the cell pellets on ice.
- Preparation of cytoplasmic fractions: Gently resuspend the cell pellets to be used for subcellular fractionation in 50 μL of 1x hypotonic Buffer (step 3.2.2). Incubate the cells on ice for 15 min to allow the cells to swell.
NOTE: The volume of hypotonic buffer used can be increased empirically depending on the cell number.- Add 0.8 - 2.5 μL (1:20 to 1:60) of detergent into each sample and vortex on the highest setting for 10 s.
- To determine the optimal concentration of detergent to use for a specific cell type to isolate nuclear and cytoplasmic fractions, perform a detergent gradient initially. A range of 1:20 to 1:60 (i.e., 2.5 μL to 0.8 μL of detergent into 50 μL of Hypotonic Buffer) should be adequate.
NOTE: If the volume of hypotonic buffer in step 3.4 is adjusted, ensure the appropriate detergent ratio is maintained. - Verify cell lysis by observing cells using a phase contrast microscope before and after addition of detergent. Whole cells appear larger with a dense, dark nucleus. The cytoplasm will appear as a bright halo around the nucleus.
NOTE: Appropriate lysis is further confirmed by using Western blotting to analyze specific proteins within the lysed fractions generated from the detergent gradient.
- To determine the optimal concentration of detergent to use for a specific cell type to isolate nuclear and cytoplasmic fractions, perform a detergent gradient initially. A range of 1:20 to 1:60 (i.e., 2.5 μL to 0.8 μL of detergent into 50 μL of Hypotonic Buffer) should be adequate.
- Once lysed, centrifuge the samples at 14,000 x g for 30 s at 4 °C.
- Carefully transfer the supernatant into a pre-chilled, labelled microfuge tube. This cytoplasmic fraction can be stored at -80 °C until required for further analysis. The remaining pellet contains the nuclear fraction (step 3.5).
NOTE: Avoid repeated freeze/thaw cycles of the samples.
- Add 0.8 - 2.5 μL (1:20 to 1:60) of detergent into each sample and vortex on the highest setting for 10 s.
- Preparation of nuclear fractions: Resuspend each nuclear pellet in 50 μL of complete lysis buffer (step 3.2.4) by pipetting up and down.
NOTE: The volume of complete lysis buffer can be adjusted empirically according to the starting cell number.- Add 2.5 μL of detergent to solubilize proteins associated with the nuclear membrane and vortex on the highest setting for 10 s. Incubate the samples on ice for 30 min.
- Vortex on the highest setting for 30 s, then centrifuge the samples at 14,000 x g for 20 min at 4 °C.
- Transfer the supernatant into a pre-chilled, labelled microfuge tube. This nuclear fraction can be stored at -80 °C until required for further analysis.
NOTE: Avoid repeated freeze/thaw cycles of the samples.
- Preparation of whole cell lysates (WCL) from CLL cells
NOTE: The preparation of the whole cell extract can be carried out at the same time as the preparation of the nuclear fractions (step 3.5).- Resuspend the whole cell extract pellets in 100 μL of complete lysis buffer (prepared in step 3.2.4) by pipetting up and down, then add 5 μL of detergent to ensure complete cell lysis. Incubate the samples on ice for 30 min.
- Vortex on the highest setting for 30 s, then centrifuge the samples at 14,000 x g for 20 min at 4 °C.
- Transfer the supernatant into a pre-chilled microfuge tube. This whole cell lysate can be stored at -80 °C until required for further analysis.
NOTE: Avoid repeated freeze/thaw cycles of the samples.
4. Downstream Analysis of Subcellular Fractions
NOTE: In this protocol, analysis of the generated cell fractions was carried out by Western blotting using standard protocols, loading equal cell numbers/lane (equivalent to ~10 µg of protein) for nuclear and cytoplasmic fractions.
- Quantification of protein trafficking between nuclear and cytoplasmic fractions: Perform quantitative Western blot analysis through quantitation of signal intensity, or densitometry using freely available Western blot analysis software.
- Importing images: Western blot images generated from different developing instruments must be imported as JPG, PNG or TIFF files. A 16-bit depth RAW file is recommended. To import an image, click the software icon and hover over Import. Then click Third-Party Images. Select the image file and click Open.
- Displaying the image: In the Image ribbon, click on the Choose button in the Display group. The Adjust Display dialog will open to enable further adjustments if necessary. Implement additional enhancements including Brightness or Contrast using the adjustable sliders on the Image LUTs tab. Use the Curves tab for finer adjustments.
- Data analysis (channel deselection): Click the Analysis ribbon. To analyze only one channel, deselect the channel(s) not being analyzed. Click on a channel’s Don’t Show Channel thumbnail in the Image LUTs, leaving only the desired channel displayed. Images imported as JPG, PNG or TIFF files may require deselection of multiple unwanted RGB channels.
- Adding shapes: To quantify signal intensity, click Add Rectangle to add a rectangle to the image. Click the center of a feature (e.g., a protein band) to place a rectangle around it. Alternatively, to manually draw a shape, choose Draw Rectangle. After adding all the desired shapes, click Select to return the cursor to the selection tool.
NOTE: Add multiple shapes in a logical order, as the data is sorted by an ID number that is generated sequentially. - Background subtraction: To subtract background noise, click the first button in the Background group and select Median from the drop-down menu. Set the border width to 3 in the Background dialogue and select the segments to be used for the background calculation. When choosing which segments to use, select segments that best represent the image background.
NOTE: Background noise can affect signal quantification, so it must be subtracted to accurately calculate signal from the shapes of interest. - Trim Signal and Trim Background - OPTIONAL: Files imported in JPG, PNG or TIFF formats may exhibit pixel saturation: highlighted/bright regions within a protein band. Trim Signal and Trim Background (Bkgnd) removes saturated pixels from analysis. To view these values, add Trim Signal and Trim Bkgnd to a table by clicking on the Columns button to the right of the table view.
NOTE: Pixel saturation may lead to unreliable quantification. Saturated pixels can only be removed if fewer than 5% of pixels within a shape are saturated.
- Adding shapes: To quantify signal intensity, click Add Rectangle to add a rectangle to the image. Click the center of a feature (e.g., a protein band) to place a rectangle around it. Alternatively, to manually draw a shape, choose Draw Rectangle. After adding all the desired shapes, click Select to return the cursor to the selection tool.
- Export Data: Click the Shapes tab above the table. For densitometry, values in the Signal column are required. Signal is the sum of the pixel intensity values (Total) for a shape minus the product of the Bkgnd and the Area. Click the Report button. Click Save As or Launch Spreadsheet.
Signal = Total – (Bkgnd x Area)
NOTE: The shapes tab provides a table of quantitative values including Signal, Total, Area and Bkgrnd. - Quantifying protein expression: Within the saved spreadsheet, calculate normalized expression of the protein of interest for each lane or variable by dividing the obtained Signal for the protein of interest by the Signal for the corresponding protein loading control band.
NOTE: Comparisons between the amounts of a normalized protein of interest across nuclear and cytoplasmic fractions cannot be directly compared because of the different loading controls used to distinguish nuclear and cytoplasmic fractions. However, comparisons within individual fractions, e.g. following drug treatment, are appropriate. - Export image for publication or presentation: Click the Images tab found above the table and then click on the image to be exported. If using the image for a slide presentation or other digital formats, click the software icon, hover over Export and click on Image for Digital Media. Save the image as a JPG, PNG or TIFF file, as required.
Representative Results
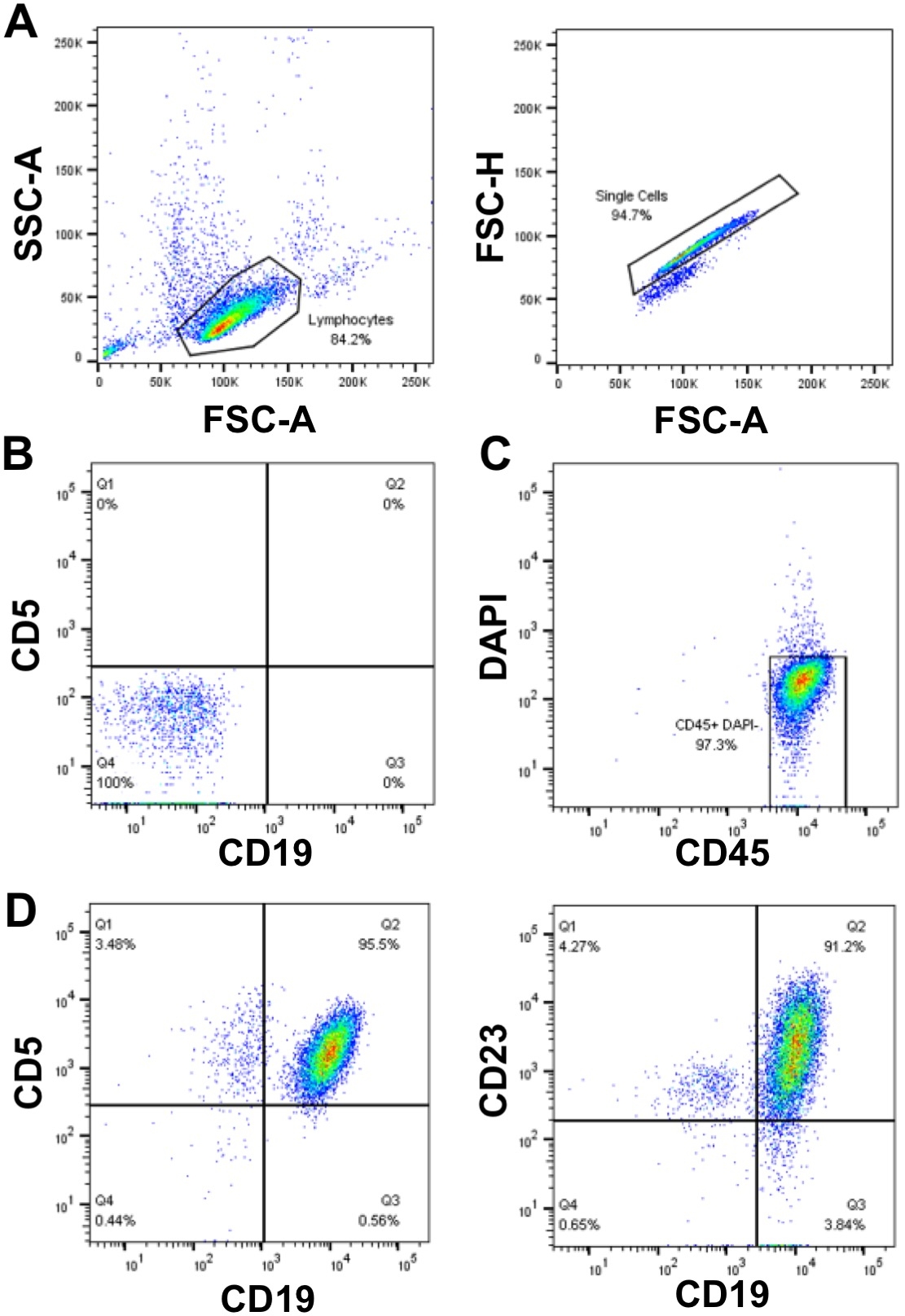
When planning experiments on primary CLL cells, if assays require a large number of cells (>50 x 106 cells), there is a preference to use freshly isolated CLL cells, rather than cryopreserved cells that require thawing, however this is not always possible. This is because the freeze/thaw process can result in the death of up to 50% of the CLL cells, although this is sample dependent. Enrichment of CLL cells with a WCC >40 x 106/mL using density centrifugation as described here (steps 1.3 – 1.5) enables a high cell recovery with high purity (≥ 95%) of primary CLL cells. In the sample shown, the WCC = 177 x 106/mL: from a 30 mL blood sample 5 x 109 cells were recovered, which represents a cell yield of 94% of total cells. Analysis of this sample by flow cytometry revealed a purity of CLL cells of >95% as indicated by the dual surface expression of CLL cell markers CD19 and CD5 after gating on FSC/SSC, single cells that were DAPI negative (viable cells) (Figure 1).
Optimization of the subcellular fractionation procedure was carried out using a range of detergent ratios (1:20 to 1:60) during the preparation of the cytoplasmic fraction (step 3.4). Thereafter, the nuclear fractions and WCLs were prepared (steps 3.5 and 3.6 respectively). Immunoblots were performed on the resultant fractions of the CLL cell line MEC1 (Figure 2A) and primary CLL cells (Figure 2B). The blots were probed for the fraction markers Lamin A/C (nuclear; 74/63 kDa) and β-tubulin (cytoplasmic; 55 kDa) to confirm successful cell fractionation. The fractionation indicates that the optimal detergent level for MEC1 cells is a 1:60 dilution (Figure 2A), compared with a 1:30 dilution being optimal for primary CLL cells (Figure 2B), as indicated by an enrichment of nuclear protein and a lack of cytoplasmic protein in the fractions and vice versa. WCLs represent the total protein and act as a positive control for antibodies used to probe the subcellular fractions. It is important to choose appropriate proteins as fraction markers: Figure 2C shows immunoblots of nuclear/cytoplasmic fractions prepared from MEC1 cells in which RNA polymerase II (Rpb1 CTD; 250 kDa) and Lamin A/C were blotted as markers of nuclear fractions, while β-tubulin and γ-tubulin (50 kDa) were used as cytoplasmic markers. It is clear that γ-tubulin is enriched in the cytoplasm however expression is evident in the nucleus, as shown previously9.
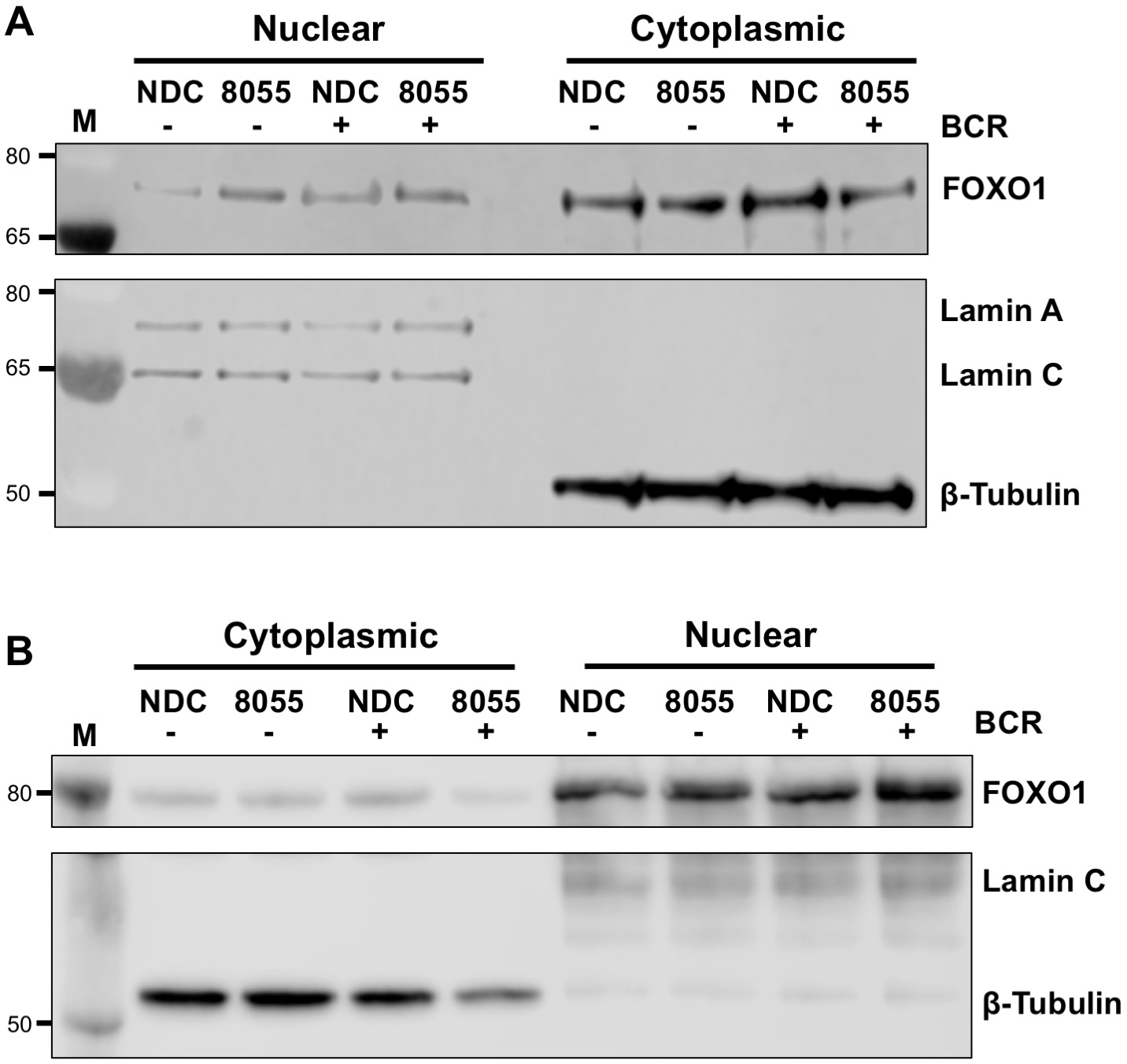
Once experimental conditions are optimized, an experiment can be performed. In the examples shown, the subcellular localization of FOXO1 in nuclear and cytoplasmic fractions was determined upon stimulation of cells with the B cell antigen receptor (BCR) in the presence or absence of the dual mTORC1/2 inhibitor AZD8055, in MEC1 cells (Figure 3A) and primary CLL cells (Figure 3B)5,10. In both examples, the generation of highly enriched nuclear and cytoplasmic fractions was achieved as indicated by the almost exclusive expression of Lamin in the nuclear fraction and β-tubulin in the cytoplasmic fractions. In both cell types, FOXO1 expression was reduced in the cytoplasm following treatment with AZD8055 compared to NDC, accompanied by an increase of FOXO1 expression in the nuclear compartment, thus demonstrating protein translocation (Figure 3). To remove the subjectivity of data interpretation, individual immunoblots from five primary CLL samples were quantified within subcellular fractions (step 4; Figure 4A), using the respective nuclear or cytoplasmic proteins as internal loading controls for each sample, and then normalizing each fraction to the unstimulated (US) no drug control (NDC) control, as indicated. The resultant graph shows trends of FOXO1 movement between the nuclear and cytoplasmic fractions, with AZD8055 reducing the levels of FOXO1 expression in the cytoplasm, while concurrently increasing expression in the nucleus. Furthermore, an elevation in cytoplasmic FOXO1 expression is evident upon BCR crosslinking.
| Tube | Tube Name | Cells/Beads | Antigen | Fluorophore |
| 1 | Unstained | Cells | NA | NA |
| 2 | Single Stain | Beads | CD5 | FITC |
| 3 | Single Stain | Beads | CD19 | PE-Cy7 |
| 4 | Single Stain | Beads | CD23 | APC |
| 5 | Single Stain | Beads | CD45 | APC-Cy7 |
| 6 | Single Stain | Cells | Viability | DAPI |
| 7 | CLL Stain | Cells | CD5, CD19, CD23, CD45 & viability | FITC, PE-Cy7, APC, APC-Cy7 & DAPI |
Table 1: Table showing the ideal set of sample tubes required for flow cytometry of CLL cells. Each experiment must include all the appropriate controls for accurate analysis of the results obtained.

Figure 1: Representative flow cytometry analysis plot of enriched CLL patients. Mononuclear-CLL cells enriched from the peripheral blood of an individual CLL patient were gated using FSC-A vs. SSC-A, and doublets were then excluded using FSC-A vs. FSC-H (A). Unstained cells (tube 1) and compensation controls (tubes 2-6) were used to set up the flow cytometer to detect cells and compensate between the fluorescent channels, thus ensuring that the fluorescence signals were detected correctly. (B) An example of negative staining (unstained cells; tube 1) in the CD19 and CD5 fluorescence channels. Live (DAPI negative) and CD45 positive cells were gated (C) and the proportion of CD19+CD5+ (95.5%) and CD19+CD23+ (91.2%) cells within the DAPI-CD45+ population was determined (D). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Optimization of nuclear/cytoplasmic fractionation. Cytoplasmic and nuclear fractions, and whole cell lysates (WCL), were prepared from cell pellets (10 – 20 x 106 cells) of the CLL cell line (A) MEC1 or (B) primary CLL cells enriched from the peripheral blood of patients as described in Step 3. Optimization of the subcellular fractionation was carried out by using a range of detergent ratios (1:20 to 1:60) when preparing the cytoplasmic fraction (as described in step 3.4). The resultant samples were immunoblotted and probed with anti-Lamin A/C (nuclear) and anti-β-tubulin (cytoplasmic) antibodies to confirm successful cell fractionation alongside WCL. Molecular weight markers are shown on the left of the blot (M). * indicates the optimal detergent conditions for cell lysis. (C) Immunoblot of nuclear and cytoplasmic fractions from MEC1 cells with control conditions (NDC) or drug treatment (8055) in the presence of absence of stimulation (+ or – BCR crosslinking respectively). Blots were probed with anti-Rbp1 CTD (clone 4H8; recognizing RNA polymerase II subunit B1), anti-Lamin A/C, anti-β-tubulin or anti-γ-tubulin (clone GTU-88) antibodies, to identify subcellular fractions. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Subcellular fractionation demonstrates the shuttling of FOXO1 between the nucleus and cytoplasm in CLL. (A) MEC-1 cells and (B) primary CLL cells were pre-treated for 30 min with 100 nM AZD8055 (8055) or left untreated (NDC) as indicated and then BCR was ligated for 1 h or left US. Nuclear and cytoplasmic fractions were then prepared and immunoblotted. Following confirmation of fractionation by probing with anti-Lamin A/C (nuclear) and anti-β-tubulin (cytoplasmic) antibodies, the effect of both drug treatment and BCR ligation was assessed on FOXO1 protein expression, using an anti-FOXO1 antibody. M indicates molecular weight marker. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: A worked example of quantitative Western blot analysis (densitometry). (A) Densitometry was performed using Western blot analysis software available online. Briefly, within the Analysis ribbon, rectangles were drawn around protein bands in the image to calculate signal intensity. Depicted is densitometry of a representative Western blot image of a CLL patient sample that underwent cytoplasmic/nuclear fractionation. Cytoplasmic and nuclear fractions are distinguished by the expression of cytoplasmic (β-tubulin) and nuclear (Lamin A/C) markers. Normalized expression of FOXO1 for a given condition can be calculated by dividing the signal obtained for FOXO1 by the corresponding signal for β-Tubulin or Lamin A/C, depending on the fraction being analyzed. Relative FOXO1 expression (relative to US vehicle control), can be calculated by dividing normalized FOXO1 expression of a given condition by the normalized FOXO1 expression of the US vehicle control of a given cellular fraction. (B) Graph showing the FOXO1 expression levels in the cytoplasmic (left) or nuclear (right) fractions normalized to US-NDC control within each cellular fraction. The red dot on the graph is the worked example shown. This data shows the average fold change in FOXO1 expression compared to US-NDC ± SEM. P values were determined by two-tailed Students paired t test. n = 5 individual CLL patient samples. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The protocol described provides a fast and efficient method for the generation of nuclear and cytoplasmic fractions from primary CLL cells, and subsequent quantification of protein trafficking between the nuclear and cytoplasmic fractions upon cell stimulation and drug treatment. The data presented demonstrates the ability to detect trafficking of specific proteins, for example, FOXO1, between the nuclear and cytoplasmic fractions, upon treatment with a dual mTOR inhibitor AZD8055 in the presence/absence of BCR crosslinking through F(ab’)2 fragment stimulation (Figure 3 and Figure 4). Coupling these experiments with quantification of Western blots from individual CLL patient samples, enables objective analyses of the data generated and demonstrates the robustness of the assay described to quantify global changes in protein localization in CLL cells isolated from patient cohorts (Figure 4). It is clear from the data that an average of five patient samples in the cytoplasmic fractions reached near significance. Given the clinical heterogeneity of CLL patients11, these analyses would ordinarily be carried out on bigger patient cohorts, and/or focused on specific prognostic subgroups of patients to gain a fuller understanding of the cellular response of CLL cells to specific drug treatments.
The data presented demonstrate the importance of choosing protein markers that exclusively reside in either the cytoplasmic or nuclear fractions, as the purity of the fractionation will be confirmed by these markers. β-tubulin was chosen for cytoplasmic fraction confirmation, and Lamin A/C as a nuclear marker. Additional proteins commonly used are GAPDH and α-tubulin to identify the cytoplasmic fraction or Brg1 (SMARCA4), TFIID and RNA Polymerase II for nuclear fraction purity4,5. However, care must be taken when choosing proteins that are highly enriched in specific fractions, and not present in both fractions (e.g., γ-tubulin) (Figure 2C)9. Indeed, GAPDH and actin while generally considered to be cytoplasmic proteins can localize to the nucleus12,13, highlighting the importance of choosing a fraction marker that does not relocate when stimulation or treatment is applied to the cells. Furthermore, it is important to confirm that the protein marker chosen is expressed in the cell of interest by running the WCL alongside the subcellular fractionations.
In the representative experiment shown, the same number of CLL cells was used for each condition (stimulation/drug treatment), and thereafter the fractionation samples were prepared immediately. Loading 10 µg of fractionated protein/lane provides sufficient material for detection of the proteins of interest. As these samples only underwent a short-term drug treatment and stimulation (up to 4 h), it was assumed that the protein level would remain the same in each sample, and a protein assay was not performed. However, if cell treatments are extended (18 - 72 h), the level of cell death or proliferation in cells may significantly alter the quality and quantity of protein extracted, dependent on the drug/cell stimulation applied, thus altering protein levels in the treated/stimulated samples. In these cases for longer term drug treatments, it is advisable to carry out protein quantification using a Bradford assay or equivalent, prior to Western blotting to ensure the same amount of protein is run in each lane of the immunoblot. The presence of detergents may interfere with specific protein assays14, this interference can be reduced by diluting cell fraction protein samples. In addition, use the complete lysis buffer as the blank, using the same dilution as in the samples being tested.
To provide supporting evidence for findings described here, parallel experiments could be performed using fluorescence microscopy to analyze the location of FOXO1 within CLL cells to enable visualization of these findings5. Furthermore, the subcellular fractions generated can also be used for enzyme activity assays or proteomics analysis in further downstream analyses.
Acknowledgements
The authors would like to thank Dr. Natasha Malik for critically reviewing the manuscript. This study was funded by a Bloodwise project grant awarded to AMM (18003). FACS analysis facilities were funded by the Howat Foundation. MWM was funded by a PhD studentship from Friends of Paul O’Gorman Leukaemia Research Centre, JC was funded by the Friends of Paul O’Gorman Leukaemia Research Centre and JH was funded by a Bloodwise project grant (18003).
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL microcentrifuge Tubes | Griener Bio one | 616201 | |
| 3 mL Pasteur Pipettes | Griener Bio one | 612398 | |
| 12 mm x 75 mm FACS Tubes | Elkay | 2052-004 | |
| 15 mL Tube | Griener Bio one | 188271 | |
| 50 mL Tube | Griener Bio one | 227261 | |
| anti-CD5 FITC antibody | BD Biosciences | 555352 | phenotypic surface marker |
| anti-CD19 PE Cy7 antibody | BD Biosciences | 557835 | phenotypic surface marker |
| anti-CD23 APC antibody | BD Biosciences | 558690 | phenotypic surface marker |
| anti-CD45 APC Cy7 antibody | BD Biosciences | 557833 | phenotypic surface marker |
| anti-β-Tubulin antibody | Cell Signaling | 2146 | cytoplasmic marker |
| anti-γ-Tubulin Mouse antibody (clone GTU-88) | Sigma-Aldrich | T5326 | |
| anti-FoxO1 (C29H4) Rabbit antibody | Cell Signaling | 2880 | |
| anti-Lamin A/C antibody | Cell Signaling | 2032 | nuclear marker |
| anti-mouse IgG, HRP-linked Antibody | Cell Signaling | 7076 | Secondary antibody |
| anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | 7074 | Secondary antibody |
| anti-Rpb1 CTD antibody (clone 4H8) | Cell Signaling | 2629 | nuclear marker |
| BDFACS Canto II | BD Biosciences | By Request | Flow Cytometer |
| DAPI Solution | BD Biosciences | 564907 | live/dead discriminator |
| DMSO | Sigma | D2650 | |
| EDTA | Sigma | EDS | |
| Fetal Bovine Serum | Thermofisher | 10500064 | |
| GraphPad Prism 6 | GraphPad Software | Software | |
| Histopaque1077 density gradient media | Sigma | H8889 | |
| HyperPAGE 10 - 190 kDa protein marker | Bioline | BIO-33066 | Molecular weight marker |
| Image Studio Lite (version 5.2.5) | LI-COR | www.licor.com | Software |
| Labnet VX100 | Fisher Scientific | Vortex | |
| Nucelar Extract Kit | Active Motif | 40010 | |
| OneComp eBeads | Thermofisher | 01-111-42 | |
| Trypan Blue Solution | Thermofisher | 15250061 | |
| PageRuler Plus Prestained Protein Ladder | Thermo Fisher Scientific | 26619 | Molecular weight marker |
| PBS Tablets | Fisher Scientific | BR0014G | |
| RosetteSep Human B Cell Enrichment Cocktail | Stem Cell Technologies | 15064 | |
| Sigma 3-16P | SciQuip | Centrifuge | |
| Sigma 1-15PK | SciQuip | Centrifuge |
References
- Kau, T. R., Way, J. C., Silver, P. A. Nuclear transport and cancer: from mechanism to intervention. Nature Reviews Cancer. 4, 106-117 (2004).
- Turner, J. G., Dawson, J., Sullivan, D. M. Nuclear export of proteins and drug resistance in cancer. Biochemical Pharmacology. 83, 1021-1032 (2012).
- Nakamura, N., et al. Forkhead transcription factors are critical effectors of cell death and cell cycle arrest downstream of PTEN. Molecular and Cellular Biology. 20, 8969-8982 (2000).
- Calnan, D. R., Brunet, A. The FoxO code. Oncogene. 27, 2276-2288 (2008).
- Cosimo, E., et al. AKT/mTORC2 inhibition activates FOXO1 function in CLL cells reducing B cell receptor-mediated survival. Clinical Cancer Research. 25, 1574-1587 (2019).
- Mahipal, A., Malafa, M. Importins and exportins as therapeutic targets in cancer. Pharmacology & Therapeutics. 164, 135-143 (2016).
- Hing, Z. A., et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia. 30, 2364-2372 (2016).
- Lapalombella, R., et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 120, 4621-4634 (2012).
- Hořejší, B., et al. Nuclear γ-tubulin associates with nucleoli and interacts with tumor suppressor protein C53. Journal of Cellular Physiology. 227, 367-382 (2012).
- McCaig, A. M., Cosimo, E., Leach, M. T., Michie, A. M. Dasatinib inhibits B cell receptor signalling in chronic lymphocytic leukaemia but novel combination approaches are required to overcome additional pro-survival microenvironmental signals. British Journal of Haematology. 153, 199-211 (2011).
- Fischer, K., Hallek, M. Optimizing frontline therapy of CLL based on clinical and biological factors. Hematology. American Society of Hematology. Education Program. 2017, 338-345 (2017).
- Butera, G., et al. Regulation of autophagy by nuclear GAPDH and its aggregates in cancer and neurodegenerative disorders. International Journal of Molecular Sciences. 20, 2062 (2019).
- Virtanen, J. A., Vartiainen, M. K. Diverse functions for different forms of nuclear actin. Current Opinion in Cell Biology. 46, 33-38 (2017).
- Friedenauer, S., Berlet, H. H. Sensitivity and variability of the Bradford protein assay in the presence of detergents. Analytical Biochemistry. 178, 263-268 (1989).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved