Generation of Orthotopic Pancreatic Tumors and Ex vivo Characterization of Tumor-Infiltrating T Cell Cytotoxicity
In This Article
Summary
This protocol describes the surgical generation of orthotopic pancreatic tumors and the rapid digestion of freshly isolated murine pancreatic tumors. Following digestion, viable immune cell populations can be used for further downstream analysis, including ex vivo stimulation of T cells for intracellular cytokine detection by flow cytometry.
Abstract
In vivo models of pancreatic cancer provide invaluable tools for studying disease dynamics, immune infiltration and new therapeutic strategies. The orthotopic murine model can be performed on large cohorts of immunocompetent mice simultaneously, is relatively inexpensive and preserves the cognate tissue microenvironment. The quantification of T cell infiltration and cytotoxic activity within orthotopic tumors provides a useful indicator of an antitumoral response.
This protocol describes the methodology for surgical generation of orthotopic pancreatic tumors by injection of a low number of syngeneic tumor cells resuspended in 5 µL basement membrane directly into the pancreas. Mice bearing orthotopic tumors take approximately 30 days to reach endpoint, at which point tumors can be harvested and processed for characterization of tumor-infiltrating T cell activity. Rapid enzymatic digestion using collagenase and DNase allows a single-cell suspension to be extracted from tumors. The viability and cell surface markers of immune cells extracted from the tumor are preserved; therefore, it is appropriate for multiple downstream applications, including flow-assisted cell sorting of immune cells for culture or RNA extraction, flow cytometry analysis of immune cell populations. Here, we describe the ex vivo stimulation of T cell populations for intracellular cytokine quantification (IFNγ and TNFα) and degranulation activity (CD107a) as a measure of overall cytotoxicity. Whole-tumor digests were stimulated with phorbol myristate acetate and ionomycin for 5 h, in the presence of anti-CD107a antibody in order to upregulate cytokine production and degranulation. The addition of brefeldin A and monensin for the final 4 h was performed to block extracellular transport and maximize cytokine detection. Extra- and intra-cellular staining of cells was then performed for flow cytometry analysis, where the proportion of IFNγ+, TNFα+ and CD107a+ CD4+ and CD8+ T cells was quantified.
This method provides a starting base to perform comprehensive analysis of the tumor microenvironment.
Introduction
This method details, from start-to-finish, the surgical procedure for generating orthotopic pancreatic tumors using a minimal amount of cellular material and the subsequent rapid dissociation of established tumors for comprehensive flow cytometry analysis of immune cell populations, including ex vivo analysis of T cell function.
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive carcinoma with only 8 % of patients surviving 5 years1. As less than 20 % of patients are eligible for surgical resection2, fresh patient samples are not readily accessible for research and thus in vivo models provide essential tools to investigate this disease. There are multiple murine models of PDAC: orthotopic, subcutaneous, transgenic, intravenous and patient-derived xenograft (PDX), extensively described here3. The orthotopic model described here allows the injection of syngeneic PDAC cells into the pancreas of immunocompetent mice. This can be performed in large cohorts of wild-type or mutant mice, and thus provides a cost-effective and consistent model for comparison of therapeutic agents. Importantly, the orthotopic model provides the cognate microenvironment for tumor cell growth and metastasizes in our hands and others4 to clinically relevant sites (e.g., liver), making it more clinically relevant than the subcutaneous or chemically-induced models. Orthotopic tumors display key features of PDAC, such as a strong desmoplastic reaction with an abundance of fibroblasts and extracellular matrix deposition5. Transgenic models of PDAC are the gold-standard of murine model and the most commonly used is the KPC model, which expresses mutant KrasG12D/+ and Trp53R172H/+ under the pancreas-specific Pdx-1-Cre promoter6. Additional KPC and other in vivo PDAC models are reviewed here7. KPC mice spontaneously develop pancreatic tumors with a disease progression that faithfully replicates features of human PDAC6. However, as for all transgenic models, the breeding programme is costly, tumor progression is variable and therefore often requires large cohorts of mice. PDX models use patient-derived tumor cells or pieces which are then grown either orthotopically or more often subcutaneously in immunocompromised mice. Xenograft models provide useful tools for screening therapeutic compounds and account for patient heterogeneity. However, they do not provide a complete immune microenvironment, thus limiting their applications8,9.
Once established, orthotopic tumors typically take around 1 month or more to grow (depending on the cell line used) and form large tumors that can be readily imaged by ultrasound or MRI to track progression and determine treatment efficacy4,5,10. However, once in exponential growth, the last phase of tumor growth can be rapid, so most treatment regimens are commenced relatively early (e.g., 14 days)11,12. The immune system plays a critical role in tumor development, including in PDAC, which is characterized by an immunosuppressive tumor infiltrate with relative paucity of T cells and frequent presence of myeloid cells13. A high presence of T cells in PDAC confers a better prognosis14,15. However, as single agents, immune checkpoint inhibitors that relieve T cell immunosuppression, such as anti-CTLA-416 and anti-PD-L117, have not shown clinical benefit in PDAC patients, most likely because the overall T cell reactivity is very low. However, agents that prime T cell responses, such as anti-CD40, can overcome anti-PD-L1/CTLA-4 resistance18,19 and vaccination with GM-CSF-secreting allogeneic PDAC vaccine (GVAX) can increase the immunogenicity of PDAC tumors20, indicating that enhancing T cell responses forms important therapeutic avenue.
Critical to an antitumoral T cell response is the recognition of tumor-derived antigens via the T-cell receptor (TCR) and the subsequent production of cytotoxic cytokines and granules. While T cell antigen-recognition can be determined by TCR sequencing, this approach is costly and time consuming. However, quantification of tumor-infiltrating T cell subsets provides a good indication of an anti-tumoral response. Further examination of T cell activity ex vivo in terms of degranulation, cytokine production and other cytotoxic factors provides a deeper functional analysis. These assays can be performed on fresh tumor samples and many parameters of T cell function can be measured rapidly by flow cytometry.
CD8+ and CD4+ T cells produce cytokines such IFNγ and TNFα to potentiate an immune response21. IFNɣ induces MHCI upregulation on target cells, induces differentiation and recruitment of immune cells and aids cell death. IFNγ production by CD8+ T cells is well-characterized to be part of an antitumoral response and correlates with tumor regression22,23. TNFα is another proinflammatory cytokine produced by both CD8+ and CD4+ T cells. It enhances TCR-dependent activation and the proliferation of T cells, aiding the anti-tumoral response. Upon TCR engagement, cytotoxic CD8+ T cells can undergo degranulation, where pre-formed secretory lysosomes containing cytotoxic molecules are released into the immunological synapse to cause target-cell degradation21. These molecules include Perforin, a protein that binds to the target cell membrane, forming pores that then disrupt membrane integrity and allow diffusion21 or endocytosis24 of other cytotoxic molecules, such as Granzyme B, directly into the cytoplasm of the target cell. Granzyme B is a protease that enacts the degradation of multiple proteins within the target cell, leading to cell death21. The release of such molecules requires exocytosis of endosomes to the cell surface, where the endosomal marker CD107a (also known as LAMP-1) is transiently incorporated into the cell membrane25.
The measurement of cytokine secretion by T cells requires their isolation by either flow-assisted cell sorting or bead-based separation assays, which cannot be readily performed on large number of samples simultaneously. However, measurement of intracellular cytokines does not require any pre-isolation steps and can be easily be performed on multiple samples at one time, allowing a higher-throughput approach. As cytokines are rapidly secreted by T cells, the intracellular levels can be undetectable and thus the T cell requires stimulation to increase basal cytokine production. To assess antigen-driven cytokine production, the antigen recognized by the TCR must be presented to the T cell by a primed APC in vitro. In cases where the antigen specificity is not known, a broad stimulation approach is required. TCR stimulation can be mimicked using anti-CD3/28 beads that provide both TCR activation and costimulation, which induces cytokine production and proliferation. However, a more cost-effective alternative is the use of PMA and ionomycin, which together broadly activate signaling pathways that lead to the synthesis and release of intracellular cytokines. Specifically, PMA activates protein kinase C (PKC) and ionomycin raises intracellular Ca2+ ions, leading to increased cell signaling. In order to preserve intracellular content of cytokines, this stimulation can be effectively combined with protein-transport inhibitors brefeldin A and monensin, which block proteins in the Golgi and thus prevent extracellular release. The use of PMA/ionomycin is a well-established method for stimulating T cells and there is a strong correlation between extracellular-released and intracellular cytokines26. Stimulation of T cells with PMA and ionomycin also increases lysosome trafficking to cell membrane and thus CD107a becomes transiently integrated on the cell surface before being recycled into the cell. By including an anti-CD107a antibody during the stimulation, it is possible to use it as a marker of degranulation activity25.
This method rapidly digests the tumors to provide a single-cell suspension. At this point, individual populations can be directly stained for flow cytometry or purified by downstream methods: flow-assisted cell sorting or magnetic-bead separation. Preparation of a single-cell suspension for flow cytometry analysis allows high-throughput analysis of multiple immune cell populations and their phenotypic markers, providing an accurate quantification of immune cell number and phenotype.
Finally, the digestion protocol described here prevents cell-surface markers loss and maintains immune cell viability, allowing immune cells to undergo further cell purification steps and culture as required. However, this method has not been tested for deriving epithelial cells from this digestion.
Protocol
Orthotopic pancreatic tumors were generated as previously described10 in accordance with the U.K. Home Office Animal and Scientific Procedures Act 1986 and the European Directive 2010/63/EU. All mice were monitored perioperatively for signs of pain or suffering, including but not limited to weight loss (> 15 % in 72 h or 20 % in any given period), piloerection, narrowing of eyes, raised gait, hunched appearance, as well of signs of wound infection including bleeding, redness and ulceration. Tumor growth was monitored by palpation, and additional clinical signs such as labored breathing, jaundice and cold extremities were also monitored in order to assess if signs of endpoint had been reached. All procedures should be carried out in sterile conditions. All reagents used prior to flow cytometry staining should be prepared in sterile conditions.
1. Preparation of tumor cells for injection
- Take an aliquot of basement membrane from -20 °C and place on ice at 4 °C overnight.
NOTE: Basement membrane concentration may vary from batch to batch; therefore, lot specific basement membrane batches must be tested in vivo to ensure reproducibility. A new batch of basement membrane is thawed on ice at 4 °C overnight then aliquoted in user-defined aliquots, on ice, and then further stored in -20 °C until required. This minimizes the pipetting and freeze-thawing when using basement membrane.- Place sterile PBS at 4 °C overnight to chill.
- Place sterile 200 µL and 1000 µL pipette tips at -20 °C overnight to chill.
- Use tumor cells that are mycoplasma free, grown for at least 2- 10 passages post-thawing and in log-phase of growth prior to harvest. This protocol uses the female murine C57BL/6 KPC-derived cell line: TB32048 provided as a generous gift by the lab of David Tuveson.
- When tumor cells are required for harvest, remove medium from the flask and wash cells twice in PBS (pre-warmed to 37 °C).
- Add 2x trypsin (pre-warmed to 37 °C) to the flask for 10 min at 37 °C (to a T175 flask, add 5 mL).
- After 10 min, add an equal volume of complete medium (10% FBS, 1x penicillin, 1x streptomycin in DMEM) to the flask and dissociate cells by gently tapping the flask and resuspending well in medium.
- Transfer the cells into a tube and centrifuge for 5 min at 300 x g and room temperature (RT).
- Remove the supernatant and resuspend cells in complete medium for cell counting.
- Centrifuge cells again for 5 min at 300 x g and RT, and remove the supernatant.
- Resuspend cells in pre-chilled PBS to achieve a concentration of 1x106 cells/mL.
NOTE: This stock concentration is prepared to achieve a final injection concentration of 1000 cells in 5 µl. We found injection of a lower number of cells in a low injection volume minimized cell leakage and therefore increased reproducibility however, tumour growth may be cell-line dependent therefore users should optimize each cell line.
- Alongside this, place a pre-aliquoted basement membrane aliquot, on ice, into the hood.
- The ratio of the final solution of basement membrane, PBS and tumor cells in PBS prepared for injection is 5:3:2. Therefore to a 500 µL aliquot of basement membrane add 300 µL of pre-chilled PBS using a pre-chilled 1000 µL pipette tip.
- Add the PBS directly to the basement membrane aliquot to minimize pipetting.
- Keep the p1000 set to 300 µL and resuspend the PBS and basement membrane, making sure to keep the tube on ice to preserve the basement membrane in liquid state.
- When finished ejecting all the basement membrane from the p1000 tip, leave the tip in the tube to allow any basement membrane/PBS to come down the pipette tip.
- After 5-10 min, eject more basement membrane from the p1000 tip back into the tube and leave the tube to sit on ice.
- Take 200 µL of resuspended tumor cells in PBS and add directly to the basement membrane using a pre-chilled 200 µL pipette tip.
- Take a fresh pre-chilled p1000 pipette tip, set the pipette at 300 µL and resuspend 30-40 times. A larger pipette tip, set on a low volume, is preferable as basement membrane can travel up the tip and touch the pipette tip filter during resuspension.
- The tumor cells are ready for injection. Keep tumor cell/basement membrane on ice during surgery.
2. Orthotopic injection of tumor cells
- Acclimate the mice in the animal facility for 7 days.
- Around 2 h prior to surgery, shave the left-hand side of abdomen and back, then administer pre-operative analgesic subcutaneously under the scruff of the neck (Buprenorphine at 50-100 µg/kg).
- Prepare surgical field, with a heat mat to lay mouse on and drapes for surrounding equipment and over mouse. Sterilize all surgical tools; prepare enough sets of tools for each mouse.
- Place the mouse in a 5% isoflurane with O2 chamber until unconscious.
- Transfer the mouse, lying on its back, onto a heat mat and maintain anesthesia using a mask, usually at a lower 2-3% isoflurane.
- Confirm deep anesthesia; as identified by loss of the pedal-withdrawal reflex when the hind paw is pinched and monitor breathing rate remains constant.
- Cover the body in drape, with only the shaved portion exposed. Ensure the mouse is securely in the anesthesia mask.
- Using a sterile cotton bud, add iodine solution in a circular motion over the shaved area: starting from the center and circling out to the edge. Repeat process again with fresh cotton bud and iodine.
- Use scalpel to make a 1 cm incision directly above the pancreas/spleen location (upper-left quadrant). Sterile scissors may also be used to make the incision, if preferred.
- Pull the skin apart using forceps. With new forceps, locate the peritoneal wall and use scissors to make another 1 cm incision through the peritoneal wall.
- Extract the pancreas, which may come with the spleen, from the body using the second pair of forceps.
- Gently invert the vial of tumor cells/basement membrane several times to mix.
- Prepare the glass syringe with 5 µL containing 1,000 tumor cells in basement membrane and place on the heat mat for a few seconds to allow it to warm.
NOTE: The brief warming of the syringe will allow the basement membrane to start solidifying, in order to make it easier to inject without leaking. However, this must be kept brief, if left too long the basement membrane will solidify completely and will not be injected. The use of a glass syringe allows a low volume to be precisely injected. - Hold the pancreas at the tail to extend it and insert the needle directly into the center of the pancreas, parallel to the pancreas itself with an effort to avoid visible blood vessels.
NOTE: The center of the pancreas has a large area and it is easiest to inject. However, the head or tail of the pancreas may also be specifically injected if preferred. - Slowly inject 5 µL of basement membrane into the pancreas and hold the needle steady in the pancreas for at least 30 s after injection to allow the basement membrane to solidify and prevent leaking. The basement membrane should be visible as a small clear bubble will have formed; however, it may not be visible.
NOTE: Larger volumes of tumor cells/basement membrane can be injected; however, the exact volume must be tested to ensure leakage does not occur. - Remove the needle from the pancreas and wait to confirm no bleeding has occurred. Gently insert the pancreas back into the abdominal cavity, taking care not to touch the basement membrane bubble.
- Pull the peritoneal wall together and perform a single suture, or two interrupted sutures if needed.
- Pull the two sides of the skin incision together and perform multiple interrupted sutures as needed or insert two surgical clips.
- Administer another subcutaneous injection of buprenorphine into the scruff.
- Transfer the mouse into a heated 37 °C cage for at least 30 min post-surgery to maintain body temperature before transferring back into a fresh cage.
- Prepare a mash diet available in the cage, to ensure rehydration and body weight.
- Re-administer post-operative analgesia as recommended and watch closely for signs of wound opening, pain or infection and weight loss. If using surgical clips, these can be removed 7-10 days later using a clip remover.
- After around 14 days the scar tissue will have healed sufficiently to begin palpating the abdomen. Monitor tumor size closely via palpation until mice reach endpoint.
- At endpoint the mouse is culled via cervical dislocation followed by decapitation. The skin and peritoneal cavity are opened using scissors and the pancreas tumor excised using forceps to hold the tumor, and scissors to remove surrounding tissue.
3. Digestion of pancreatic tumors
- Place the dissected pancreatic tumor, metastatic site tumors, or healthy pancreatic tissue in ice-cold PBS, and store on ice.
- Use forceps to transfer the tumor onto a Petri dish.
- Add 5.0 mL of digestion medium (2 mg/mL Collagenase, 0.025 mg/mL DNase RPMI) into a 50 mL tube; store on ice to prevent enzyme activity commencing.
NOTE: This protocol uses Collagenase Type V, which has an activity of ≥1 units/mg FALGPA and > 125 collagen digestion units (CDU)/mg solid. Collagenase and DNase aliquots can be stored at -20 °C and thawed on ice before use. When both are completely solubilized in sterile RPMI, they can be passed through a 0.2 µm filter to remove contaminants. Collagenase must be completely solubilized before filtering to avoid loss of material. - Take a small aliquot of this solution to cover the tumor on the Petri dish.
- Use sterile scalpel and forceps to cut the tumor into small pieces, roughly less than 3 mm in length.
- Scrape the tumor pieces into the tube and gently invert the tube until all the pieces are submerged in digestion media. Store on ice if other tumor samples need to be prepared in a batch.
- Transfer onto a shaking device for 20 min at 37 °C. Make sure all pieces of tumor are submerged and not stuck to the edge of the tube. If shaking is not possible, then vortex the sample every 5 min to aid digestion.
4. Preparation of single-cell suspension from digested tumor
- Immediately after the digestion step, place the tube on ice to slow enzyme activity.
- Add EDTA to achieve a final concentration of 20 mM and briefly vortex sample to mix. This will further slow enzyme activity.
- Open the tube and rinse any tumor digest off the lid of the tube with fresh RPMI medium.
- Prepare a 70 µm strainer (the µm size of the strainer can be altered as desired) on a 50 mL open tube, on ice.
- Pre-wet the filter with medium.
- Resuspend the digested cells and wash the sides of the tube using a 25 mL stripette, or larger. The wider opening of the stripette is important to allow the thick digest to pass easily.
- Transfer all of the digest, using the 25 mL stripette, onto the strainer.
- Mash the tumor on top of the filter using a 1 mL syringe plunger. Mash only directly up and down to minimize shear stress to cells.
- Continuously wash cells through the strainer with RPMI. Make sure to wash with enough force to push cells through.
- If there is still material to mash, but the RPMI stops flushing through, the strainer will be saturated. Therefore, transfer the sample into a new filter and continue.
NOTE: Eventually only extracellular matrix components will remain in the filter, all single cells should have passed through.
- Centrifuge the tube for 5 min at 300 x g and 4 °C.
- Carefully resuspend the cell pellet in complete RPMI and pass directly through another filter to remove any extracellular matrix or large cell clumps that cannot be adequately resuspended.
- At this point, if no stimulation is required, immediately stain the isolated cells for flow cytometry analysis by skipping to Step 6.1. Alternatively, resuspend them in freezing medium (10% DMSO in FBS) and store at -80 °C followed by long-term storage in liquid nitrogen.
NOTE: The freezing step can allow purification of immune cells at a later date; however, the quantification of immune cell subsets may require optimization to confirm that cell numbers and phenotype is not affected by the freeze/thaw process. The ex vivo T cell stimulation is best performed on fresh tumor samples. At this point the sample can be further purified by bead-based dead-cell removal or immune cell enrichment assays if required.
5. Preparing cells for ex vivo stimulation
- Count the cells to achieve a concentration of 2 x 106/ 100 µL in complete medium (RPMI 10 % FBS, 1X penicillin and 1X streptomycin).
NOTE: The high number of total cells plated ensures that there will be adequate T cells within this sample to analyze. However, the number can be scaled up or down depending on sample availability and the rare nature of T-cell subsets of interest.- Plate 100 µL of cells in a U-bottomed 96-well plate.
- Add 100 µL of complete medium containing a 2x preparation of PMA/ ionomycin (to achieve a final concentration 0.081 µM and 1.34 µM, respectively, as recommended by the manufacturer).
NOTE: If measuring degranulation/exocytosis, also include here a fluorescently conjugated anti-mouse CD107a in the media. A control sample that does not contain CD107a must also be performed. - Place in 37 °C incubator with 5% CO2 for 1 h.
- Add 20 µL of a 10x preparation of brefeldin A and monensin (to achieve a final concentration 1.06 µM and 2.0 µM, respectively (as recommended by the manufacturer) in complete media.
NOTE: Brefeldin A and monensin are protein transport inhibitors and thus block extracellular release of cytokines, etc. permitting their detection by flow cytometry. If measuring cytokine release into the supernatant by ELISA or similar methods – then this step can be skipped. - Place the plate in a 37 °C incubator with 5% CO2 for further 4 h.
6. Extracellular and intracellular staining for flow cytometry
- Remove the plate and resuspend each well to transfer all material to a V-bottomed plate, placed on ice.
NOTE: Epithelial cells, macrophages and other adherent cells may not be fully retrieved by resuspending. However as the downstream analysis is only on T cells, this is not a problem.- 6.1.1 Centrifuge the plate for 5 min at 300 x g and 4 °C (use these conditions for subsequent steps unless stated).
- Remove the supernatant by flicking the plate upside down in one sharp movement.
- Resuspend in 50 µL of a fixable viability dye, prepared in ice-cold PBS. When resuspending, set the pipette to a lower volume to avoid making bubbles.
- Incubate for 20 min at 4 °C, in the dark.
- Wash step: Add 100 µL of FACS buffer, centrifuge and remove the supernatant.
- Resuspend each well with 50 µL of anti-CD16/CD32 (2.5 µg/mL) in FACS buffer (0.5 % BSA, 2.0 mM EDTA in PBS) to block non-specific binding of detection antibodies to Fc receptors.
- Incubate for 15 min at 4 °C, in the dark.
- Add directly to each well a 2x mastermix of fluorochrome-conjugated anti-mouse CD45, CD3, CD4 and CD8 (further extracellular markers can be added as desired) in FACS buffer.
- Incubate for 30 min at 4 °C, in the dark.
- Wash step: Add 100 µL of FACS buffer, centrifuge and remove the supernatant.
- Add 100 µL of 1x Intracellular (IC) fixation buffer and incubate for 30 min at RT, in the dark.
- Prepare the centrifuge at RT.
- Add 100 µL of FACS buffer, centrifuge for 5 min at 300 x g and RT and remove the supernatant. Repeat with 1x permeabilization buffer and centrifuge for 5 min at 300 x g; then remove the supernatant.
- Add 50 µL of 1x mastermix of fluorochrome-conjugated anti-mouse IFNγ, TNFα, and other intracellular markers prepared in 1x permeabilization buffer.
- Incubate for 1 h at RT, in the dark.
- Add 100 µL of permeabilization buffer to wash. Then centrifuge for 5 min at 300 x g and RT and remove the supernatant.
- Add 100 µL of FACS buffer to wash, centrifuge for 5 min at 300 x g and RT and remove the supernatant.
- After this final centrifugation, resuspend cells in a volume compatible for the flow cytometer. It may vary depending on the size of FACS tubes.
- Transfer this volume into appropriate FACS tubes for acquisition.
- Cover from light and store in the fridge and acquire the samples within 24 h.
Representative Results
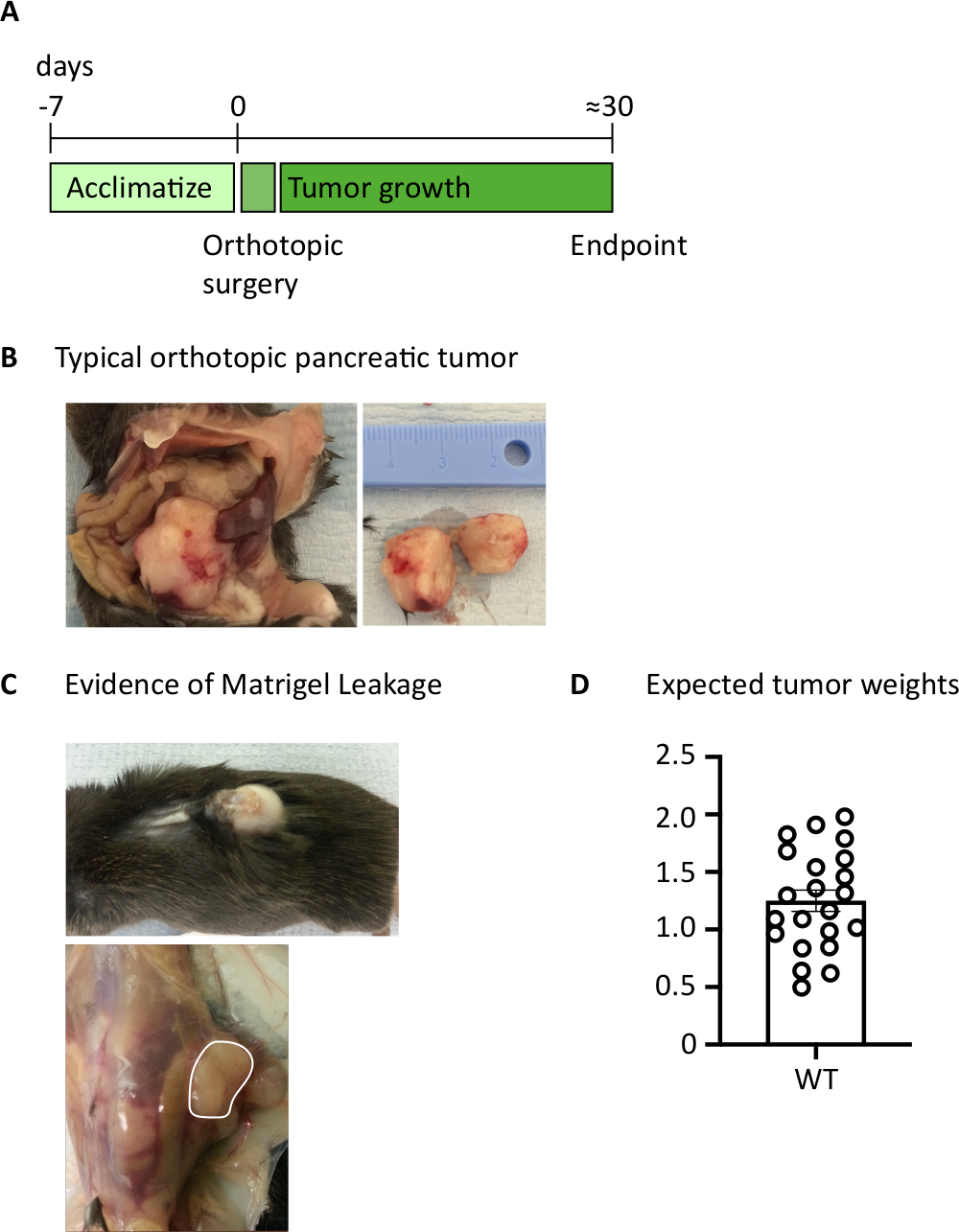
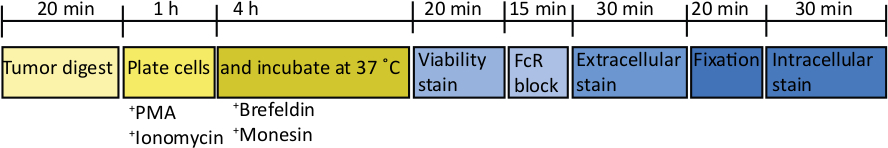
After injecting 1000 TB32048 cells into the pancreas, orthotopic tumors take approximately 30 days to develop (Figure 1A,B). Basement membrane leakage during surgery can cause large tumors to form directly on the peritoneal wall, which are prominently visible through the skin (Figure 1C). We would remove these mice from the study. However, with good surgical skills the incidence of leakage is minimized. Orthotopic tumors harvested at endpoint can grow to a substantial size in C57BL/6 wild-type mice (Figure 1D). Harvested orthotopic tumors require digestion in collagenase/ DNase for 20 min in order to achieve a single-cell suspension (Figure 2). At this point, tumor-derived cells can be plated in a U-bottomed plate at 2 x 106 cells/well. The number of cells plated can be altered depending on the prevalence of T cells within the sample; the cell number can be lowered if T cells are at a high density. Control spleen or lymph node samples can also be plated at this point for stimulation. Each well is stimulated with PMA and ionomycin for 5 h and after 1 h incubation, brefeldin A and monensin are added in order to block extracellular release of cytokines (Figure 2). After the incubation, samples are stained for extracellular epitopes and intracellular cytokines for analysis by flow cytometry (Figure 2).
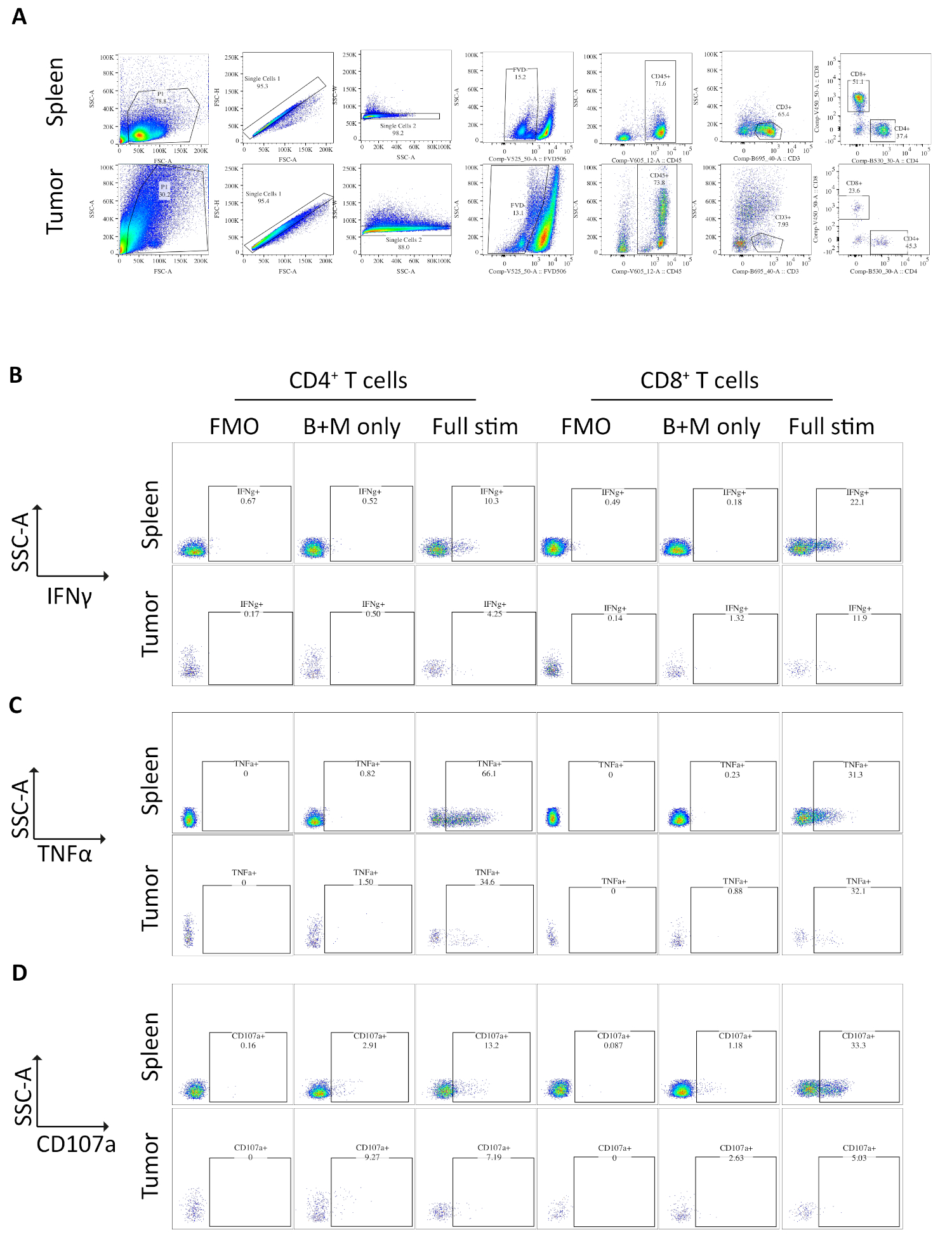
Samples of spleen and tumors from mice bearing orthotopic tumors were analyzed by flow cytometry. The gating strategy used in flow cytometry analysis for the spleen and orthotopic tumors excludes debris using FSC-A, SSC-A, doublets by FSC-A/FSC-H and SSC-A/SSC-W, then dead or apoptotic cells as positive for fixable viability dye (Figure 3A). Immune cells are then gated on as CD45+, and T cells further gated on as CD3+ from which CD4+ and CD8+ subsets are defined (Figure 3A). A fluorescence minus one (FMO) is performed to determine background fluorescence for gating and a brefeldin A/monensin only control is performed to determine basal production of cytokines (Figure 3B-D).
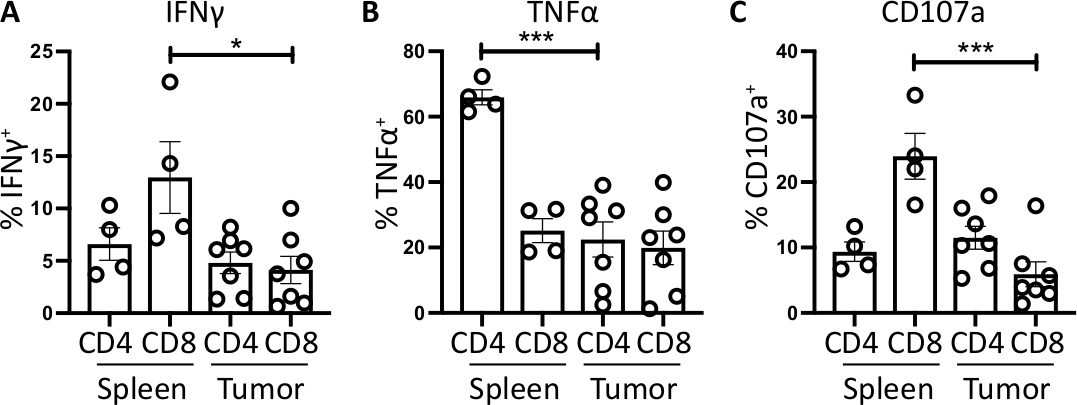
For IFNγ, incubation with brefeldin A/ monensin resulted in no increase in IFNγ over FMO control in both spleen and tumor samples. However, the addition of PMA and ionomycin increased the% of intracellular IFNɣ detectable in both splenic and tumor-derived CD4+ and CD8+ T cells.
Splenic CD4+ and CD8+ T cells, used as a positive control, have a relatively higher IFNγ production than tumor-infiltrating T cell subsets, with an average of 6.60 ± 1.5 % and 12.97 ± 3.4 % compared to 4.81 ± 1.0 % and 4.13 ± 1.3 %, indicating immunosuppression occurs within the tumor (Figure 3B and 4A). Using the same strategy for TNFα, we visualized that a high percentage of splenic CD4+ T cells are positive for intracellular TNFα (65.93 ± 2.3%), compared to tumor-infiltrating CD4+ T cells (22.45 ±5.4%). Splenic and tumor-infiltrating CD8+ T cells produce similar levels of TNFα (25.15 ± 3.7 % and 19.91 ± 5.1 %, respectively) (Figure 3C, Figure 4B).
Finally, CD107a is an endosomal marker that is expressed transiently on the cell surface during the exocytosis of cytotoxic granules and cytokines, as such, it is used as a surrogate marker for cytotoxicity. The benefit of staining for CD107a during the stimulation is that all transiently cell-surface expressed CD107a will be captured by the fluorescent-antibody. The basal levels of CD107a are shown in brefeldin A/monensin only treated cells. For splenic CD8+ T cells, stimulation with PMA/ ionomycin increases the level of CD107a detected, with the strongest upregulation in CD8+ cells which were 23.95 ± 3.5% CD107a+, compared to 5.8 ± 1.9 % in tumor-infiltrating CD8+ cells, indicating splenic CD8+ had a greater rate of degranulation. On the other hand, splenic and tumor-infiltrating CD4+ expressed comparable levels of CD107a 9.37 ± 1.5 % and 11.50 ± 1.8 % (Figure 3D and 4C).
Overall these results highlight that orthotopic tumors can be generated from the injection of a very low number (1,000) of tumor cells into the pancreas. These tumors can be rapidly digested for the isolation of T cells for ex vivo stimulation. Detection of intracellular cytokines is possible and highlights the basal level of immunosuppression of infiltrating T cells, compared to T cells in secondary lymphoid organs.

Figure 1: Generation of orthotopic pancreatic tumors. (A) Schedule of in vivo experiments. (B) The macroscopic appearance of orthotopic tumors within the abdominal cavity (left) and after excision (right) where the tumor shown has been cut in half. (C) Evidence of basement membrane leakage during surgery can cause tumors to develop which are visible through the skin (upper photo) and form on the peritoneal wall (lower photo). (D) Orthotopic pancreatic tumor weights harvested from mice which had reached endpoint (n=22). Each data point represents an individual mouse, bar graph shows mean ± SEM. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Schematic of processing orthotopic tumors for ex vivo T cell stimulation. After harvesting, pancreatic tumors are rapidly digested in Collagenase (2 mg/mL) and DNase (0.025 mg/mL) for 20 min at 37 °C. Following this, cells are resuspended at 2 x 106/mL in complete RPMI media and plated in a U-bottomed plate. A stimulation cocktail of PMA and ionomycin is added for 5 h, at which point the anti-mouse CD107a antibody can also be added to the culture. After 1 h incubation the intracellular transport blockers, brefeldin A and monensin, are added. After ex vivo stimulation the cells are transferred into a v-bottomed plate for staining with the fixable viability dye (in PBS) for 20 min 4 °C. Cells are washed in FACS buffer and incubated in anti-CD16/32 (FcR block) for 15 min (in FACS buffer) and then incubated with extracellular fluorescent-conjugated antibodies for a further 30 min (in FACS buffer). Cells are washed again in FACS buffer and resuspend in intracellular fixation buffer for 20 min. After this, cells are washed once in FACS buffer and once in 1x permeabilization buffer. Cells are resuspended for 1 h at RT in intracellular fluorescent-conjugated antibodies for 1 h (in 1x permeabilization buffer). Cells are washed once in 1x permeabilization buffer and once in FACS buffer before resuspending in FACS buffer for acquisition on the flow cytometer within 24 h. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Flow cytometry analysis of ex vivo stimulated spleen- and tumor-derived T cells. (A) Flow cytometry gating strategy used for spleen (positive control) and orthotopic tumors samples. Cells are discriminated from debris using FSC-A/SSC-A and single cells are further isolated using FSC-A/FSC-H and SSC-A/SSC-W. Dead or apoptotic cells are excluded using the fixable viability dye -FVD506 and immune cells are gated on by CD45+. Following this CD3+ T cells and CD4+ and CD8+ subsets are defined. Data was acquired on a BD Fortessa. (B) The gating strategy used to quantify IFNγ+ CD4+ and CD8+ T cells. A fluorescent minus one (FMO) control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal cytokine production. The fully stimulated sample is then used to calculate the % IFNγ+ T cells. (C) The gating strategy used to quantify TNFα+ CD4+ and CD8+ T cells. An FMO control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal cytokine production. The fully stimulated sample is then used to calculate the % TNFα+ T cells. (D) The gating strategy used to define CD107a+ CD4+ and CD8+ T cells. An FMO control is used on fully stimulated samples (PMA/ionomycin/brefeldin A/monensin) to determine the background fluorescence. A brefeldin A/monensin only control (B+M only) is used to determine the basal degranulation. The fully stimulated sample is then used to calculate the % CD107a+ T cells. All flow cytometry data was analyzed on FlowJo Version 10.6.1. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Quantification of ex vivo spleen- and tumor-derived T cell activity. The proportion of CD4+ and CD8+ T cells positive for (A) IFNγ+ (B) TNFα+ and (C) CD107a+ was quantified in the spleen (n=4) and tumor (n=7) of orthotopic-tumor bearing mice. Each data point represents an individual mouse and error bars display mean ± SEM. Statistical significance was tested using an unpaired t-test where * = p<0.05 and *** = p<0.001. All data was analyzed using Prism 8. The data in this figure has been modified from previously published work10. Please click here to view a larger version of this figure.
{kind=link}
Discussion
In vivo models of pancreatic cancer provide invaluable tools to understand disease progression and assess new therapeutics targets3. The orthotopic model in particular is a cost-effective and reproducible model that can be applied in large cohorts of mice simultaneously4,27. The orthotopic model also provides the cognate microenvironment and intact immune system for tumor growth, making it more appropriate than the subcutaneous and PDX-models. However, we have found that some elements of immune infiltration can differ between the orthotopic model and KPC mice, the gold-standard murine model10. One reason for this could be the accelerated tumor growth seen in the orthotopic model. Further differences in the density of immune cell subsets have been described between the orthotopic and subcutaneous models3,28. Therefore, although the transgenic KPC model is more costly and variable6, key findings should be verified in a small cohort of KPC mice where possible.
The preparation of tumor cells for the orthotopic surgery is a critical step in the protocol. Cells should always be in the log phase of growth and mycoplasma- and infection-free. Orthotopic surgery should be postponed if there are any concerns over tumor cell growth. The use of basement membrane improves tumor incidence rate over injecting cells without it29 and reduces cell leakage and thus peritoneal spread27. However, once suspended in basement membrane, tumor cells should be rapidly injected (within 2 hours) to avoid any cell loss. The number of tumor cells required to generate tumors is likely to be cell-line dependent, and a range of cell numbers should be tested (e.g., from 100 – 100,000) which may also determine the time to reach endpoint. It is likely there will be a margin of error when preparing 1,000 cells per mouse for injection; therefore, if multiple days of surgery are required, treatment of groups should be spread equally across days to control for batch effects. Most surgical steps can be modified depending on preferences; however, care must be taken not to disturb the basement membrane when replacing the pancreas in the abdominal cavity or closing the peritoneal wall. Basement membrane leakage can cause tumor cell growth on the peritoneal wall, which form rapidly and can result in having to sacrifice the animal earlier.
Ideally, pancreatic tumors should be rapidly digested post-harvest and prepared for ex vivo stimulation immediately. However, this might not be possible if there is a large batch of tumors to harvest, in this case tumors should be kept on ice and digested in batches. The type, concentration and length of exposure to digestive enzymes have all been shown to affect a large number of surface molecules on immune cells30,31,32. The digestion time is also deliberately short to limit cell death33. Digested cells can be frozen down in freezing medium for long-term storage; however, some cell loss will occur when thawing. The digestion process can be less than optimal if the tumor pieces are not sufficiently diced before collagenase incubation and this will be evident as hard tumor pieces will remain in the filter after digestion. The collagenase concentration can be lowered if working with healthy pancreas or early-stage tumors; reports on extracting healthy pancreatic ductal cells use significantly lower concentrations34. A high degree of epithelial cell death is to be expected during the digestion; however, immune cells should tolerate the process well. Alternative methods exist to isolate viable epithelial cells for organoid growth35 or to preserve tissue architecture36.
Modifications to the stimulation protocol can be made easily, depending on the desired read-out and immune cell analyzed (e.g., macrophages or B cells). The use of pan-stimulation reagents PMA/ionomycin does not discriminate for TCR-antigen specificity, making it useful when the antigen is not known. However, the production of IFNɣ is closely associated with TCR engagement37 and both IFNɣ and TNFα production are critical in PDAC antitumoral responses38. PMA/ionomycin stimulation reflects the maximal capacity of T cells to produce cytokines, which might or might not be produced by the T cells within the tumor microenvironment. Endogenous production can be measured without the need for stimulation; however, levels may be far lower or undetectable. There are alternative methods to stimulate T cells: anti-CD3/28-coated beads, which also do not require antigen or indeed other immune cell populations. The benefit of this method is allowing quantification of cytokine production by specific T cell subsets without the need for separation methods. Other markers of cytotoxicity (granzyme B and Perforin A), activity (IL-2) or immunosuppression (IL-10) can also be added21. However, high-quality flow cytometry antibodies are not available to detect all cytokines and factors of interest. Therefore, if there are other applications such as ELISA required the stimulation can be performed without the inclusion of brefeldin A/monensin, allowing cytokine release into the supernatant. However, of note, this will permit total cell cytokine release and it will not be possible to determine which cell populations contributed.
IFNγ production is a dominant feature of an antitumoral T cell response, often used as a substitute for TCR-antigen recognition37,38. Other in vivo methods that more accurately define antigen-specific responses utilize tumor cells expressing a known antigen, such as Ovalbumin or SV40. The universal antigen can then be used ex vivo to test T cell recognition or in combination with a TCR-restricted host mouse. Alternatively, where the antigen is unknown, quantification of T cell clonal expansion can be performed by bulk-TCR sequencing, or more recently single-cell TCR sequencing39,40. To fully understand the state of the intratumoral T cell response, markers indicative of exhaustion or inhibitory receptors should also be measured including: CTLA-4, PD-1, LAG-3, TIM3, 2B4. As well as markers of effector T cells (CD44hi, CD62lo) and proliferative activity, Ki67+ or CSFE dilution41,42,43,44. Overall, the orthotopic model provides a useful platform to rapidly test therapeutic strategies, in particular that may modulate the antitumoral T-cell response, that can be then validated on a smaller cohort of transgenic, KPC, mice.
Acknowledgements
We would like to thank the Animal Technician Service and Dr. Alzbeta Talarovicova (Barts Cancer Institute, Queen Mary University of London, London, United Kingdom) for their assistance during the orthotopic surgery. We would also like to thank Dr. Jennifer Morton (Beatson Institute for Cancer Research, Glasgow, United Kingdom) for her guidance in surgical technique, Dr. Cristina Ghirelli (Barts Cancer Institute, Queen Mary University of London, London, United Kingdom) for her advice on tumor digestion and Dr. Fabienne McClanahan (Barts Cancer Institute, Queen Mary University of London, London, United Kingdom) for her advice regarding ex vivo T cell stimulation protocols. We also would like to thank the Medical Research Council (MRC), Pancreatic Cancer Research Fund (PCFR) and Ovarian Cancer Action who funded this research.
Materials
| Name | Company | Catalog Number | Comments |
| 6/0 gauge coated vicryl absorbable sutures | Ethicon | W9500T | |
| 70 μm pore-size cell strainer | Fisher Scientific | 11597522 | |
| 9 mm Clay Adams clips | VetTech Solutions | IN015A | |
| anti-CD107a PE (clone 1D4B) | Biolegend | 121612 | 1:100 to culture media |
| anti-CD16/CD32 | BD Biosciences | 553142 | Use at final dilution 1:200 |
| anti-CD3 PerCP eFluor710 (clone 17A2) | Biolegend | 46-0032 | Use at final dilution 1:50 |
| anti-CD4 FITC (clone GK1.5) | eBioscience | 11-0041 | Use at final dilution 1:100 |
| anti-CD45 Brilliant Violet 605 (clone 30-F11) | Biolegend | 103140 | Use at final dilution 1:200 |
| anti-CD8 Brilliant Violet 421 (clone 53-6.7) | Biolegend | 100738 | Use at final dilution 1:100 |
| anti-IFN-gamma PE/Cy7 (clone XMG1.2) | Biolegend | 505826 | Use at final dilution 1:50 |
| anti-TNF-alpha Alexa Fluor 647 (clone MP6-X) | Biolegend | 506314 | Use at final dilution 1:50 |
| BD Matrigel Basement Membrane Matrix High Concentration | BD Biosciences | 354248 | Aliquot on ice and store in -20 °C |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | A4503 | |
| Cell Stimulation Cocktail (500x) (phorbol 12-myristate 13-acetate (PMA) and ionomycin) | eBioscience | 00-4970-03 | 1x Final concentration PMA 0.081 μM, ionomycin 1.34 μM |
| Clay Adams Autoclip Applier | VetTech Solutions | IN015B | |
| Clay Adams Autoclip remover | VetTech Solutions | IN015B | |
| Collagenase Type V from Clostridium histolyticum | Sigma-Aldrich | C9263 | 2 mg/mL in media |
| Dimethyl sulphoxide (DMSO) | Sigma-Aldrich | D2650-100mL | |
| DMEM High glucose (4.5 g/L) with L-Glutamine | PAA | E15-810 | |
| DNase (Deoxyribonuclease I from bovine pancreas Type II-S) stock 10 mg/mL in 0.15 M NaCl | Sigma-Aldrich | D4513 | Final concentration in digestion media 0.025 mg/mL |
| Fixable Viability Dye 506 (FVD506) | eBioscience | 65-0866 | Use at 1:200 in PBS |
| Foetal calf-serum (FCS) | GE Healthcare | A15-104 | 10% in RPMI |
| Hamilton syringe 700 series, 25 μL volume, 22s gauge needle bevel tip | Fisher Scientific | 10100332 | |
| Intracellular Fixation buffer and Intracellular Permeabilisation Buffer | eBioscience | 88-8824-00 | Dilute permeabilisation buffer to 1x in H2O |
| Penicillin/streptomycin | PAA | 15140122 | 100 units/mL Penicillin, 100 μg/mL Streptomycin |
| Protein transport inhibitor cocktail (500x) (brefeldin A and monesin) | eBioscience | 00-4980-03 | 1x Final concentration Brefeldin A 10.6 μM, monensin 2 μM |
| RPMI-1640 (containing 0.3 g/L Glutamine) | Sigma-Aldrich | R8758 | |
| Surgical Scalpel Blade No.10 | Swann-Morton | 0501 | |
| Trypsin-EDTA Solution 10x | Sigma-Aldrich | 594-18C | Trypsin (0.1%) EDTA (0.4%) final concentration |
| U-bottomed 96 microwell plate | VWR | 734-2080 | |
| Universal Cotton Tipped Applicators - 6 inch x 100 | Medisave | UN982 | |
| V-bottomed 96 microwell plate | VWR | 735-0184 |
References
- Siegel, R. L., Miller, K. D., Jemal, A. Cancer statistics. CA: A Cancer Journal for Clinicians. 68 (1), 7-30 (2018).
- Conroy, T., et al. Current standards and new innovative approaches for treatment of pancreatic cancer. European Journal of Cancer. 57, 10-22 (2016).
- Lee, J. W., Komar, C. A., Bengsch, F., Graham, K., Beatty, G. L. Genetically engineered mouse models of pancreatic cancer: The KPC model (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre), its variants, and their application in immuno-oncology drug discovery. Current Protocols in Pharmacology. 2016, (2016).
- Tseng, W. W., et al. Development of an Orthotopic Model of Invasive Pancreatic Cancer in an Immunocompetent Murine Host. Clinical Cancer Research. 16 (14), 3684-3695 (2010).
- Majumder, K., et al. A Novel Immunocompetent Mouse Model of Pancreatic Cancer with Robust Stroma: a Valuable Tool for Preclinical Evaluation of New Therapies. Journal of Gastrointestinal Surgery. 20 (1), 53-65 (2016).
- Hingorani, S. R., et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 7 (5), 469-483 (2005).
- Herreros-Villanueva, M., Hijona, E., Cosme, A., Bujanda, L. Mouse models of pancreatic cancer. World Journal of Gastroenterology. 18 (12), 1286-1294 (2012).
- Witkiewicz, A. K., et al. Integrated Patient-Derived Models Delineate Individualized Therapeutic Vulnerabilities of Pancreatic Cancer. Cell Reports. , (2016).
- Nicolle, R., et al. Pancreatic Adenocarcinoma Therapeutic Targets Revealed by Tumor-Stroma Cross-Talk Analyses in Patient-Derived Xenografts. Cell Reports. , (2017).
- Spear, S., et al. Discrepancies in the Tumor Microenvironment of Spontaneous and Orthotopic Murine Models of Pancreatic Cancer Uncover a New Immunostimulatory Phenotype for B Cells. Frontiers in Immunology. 10, 542 (2019).
- Zhu, Y., et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Research. , (2014).
- Lee, J. J., Huang, J., England, C. G., McNally, L. R., Frieboes, H. B. Predictive Modeling of In vivo Response to Gemcitabine in Pancreatic Cancer. PLoS Computational Biology. , (2013).
- Clark, C. E., et al. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Research. 67 (19), 9518-9527 (2007).
- Fukunaga, A., et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 28 (1), 26-31 (2004).
- Tewari, N., et al. The presence of tumor-associated lymphocytes confers a good prognosis in pancreatic ductal adenocarcinoma: an immunohistochemical study of tissue microarrays. BMC Cancer. 13 (1), 436 (2013).
- Royal, R. E., et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma. Journal of Immunotherapy. 33 (8), 828-833 (2010).
- Brahmer, J. R., et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. New England Journal of Medicine. 366 (26), 2455-2465 (2012).
- Winograd, R., et al. Induction of T-cell Immunity Overcomes Complete Resistance to PD-1 and CTLA-4 Blockade and Improves Survival in Pancreatic Carcinoma. Cancer Immunology Research. 3 (4), 399-411 (2015).
- Beatty, G. L., et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science. 331 (6024), 1612-1616 (2011).
- Lutz, E. R., et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunology Research. 2 (7), 616-631 (2014).
- Barry, M., Bleackley, R. C. Cytotoxic T lymphocytes: all roads lead to death. Nature Reviews. Immunology. 2 (6), 401-409 (2002).
- Mojic, M., Takeda, K., Hayakawa, Y. The dark side of IFN-γ: Its role in promoting cancer immunoevasion. International Journal of Molecular Sciences. , (2018).
- Castro, F., Cardoso, A. P., Gonçalves, R. M., Serre, K., Oliveira, M. J. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Frontiers in Immunology. , (2018).
- Thiery, J., et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nature Immunology. , (2011).
- Betts, M. R., et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. Journal of Immunological Methods. 281 (1-2), 65-78 (2003).
- Schuerwegh, A. J., De Clerck, L. S., Bridts, C. H., Stevens, W. J. Comparison of intracellular cytokine production with extracellular cytokine levels using two flow cytometric techniques. Cytometry. 55 (1), 52-58 (2003).
- Partecke, L. I., et al. A syngeneic orthotopic murine model of pancreatic adenocarcinoma in the C57/BL6 mouse using the panc02 and 6606PDA cell lines. European Surgical Research. , (2011).
- An, X., et al. Immunophenotyping of Orthotopic Homograft (Syngeneic) of Murine Primary KPC Pancreatic Ductal Adenocarcinoma by Flow Cytometry. Journal of Visualized Experiments. (140), (2018).
- Jiang, Y. J., et al. Establishment of an orthotopic pancreatic cancer mouse model: Cells suspended and injected in Matrigel. World Journal of Gastroenterology. , (2014).
- Autengruber, A., Gereke, M., Hansen, G., Hennig, C., Bruder, D. Impact of enzymatic tissue disintegration on the level of surface molecule expression and immune cell function. European Journal of Microbiology & Immunology. 2 (2), 112-120 (2012).
- Trapecar, M., et al. An Optimized and Validated Method for Isolation and Characterization of Lymphocytes from HIV+ Human Gut Biopsies. AIDS Research and Human Retroviruses. , (2017).
- Liu, Q., et al. Effects of enzymatic digestion, cell culture and preservation conditions on surface CD62L expression of primary murine CD3+CD4+T cells. Biomedical Research (India). 29 (10), 2153-2159 (2018).
- Seaman, S. A., Tannan, S. C., Cao, Y., Peirce, S. M., Lin, K. Y. Differential effects of processing time and duration of collagenase digestion on human and murine fat grafts. Plastic and Reconstructive Surgery. , (2015).
- Huch, M., et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO Journal. , (2013).
- Boj, S. F., et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. , (2015).
- Misra, S., et al. Ex vivo organotypic culture system of precision-cut slices of human pancreatic ductal adenocarcinoma. Scientific Reports. , (2019).
- Moran, A. E., Polesso, F., Weinberg, A. D. Immunotherapy Expands and Maintains the Function of High-Affinity Tumor-Infiltrating CD8 T Cells In Situ. The Journal of Immunology. , (2016).
- Stromnes, I. M., et al. T Cells Engineered against a Native Antigen Can Surmount Immunologic and Physical Barriers to Treat Pancreatic Ductal Adenocarcinoma. Cancer Cell. , (2015).
- Singh, M., et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nature Communications. 10 (1), 3120 (2019).
- Jiang, N., Schonnesen, A. A., Ma, K. Y. Opinion Ushering in Integrated T Cell Repertoire Profiling in Cancer. Trends in Cancer. 5, 85-94 (2019).
- Schietinger, A., et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 45 (2), 389-401 (2016).
- Raghav, S. K., et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. Journal of Clinical Investigation. , (2011).
- Gros, A., et al. PD-1 identifies the patient-specific in filtrating human tumors. The Journal of Clinical Investigation. , (2014).
- Miller, B. C., et al. Subsets of exhausted CD8 + T cells differentially mediate tumor control and respond to checkpoint blockade. Nature Immunology. , (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved