Isolation and 3D Collagen Sandwich Culture of Primary Mouse Hepatocytes to Study the Role of Cytoskeleton in Bile Canalicular Formation In Vitro
In This Article
Summary
Presented here is a protocol for the isolation of mouse hepatocytes from adult mouse livers using a modified collagenase perfusion technique. Also described is the long-term culture of hepatocytes in a 3D collagen sandwich setting as well as immunolabeling of the cytoskeletal components to study bile canalicular formation and its response to treatment.
Abstract
Hepatocytes are the central cells of the liver responsible for its metabolic function. As such, they form a uniquely polarized epithelium, in which two or more hepatocytes contribute apical membranes to form a bile canalicular network through which bile is secreted. Hepatocyte polarization is essential for correct canalicular formation and depends on interactions between the hepatocyte cytoskeleton, cell-cell contacts, and the extracellular matrix. In vitro studies of hepatocyte cytoskeleton involvement in canaliculi formation and its response to pathological situations are handicapped by the lack of cell culture, which would closely resemble the canaliculi network structure in vivo. Described here is a protocol for the isolation of mouse hepatocytes from the adult mouse liver using a modified collagenase perfusion technique. Also described is the production of culture in a 3D collagen sandwich setting, which is used for immunolabeling of cytoskeletal components to study bile canalicular formation and its response to treatments in vitro. It is shown that hepatocyte 3D collagen sandwich cultures respond to treatments with toxins (ethanol) or actin cytoskeleton altering drugs (e.g., blebbistatin) and serve as a valuable tool for in vitro studies of bile canaliculi formation and function.
Introduction
Hepatocytes, the central cellular structures of the liver that are responsible for its metabolic functions, are uniquely polarized epithelial cells. Their polarization, appearing in mammals shortly after birth, results in formation of the biliary canalicular network and is essential for proper bile secretion. Apical membranes of hepatocytes collectively form bile canaliculi, whereas basal membranes remain in contact with the endothelium of sinusoids. The loss of hepatocyte polarization leads to redistribution of bile transporters and results in pathological processes connected with bile retention in the liver (i.e., cholestasis).
The establishment and maintenance of hepatocyte polarization and the development of bile canaliculi entail complex mechanisms. The underlying processes depend on collective interplay among the hepatocyte cytoskeleton, cell-cell contacts, and interactions with the extracellular matrix1. The hepatocyte cytoskeleton consists of all three filament networks, the actin cytoskeleton, microtubules, and intermediate filaments, which provide structural support for canalicular formation. The differential role of cytoskeletal components in the regeneration and maintenance of bile canalicular networks has been previously illustrated in vitro in 3D collagen-sandwich hepatocyte cultures2.
Actin microfilaments and microtubules are important during the initial stages of hepatocyte membrane polarization at the sites of canaliculus generation2. The actin cytoskeleton establishes the structure and function of bile canaliculi, forming membrane-associated microfilaments and the circumferential ring, thus supporting the canalicular architecture and inserting the actin cytoskeleton into tight and adherens junctions3. The ring of keratin intermediate filaments outside the actin cytoskeleton further stabilizes the canalicular structure3.
The importance of proteins in hepatocyte junctional complexes in the organization of bile canaliculi architecture has been well-documented in several knock-out mouse models, which show distorted canaliculi in mice lacking both tight and/or adherens junctional proteins4,5,6. The deletion of the adherens junction protein α-catenin has been shown to lead to disorganization of the hepatocyte actin cytoskeleton, dilatation of bile canalicular lumens, leaky tight junctions, and effectively to a cholestatic phenotype4. Moreover, in vitro studies have shown the importance of adherens junction components E-cadherin and β-catenin in remodeling of the hepatocellular apical lumen and protein trafficking7.
Strikingly, ablation of the cytoskeleton crosslinking protein plectin, which is the major keratin organizer, has revealed phenotypes comparable to those linked to the actin cytoskeleton8. This suggests a critical role of keratin intermediate filaments in supporting of the canalicular structure. In vitro studies utilizing 3D hepatocyte collagen sandwiches have also shown the importance of the AMP-activated protein kinase and its upstream activator LKB1 in bile canalicular network formation9. These findings were then further confirmed by subsequent in vivo studies10,11. Thus, it has become clear that in vitro studies are necessary to further the understanding of signaling processes involved in the establishment of hepatocyte polarization, proper canalicular network formation, and bile secretion.
A major challenge in studying processes connected with bile canalicular formation and its response to pathological situations in vitro is using a cell culture conditions that closely resemble the situation in vivo12. Freshly isolated primary hepatocytes are not polarized; thus, they lose their function, morphology, and functional bile canaliculi in 2D culture conditions (e.g., changes in gene regulation, polarization, and de-differentiation13,14,15). Despite this fact, freshly isolated hepatocytes most closely reflect the nature of the liver in vivo, unlike liver-derived cell lines16. Even though they have been used in the past, immortalized cell lines do not exert the epithelial-like characteristic morphology of hepatocytes, and the bile canalicular lumens formed by these cells resemble liver canaliculi poorly7. Recently, 3D cultures of primary hepatocytes, from both mice and rats, have become a useful tool to investigate processes involved in bile canalicular network formation in vitro9. Primary hepatocytes cultured between two layers of collagen (referred to as a 3D collagen sandwich culture) can repolarize in several days. Because of high technical demands required when culturing mouse hepatocytes in 3D collagen sandwiches, here we present a complex protocol to isolate, to cultivate, and to immunolabel mouse hepatocytes embedded in 3D collagen sandwiches in order to characterize the involvement of cytoskeletal components during bile canalicular formation.
Protocol
All animal experiments were performed in accordance with European Directive 2010/63/EU and they were approved by the Czech Central Commission for Animal Welfare.
1. Materials
- House animals under specific pathogen-free conditions according to the guidelines of the Federation for Laboratory Animal Science Associations with free access to regular chow and drinking water. House animals under a 12 h/12 h dark/light cycle. For hepatocyte isolation and culture, use 8–12 week-old animals.
- Stock solutions A and B
- Prepare stock solution A and stock solution B according to Table 1 and Table 2, respectively, in advance. Dissolve all components in 1 L of distilled H2O (dH2O).
- Adjust the pH of solutions to 7.2 and filter the solutions through a 0.2 μm filter. Both solutions can be stored at 4 °C for up to 6 months.
- Stock solution C
- Prepare solution C on the day of primary hepatocyte isolation.
- Add all components according to Table 3, fill to a 50 mL total volume with dH2O, and dissolve. Adjust the pH to 7.3.
- Aliquot 50 mL of the solution in 50 mL tubes. Use one tube per animal. Place all required aliquots in a pre-warmed water bath (37 °C).
- Stock solution D
- Prepare solution D on the day of primary hepatocyte isolation.
- Add all components according to Table 4, fill to a 30 mL total volume with dH2O, and dissolve. Adjust the pH to 7.3.
- Aliquot 30 mL of the solution in 50 mL tubes. Add collagenase I (5 mg/30 mL) into solution D. Use one aliquot per animal. Place all aliquots in a pre-warmed water bath (37 °C).
- Stock solution E
- Prepare solution E on the day of primary hepatocyte isolation.
- Add all components according to Table 5, fill to a 50 mL total volume with dH2O, and dissolve. Adjust the pH to 7.3.
- Aliquot 50 mL of the solution in 50 mL tubes. Add albumin V (0.65 g/50 mL) into solution E. Use one aliquot per animal. Place all aliquots in a pre-warmed water bath (37 °C).
2. Preparation of collagen sandwiches
- One day before primary hepatocyte isolation, prepare the first layer of the collagen I sandwich.
NOTE: Work on ice and use pre-chilled solutions, tips, plates, and tubes to minimize unwanted gelation of collagen I. - Neutralize the required amount of collagen I (from rat tail) according to the manufacturer's protocol. A volume of 100 µL of neutralized collagen (1.5 mg/mL) per experimental sample (3.5 cm dish) is required. To prepare 1 mL of neutralized collagen (1.5 mg/mL), add 100 μL of 10x DMEM, 1.5 μL of 1M NaOH, and 48.5 μL of dH2O into 500 μL of collagen (stock concentration = 3 mg/mL). Check the pH of neutralized collagen with litmus paper (the pH should be ~7.5).
- Disperse 100 μL of neutralized collagen solution evenly using a pre-chilled 200 μL tip over the surface of a 3.5 cm dish set on ice.
- Incubate overnight under standard culture conditions (incubator with 5% CO2 at 37 °C). On the day of primary hepatocyte isolation, add 1 mL of pre-warmed (37 °C) PBS to the first collagen layer. Allow the collagen to rehydrate for 2-3 h at 37 °C.
3. Equipment set up
- Prepare all equipment as listed in Table of Materials and set up as shown in Figure 1.
4. Surgical procedure
- Anesthetize the mouse by intramuscular injection of tiletamine (60 mg/kg of body weight), zolazepam (60 mg/kg), and xylazine (4.5 mg/kg). After several minutes, confirm proper anesthetization by the toe pinch. If animal does not respond to the pinch, proceed to 4.2.
- Place the anesthetized mouse on a dissection mat and tape the lower and upper extremities to fix the mouse in a supine position. Swab the abdomen with 70% ethanol and open the abdomen with a V-shape incision from the pubic area to front legs. Fold the skin over the chest to uncover the abdominal cavity. Place the dissecting mat under a dissecting microscope.
- Bend an insulin syringe needle (30 G) to a 45° angle. Expose the inferior vena cava (IVC) by moving the intestines and colon in the caudal direction.
- Fill 2.5 mL of pre-warmed (37 °C) solution C into a 2 mL syringe with a cannula. Ensure that there are no air bubbles in the cannula or syringe.
- Before cannulation of the IVC, reposition the liver lobes by pressing them up to the diaphragm with a wet (PBS) cotton swab. Place a silk suture around the IVC just below the liver (Figure 2A,B).
- Inject 10 µL of heparin (5000 U/mL) into the portal vein using an insulin syringe with a 30 G needle bent at a 45° angle (Figure 2C).
- To cannulate the liver, make a small incision with microsurgical scissors to the IVC directly next to the liver (below the suture; Figure 2D) that is large enough to insert the cannula. Secure the cannula in the position using sutures and two surgical knots (Figure 2E).
- Cut the portal vein (Figure 2F) to allow the perfusion buffers to flow out from the liver to prevent expansion of the liver.
- Manually perfuse the liver with 1.5 mL of pre-warmed solution C by slowly pressing the syringe connected to the cannula (this should take ~15 s). The removal of blood from the liver by discoloration of the liver can now be observed.
- Pre-fill a peristaltic pump with pre-warmed (37 °C) solution C. Check the perfusion apparatus and ensure that there are no air bubbles in the system.
- Cautiously disconnect the cannula from the syringe and connect it to the tubing of the peristaltic pump running at flow rate of 2.5 mL/min. Work quickly but carefully and ensure that the cannula remains in position and that no bubbles enter the tubing or cannula.
- Perfuse the liver with solution C for 2 min (5 mL of solution C). Change to solution D and continue the perfusion for an additional 10 min (25 mL of solution D).
- Once the liver has been perfused, remove it from the abdominal cavity. The liver will now be very fragile and pale in color (Figure 3).
5. Isolation of primary hepatocytes
- Remove the liver from the mouse. The cannula will still be tied to the liver, so use the forceps to lift the cannula with the liver, and carefully cut off all fascia connections. Transfer the liver to a 50 mL tube containing 20 mL of pre-warmed solution E.
- Hold the cannula with the liver using forceps and disassociate the tissue by rubbing the liver around the wall of the tube to transfer the isolated hepatocytes into buffer. Keep the isolated cells on ice.
NOTE: Isolated primary hepatocytes are viable for several hours while kept on ice. Users can repeat steps 4.1 through 5.2 isolating primary hepatocytes from another donor mouse, if necessary, or proceed to the next step. - Place a 70 µm nylon cell strainer on top of a 50 mL tube and filter the isolated cells.
- Centrifuge the tube at 50 x g and 4 °C for 5 min. Aspirate the supernatant. To remove dead cells and increase the percentage of viable cells, resuspend the pellet in 20 mL of 40% percol in DMEM.
- Centrifuge the tube at 50 x g and 4 °C for 5 min. Aspirate the supernatant containing dead cells and resuspend the pellet in 20 mL of solution E.
- Centrifuge tubes at 50 x g and 4 °C for 5 min. Aspirate the supernatant and resuspend the pellet in 10 mL of solution E.
- Check the primary hepatocyte yield and viability using trypan blue staining. Count the cell number using a Neubauer cell counting chamber and adjust the cell concentration to 3.75 x 105 viable cells/mL. Keep the cells on ice.
6. Cultivation of primary hepatocytes in 3D collagen sandwiches
- Prepare hepatocyte culture medium (HCM). Add 15 µL of glucagon (1 mg/mL), 15 µL of hydrocortisone (50 mg/mL), and 40 µL of insulin (10 mg/mL) to 50 mL of complete medium (DMEM, high glucose, 10% FBS, 1% penicillin-streptomycin).
- Evenly disperse 2 mL (5 x 105 cells/mL) of viable primary hepatocytes in a pre-coated 3.5 cm dish. Incubate the cells with 5% CO2 at 37 °C for 3 h. Important: Evenly distribute the cells by tilting the dish in all directions just before putting the dish into incubator.
- Prepare neutralized collagen I (100 µL/3.5 cm dish; i.e., a sufficient volume for all dishes as described above [step 2.1]). Keep on ice till use.
- After 3 h, carefully remove the medium and unattached cells and add 100 µL of neutralized collagen I to each 3.5 cm dish to form the top layer of collagen sandwich on cells. Incubate the collagen sandwich under standard cell-culture conditions (5% CO2 at 37 °C) for 1 h. After a 1 h incubation, carefully add 2 mL of HCM.
- After 24 h of culture, change the HCM medium for a fresh one.
- Culture for 3–8 days, depending on the formation of bile canaliculi. Check the culture every day under a microscope (Figure 4). Change the HCM every second day.
7. Immunolabeling of primary hepatocytes in 3D collagen sandwiches
- Remove the media from the hepatocyte sandwiches, then wash carefully with pre-warmed PBS. Fix the sandwich cultures with 1 mL of 4% paraformaldehyde in PBS for 30 min at room temperature (RT).
- After fixation, wash the sandwiches 3x for 10 min in 2 mL of PBS + 0.1% Tween 20 (PBS-T).
- Permeabilize cells with 1 mL of 0.1 M glycine, 0.2% Triton X-100 in PBS at RT for 1 h. Wash 3x for 10 min in PBS-T.
- Gently disturb the top layer of collagen using a 10 μL loading tip connected to a vacuum aspiration to ensure sufficient antibody penetration.
- Block with 1 mL of 5% BSA in PBS-T (i.e., blocking solution) for 2 h. Incubate with primary antibodies diluted in blocking solution overnight at 37 °C. Wash 3x for 15 min in PBS-T.
- After washing, incubate with secondary antibody at 37 °C for 5 h. Wash 3x for 15 min in PBS-T followed by 1x wash with distilled H2O.
- Mount with anti-fade mounting media (see Table of Materials) for microscopy.
Representative Results
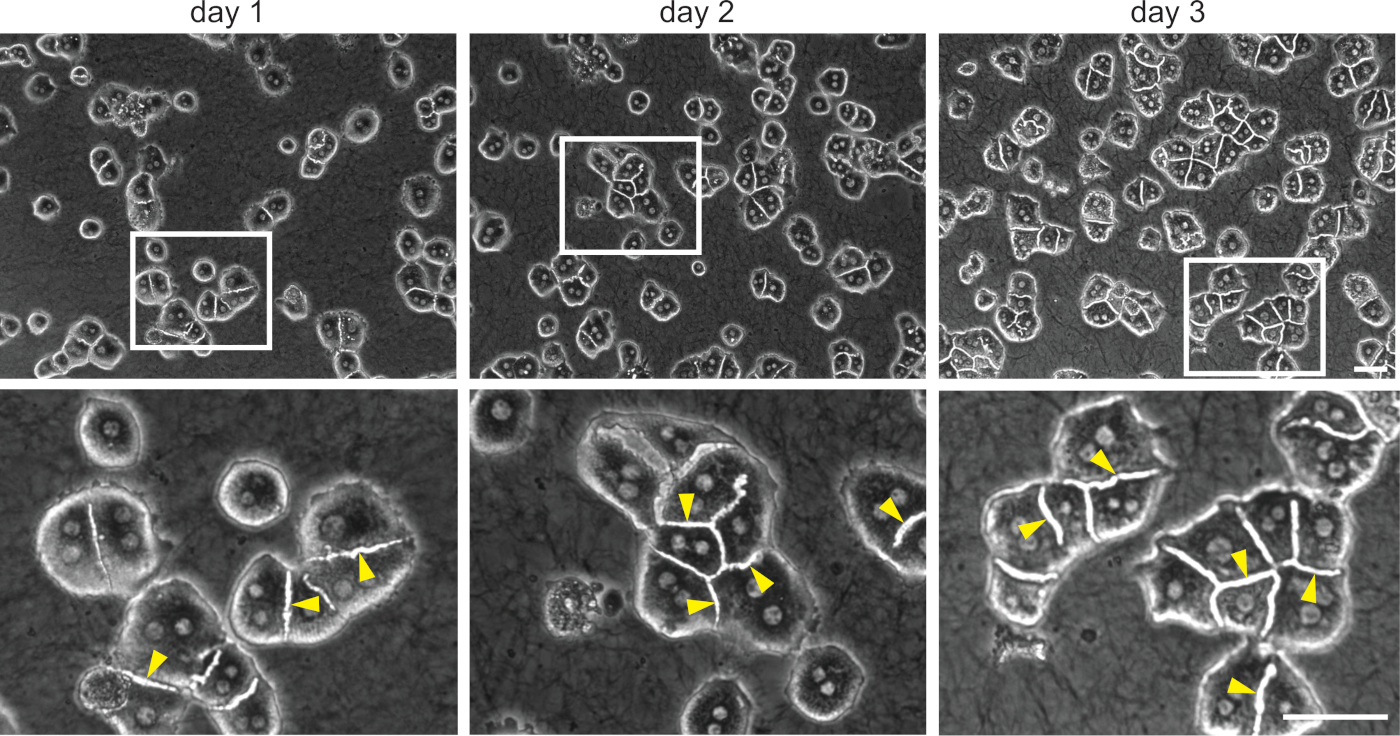
Mouse primary hepatocytes were isolated and seeded in 3D collagen sandwiches. Bile canaliculi between two adjacent cells started to form within several hours after seeding. Cells formed clusters and self-organized in an approximately regular network of bile canaliculi within 1 day (Figure 4). Within 3–6 days, clusters of 5–10 cells were usually observed, with fully polarized hepatocytes forming a canalicular network (Figure 4).
Treatment of primary mouse hepatocytes in 3D collagen sandwiches with either toxin (ethanol) or cytoskeleton-altering drugs (e.g., blebbistatin, okadaic acid) resulted in changes in the hepatocyte cytoskeleton, canaliculi width, shape, and number of bile canaliculi illustrated by immunolabeling with an antibody to keratin 8 (the most abundant keratin in hepatocytes), phalloidin (visualizing F-actin), and antibody to tight junction protein zonula occludens-1 (ZO-1; Figure 5).
Ethanol treatment had only a mild effect on organization of keratin 8; however, it increased the tortuosity (as seen from F-actin staining) and distribution of bile canalicular widths (Figure 5). The signal intensity of ZO-1 staining was decreased in ethanol-treated bile canaliculi compared to untreated controls, suggesting a loss of tight junctions after ethanol treatment. The inhibition of actomyosin contractility with blebbistatin significantly affected the shape and number of bile canaliculi. The regular canalicular network was reorganized, compared to untreated hepatocytes, into disorderly shaped bile canaliculi with an increased incidence of thick rounded bile canaliculi instead of thin long ones (as seen in the histogram of canalicular widths).
Additionally, treatment with okadaic acid (OA) inhibiting phosphatases strongly affected the physical properties of keratins, as previously shown8,17. OA changes the solubility of keratin filaments; thus, the treatment resulted in profound reorganization of the keratin meshwork, which collapsed into large perinuclear aggregates. Both F-actin and tight junction protein ZO-1 were not localized into any particular structures, suggesting almost complete disappearance of organized bile canaliculi and a complete loss of hepatocyte polarity. The remaining bile canaliculi were significantly narrowed compared to untreated controls, as seen in the canalicular width histogram (Figure 5).
To correlate microscopic observations of changes in the hepatocyte cytoskeleton with the hepatocellular biochemical response to treatment, the protocol also measured levels of alanine aminotransferase (ALT) and aspartate transaminase (AST) (two liver enzymes that are commonly used as hepatocellular injury markers) in supernatant from the 3D collagen sandwiches (Figure 6)18. Ethanol treatment significantly elevated the levels of both ALT and AST, suggesting severe hepatocellular injury. Blebbistatin treatment did not lead to any considerable changes in both ALT and AST levels compared to okadaic acid treatment, which triggered mild biochemical changes with increased levels of ALT, but no change in levels of AST. Thus, biochemical markers of hepatocellular injury measured in vitro from hepatocyte supernatant correlate with the cytoskeletal changes observed by immunostaining.

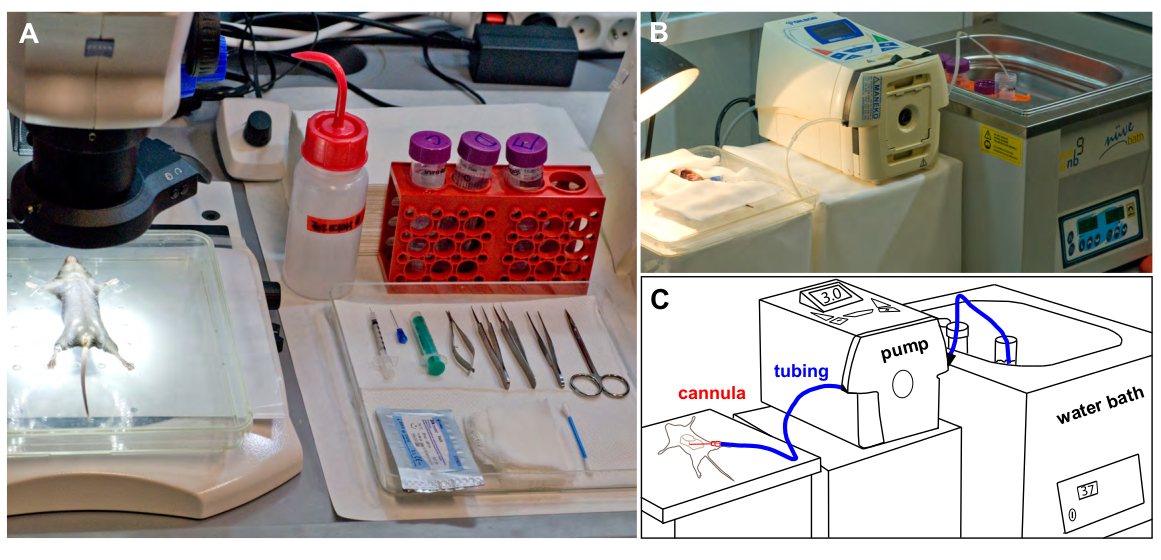
Figure 1: Experimental set up. (A) Mouse fixed in a supine position, is placed under a dissecting microscope before the surgery. All surgical instruments required are placed on a tray. (B) The perfusion suite during mouse liver perfusion showing silicone tubing connecting the reservoir with warm buffer and the perfused mouse. (C) Schematic representation of B. Please click here to view a larger version of this figure.
{kind=link}

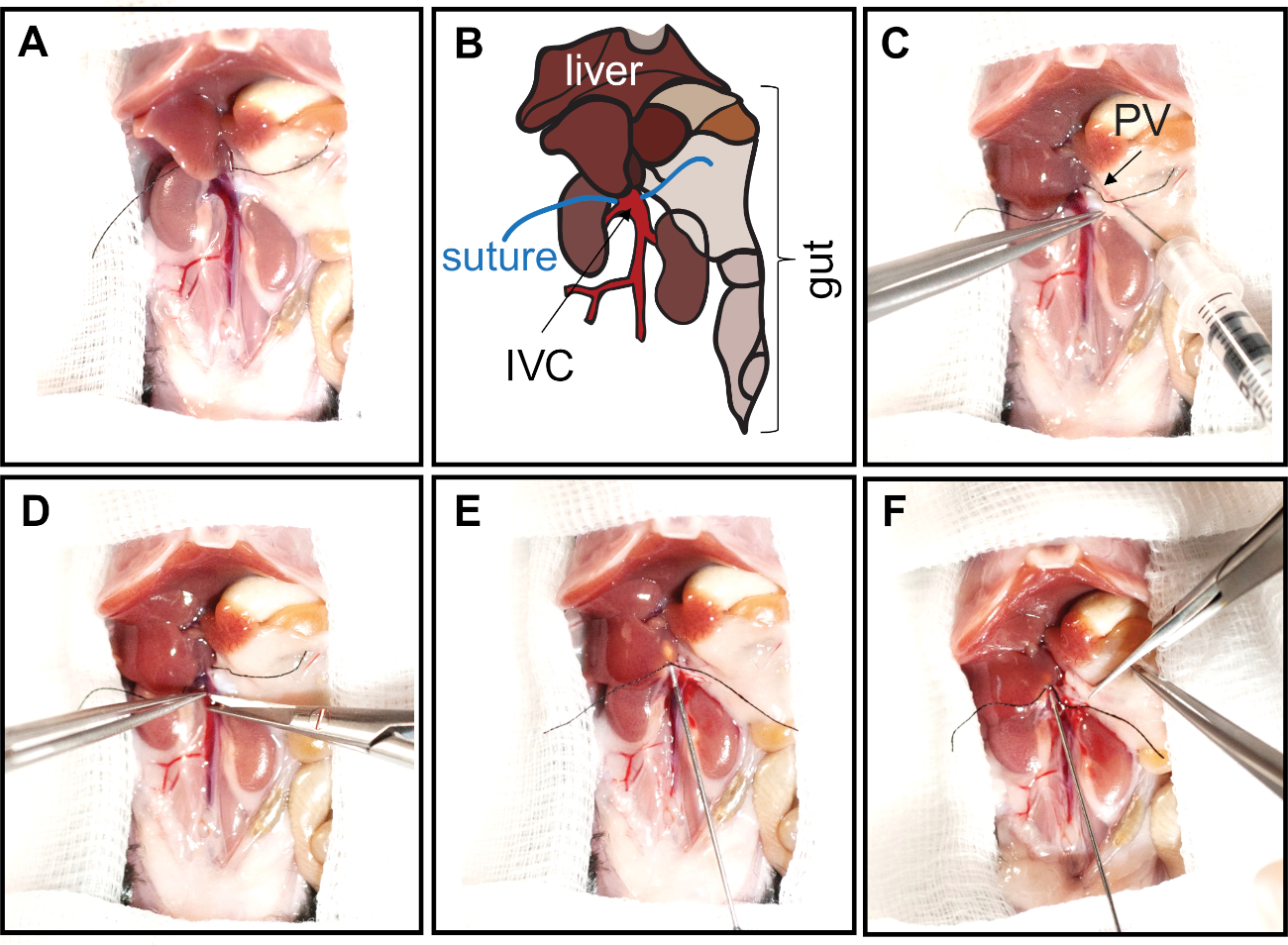
Figure 2: Opening of the abdominal cavity and cannulation of the IVC. The abdomen is opened with a V-shape incision from the pubic area to front legs. The skin is folded over the chest to expose and enlarge the abdominal cavity. To expose the IVC, the intestines and colon are carefully moved caudally. (A, B) Prior to cannulation of the IVC, liver lobes should be repositioned by pressing them upwards to the diaphragm with a PBS-wetted cotton swab. The IVC is then carefully separated from surrounding tissues, and a silk suture is placed around the IVC in close proximity of the liver. Panel B represents schematics of the abdominal cavity shown in panel A. The liver lobes, gut, inferior vena cava (IVC, red), and sutures are indicated. (C) Heparin is injected into the portal vein (PV, arrow) with an insulin syringe (30 G needle bent at 45° angle). (D) To cannulate the liver, the IVC is incised directly next to the liver (below the suture). (E) The cannula is inserted and secured with sutures by tying two surgical knots. (F) The portal vein is fully cut to allow free buffer outflow, preventing liver expansion. Please click here to view a larger version of this figure.
{kind=link}

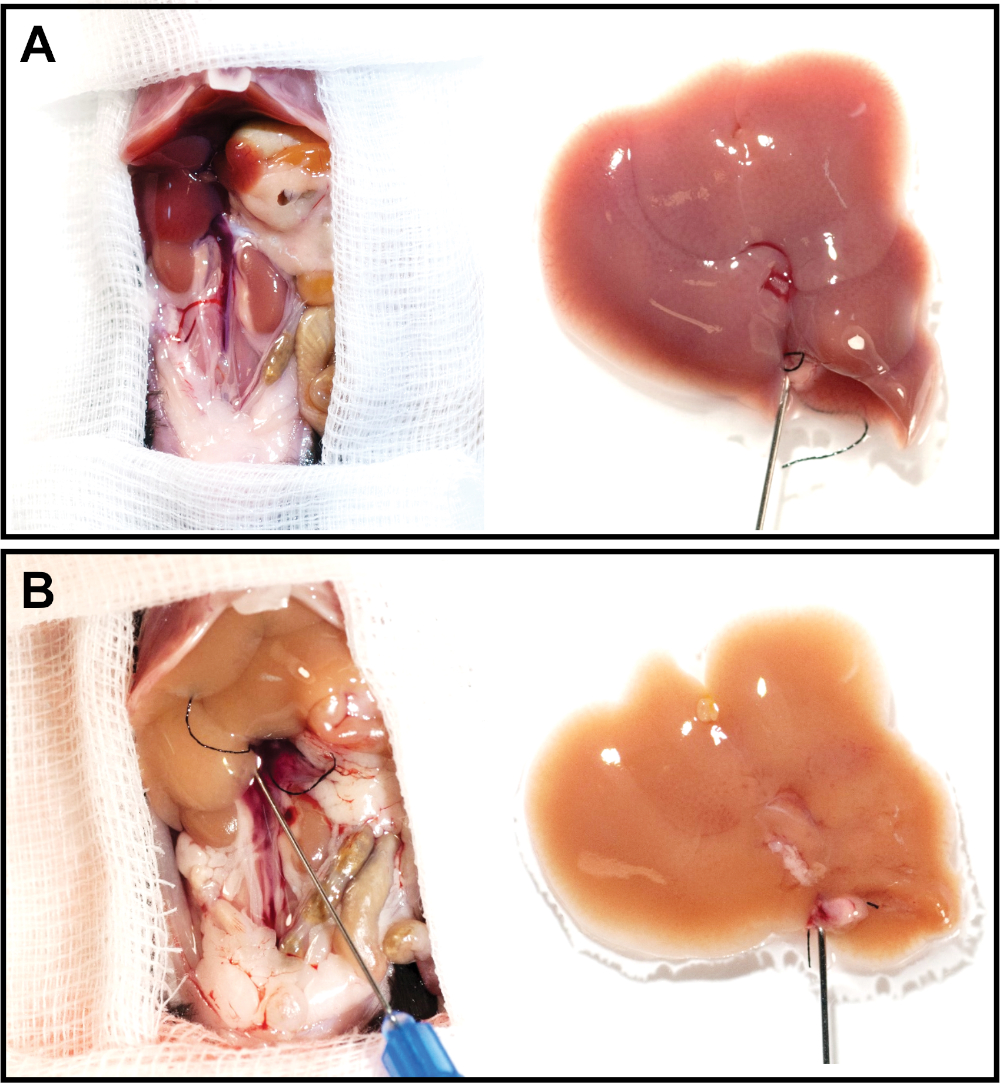
Figure 3: Representative liver images before and after perfusion. (A, B) The cannulated liver was resected and perfused for 12 min at a flow rate of 2.5 mL/min. Note the significantly discolored liver after perfusion (B) compared to the freshly resected liver (A). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: 3D collagen sandwich culture of primary mouse hepatocytes. Representative bright-field images of mouse primary hepatocytes cultured for 1, 2, and 3 days in 3D conditions. It should be noted that larger clusters of highly organized cells are formed after 3 days in culture. Boxed areas show ~3x magnified images. Arrowheads indicate the bile canaliculi. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Evaluation of the morphological response to toxic stress by immunofluorescent microscopy. Primary mouse hepatocytes cultured in 3D collagen sandwiches were treated with toxins (ethanol, blebbistatin, or okadaic acid) on day 3 of culture. Fixed cells were stained to visualize cytoskeletal components: keratin 8 (green), F-actin (red), and zonula occludens-1 (ZO-1, magenta) by immunofluorescence. The toxic treatment led to disorganization of visualized cytoskeletal components, and it reduced the number and increased the tortuosity of bile canaliculi. Canalicular widths were measured in both untreated and treated hepatocytes and are depicted as histograms of widths distribution. Arrowheads indicate the bile canaliculi. Scale bar = 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Biochemical analysis of the response of 3D hepatocyte collagen sandwiches to toxic injury in vitro. ALT and AST, well-established markers of hepatocellular injury, were measured in supernatant from 3D hepatocyte collagen sandwiches treated with toxins (ethanol, blebbistatin, and okadaic acid). ALT and AST were elevated in treated cells compared to untreated ones. Data are reported as arithmetic means ± SEM. Please click here to view a larger version of this figure.
{kind=link}
| Stock Solution A (10x) | |
| Reagent | Final concentration (g/liter) |
| NaCl | 80 |
| KCl | 4 |
| MgSO4·7H2O | 1.97 |
| Na2HPO4·2H2O | 0.598 |
| KH2PO4 | 0.6 |
Table 1: Stock solution A recipe.
| Stock Solution B (10x) | |
| Reagent | Final concentration (g/liter) |
| NaCl | 69 |
| KCl | 3.6 |
| KH2PO4 | 1.30 |
| MgSO4·7H2O | 2.94 |
| CaCl2 | 2.772 |
Table 2: Stock solution B recipe.
| Solution C | |
| Reagent | |
| Stock Solution A (10x) | 5 mL |
| NaHCO3 | 0.1094 g |
| EGTA | 0.0095 g |
| dH2O | to 50 mL |
Table 3: Stock solution C recipe.
| Solution D | |
| Reagent | |
| Stock solution A (10x) | 3 mL |
| NaHCO3 | 0.065 g |
| CaCl2 | 0.0125 g |
| dH2O | to 30 mL |
Table 4: Stock solution D recipe.
| Solution E | |
| Reagent | |
| Stock solution B (10x) | 5 mL |
| NaHCO3 | 0.1 g |
| glucose | 0.045 g |
| dH2O | to 50 mL |
Table 5: Stock solution E recipe.
Discussion
The use of mouse primary hepatocyte cultures is important for in vitro studies to better understand the signaling processes involved in the establishment of hepatocyte polarization, proper canalicular structure formation, and bile secretion. The challenges in isolation and long-term culture of mouse primary hepatocytes in 2D culture have driven the invention of several technical approaches with increased isolation effectivity and longevity of isolated cells, each with several advantages and disadvantages. It is now widely accepted that 2D cultures of primary hepatocytes mimic only limited number of attributes of liver biology for a short period of time. Thus, 3D cultivation in a collagen sandwich arrangement is widely replacing the 2D conditions, particularly when focused on function of the cytoskeleton in liver biology (e.g., toxic drug effects or spatial organization of bile transport).
Since the 1980s, several protocols for isolation of mouse hepatocytes with various modifications have been described. The two-step collagenase perfusion approach has become widely used in many laboratories. The addition of gradient centrifugation into the isolation protocol allows the removal of dead cells19,20 and significantly increases the number of viable cells (here, routinely to ~93%). Even though this step extends the handling time of the cells and results in reduced cell numbers21, this step is viewed as necessary in the 3D collagen sandwich culture for proper bile canalicular network formation. Additionally, it is more important to proceed quickly and accurately during perfusion steps, which shortens the time the cells are handled.
Other important factors that increase the viability of the cells and their ability to form canalicular networks in 3D is the usage of freshly prepared solutions and avoidance of bubbles during perfusion. Therefore, solutions should be prepared on the day of mouse hepatocyte isolation, and the peristaltic pump and tubing should be checked when changing solutions. If the protocol is closely followed, the isolation of primary hepatocytes should be successful with a high yield of viable cells.
Another critical factor in long-term 3D hepatocyte culture is the initial source of primary hepatocytes that are used. It is important to use animals that are 8–12 weeks old, which serve as optimal donors of hepatocytes. The use of hepatocytes from older animals was not as successful in long-term culture, as these hepatocytes changed their morphology more often, depolarized, and ceased to form canalicular networks. Also, plating hepatocytes on properly neutralized collagen gel formed from relatively highly concentrated solution is a vital step. In most protocols, concentrations of about 1 mg/mL are used. After many optimizations, a concentration of 1.5 mg/mL is optimal for long-term hepatocyte cultivation and provides highly organized hepatocytes with formed bile canaliculi.
This easy-to-follow protocol allows long-term cultivation of primary mouse hepatocytes. Representative results demonstrate a broad spectrum of use for 3D cultured primary mouse hepatocytes when studying the role of cytoskeletal components in bile canaliculi formation.
Acknowledgements
This work was supported by the Grant Agency of the Czech Republic (18-02699S); the Grant Agency of the Ministry of Health of the Czech Republic (17-31538A); the Institutional Research Project of the Czech Academy of Sciences (RVO 68378050) and MEYS CR projects (LQ1604 NPU II, LTC17063, LM2015040, OP RDI CZ.1.05/2.1.00/19.0395, and OP RDE CZ.02.1.01/0.0/0.0/16_013/0001775); Charles University (personal stipend to K.K.), and an Operational Program Prague–Competitiveness project (CZ.2.16/3.1.00/21547). We acknowledge the Light Microscopy Core Facility, IMG CAS, Prague, Czech Republic (supported MEYS CR projects LM2015062 and LO1419) for support with the microscopy imaging presented.
Materials
| Name | Company | Catalog Number | Comments |
| 35 mm TC-treated culture dish | Corning | 430165 | |
| 50 mL Centrifuge tubes, SuperClear, Ultra High Performance | VWR | 525-0156 | |
| 70 µm nylon cell strainer | Biologix | 15-1070 | |
| Albumin Fraction V | ROTH | 8076.4 | |
| Arteriotomy Cannula (1mm) | Medtronic | 31001 | |
| Calcium Chloride | Sigma-Aldrich | P5655 | |
| Collagen I. from Rat tail (3mg/ml) | Corning | 354249 | |
| Collagenase (from Clostridium Hystolyticum) | Sigma-Aldrich | C5138 | |
| D(+) glucose monohydrate | ROTH | 6780.1 | |
| Dissecting microscope | Zeiss | Stemi 508 | |
| DMEM, high glucose | Sigma-Aldrich | D6429 | |
| EGTA | ROTH | 3054.2 | |
| FBS | Gibco | 10270-106 | |
| Glucagon | Sigma-Aldrich | G-2044 | |
| Glycine | Pufferan | G 05104 | |
| Heparin (5000 U/mL) | Zentiva | 8594739026131 | |
| Hydrocortisone | Sigma-Aldrich | H0888 | |
| Insulin | Sigma-Aldrich | I9278 | |
| Insulin syringe (30G) | BD Medical | 320829 | |
| Magnesium Sulphate Heptahydrate | ROTH | T888.1 | |
| microsurgical forceps | FST | 11252-20 | |
| microsurgical forceps | FST | 11251-35 | |
| microsurgical scissor | FST | 15025-10 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 | |
| Percoll | Sigma-Aldrich | P1644 | |
| Peristaltic Pump Minipuls Evolution | Gilson | F110701, F110705 | |
| Potassium Chloride | ROTH | 6781.1 | |
| Potassium Phosphate monobasic | Sigma-Aldrich | P5655 | |
| ProLong Gold Antifade Mountant | Thermo Fischer Scientific | P10144 | |
| Refrigerated centrifuge | Eppendorf | 5810 R | |
| Round cover glasses, 30 mm, thickness 1.5 | VWR | 630-2124 | |
| Silk braided black | Chirmax | EP 0.5 – USP 7/0 | |
| Sodium Chloride | ROTH | 3957.1 | |
| Sodium Hydrogen Carbonate | Sigma-Aldrich | 30435 | |
| Sodium hydroxide | Sigma-Aldrich | 221465 | |
| Sodium Hydroxide | Sigma-Aldrich | S8045 | |
| Sodium Phosphate Dibasic Dihydrate | Sigma-Aldrich | 30435 | |
| Syringe 2 ml | Chirana | CH002L | |
| Triton X-100 | Sigma-Aldrich | X100 | |
| Tween 20 | Sigma-Aldrich | P1379 | |
| Water bath | Nuve | NB 9 | |
| Whatman membrane filters nylon, pore size 0.2 μm, diam. 47 mm | Sigma-Aldrich | WHA7402004 | |
| Whatman pH indicator papers, pH 6.0-8.1 | GE Healthcare Life Sciences | 2629-990 | |
| Zoletil | Vibrac | VET00083 |
References
- Gissen, P., Arias, I. M. Structural and functional hepatocyte polarity and liver disease. Journal of Hepatology. 63 (4), 1023-1037 (2015).
- LeCluyse, E. L., Fix, J. A., Audus, K. L., Hochman, J. H. Regeneration and maintenance of bile canalicular networks in collagen-sandwiched hepatocytes. Toxicology In Vitro. 14 (2), 117-132 (2000).
- Tsukada, N., Ackerley, C. A., Phillips, M. J. The structure and organization of the bile canalicular cytoskeleton with special reference to actin and actin-binding proteins. Hepatology. 21 (4), 1106-1113 (1995).
- Herr, K. J., et al. Loss of alpha-catenin elicits a cholestatic response and impairs liver regeneration. Scientific Reports. 4, 6835 (2014).
- Yeh, T. H., et al. Liver-specific beta-catenin knockout mice have bile canalicular abnormalities, bile secretory defect, and intrahepatic cholestasis. Hepatology. 52 (4), 1410-1419 (2010).
- Pradhan-Sundd, T., et al. Dual catenin loss in murine liver causes tight junctional deregulation and progressive intrahepatic cholestasis. Hepatology. 67 (6), 2320-2337 (2018).
- Theard, D., Steiner, M., Kalicharan, D., Hoekstra, D., van Ijzendoorn, S. C. Cell polarity development and protein trafficking in hepatocytes lacking E-cadherin/beta-catenin-based adherens junctions. Molecular Biology of the Cell. 18 (6), 2313-2321 (2007).
- Jirouskova, M., et al. Plectin controls biliary tree architecture and stability in cholestasis. Journal of Hepatology. 68 (5), 1006-1017 (2018).
- Fu, D., Wakabayashi, Y., Ido, Y., Lippincott-Schwartz, J., Arias, I. M. Regulation of bile canalicular network formation and maintenance by AMP-activated protein kinase and LKB1. Journal of Cell Science. 123, 3294-3302 (2010).
- Woods, A., et al. LKB1 is required for hepatic bile acid transport and canalicular membrane integrity in mice. Biochemical Journal. 434 (1), 49-60 (2011).
- Porat-Shliom, N., et al. Liver kinase B1 regulates hepatocellular tight junction distribution and function in vivo. Hepatology. 64 (4), 1317-1329 (2016).
- Sarnova, L., Gregor, M. Biliary system architecture: experimental models and visualization techniques. Physiological Research. 66 (3), 383-390 (2017).
- Talamini, M. A., Kappus, B., Hubbard, A. Repolarization of hepatocytes in culture. Hepatology. 25 (1), 167-172 (1997).
- Bhandari, R. N., et al. Liver tissue engineering: a role for co-culture systems in modifying hepatocyte function and viability. Tissue Engineering. 7 (3), 345-357 (2001).
- Godoy, P., et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Archives of Toxicology. 87 (8), 1315 (2013).
- Wilkening, S., Stahl, F., Bader, A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metabolism and Disposition. 31 (8), 1035-1042 (2003).
- Strnad, P., Windoffer, R., Leube, R. E. In vivo detection of cytokeratin filament network breakdown in cells treated with the phosphatase inhibitor okadaic acid. Cell and Tissue Research. 306 (2), 277-293 (2001).
- Chalupsky, K., et al. ADAM10/17-Dependent Release of Soluble c-Met Correlates with Hepatocellular Damage. Folia Biologica. 59 (2), 76-86 (2013).
- Li, W. C., Ralphs, K. L., Tosh, D. Isolation and culture of adult mouse hepatocytes. Methods in Molecular Biology. 633, 185-196 (2010).
- Horner, R., et al. Impact of Percoll purification on isolation of primary human hepatocytes. Scientific Reports. 9 (1), 6542 (2019).
- Severgnini, M., et al. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology. 64 (2), 187-195 (2012).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved