Obtaining High-Quality Transcriptome Data from Cereal Seeds by a Modified Method for Gene Expression Profiling
In This Article
Summary
A method for transcriptome profiling of cereals is presented. The microarray-based gene expression profiling starts with the isolation of high-quality total RNA from cereal grains and continues with the generation of cDNA. After cRNA labelling and microarray hybridization, recommendations are given for signal detection and quality control.
Abstract
The characterization of gene expression is dependent on RNA quality. In germinating, developing and mature cereal seeds, the extraction of high-quality RNA is often hindered by high starch and sugar content. These compounds can reduce both the yield and the quality of the extracted total RNA. The deterioration in quantity and quality of total RNA can subsequently have a significant impact on the downstream transcriptomic analyses, which may not accurately reflect the spatial and/or temporal variation in the gene expression profile of the samples being tested. In this protocol, we describe an optimized method for extraction of total RNA with sufficient quantity and quality to be used for whole transcriptome analysis of cereal grains. The described method is suitable for several downstream applications used for transcriptomic profiling of developing, germinating, and mature cereal seeds. The method of transcriptome profiling using a microarray platform is shown. This method is specifically designed for gene expression profiling of cereals with described genome sequences. The detailed procedure from microarray handling to final quality control is described. This includes cDNA synthesis, cRNA labelling, microarray hybridization, slide scanning, feature extraction, and data quality validation. The data generated by this method can be used to characterize the transcriptome of cereals during germination, in various stages of grain development, or at different biotic or abiotic stress conditions. The results presented here exemplify high-quality transcriptome data amenable for downstream bioinformatics analyses, such as the determination of differentially expressed genes (DEGs), characterisation of gene regulatory networks, and conducting transcriptome-wide association study (TWAS).
Introduction
The transcriptome represents the complete set of ribonucleic acid (RNA) transcripts expressed by the genome of an organism at a given time and in particular environmental and growth conditions. Each cell has its individual transcriptome, which reflects its current physiologic and metabolic state. A collection of cells derived from a similar tissue or organ is used in a typical transcriptome study, but single-cell and spatially-resolved transcriptomics are getting popular1. Transcriptomic analyses start with the extraction of the total RNA from a selected tissue at a specific time point, and in defined growth conditions. For this purpose, we are recommending the use of a newly developed method for the extraction of total RNA from samples high in starch or sugar content, such as cereal seeds2. The comparison of transcriptomes among different samples results in the identification of RNA molecules with different abundance. These RNA molecules are considered as differentially expressed genes (DEGs). The abundance of transcripts derived from specific marker genes can then be used to estimate the developmental status or determine the response of an organism to environmental fluctuations. Genes with no detectable changes in their transcript abundance across the developmental time points under study are often used as reference or housekeeping genes.
The RNA is typically detected and quantified by various methods, such as Northern blotting and quantitative reverse transcriptase polymerase chain reaction (qRT-PCR), but current high-throughput transcriptomics methods rely heavily on nucleic acid hybridization using microarray technology as well as RNA sequencing (RNA-Seq). RNA-Seq is very popular at present because it provides several advantages for high throughput transcriptomic applications as reviewed elsewhere3,4. Although an older technology, gene expression profiling using microarray chips is still widely used because it is a more established technology, which requires less of a background in bioinformatics. Compared to RNA-Seq, the data sets generated from microarray experiments are smaller and easier to analyze. In addition, it is more cost effective, especially if dealing with large sample numbers. In our laboratory, we routinely use transcriptomic analyses using the microarray technology to determine the role of central regulatory hubs that govern the molecular networks and pathways involved in the growth, development and metabolism of cereal grains5,6,7,8,9. We also routinely use it to conduct of genome-wide gene expression profiling studies to obtain a mechanistic understanding of the response of cereal grains to abiotic stresses10, as well as in the conduct of transcriptome-wide association (TWAS) and linkage mapping studies to identify genes responsible for cereal grain quality and nutrition11,12. Other groups have also used the microarray technology in providing development-specific gene expression atlas in barley13, rice14,15,16, sorghum17, and wheat18.
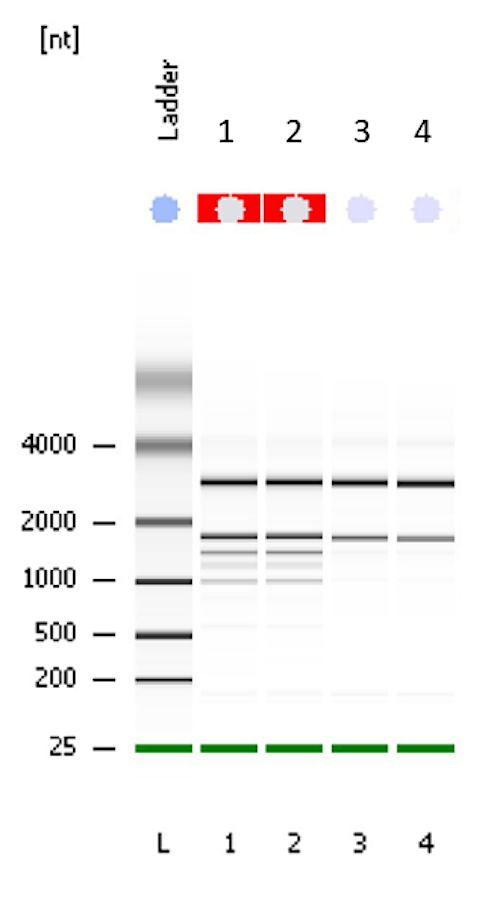
The purpose of this publication is to provide a brief textual summary and a detailed visual description of the method we currently employ in our laboratory for the transcriptomic profiling of cereal grains using an Agilent microarray platform. Please note that other microarray platforms are available, but will not be covered in this method. We begin the protocol by presenting a detailed description of RNA extraction from developing or germinating cereal seeds. Based on our experience, obtaining a high quality and high quantity transcriptome amenable to downstream transcriptomic analyses is often the bottleneck when using cereal seed tissues. We have tried several commercially available RNA extraction kits, but none have provided satisfactory results. Hence, we developed a chemical extraction protocol to obtain a crude RNA extract that is then subjected to column purification using a commercially available kit. Using this method, we routinely and reproducibly obtain high quality RNA2 (Figure 1), which can be used for different downstream applications to generate a transcriptome profile.
Protocol
1. Total RNA extraction and purification
NOTE: Always work under the fume hood as this method involves the use of harmful volatile organic solvents. Pre-warm a microfuge tube of nuclease-free water in 50 °C heat block before commencing the experiment. This nuclease-free water will be used to elute the total RNA from spin column in Step 1.8.
- Separately ground the samples, each with at least three biological replicates, into a fine powder using a sterilized mortar and pestle that are pre-cooled in liquid nitrogen.
- Scoop the powdered samples using a small metal spatula dipped in liquid nitrogen and place the powdered samples in 2.0 mL nuclease-free microfuge tubes, also pre-dipped in liquid nitrogen. Immediately store the samples in ultralow temperature freezer (-80 °C) until the day allocated for batch RNA extraction (STOP POINT).
NOTE: Seed samples can be ground into fine powder with or without dehusking. For rice, we routinely grind the samples without dehusking because the husk contains silica that can aid during the grinding process.
CAUTION: This step must be done fast and under strictly cryogenic conditions. One option for automation is to grind the samples using a cryogenic ball mill to minimize RNA degradation and quality deterioration.
- Scoop the powdered samples using a small metal spatula dipped in liquid nitrogen and place the powdered samples in 2.0 mL nuclease-free microfuge tubes, also pre-dipped in liquid nitrogen. Immediately store the samples in ultralow temperature freezer (-80 °C) until the day allocated for batch RNA extraction (STOP POINT).
- Obtain a crude RNA extract using approximately 200 mg of the original powdered sample. Individually add each sample to a nuclease-free microfuge tube containing 750 µL of RNA Extraction Buffer (100 mM Tris, pH 8.0, 150 mM LiCl, 50 mM EDTA, 1.5% SDS, 1.5% 2-mercaptoethanol) and 500 µL of phenol:chloroform:isoamyl (25:24:1).
- Mix all samples at the same time for 5 min at room temperature using a multi-tube vortex mixer set at high speed. Immediately keep all tubes on ice for two min.
CAUTION: Always work inside the fume hood, especially for all steps involving phenol, chloroform and other organic solvents. Ideally, use a wide fume hood that can accommodate one benchtop centrifuge, vortex mixer, multi-tube vortex mixer, and multi-tube rotary mixer.
- Mix all samples at the same time for 5 min at room temperature using a multi-tube vortex mixer set at high speed. Immediately keep all tubes on ice for two min.
- Separate the crude RNA extract from the debris by centrifugation at 14,000 x g for 10 min. Do all centrifugation steps in the same settings for this section.
- Transfer approximately 800 µL of supernatant to a new 2.0 mL microfuge tube containing 400 µL of 1.2 M NaCl and 700 µL of isopropanol. Mix the solution gently by inversion 5x.
- Precipitate the RNA by incubating in a regular (-20 °C) or ultralow temperature freezer (-80 °C), for at least 1 h (or overnight).
STOP or PAUSE point: just keep the samples in regular -20 °C freezer to pause for a few hours. For overnight or a longer stop point, store the samples in ultralow temperature freezer (-80 °C).
- Obtain a crude RNA pellet by centrifugation at 18,800 x g for 15 min. After discarding the supernatant, purify the crude RNA pellet by column purification using a Mini Kit (Table of Materials).
- Dissolve the pellet in freshly prepared 450 µL of RLT Buffer (prior to use, add 100 µL of β-mercaptoethanol per 100 mL of RLT buffer, prepared fresh daily). Enhance the dissolution of all pellets by mixing all tubes at room temperature in a multi-tube vortex mixer for at least 5 min.
- Purify the dissolved RNA pellet by passing through the purple mini spin column with a 2 mL collection tube. Centrifuge at room temperature for 1 min at 9,000 x g and discard the purple mini spin column.
- Add 0.5 volume of absolute ethanol (~225 µL) to the cleared lysate in the 2 mL collection tube. Mix the solution using the same plastic tip by pipetting up and down a few times.
- Collect the purified RNA by immediately transferring the solution to a pink mini spin column with a 2 mL collection tube. Centrifuge at 9,000 x g for 15 s to collect the RNA onto the silica membrane of the pink min spin column. Discard flow through and wash the RNA with 350 µL of Buffer RW1 by centrifugation at 9,000 x g for 15 s.
- Remove contaminating genomic DNA by adding 80 µL of diluted DNase 1 (50 µL of frozen DNase stock + 350 µL of RDD using the DNase set) directly onto the membrane and digest by incubating at room temperature for at least 15 min (PAUSE POINT).
- Wash off the DNase 1 by adding 350 µL of RW1 and spinning down at 9,000 x g for 15 s. Repeat twice, but this time wash with 500 µL of Buffer RPE. Transfer to a new 2 mL collection tube and spin at 9,000 x g for 2 min to dry.
- Transfer the dried spin column to a new 1.5 mL nuclease-free collection tube. Begin the elution process for the purified RNA extract by adding 50 µL of nuclease-free water (prewarmed at 50 °C) and incubating for 3 min at 50 °C heat block.
- After incubation, elute the total RNA extract by centrifuging at 9,000 x g for 1 min. Immediately place all samples on ice.
- Determine the quantity and quality of the total RNA extract by running appropriate dilutions (usually 1:10) in Nanodrop and BioAnalyzer using their respective manufacturer protocols. RNA extracts should at least have a RIN value of 8.0 and a concentration of 50 ng/µL. Figure 1 shows a typical result for RNA extracts obtained from barley leaf and seeds.
STOP POINT: Store the samples in -80 °C until use.
2. cDNA synthesis followed by cRNA transcription and labelling
NOTE: This step is suitable for processing 24 samples simultaneously. It is suggested that the entire step is conducted continuously in one day, but optional stop and pause points are identified. Be sure to pre-warm three microfuge tube heat blocks at 80 °C, 65 °C, and 37 °C before commencing the steps below. These temperature settings will be changed as noted in the steps below. For instance, the 37 °C heat block will be set to 40 °C after the Spike Mix is prepared (or if it has already been previously prepared). Prepare one-color Spike Mix, T7 Promoter Mix and cDNA master mix using the Low Input Quick Amp Gene Expression Labeling Kit (see Table of Materials) based on the manufacturer's instructions. This preparation is dependent on the amount of starting RNA extracts, which usually range from 10 to 200 ng for total RNA or 5 ng for PolyA RNA. We routinely use 50 ng total RNA as starting material for microarray hybridization2 and for the method that will be described below. In preparing a master mix for 24 samples, add 2 extra reactions to account for pipetting variations. Always use nuclease-free microfuge tubes and nuclease-free water in this step.
- Due to high yield, typically dilute the RNA extract 100-fold to fit within the 50-100 ng range. Based on the RNA quantification results in Step 1.9, make a 1:100 dilution by adding 1 µL of purified RNA extract to 99 µL of nuclease-free water in a 1.5 mL microfuge tube. Determine the actual concentration of this dilution using a Nanodrop.
- Make a second dilution of each total RNA sample in 1.5 mL microfuge tube to make 50 ng of total RNA in a final volume of 1.5 µL. Keep all samples on ice.
- Prepare the Spike Mix from based on manufacturer's instructions. This step is also summarized in Püffeld et al. 20192. Use a 37 °C heat block for this. Store the first and second dilution of the one-color spike mix positive controls in an ultralow temperature freezer (-80 °C) and only go through eight repeated freeze/thaw cycles.
- Prepare the third and fourth dilution fresh daily. Thaw and store the third and fourth dilution of spike mix on ice before use.
NOTE: Convert the temperature of the 37 °C heat block to 40 °C when the Spike Mix had been prepared (or if it was previously prepared).
- Prepare the third and fourth dilution fresh daily. Thaw and store the third and fourth dilution of spike mix on ice before use.
- Prepare the T7 Promoter Mix and store on ice prior to use. The following is sufficient for 24 samples:
20.8 µL of T7 promoter primer
+ 26.0 µL of nuclease-free water
= 46.8 µL of total volume T7 Promoter Mix - Add 1.8 µL of T7 Promoter Primer Mix (Step 2.3) into each microfuge tube containing 1.5 µL of 50 ng RNA sample from Step 2.1. Mix the components properly by pipetting several times. Denature the RNA template-primer mix by incubating in 65 °C heat block for 10 min. Immediately place the microfuge tubes on ice in preparation for the next step.
- While denaturing the template-primer mix (Step 2.4), pre-warm the 5x First Strand Buffer at 80 °C for at least 4 min. Quickly prepare the cDNA synthesis Master Mix from the Low Input Quick Amp Labelling Kit (see Table of Materials) based on manufacturer's instructions. The following is sufficient for 24 reactions:
52.0 µL of Pre-warmed 5x First Strand Buffer (Step 2.5)
+ 26.0 µL of 0.1M DTT
+ 13.0 µL of 10 mM dNTP mix
+ 31.2 µL of RNase Block Mix
= 122.2 µL of total volume cDNA synthesis Master Mix- Thaw each component on ice. Mix each component by gentle pipetting. Keep the Master Mix at room temperature prior to use.
CAUTION: The RNase Block Mix should be immediately placed back in the -20 °C freezer after use.
- Thaw each component on ice. Mix each component by gentle pipetting. Keep the Master Mix at room temperature prior to use.
- Remove the RNA template-primer mix on ice (Step 2.4) and centrifuge briefly at room temperature to spin down all the contents to the bottom of the microfuge tube. Dispense 4.7 µL of the cDNA Master Mix (Step 2.5) to each tube, mixing carefully by pipetting up and down. The total volume when all components are added should be 8.0 µL.
- Synthesize the cDNA for 2 h in 40 °C heat block. During the 2 h incubation, shift the temperature of the 65 °C heat block to 70 °C in preparation for heat inactivation (Step 2.7).
OPTIONAL PAUSE POINT: Store the samples at -80 °C overnight. The experiment can be continued the next day after heat inactivation (Step 2.7).
- Synthesize the cDNA for 2 h in 40 °C heat block. During the 2 h incubation, shift the temperature of the 65 °C heat block to 70 °C in preparation for heat inactivation (Step 2.7).
- Incubate each tube in 70 °C heat block for 15 min to heat-inactivate the RNase Block Mix. Transfer the tubes on ice immediately and incubate for at least 5 min.
- While the samples are on ice for 5 min (Step 2.7), quickly prepare the Transcription Master Mix. All components can be combined at room temperature but thaw components on ice. The following is sufficient for 24 samples:
19.5 µL of nuclease-free water
+ 83.2 µL of 5x Transcription Buffer
+ 15.6 µL of 0.1M DTT
+ 26.0 µL of NTP mix
+ 5.5 µL of T7 RNA Polymerase Blend
+ 6.2 µL of Cyanine 3-CTP (Cy3)
= 156.0 µL of total volume Transcription Master Mix
CAUTION: The T7 RNA Polymerase Blend should be kept in the -20 °C freezer and placed back immediately after use. Moreover, Cy3 is light sensitive. Hence, mixing (Step 2.9) and dispensing (Step 2.10) should be done in low light conditions. For this, we usually turn off any light directly above the lab bench. - As the tubes were subjected to abrupt heating and cooling (Step 2.7), briefly spin down the contents of each sample using a microcentrifuge to collect all the liquid at the bottom of each microfuge tube. Add 6 µL of Transcription Master Mix (Step 2.8) by gently pipetting up and down. The total volume of this reaction should be 16 µL at this stage. Incubate the tubes at 40 °C for 2 h to generate the Cy3-labeled cRNA.
OPTIONAL STOP POINT: The tubes can be stored at -80 °C after transcription. - While generating the cRNA (Step 2.9), set the temperature of the heat block to 55 °C. Add at least one microfuge tube of nuclease-free water in the heat block. This nuclease-free water will be used for the elution of purified cRNA.
- After cRNA transcription and labeling, purify the labelled cRNA using a RNeasy Mini Kit as detailed in Step 3.
3. cRNA Purification
- Purify the labelled cRNA using a RNeasy Mini Kit (see Table of Materials). Prepare the buffers based on manufacturer's instructions. For instance, Buffer RPE is supplied in the kit in concentrated form. Prior to use, add 4 volumes of molecular biology grade absolute ethanol (96-100%).
- Add 84 µL of nuclease-free water to each sample to adjust the total volume to 100 µL. Then add 350 µL of Buffer RLT and 250 µL of absolute ethanol to each tube. Mix thoroughly by pipetting.
- Transfer 700 µL of each mixture onto an the mini spin column with a 2 mL collection tube. Collect the labelled cRNA onto the membrane by centrifugation at 4 °C for 30 s at 7,534 x g. Discard the flow-through liquid.
- Wash each sample in 500 µL of Buffer RPE. Centrifuge and discard flow-through as in the previous step. Repeat this step once, then move to the next step.
- Transfer the mini spin column into a new collection tube. Dry the samples by centrifugation as in Step 3.3.
- Transfer the mini spin column into a nuclease-free 1.5 mL microfuge tube that is supplied with the kit. Elute each labelled cRNA sample by adding 30 µL of nuclease-free water (pre-warmed at 55 °C) directly into the membrane filter. Enhance elution by incubating in 55 °C heat block for 60 s.
- Collect the labelled cRNA by centrifugation at room temperature for 30 s at 7,535 x g. Discard the spin column and close each microfuge tube. Immediately place each tube on ice.
OPTIONAL STOP POINT: The samples can be stored at -80 °C after elution. - Quantify the cRNA with Nanodrop using the microarray feature. Set the sample to RNA-40. Obtain the following values and record in a spreadsheet: cyanine 3 dye concentration (pmol/µL), RNA absorbance ratio (260 nm/280 nm), and cRNA concentration (ng/µL).
STOP POINT: Immediately store the cRNA samples at -80 °C after reading. - Compute the cRNA yield and the specific activity as detailed in Püffeld et al. 20192.
4. Microarray hybridization and scanning
NOTE: This step only takes 3-4 h and hence hybridization of 24 samples can be started after lunch. One operator can comfortably run up to 4 slides (32 samples). The morning of the following day is allocated for washing and scanning of microarray slides. Additional runs can then be performed in the afternoon of the second day. This step is repeated until all samples are hybridized and scanned. It is highly recommended to use color-free, powder-free latex gloves for handling and processing the slides to ensure that the samples are not contaminated with colored pigments that can interfere with the microarray analyses. Aside from commercially available microarrays, the following custom arrays are designed by our group and available for order from Agilent: Order Code 028827 for Barley (Hordeumvulgare), 054269 for Rice (Oryzasativa subs. Japonica), 054270 for Rice (Oryzasativa subs. Indica) and 048923 for Wheat (Triticumaestivum).
- Perform microarray hybridization using the Gene Expression Hybridization Kit (see Table of Materials). Prepare 10x Blocking Agent according to manufacturer's specification2. Aliquot the 10x Blocking Agent into 200 µL portions and store at -20 °C until use. Each aliquot is enough for up to 40 hybridizations and is stable up to 2 months.
- On the day allocated for microarray hybridization, thaw one 200 µL aliquot of 10x Blocking Agent on ice. In addition, pre-warm the hybridization oven to 65 °C and pre-warm a heat block to 60 °C for use during cRNA fragmentation.
- Prepare the Fragmentation Mix for each sample as described by the manufacturer2. In our lab, we routinely use 600 ng of linearly amplified Cy3-labeled cRNA for hybridization in an 8-pack microarray. In this case, the list below summarizes the components of the Fragmentation Mix per sample:
600 ng of linearly amplified cRNA, cyanine 3-labeled
+ 5 µL of 10x Blocking Agent
Adjust volume to 24 µL with nuclease-free water
+ 1 µL of 25x Fragmentation Buffer
= 25 µL of total volume Fragmentation Mix- Mix samples gently using a vortex mixer. Spin all samples briefly using a microcentrifuge.
- Incubate all the samples in the 60 °C heat block for exactly 30 min. Immediately cool each tube on ice for one min then quickly proceed to the next step.
- To fully stop the fragmentation reaction, add 2x GEx Hybridization Buffer HI-RPM to each tube. Mix gently by pipetting up and down, taking great care not to introduce any bubbles during mixing. We routinely use 25 µL of Hybridization Buffer to stop the fragmentation reaction for the 8-pack microarray format.
- Centrifuge all tubes for 1 min at 15,750 x g in ambient room temperature. Quickly place all tubes on ice and load each sample as fast as possible.
- Before leaving the lab for the 17 h overnight incubation, prepare the hybridization assembly2 as detailed in the video.
- CRITICAL STEP: Transfer each hybridization mix and dispense slowly in the center of each gasket well, observing great care not to introduce any bubbles during dispensing. We routinely use 44 µL of hybridization mix for the 8-pack microarray format. Also place 44 µL of 1x Hybridization Buffer in any unused wells.
- Immediately put the microarray slide with correct orientation on top of the gasket slide. This needs to be done very carefully in order not to spill any liquid. Close the hybridization assembly tightly and rotate it to check if no permanent air bubbles have been introduced. All bubbles should be moving within the gasket slide.
- Put the hybridization chamber assembly in the hybridization oven rotator. If using the 2x GEx Hybridization Buffer, set the rotation speed to 10 rpm and hybridize at 65 °C for exactly 17 h.
- Wash hybridized microarrays using the Gene Expression Wash Buffer Kit (see Table of Materials). Prepare the Gene Expression Wash Buffers 1 and 2 based on manufacturer's instructions2. Add 2 mL of 10% Triton X-102 to both buffers (purely optional but highly recommended to reduce the incidence of microarray wash artifacts)2.
- Prepare three wash chamber assemblies as detailed in the previous publication2. Details of each wash chamber is tabulated below (Table 1).
- Aliquot 500 mL of Wash Buffer 1 in a 1 L reagent bottle and leave at ambient room temperature. Prepare an extra 1 L reagent bottle and label with "Wash Buffer 1 Reuse" to save the wash buffer from the first chamber. Additionally, aliquot 500 mL of Wash Buffer 2 in a reagent bottle and incubate in a 37 °C water bath overnight.
NOTE: Do not leave the lab without preparing Steps 4.9 to 4.11. They are necessary for the washing steps the next day. - The next day, prepare the wash buffers as detailed in Table 2. Fill each dish to three fourths of their corresponding buffer. Each wash buffer is good for up to 4-5 slides.
- After exactly 17 h of hybridization, get one hybridization chamber and disassemble on the lab bench lined with lint-free paper as detailed in the previous publication2.
- CRITICAL STEP: Transfer the microarray sandwich to Dish 1. Ensure that the microarray barcode is facing up in a slanted position, and avoid submerging the entire slide in the buffer. Handle each microarray slide from their ends and avoid touching the active side of the slide.
- Separate the two glass slides using a forceps2. Let the gasket slide drop gently into the bottom of Dish 1, while at the same time ensuring that the microarray slide is held firmly for the next step.
- CRITICAL STEP: Slowly lift the microarray slides sideways and immediately transfer into the microarray rack in Dish 2 as detailed in the previous publication2. It is critical that the microarray slides are minimally exposed to air.
- Repeat Steps 4.13 and 4.14 until the eight slides are in the rack. Distribute the microarray slides evenly along the rack. This step can be done by one operator unassisted for up to 4 slides. Attach the rack holder and transfer the entire Dish 2 setup to the first magnetic stirrer. Stir gently for exactly 1 min.
- While stirring for 1 min (Step 4.14), transfer Dish 3 from the mini 37 °C incubator and place on top of the second magnetic stirrer. Gently place Wash Buffer 2 (from the 37 °C water bath) on Dish 3. Avoid bubble formation while pouring.
- After 1 min stirring is done (Step 4.14), gently and slowly lift the slide rack from Dish 2 and transfer to Dish 3. Remove the rack holder and stir for exactly 1 min.
- Slowly and gently lift the slide rack from Dish 3. Be sure to avoid the formation of buffer droplets2. Dry the sides of each slide by gently touching on lint-free paper. Place each blot-dried slide in a slide box and repeat step 4.16 until all microarray slides are in the slide box. Dry for approximately 15 min.
OPTIONAL STOP POINT - Place each microarray slide in a slide holder, ensuring that the barcode is facing up.
NOTE: The ozone levels inside the microarray room should be 50 ppb (100 µg/m3) or less. This is purely optional but is highly recommended: use an Ozone Barrier Slide Cover to avoid ozone-induced degradation of cyanine dyes. - Place the assembled slide holders into the scanning carousel. Load the samples in sequence, based on barcode number. Scan the slides immediately using a microarray scanner (see Table of Materials), as detailed in the previous publication2.
- After the run, subject the data to feature extraction and QC validation, as detailed in the previous publication2.
Representative Results
This method is optimized for extracting cereal seed samples containing significant amounts of contaminating starch or sugars. It is designed to extract total RNA from 24 seed samples per day. It should be conducted continuously in one day, but optional stop and pause points are identified throughout the protocol. Alternatively, the reader can use their preferred RNA extraction kits or manual chemical extraction method. However, based on our previous experience, commercially available plant RNA extraction kits are not appropriate for seeds due to significant amounts of starch, proteins, sugar and/or lipid contamination. In the described method, a chemical extraction of crude RNA is followed by column purification using a commercial RNA extraction kit. This typically provides RNA with higher quality and yield. Figure 1 shows the result of a BioAnalyzer run to test the quality of RNA extraction using the method described here. Results are presented for barley leaf (Samples 1 and 2 representing low starch content samples) and barley seeds (Samples 3 and 4 representing high starch content samples). The RNA integrity (RIN) value for all samples is 10.
Figure 1 shows a representative gel of the purified RNA extract, where the quality was tested using a Bioanalyzer. Samples 1 and 2 are typical results for low starch content samples such as barley leaves, where additional rRNA bands from chloroplasts are evident. Samples 3 and 4 are representative results for high starch content samples such as barley seeds, showing 18S and 28S rRNA. Please note that no automated RIN value can be calculated for green plant tissues such as leaf samples, due to chloroplast rRNA. However, the integrity and high quality can be visually ascertained by to the absence of any degradation products, which typically appears as low molecular smear. The RIN value for high-starch seeds samples such as barley seeds is typically 10 using the protocol described in this paper.
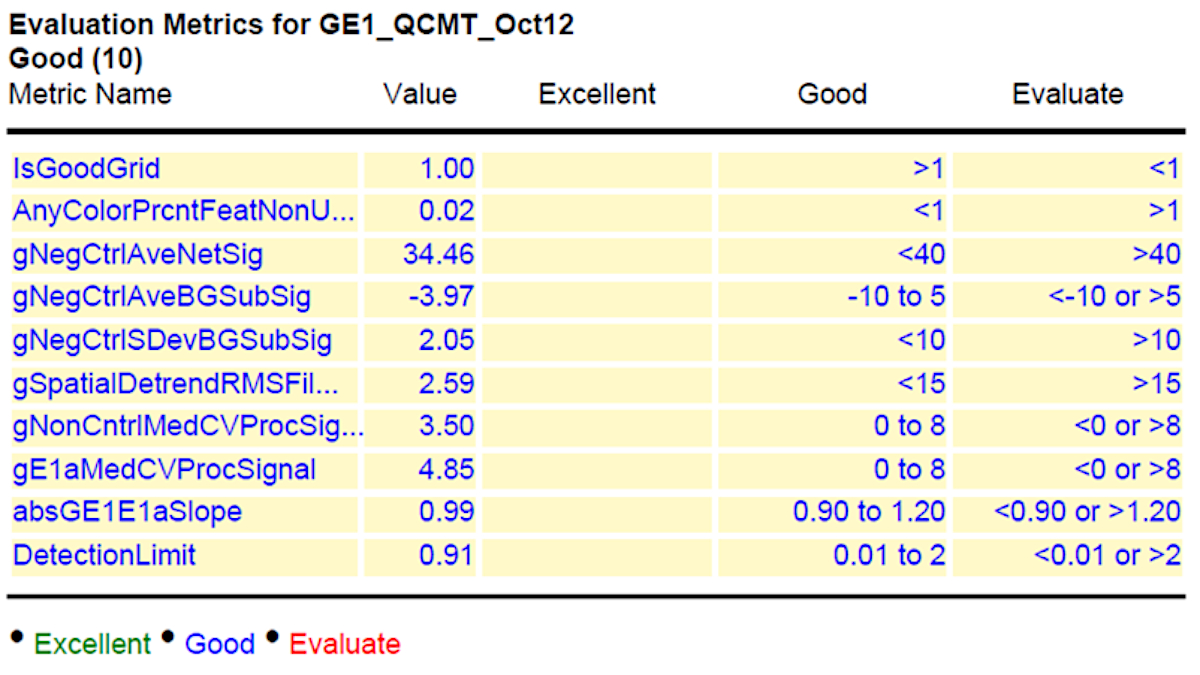
In addition, we also present here a representative data of a time course experiment of two elite barley inbred lines (Sofiara and Victoriana) used for analysis of malting quality19. During the malting process, starch is converted into sugar. Therefore, the samples represent tissues with varying proportions of starch and sugar contents. As the industrial malting process is similar to the germination process, the transcriptomes of two barley lines, differing in their malting quality, were analyzed. RNAs were extracted from germinating seeds at 2, 24, 48, 72, 120, 144 and 196 h after imbibition in biological triplicates. The RNA preparation and hybridization to the customized barley microarray chip were performed as described above. Figure 2 indicates the normalized grid read out from the microarray hybridization given in the quality control (QC) report (Figure 3) and the histogram plot for detected signals. The grid gives the example of derived signals from each corner of the chip, including background and spike-in read out dots used for calibration. The histogram indicates the deviation of detectable dots with respective signal intensities. A successful hybridization gives a broad Gaussian-shaped curve with only minor outliers as shown in the figure. Failed hybridizations can result in a strong shift towards one side ("green monsters").
Lastly, a representative result of the acceptable values is shown in column 4 of Figure 3. To indicate the reliability of the performed experiments, the results are further evaluated using the GeneSpring software. The collected data are presented as principal component analysis (PCA). The PCA integrates the values of selected dots (genes) as vector. The number of evaluated dots that are used can vary from several hundred to the entire chip and is dependent on the software used. Each chip (sample) results in one value (vector) that resulted from the integrated signal intensities for the analyzed dots. Therefore, the relative position in the graph (PCA) indicates the similarity of the samples to each other. The closer the samples are, the more similar they are. Technical replicates should be closer together than biological ones, and biological replicates of a sample should cluster closer together, than samples from different time points, tissues or conditions.

Figure 1: Electrophoresis file run summary obtained after checking the quality of RNA with Bioanalyzer. Samples 1 and 2 are barley leaf tissue as indicated by additional ribosomal bands from chloroplasts. Samples 3 and 4 are barley seed tissue with 18S and 28S rRNA bands shown. A RIN factor is not always calculated for green tissues such as leaf but according to the gel, the quality of RNA is very good. The RIN value for samples 3 and 4 is 10. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: QC Report from successful barley microarray hybridization. A + indicates detected signals on the grid from all corners of the chip. The histogram shows the number of signals categorized according to signal intensity (fluorescence) as logarithm after background subtraction. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Summary of quality control (QC) after hybridization and scanning. The values for the hybridized slide are given in column 2 (value) and the range of acceptable values is shown in column 4. Please click here to view a larger version of this figure.
{kind=link}
| Wash Chamber Assembly | Content and Label | Purpose |
| Dish 1 | Empty, leave on lab bench until next day | Fill with Wash Buffer 1 the next day, used to disassemble the microarray slides |
| Dish 2 | Add one microarray slide rack and a small magnetic stir bar; label with "Wash Buffer 1", leave on lab bench until next day | Used to wash the microarray slides with Wash Buffer 1 the next day |
| Dish 3 | Add one small magnetic stir bar, label with "Wash Buffer 2" and place in a 37 °C mini incubator. | Used to wash the microarray slides with Wash Buffer 2 the next day inside the 37 °C mini incubator |
Table 1: Preparation of the wash chamber assemblies.
| Steps | Dish | Wash Buffer | Temperature | Time |
| Disassembly | 1 | 1 | Ambient | As fast as possible |
| (Step 4.13) | ||||
| First wash | 2 | 1 | Ambient | 1 min |
| (Step 4.14) | ||||
| Second wash | 3 | 2 | 37 °C | 1 min |
| (Step 4.15) |
Table 2: Incubation temperature and time for wash chamber assemblies.
Discussion
The described method provides highly reproducible results for high-yielding and high-quality RNA extracts (Figure 1). Based on our experience, we recommend three biological replicates for one genotype, stage or condition for analysis. To be able to detect statistically meaningful differences, the general abundance of mRNA must be considered. Please note, however, that a starting amount of 200 mg tissue samples is required in triplicates for the RNA extraction step. Hence, for experiments that have a limited amount of samples such as genetically modified plant materials, this method may not be appropriate.
During microarray hybridization, the following steps are most critical: (1) loading the samples to the gasket slide (Step 4.7) and (2) washing the arrays after hybridization (Steps 4.13 and 4.14). Sample loading needs to be done very carefully to avoid bubble formation and liquid spilling. Caution is necessary when putting the microarray slide on the gasket slide containing the loaded samples: the DNA-side (with spotted probes) should be facing down to allow hybridization. For washing the microarrays, the second wash step is especially critical: here, removing the slides from wash buffer 2 needs to be performed very slowly (10 s) to avoid buffer artefacts on the slides, which can interfere with data acquisition and analyses.
In order to receive results from the hybridization experiment that are eligible for down-stream bioinformatics analysis, one should follow a few important criteria. The QC report, which is generated after the performance of the protocol above, gives a good idea of what should be improved, and which data are in a usable condition (Figure 3). The first information received is the visualized grid (Figure 2). Here, one can directly see if all areas for the fluorescence read-out are properly aligned. If there is a visually detectable shift, this will influence all other values (Background, signal intensity, etc.) and the read out of this chip cannot be used. On the overview (Spatial Distribution of all Outliers), one can easily spot any contamination (e.g., dust or hair), which affected the outcome. Dirt can be removed by softly blowing and rescanning the chip. The histogram of signal plot as mentioned above should result in a broad Gauss-shaped curve, with only minor outliers (Figure 2). Depending on the tissue analyzed (Figure 1), this curve can look different and can be appearing to have two peaks, or one predominant peak with a shoulder. This will not influence the quality of data. Only a strong shifted peak, or high signals on the edges indicate problems, such as "green monsters" (too high fluorescence intensities), which cannot be removed by washing and should be reported to the chip manufacturer. Also, the "Spatial Distribution graph for Median Signals" should vary around a common value. This should be in the range between 40 and 100, depending on the general intensity readout of the chip analyzed. Another important quality control is the evaluation of the spike in data: The log-graph for the spike in signals should result in a linear and regular line. Finally, the table of "evaluation metrics" gives a good and reliable view of the received results. Here the manufacturer provides a range of values which can be classified as Excellent, Good and Evaluate (Figure 3). According to our experience, some of the values cannot always be applied for all chips. For rice custom chips, the "nonControl value" was frequently out of range but does not significantly affect further data processing. In addition, the range for "NegControl" might be extended, based on tissue, organism and general output of the Chip to <80. The range for evaluation for value E1 med CV might be extended to <9, instead of <8 according to our experience. Following this, a proper evaluation of all chip read out data is possible. In general, one should always keep in mind that independent biological replicates always give the most reliable result.
The advantage of this technique compared to other methods lies in the fact that it represents a cost-effective, high-throughput method for transcriptional profiling. Moreover, the downstream data analysis pipeline is easier and more established. Despite advantages, there are certain limitations of this technique, such as the number of genes that can be analyzed. The microarray can only facilitate the analysis of genes previously spotted probes on the array. Therefore, this technique is only applicable to plant species with sequenced genomes and fully annotated genes. We envision that the microarray-based transcriptomics will continue to be very useful and relevant not only for gene expression profiling but also for other downstream bioinformatics pipelines such as gene regulatory network analyses and transcriptome-wide association study.
Acknowledgements
This work has been supported under the CGIAR thematic area Global Rice Agri-Food System CRP, RICE, Stress-Tolerant Rice for Africa and South Asia (STRASA) Phase III, and IZN (Interdisciplinary Centre for Crop Plant Research, Halle (Saale), Germany. We thank Mandy Püffeld for her excellent technical assistance and Dr. Isabel Maria Mora-Ramirez for sharing information and experience with the wheat chip. We thank Dr. Rhonda Meyer (RG Heterosis, IPK Gatersleben, Germany) for comments and critically reading the manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| ß-mercaptoethanol | Roth | 4227.3 | Add 300 ul to 20 mL RNA Extraction Buffer immediately prior to use |
| Bioanalyzer 2100 | Agilent Technologies | To determine quality and quantity of RNA extract | |
| Crushed ice maker | Various brands | To keep samples on ice during sample processing | |
| Dewar flask | Various brands | Used as liquid nitrogen container | |
| Ethanol, absolute | Roth | 9065.1 | |
| Gene Expression Hybridization Kit | Agilent Technologies | 5188-5242 | Kit components: 2X Hi-RPM Hybridization Buffer 25X Fragmentation Buffer 10X Gene Expression Blocking Agent |
| Gene Expression Wash buffer Kit | Agilent Technologies | 5188-5325 | Kit components: Gene Expression Wash Buffer 1 Gene Expression Wash Buffer 2 Triton X-102 |
| Heat block | Various brands | It is recommended to have at least one heat block for 1.5 ml tubes and another one for 2.0 ml tubes | |
| Hybridization oven | Agilent | G2545A | Stainless-steel oven designed for microarray hybridization From Sheldon Manufacturing, used to hybridize the sample in microarray overnight. |
| Hybridization gasket slide kit | Agilent | G2534-60014 | |
| iQAir air cleaner | To ensure that the experiment is conducted in low-ozone area | ||
| Isopropanol | Roth | 9866.5 | |
| Lint-free paper, Kimwipes | Kimberly-Clark Professional | KC34155 | |
| Low Input Quick Amp Labeling Kit | Agilent Technologies | 5190-2305 | Kit components: T7 Primer 5x First Strand Buffer 0.1M DTT 10 mM dNTP Mix AffinityScript RNAse Block Mix 5x Transcription Buffer NTP Mix T7 RNA Polymerase Blend Nuclease-free water Cyanine 3-CTP |
| Metal spatula, small | Ensure that the small metal spatula can fit in the microfuge tubes to ensure easy scooping of samples. | ||
| Microarray scanner | Agilent Technologies | Agilent SureScan or Agilent C microarray scanner are recommended | |
| Microfuge tubes, nuclease-free | Various brands | 2.0 mL and 1.5 mL volume | |
| Microcentrifuge | Eppendorf | 5810 R | It is recommended to have at least one ambient and cold temperature microfuge with rotors that can hold 24 each of 1.5 ml and 2.0 ml microfuge tubes |
| Mini incubator | Labnet | I5110-230V | Any small incubator will do. |
| Mortar and pestle | Any small ceramic or marble mortar and pestle that can withstand cryogenic grinding using liquid nitrogen. | ||
| NaCl | Sigma-Aldrich | S7653-1KG | |
| Nanodrop 1000 | Peqlab / Thermofisher | ||
| Nuclease-free water | Biozym | 351900302 | |
| One-Color RNA Spike-In Kit | Agilent | 5188-5282 | Kit components: One color RNA Spike-Mix Dilution Buffer |
| Ozone barrier slide cover kit | Agilent | G2505-60550 | Optional but highly recommended |
| PCR tube, 0.2 mL, RNase-free | Stratagene | Z376426 | |
| Phenol:chloroform:isoamyl mixture (25:24:1) | Roth | A156.2 | |
| Pipette tips, nuclease free, filter tips | Various brands | To accommodate 2, 20, 20, 100, 200 and 1000 ul volumes | |
| Pipettor set | Various brands | For 2, 20, 20, 100, 200 and 1000 ul volumes | |
| RNA Extraction Buffer | 10 mM Tri-HCl pH 8.0 150 mM LiCl 50 mM EDTA 1.5% EDTA 15 ul/mL β-mercaptoethanol | We routinely use Roth or Sigma chemicals; Add β-mercaptoethanol fresh daily | |
| RNase-free DNase set | Qiagen | 79254 | |
| RNeasy Mini Kit | Qiagen | 74104 | Kit components: RNeasy mini spin column (pink) 1.5 ml collection tubes 2 ml collection tubes Buffer RLT Buffer RW Buffer RPE Nuclease-free water |
| RNeasy Plant Mini Kit | Qiagen | 74904 | Kit components: RNeasy mini spin column (pink) QIAshredder spin column (purple) 1.5 ml collection tubes 2 ml collection tubes Buffer RLT Buffer RLC Buffer RW1 Buffer RPE Nuclease-free water |
| Slide holders for DNA microarray scanner | Agilent | G2505-60525 | |
| Slide staining dish with removable rack | DWK Life Sciences 900200 | Fisher Scientific 08-812 | We recommend DWK Life Sciences Wheaton™ Glass 20-Slide Staining Dish with Removable Rack (Complete with dish, cover, and glass slide rack) |
| Sodium chloride | Roth | P029.1 | |
| Ultralow temperature freezer | Various brand | Capable of storing sample at -80 °C | |
| Vortex mixer | Various brands | At least one for single tube vortex-mixer and another one for that can vortex-mix multiple tubes |
References
- Wang, X., et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science. 361 (6400), (2018).
- Püffeld, M., Seiler, C., Kuhlmann, M., Sreenivasulu, N., Butardo, V. M. Analysis of Developing Rice Grain Transcriptome Using the Agilent Microarray Platform. Methods in Molecular Biology. 1892, 277-300 (2019).
- Martin, L., Fei, Z., Giovannoni, J., Rose, J. Catalyzing plant science research with RNA-seq. Frontiers in Plant Science. 4 (66), (2013).
- Hrdlickova, R., Toloue, M., Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdisciplinary Reviews. RNA. 8 (1), (2017).
- Radchuk, V. V., et al. Spatiotemporal profiling of starch biosynthesis and degradation in the developing barley grain. Plant Physiology. 150 (1), 190-204 (2009).
- Sreenivasulu, N. Systems biology of seeds: deciphering the molecular mechanisms of seed storage, dormancy and onset of germination. Plant Cell Reports. 36 (5), 633-635 (2017).
- Sreenivasulu, N., Wobus, U. Seed-development programs: a systems biology-based comparison between dicots and monocots. Annual Review of Plant Biology. 64, 189-217 (2013).
- Butardo, V. M., et al. Systems Genetics Identifies a Novel Regulatory Domain of Amylose Synthesis. Plant Physiology. 173 (1), 887-906 (2017).
- de Guzman, M. K., et al. Investigating glycemic potential of rice by unraveling compositional variations in mature grain and starch mobilization patterns during seed germination. Scientific Reports. 7 (1), 5854 (2017).
- Sreenivasulu, N., Sunkar, R., Wobus, U., Strickert, M. Array platforms and bioinformatics tools for the analysis of plant transcriptome in response to abiotic stress. Methods in Molecular Biology. 639, 71-93 (2010).
- Anacleto, R., et al. Integrating a genome-wide association study with a large-scale transcriptome analysis to predict genetic regions influencing the glycaemic index and texture in rice. Plant Biotechnology Journal. , (2018).
- Pietsch, C., Sreenivasulu, N., Wobus, U., Roder, M. S. Linkage mapping of putative regulator genes of barley grain development characterized by expression profiling. BMC Plant Biology. 9, 4 (2009).
- Druka, A., et al. An atlas of gene expression from seed to seed through barley development. Functional & Integrative Genomics. 6 (3), 202-211 (2006).
- Fujita, M., et al. Rice Expression Atlas In Reproductive Development. Plant and Cell Physiology. 51 (12), 2060-2081 (2010).
- Wang, L., et al. A dynamic gene expression atlas covering the entire life cycle of rice. The Plant Journal. 61 (5), 752-766 (2010).
- Yamakawa, H., Hakata, M. Atlas of rice grain filling-related metabolism under high temperature: Joint analysis of metabolome and transcriptome demonstrated inhibition of starch accumulation and induction of amino acid accumulation. Plant and Cell Physiology. 51 (5), 795-809 (2010).
- Shakoor, N., et al. A Sorghum bicolor expression atlas reveals dynamic genotype-specific expression profiles for vegetative tissues of grain, sweet and bioenergy sorghums. BMC Plant Biology. 14, 35 (2014).
- Yu, Y. L., et al. Transcriptome analysis reveals key differentially expressed genes involved in wheat grain development. Crop Journal. 4 (2), 92-106 (2016).
- Kochevenko, A., et al. Identification of QTL hot spots for malting quality in two elite breeding lines with distinct tolerance to abiotic stress. BMC Plant Biology. 18 (1), 106 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved