イニチオ熱化学による分子クラスターの大気濃度の計算
要約
弱結合分子クラスターの大気濃度は、遺伝的アルゴリズムと半実証的およびab initio量子化学を利用した多段階構成サンプリング方法論を通じて見られる低エネルギー構造の熱化学的特性から計算することができる。
要約
大気エアロゾルの形成と成長の計算研究では、ガス相電子構造と振動周波数計算から得ることができる正確なギブス自由エネルギー表面が必要です。これらの量は、ジオメトリが潜在的なエネルギーサーフェスの最小値に対応する大気クラスターに対して有効です。最小エネルギー構造のギブス自由エネルギーは温度および圧力のようないろいろな条件の下でクラスターの大気の集中を予測するために使用することができる。遺伝的アルゴリズムベースの構成サンプリングに基づいて構築された計算上の安価な手順を提示し、その後で一連のますます正確なスクリーニング計算を行います。この手順は、半経験的モデルを使用して大規模な構成セットの幾何学を生成および進化させることから始まり、結果として生じる一意の構造を一連の高レベルの ab initio理論レベルで洗練します。最後に、熱力学的補正は、生成された最小エネルギー構造のセットに対して計算され、ギブズの形成、平衡定数、大気濃度の自由エネルギーを計算するために使用されます。この手順を、周囲条件下での水和グリシンクラスターの研究に応用して紹介する。
概要
気候変動の大気研究における最も不確実なパラメータは、雲粒子が入ってくる日射量をどの程度反射させるかです。気体に懸濁した粒子状物質であるエアロゾルは、入射放射線を散乱する雲結縮核(CCN)と呼ばれる雲粒子を形成し、大気1の吸収およびその後の加熱を防止する。この正味冷却効果を詳細に理解するには、エアロゾルのCCNへの成長を理解する必要があり、その結果、小分子クラスターがエアロゾル粒子に成長するのを理解する必要があります。最近の研究では、エアロゾル形成は直径3nm以下の分子クラスターによって開始される2;しかし、このサイズレジームは、実験技術33、44を使用してアクセスすることは困難です。したがって、この実験上の限界を克服するために、計算モデリング手法が望まれている。
以下に説明するモデリング手法を使用して、水和されたクラスターの成長を分析できます。我々は、生物前環境における小さな構成物質からの大きな生体分子の形成における水の役割に関心を持っているので、グリシンによるアプローチを示す。これらの研究課題に対処するために必要な課題とツールは、大気エアロゾルと前,核クラスタ,55、6、7、8、9、10、11、12、13、14、156,7,の研究に関与するものと非常によく似ています。8,910,1112,13,14,15ここでは、単離されたグリシン分子から始まる水和グリシンクラスターを調べ、続いて最大5つの水分子を段階的に添加した。最終目標は、大気圏のGly(H2O)n=0-5クラスターのn=0-5平衡濃度を、海面で室温、相対湿度(RH)100%を計算することです。2
これらのサブナノメートル分子クラスターの少数は、他の蒸気分子を追加するか、既存のクラスター上で凝固することによって、メタスタブルクリティカルクラスター(直径1〜3nm)に成長します。これらの重要なクラスターは、はるかに大きな(最大50〜100 nm)雲凝縮核(CCN)の形成につながる良好な成長プロファイルを有し、雲の降水効率に直接影響を及ぼし、入射光を反射する能力も直接影響を及ぼします。したがって、分子クラスターの熱力学とその平衡分布を理解することは、エアロゾルが地球規模の気候に与える影響をより正確に予測する必要があります。
エアロゾル形成の記述モデルには、分子クラスター形成の正確な熱力学が必要です。分子クラスター形成の正確な熱力学の計算では、最も安定した構成の同定が必要であり、これは、クラスターの潜在的エネルギー表面(PES)16上のグローバルおよび局所的なミニマを見つけることを含む。このプロセスは、構成サンプリングと呼ばれ、分子動力学(MD)17、18、19、20、モンテ,20カルロ18,19(MC)21、22、および遺伝的21,アルゴリズム17,(GA)23、24、25に基づくもの23を含む様々な技術,24を通じて達成することができる。2225
高い理論レベルで大気水和物の構造と熱力学を得るために、長年にわたって異なるプロトコルが開発されてきました。これらのプロトコルは、(i)構成サンプリング法、(ii)構成サンプリングで使用される低レベル法の性質、および(iii)後のステップで結果を洗練するために使用される高レベルのメソッドの階層の選択が異なった。
構成サンプリング法には、化学的直感26、ランダムサンプリング27、28、28分子動力学(MD)29、30、洗面体ホ29,30ッピング(BH)31、および遺伝的アルゴリズム(GA)24、25、32が含まれていた32。24,25,2731これらのサンプリング方法で使用される最も一般的な低レベルの方法は、力場またはPM6、PM7およびSCC-DFTBのような半経験的モデルである。これらはしばしば、ヤコブのはしご33の高いラングからますます大きなベーシスセットとより信頼性の高い機能を持つDFT計算が続きます。場合によっては、MP2、CCSD(T)、コスト効率の高い DLPNO-CCSD(T)34、,35など、より高いレベルの波動関数方式が続きます。
Kildgaardら36は、フィボナッチ球体37の小さな水和または非水和クラスターの周りに水分子を添加し、より大きなクラスターの候補を生成する体系的な方法を開発した。非物理的な候補と冗長な候補は、近接接触しきい値と異なる適合者間のルート平均平方距離に基づいて削除されます。PM6セミ経験的手法とDFTと波関数法の階層を用いた後の最適化は、高い理論レベルで低エネルギー適合者のセットを得るために使用される。
人工的なハチコロニー(ABC)アルゴリズム38は、最近、ABCluster39と呼ばれるプログラムで分子クラスターを研究するためにZhangらによって実施された新しい構成サンプリングアプローチである。Kubeckaら40は、構成サンプリングのためにABClusterを使用し、続いて、緊密結合GFN-xTB半実証法41を用いた低レベルの再最適化を行った。さらに、DFT法を用いて構造とエネルギーを改良し、その後にDLPNO-CCSD(T)を用いた最終的なエネルギーを使用しました。
方法に関係なく、構成サンプリングは、ペスのポイントのランダムまたは非ランダムに生成された分布から始まります。各点は、問題の分子クラスターの特定の幾何学に対応し、サンプリング法によって生成されます。次に、PESの「下り坂」方向に従うことによって、各ポイントに最も近い局所最小値が見つかります。このように見つかったミニマのセットは、少なくともしばらくの間、安定している分子クラスターの幾何学に対応する。ここで、PESの形状と、表面上の各点におけるエネルギーの評価は、より正確な物理的記述がより計算的に高価なエネルギー計算をもたらすシステムの物理的記述に敏感であろう。我々は、特に、サンプリングポイントの初期セットを生成するために25、グローバルな最適化と構成サンプリングの問題42、43、44、4543,44,の42様々なに適用されているOGOLEM 25プログラムで実装されたGAメソッドを使用します。45PESは、MOPAC2016プログラム47に実装されたPM7モデル46で説明する。この組み合わせは、MDおよびMC法に比べてより多様なポイントを生成し、PESのより詳細な記述よりもローカルミニマを迅速に検出するため採用されています。
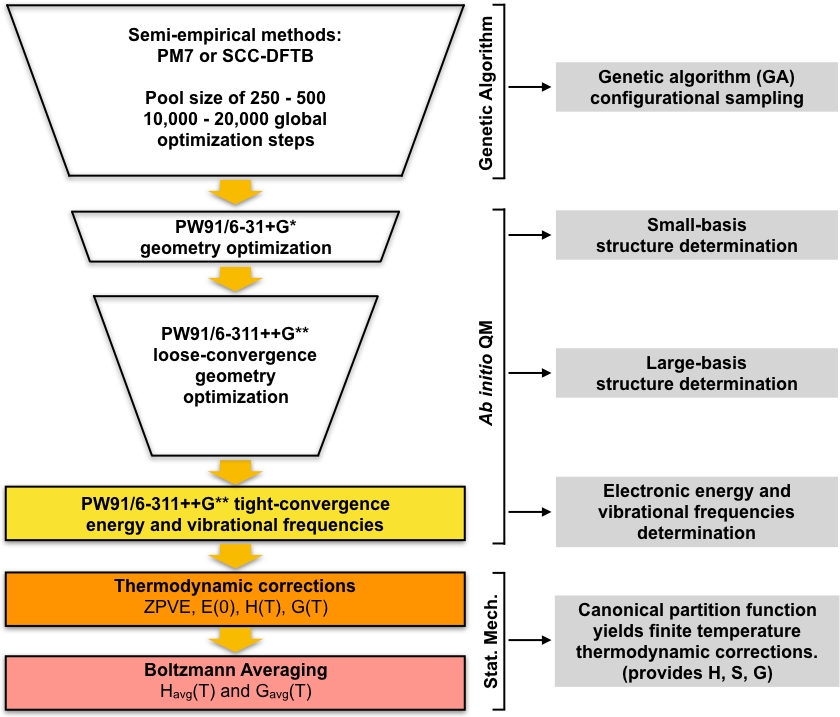
GA 最適化されたローカル ミニマのセットは、一連のスクリーニングステップの開始ジオメトリとして取られ、低位置の最小エネルギーのセットにつながります。プロトコルのこの部分は、密度機能理論(DFT)を使用して、小さな基本セットを使用して、ユニークなGA最適化構造のセットを最適化することから始まります。この最適化のセットは、一般的に、GA最適化半経験的構造と比較して、より詳細にモデル化される一意のローカル最小構造の小さなセットを与えます。次に、より大きな基準セットを使用して、この小さな構造体のセットに対してDFT最適化の別のラウンドが実行されます。繰り返しますが、このステップは一般的に、小さな基礎DFTステップと比較してより詳細にモデル化されるユニークな構造の小さなセットを与えます。その後、固有の構造の最終的なセットは、より緊密な収束に最適化され、調和振動周波数が計算されます。このステップの後、我々は大気中のクラスターの平衡濃度を計算するために必要なすべてを持っています。全体的なアプローチは図1に図示されています。ガウシアン49DFT実装におけるPW9148一般化勾配近似(GGA)交換相関関数と、Pople50ベーシスセット(小ベーシスステップでは6-31+G*、大ベーシスステップでは6-311++G**)の2つのバリエーションを使用します。交換相関機能とベーシスセットのこの特定の組み合わせは、大気クラスター51、52,52のための形成の正確なギブス自由エネルギーを計算する上での以前の成功のために選ばれました。
このプロトコルは、ポータブル バッチ システム53 (PBS)、MOPAC2016 (http://openmopac.net/MOPAC2016.html) 47、OGOLEM (https://www.ogolem.org)25、ガウス 09 (https://gaussian.com)49、および特定のインストール49手順に従ってインストールされた OpenBabel54 (http://openbabel.org/wiki/Main_Page) ソフトウェアを使用して、高性能コンピューティング (HPC) クラスターにアクセスできるものとします。このプロトコルの各ステップでは、社内シェルと Python 2.7 スクリプトのセットも使用しており、ユーザーの$PATH環境変数に含まれるディレクトリに保存する必要があります。上記のすべてのプログラムを実行するために必要なすべての環境モジュールと実行権限も、ユーザーのセッションにロードする必要があります。GA コード (OGOLEM) と準経験コード (MOPAC) によるディスクとメモリの使用量は、最新のコンピュータ リソース標準では非常に小さいものです。OGOLEM/MOPAC の全体的なメモリとディスク使用量は、使用するスレッドの数によって異なり、ほとんどの HPC システムの機能に比べてリソースの使用量は小さくなります。QM法のリソースニーズは、クラスターのサイズと使用される理論のレベルによって異なります。このプロトコルを使用する利点は、理論のレベルを変えて、低エネルギー構造の最終セットを計算できることです。
ユーザーのローカル コンピュータは、わかりやすくするため、"ローカル コンピュータ" と呼ばれ、アクセスできる HPC クラスタは "リモート クラスタ" と呼ばれます。
プロトコル
1. 分離されたグリシンと水の最小エネルギー構造を見つける
注:ここでの目標は2倍です:(i)遺伝的アルゴリズム構成サンプリングで使用するための孤立した水とグリシン分子の最小エネルギー構造を取得し、(ii)大気濃度の計算に使用するためにこれらの分子の気相エネルギーに熱力学的補正を計算することです。
- ローカル コンピュータで、アボガドロの新しいセッションを開きます。
- [ビルド] > [挿入] > [ペプチド] をクリックし、[ペプチドの挿入]ウィンドウからGlyを選択して、視覚化ウィンドウでグリシン モノマーを生成します。
- [拡張機能] > [ガウス]をクリックし、テキスト ボックスの最初の行を編集して '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt (タイト,maxcyc=300) freq' を読み取ります。[生成] をクリックし、入力ファイルをglycine.comとして保存します。
- 分子が55を行うように、分子が大きな立体構造の柔軟性を持っている場合、グローバル最小構造および他の低地の適合体を識別するために立体構造解析を行うことが重要であることに注意してください。OpenBabel54は、さまざまなアルゴリズムとクイックフォースフィールドを利用した堅牢な立体構造検索ツールを提供します。適合者は、GA とその後の計算中にリラックスして相互変換することが許可されますが、それぞれが異なるコンフォーマーから始まる複数の GA 計算を実行する必要がある場合があります。
- ローカル コンピュータで、アボガドロで新しいセッションを開きます。
- [ビルド] > [挿入] > [フラグメント] をクリックし、[フラグメントの挿入] ウィンドウから 「水」 を検索して、水の座標を取得します。
- [拡張機能] > [ガウス]をクリックし、テキスト ボックスの最初の行を編集して '# pw91pw91/6-311++G** int(Acc2E=12,UltraFine) scf(conver=12) opt (タイト,maxcyc=300) freq' を読み取ります。[生成] をクリックし、 [ 入力ファイルをwater.com] として保存します。
- 2 つの.comファイルをリモート クラスターに転送します。リモート クラスタにログインしたら、バッチ送信スクリプトで Gausssian 09 を呼び出して計算を開始します。計算が終了したら、OpenBabel を呼び出して最小エネルギー構造のデカルト座標(.xyzファイル) を抽出します。グリシンの場合、実行するコマンドは次のとおりです。
オバベル -ig09 グリシン.ログ -oxyz > グリシン.xyz
これらの 2 つの.xyzファイルは、次の手順で GA 構成サンプリングで使用されます。
2. Gly(H2O)n=1-5クラスタの遺伝的アルゴリズムベースの構成サンプリング
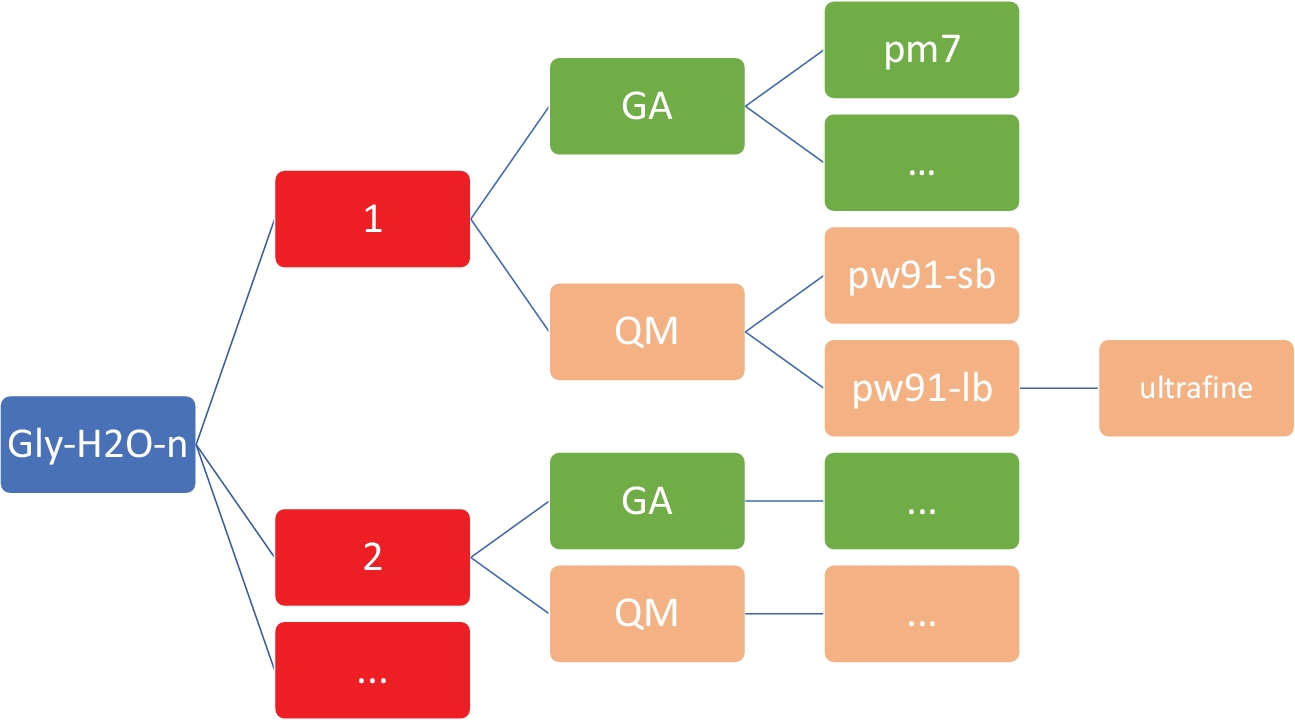
注:ここでの目標は、MOPAC47で実装されたPM746モデルを使用して、低い理論の半経験レベルでGly(H2O)n=1-5のための低エネルギー構造のセットを取得することです。2n=1-5図 2に示すように、作業ディレクトリが正確な構成と構造を持つことが不可欠です。これは、カスタムシェルとPythonスクリプトが失敗することなく動作することを保証するためです。
- 必要なすべてのスクリプトをリモート クラスタにコピーし、その場所を$PATH
- すべてのスクリプトとテンプレートファイルをフォルダ(例:スクリプト)に配置し、リモートクラスタにコピーします。
- すべてのスクリプトが実行可能であることを確認します。
- ターミナルに次のコマンドを入力して、$PATH環境変数にスクリプトディレクトリの場所を追加します。スクリプトのデフォルトの場所は$HOME/JoVE-demo/scriptsに設定されていますが、スクリプトを含むディレクトリを指す$SCRIPTS_HOMEという環境変数を定義し、$SCRIPTS_HOMEをパスに追加できます
- バッシュシェル:
エクスポートSCRIPTS_HOME=/パス/スクリプト
エクスポートパス=${SCRIPTS_HOME}:${パス} - Tcsh/Csh シェル:
/パス/スクリプトのSCRIPTS_HOMEを設定します。
パス ${SCRIPTS_HOME}
- バッシュシェル:
- リモート クラスターで、GA 計算を設定して実行します。
- gly-h2o-nというディレクトリを作成し、nは水分子の数です。
- 遺伝的アルゴリズム計算を実行するには、gly-h2o-nディレクトリの下にGAというサブディレクトリを作成します。
- OGOLEM 入力ファイル (例: pm7.ogo)、モノマーデカルト座標 (g. g. glycine.xyz, water.xyz) および PBS バッチサブミッション スクリプト (例: run.pbs) をGAディレクトリにコピーします。
- OGOLEM 入力ファイルとバッチ送信ファイルに必要な変更を加えます。
- 計算を送信します。計算が開始されると、OGOLEM は GA ディレクトリ内の OGOLEM 入力ファイル (例えば pm7) のプレフィックスとして名前が付けられた新しいディレクトリを作成し、そこに新しく生成された座標を格納します。
- 計算が完了したら、エネルギーと回転定数をコンパイルし、その情報を使用して、どれが一意の低エネルギー構造かを判断します。
- ディレクトリをgly-h2o-n/GA/pm7に変更し、
- 次のコマンドを使用して、エネルギーを抽出し、GA 最適化クラスターの回転定数を計算します。
ゲットロットコンスト-GA.csh N 0 99
ここで、N は分子クラスター内の原子の数で、'0 99' は GA プール サイズが 100 で、インデックスは 0 ~ 99 で実行されることを示します。これにより、ROTCONSTSDATA_Cと呼ばれるファイルが生成され、GA 最適化されたすべてのクラスター構成、それらのエネルギー、および回転定数のソートされたリストが含まれます。 - 次のコマンドを実行します。
similarityAnalysis.py pm7 rotConstsData_C
ここで、pm7 はファイル命名ラベルとして使用され、一意の GA 最適化クラスターを検索して保存します。これにより、ユニークな GA 最適化構成のソートされたリストを含むuniqueStructures-pm7.dataというファイルが生成されます。これは、理論のPM7レベルで最適化されたGly(H2O)nクラスタnの固有の局所最小構造のリストであり、これらの構造はDFTを使用して洗練される準備ができています。2
- gly-h2o-n/GAディレクトリに移動し、結合 GA.cshスクリプトを使用して、複数の比較可能な GA 実行の結果を結合します。構文は次のとおりです。
GA.csh <ラベル>
この特定の場合、コマンドは次のとおりです。
コンバイン GA.csh pm7 pm7
gly-h2o-n/GAディレクトリに 'uniqueStructures-pm7.data' という名前の新しい一意の構造体リストが生成されます。
3. 小さな基準セットを使用したQM法による絞り込み
注: ここでの目標は、Gly(H2O)n=1-5クラスタ構造のより小さいかつ正確な集合を得るために、より良い量子力学的記述を使用して、Gly(H2O)n=1-5クラスタの構成サンプリングを改良することです。このステップの開始構造は、ステップ 2 の出力です。
- 小さい基準セットの DFT 計算を準備して実行します。
- gly-h2o-nディレクトリの下にQMというサブディレクトリを作成します。QM ディレクトリの下にpw91-sbという名前の別のサブディレクトリを作成します。
- gly-h2o-n/GAディレクトリからQM/pw91-sbディレクトリに一意の構造体リスト (uniqueStructures-pm7.data) をコピーします。
- ディレクトリをそのgly-h2o-n/QM/pw91-sbに変更します。
- 次のコマンドを使用して、小さな基本セット DFT 構成サンプリング スクリプトを実行します。
run-pw91-sb.csh 固有の構造-pm7.data sb キュー 10
ここで、sb はこの計算セットのラベルであり、QUEUE はコンピューティング・クラスター上の優先キューであり、10 は 10 個の計算を 1 つのバッチ・ジョブにグループ化することを示します。このスクリプトは Gaussian 09 の入力を自動的に生成し、すべての計算を送信します。「QUEUE」に「テスト」と入力して、ドライ・ランを実行します。
- 送信された計算が完了したら、結果を抽出して分析します。
- 次のコマンドを使用して、エネルギーを抽出し、小さい基本に最適化されたクラスターの回転定数を計算します。
を取得します。
ここで pw91 は PW91 密度機能が使用されたことを示し、N はクラスター内の原子の数です。これにより、 という名前のファイルrotConstsData_C作成されます。 - 次に、コマンドを使用して固有の構造を識別します。
similarityAnalysis.py sb rotConstsData_C
sb はファイル命名ラベルとして使用されます。ファイルuniqueStructures-sb.dataに保存された PW91/6-31+G* レベルの理論で最適化された固有の構成のリストが表示されます。
- 次のコマンドを使用して、エネルギーを抽出し、小さい基本に最適化されたクラスターの回転定数を計算します。
- gly-h2o-n/QMディレクトリに移動し、結合 QM.cshスクリプトを使用して、複数の同等の QM 実行の結果を結合します。構文は次のとおりです。
QM.csh <ラベル> <ディレクトリのリストを QM calcs> と結合する
この特定の場合、コマンドは次のとおりです。
コンバイン QM.csh sb pw91-sb
gly-h2o-n/QMディレクトリに 'uniqueStructures-sb.data' という名前の新しい一意の構造体リストが生成されます。
4. 大ベーシスセットを用いたQM法によるさらなる改良
注: ここでの目標は、より良い量子力学的記述を使用して、Gly(H2O)n=1-5クラスタの構成サンプリングをさらに改良することです。このステップの開始構造は、ステップ 3 の出力です。
- より大きな基準セットを使用して、より信頼性の高い計算を送信します。
- QMディレクトリの下にpw91-lbというサブディレクトリを作成します。
- 一意の構造体リスト (uniqueStructures-sb.data) をgly-h2o-n/QMディレクトリからgly-h2o-n/QM/pw91-lbディレクトリにコピーし、そのディレクトリに変更します。
- 次のコマンドを使用して、大規模な DFT 構成サンプリング スクリプトを実行します。
ラン-pw91-lb.cshユニークストラクチャス-sb.データlbキュー10
ここで lb はこの計算セットのラベルであり、QUEUE はコンピューティング・クラスター上の優先キューであり、10 は 10 個の計算を 1 つのバッチ・ジョブにグループ化することを示します。このスクリプトは Gaussian 09 の入力を自動的に生成し、すべての計算を送信します。「QUEUE」に「テスト」と入力して、ドライラン・テストを実行します。
- 送信された計算が完了したら、データを抽出して分析します。
- 次のコマンドを使用して、大ベーシス最適化クラスターの回転定数を計算します。
を取得します。
ここで pw91 は PW91 密度機能が使用されたことを示し、N はクラスター内の原子の数です。 - 次に、コマンドを使用して固有の構造を識別します。
similarityAnalysis.py lb rotConstsData_C
ここで lb はファイル命名ラベルとして使用されます。これで、ファイルuniqueStructures-lb.dataに保存された理論の PW91/6-311++G** レベルで最適化された固有の構成のリストが作成されました。
- 次のコマンドを使用して、大ベーシス最適化クラスターの回転定数を計算します。
5. 最終エネルギーと熱力学補正計算
注:ここでの目標は、Gly(H2O)n=1-5クラスタの振動構造とエネルギーを、大きな基準セットと超微細積分グリッドを使用して取得し、所望の熱化学補正を計算することです。
- 前の手順の結果から、より信頼性の高い計算を送信します。
- QM/pw91-lb ディレクトリの下に ultrafine というサブディレクトリを作成します。次に、QM/pw91-lb ディレクトリから QM/pw91-lb/ultrafineディレクトリに一意の構造体リスト (uniqueStructures-lb.data) をコピーし、そのディレクトリに変更します。 QM/pw91-lb/ultrafine
- 次のコマンドを使用して、超細かい大規模ベースの DFT スクリプトを送信します。
ラン pw91-lb-ultrafine.csh ユニークストラクチャーズ-lb.data uf キュー 10
uf はこの計算セットのラベルであり、QUEUE はコンピューティング・クラスタ上の優先キューであり、10 は 10 個の計算を 1 つのバッチ・ジョブにグループ化することを示します。このスクリプトは Gaussian 09 の入力を自動的に生成し、すべての計算を送信します。「QUEUE」に「テスト」と入力して、ドライラン・テストを実行します。
- 送信された計算が完了したら、データを抽出して分析します。
- 次のコマンドを使用して、エネルギーを抽出し、大ベーシス最適化クラスターの回転定数を計算します。
ゲットロットコンスト-dft-lb-ウルトラファイン.csh pw91 N
ここで pw91 は PW91 密度機能が使用されたことを示し、N はクラスター内の原子の数です。 - 次に、コマンドを使用して固有の構造を識別します。
similarityAnalysis.py・uf rotConstsData_C
uf はファイル命名ラベルとして使用されます。これで、ファイルuniqueStructures-uf.dataに保存された PW91/6-311++G** レベルで最適化された固有の構成のリストが作成されました。
- 次のコマンドを使用して、エネルギーを抽出し、大ベーシス最適化クラスターの回転定数を計算します。
- 熱力学補正の計算に必要な情報の最終抽出を実行します。この情報を使用して、熱力学の補正を計算します。
- 最終的な電子エネルギー、回転定数、振動周波数を抽出し、次のコマンドを使用して熱力学の補正を計算します。
ランサーモpw91.cshユニークストラクチャス-uf.データ - コマンド ライン出力をコピー/貼り付け、gly-h2o-n.xlsx' という名前の Excel スプレッドシートの "Raw_Energies" シートに貼り付けます。これは、モノマー(グリシンと水)と各水和物の最も低いエネルギーメンバー(gly-h2o-n、n=1,2、..)のためにこれを行う必要があります。
- gly-h2o-n.xlsx' スプレッドシートの最初のシートに生のエネルギーが追加されると、後続の'Binding_Energies' と 'Hydrate_Distribution' のシートが自動的に更新されます。特に、Hydrate_Distributionシートは、異なる温度(例えば298.15K)、相対湿度(20%、50%、100%)で水和物の平衡濃度を生成します。水の初期濃度([H2O])とグリシン([グリシン])。これらの計算の背後にある理論は、次のステップで説明されています。
- 最終的な電子エネルギー、回転定数、振動周波数を抽出し、次のコマンドを使用して熱力学の補正を計算します。
6. 海面で室温でのGly(H2 O)n=0-5クラスタの大気濃度の計算2
注: これは、前のステップで生成された熱力学データをスプレッドシートにコピーし、シーケンシャルハイドレーションの自由なギブズエネルギーを計算することによって達成されます。次に、ギブスの自由エネルギーを使用して、各順次水和の平衡定数を計算します。最後に、一連の線形方程式が、一定のモノマー、温度および圧力の濃度に対する水和物の平衡濃度を得るために解かれる。
- 以下に示すように、グリシンの順次水和のための化学平衡のシステムを設定して開始します。

- Kn = e-Δ Gn/(kBT)を使用して平衡定数 K nを計算し、nは水和のレベル、ΔGnは n回目の水和反応のギブス自由エネルギー変化、kBはボルツマン定数、Tは温度です。

- 質量の保存のための式を設定し、水和されたグリシンクラスターと非水和されたグリシンクラスターの平衡濃度の合計が単離されたグリシン[Gly]0の初期濃度に等しいという仮定を用いて設定する0。平衡定数式の代数的な再配置を使用して、6つの連立方程式のこのシステムを書き換え、

- 上記の方程式の系を解いて、大気中のグリシン濃度0に対する実験値56、57、5857,を用いてGly(H2O)n=0-5の平衡濃度を得て、 [Gly] 0 = 2.9 x 106n = 0-5 cm-3,および200%相対湿度と温度298.15K59で大気中の水濃度59と256、[H2O]=7.7 x 1058 217cm-3。 -3
結果
このプロトコルの最初の結果セットは、構成サンプリング手順によって見つかったGly(H2O)n=1-5の低エネルギー構造のセットである必要があります。これらの構造は、PW91/6-311++G**理論レベルで最適化されており、本論文の目的に合わせて正確であると想定されています。PW91/6-311++G**が一貫してこれらのクラスタの結合エネルギーを過小評価または過大評価していることを示唆する証拠はありません。MP2/CBS32および[DLPNO-]CCSD(T)/CBS60、61,61の推定値および実験52に対する結合エネルギーを予測する能力は、多くの変動を示している。同じことが他のほとんどの密度機能にも当てはまります。一般的に、n = 1 ~ 5 の各値は、最低エネルギー構造の約 5 kcal mol-1内の一握りの低エネルギー構造を生成する必要があります。ここでは、簡潔にするために、run-thermo-pw91.cshスクリプトによって生成される最初の構造に焦点を当てます。図3はGly(H22O)n=0-5クラスタの最も低い電子エネルギー異性体を示しています。水分子の数が増えるにつれて水素結合ネットワークが複雑化し、ほとんど平面ネットワークからn = 5の3次元ケージ状構造にさえ行くことがわかります。このテキストの残りの部分では、これらの5つの特定のクラスタに対応するエネルギーと熱力学量を使用しています。
表1は、プロトコルを実行するために必要な熱力学量を示しています。表2は、電子エネルギー、振動ゼロ点補正、および3つの異なる温度での熱力学的補正が印刷されるrun-thermo-pw91.cshスクリプトの出力例を示す。各クラスター(行)について、E[PW91/6-311++G**]は、ハーツリー単位の超微細積分格子で計算されたPW91/6-311++G**レベルのガス相電子エネルギーと、kcal mol-1単位のゼロ点振動エネルギー(ZPVE)に対応しています。各温度で、216.65 K、273.15 K、および298.15 K、熱力学補正が挙げられており、kcal mol -1単位で形成のエンタルピーSであるδ H、カルモル-1単位での-1形成のエントロピー、およびGGibbGibbの自由なエネルギーがkcalmol-1の単位で形成される。表3は、ギブズの自由エネルギー変化の総量の計算例と、順次水和に関する例を示す。反応に対する水和の総ギブス自由エネルギー変化の計算例

電子エネルギー EPW91の計算から始まる

ここで、EPW91[Gly∙(H2O)]は表2列Cから取り出され、EPW91[Gly]とEPW91[H2O]2は表1列Bから取られます。次に、反応のゼロ点振動エネルギーの変化を含めることによりガス相エネルギー変化ΔE(0)全体を算出します。

をクリックして列 D を取得します。ここで、表3列CからΔEPW91/6−311++G**を取り、表2列DからE ZPVE[Gly∙(H2 O)]、表1カラムCからEZPVE[Gly]およびEZPVE[H2O]を取る。H2ZPVE2簡潔にするために、室温クラスターに移るので、216.65 K と 273.15 K のデータはスキップします。室温では、ガス相エネルギー変化を補正することで、反応ΔHのエンタルピー変化を計算します。

ここで、表3列DからΔ(0)、ΔH[Gly∙(H2O)]は表2列Kから取り出し、ΔH[Gly]およびH2HΔH[H2O]はH2表1列Jから取り出されます。H最後に、我々は、反応ΔGの自由エネルギー変化を算出した

ここで表3列IからΔが取られ、S[Gly∙(H2O)]は表2列Lから取られ、S[Gly]と2-1-1SS[H2O]は表1列Kから取り出されます。H2
ステップ 6 に示すように、水和グリシンの大気濃度を計算するために必要な量を得ました。結果は表 4に示すデータと似ているはずですが、小さな数値の違いが予想されます。表4は、ステップ6.2の6つの方程式の系の定式から1つの行列式とその後の解に見られる平衡水和物濃度を示す。まず、方程式のシステムが次のように書くことができるという事実を認めることから始めます。

ここでKnはグリシンの順次水和のための平衡定数、wは大気中の水濃度、gは大気中の単離されたグリシンの初期濃度、gnnはGly(H2O)nの平衡濃度である。2n上記の式をAx = bと書き換えた場合、x = A-1bが得られます。この逆関数は、表 4に示すように組み込みのスプレッドシート関数を使用して簡単に計算でき、最終的な結果を得ることができます。
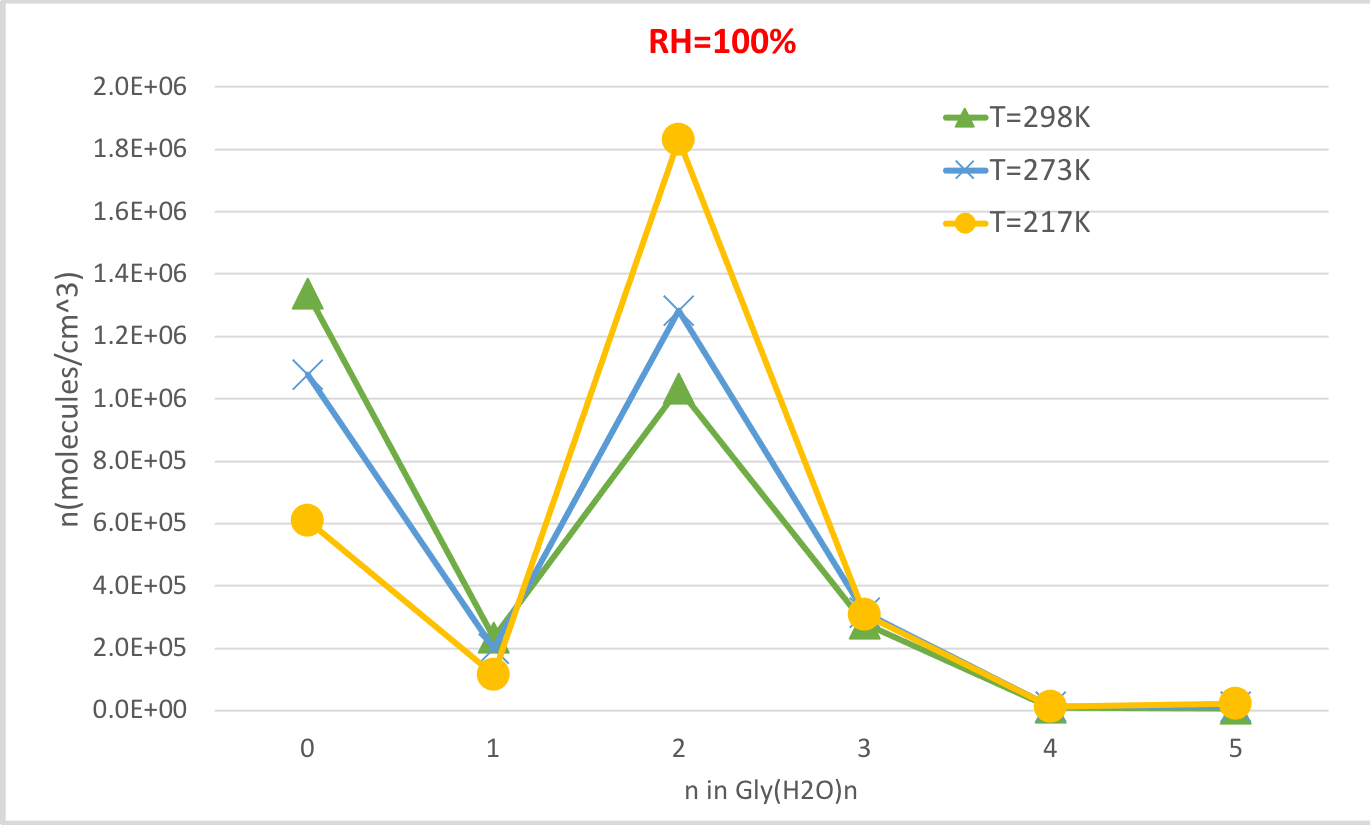
図4は、相対湿度100%および雰囲気圧力100%の温度の関数として表4で算出した水和グリシンの平衡濃度を示す。温度が298.15Kから216.65Kに低下すると、水分のないグリシン(n=0)の濃度が低下し、水和グリシンの濃度が増加することを示しています。グリシン二水和物(n=2)は、特に温度の低下に伴って劇的に増加し、他の水和物の濃度の変化は目立ちません。温度と水和物濃度の間のこれらの逆相関は、より低い温度での水和のギブス自由エネルギーを下げることが水和物の形成を支持するという期待と一致する。
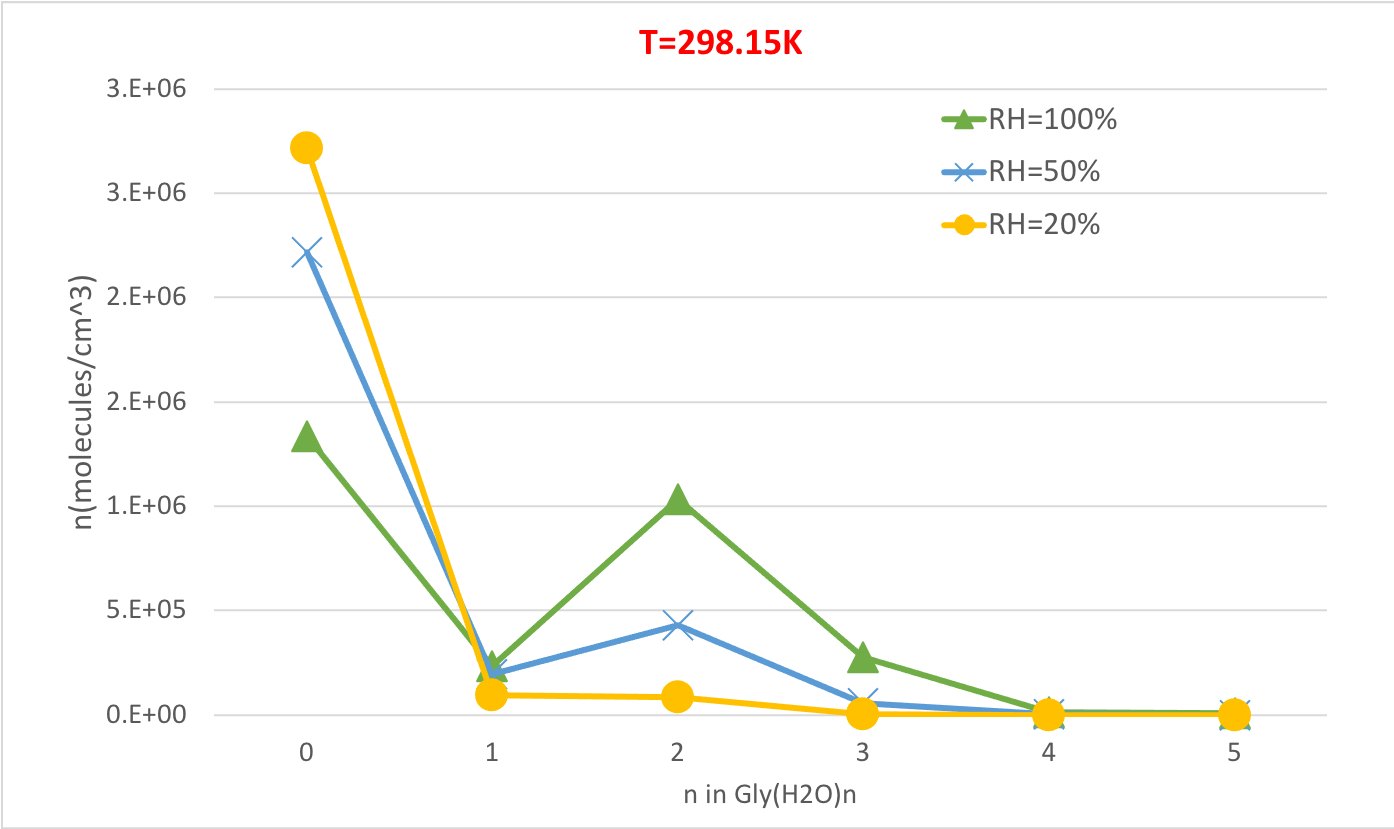
図5は、298.15Kおよび1雰囲気圧でのグリシン水和物の平衡濃度の相対湿度依存性を示す。RHが20%から100%に増加するにつれて、水分補給物(n>0)の濃度が、水分補給されていないグリシン(n=0)を犠牲にして増加することを明確に示しています。もう一度、水和物の相対湿度と濃度の間の直接的な相関関係は、より高いRHでより多くの水分子の存在が水和物の形成を促進するという考えと一致する。
提示されたように、このプロトコルは、大気中の水和グリシン集団の質的理解を与える。1立方センチメートル当たり290万分子の単離されたグリシンの初期濃度を仮定すると、水分のないグリシン(n=0)は、T=216.65KおよびRH=100%を除くほとんどの条件下で最も豊富な種であることがわかります。3つの温度すべてで最も低い連続ギブ自由エネルギーを有する水和物(n=2)は、ここで考えられる条件で最も豊富な水和物である。一水和物(n=1)およびより大きな水和物(n≥3)は、ごくわずかな量で見つかると予測されています。図3の検査では、n=1~4個のクラスターの存在量は、クラスターの水素結合ネットワークにおける安定性と歪みに関連する可能性があります。これらのクラスターは、様々な水素結合リング構造に近い幾何学的な中で、グリシンのカルボン酸部分に水素が結合した水分子を有しており、特に安定している。

図1:現在の手順の概要。遺伝的アルゴリズム(GA)によって生成された推測構造の大規模なプールは、一連の収束構造が得られるまで、一連のPW91幾何最適化によって洗練されます。これらの構造の振動周波数は計算され、ギブズの自由な生成エネルギーを計算するために使用され、周囲の条件下でクラスターの平衡濃度を計算するために使用されます。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:各クラスタの代表的なディレクトリ構造このプロトコルに含まれる社内スクリプトでは、上に示したディレクトリ構造が必要です。gly-h2o-nの n ごとに、次のサブディレクトリがあります: GA/pm7 ディレクトリを持つ遺伝的アルゴリズムの GA、 PW91/6-31+G*のQM/pw91-sb、PW91/6-311++G**のQM/pw91-lb、超微細統合グリッドの最適化と最終的な振動計算のためのQM/pw91-lb/ultrafineを用いた量子力学のQM。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:Gly(H2 O)n=0-5の代表的な低エネルギー構造2これらのクラスターは、PW91/6-311++G** 理論レベルで最適化された電子エネルギーグローバルミニマでした。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:Gly(H2O)n=0-5の温度依存性(相対湿度100%、気圧1)を示す。2n=0-5 水和物の濃度は、分子cm-3の単位で与えられる。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図5:298.15 Kおよび21気圧圧としてのGly(H2O)n=0-5の相対湿度依存性。水和物の濃度は、分子cm-3の単位で与えられる。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
| E[PW91/6-311++G**] | 216.65 K | 273.15 K | 298.15 K | ||||||||
| LB-UF | ZPVE | Δ H | S | Δ G | Δ H | S | Δ G | Δ H | S | Δ G | |
| 水 | -76.430500 | 13.04 | 1.72 | 42.59 | 5.54 | 2.17 | 44.44 | 3.08 | 2.37 | 45.14 | 1.96 |
| グリシン | -284.434838 | 48.55 | 2.65 | 69.53 | 36.14 | 3.70 | 73.81 | 32.09 | 4.22 | 75.61 | 30.22 |
表1:モノマーエネルギー電子エネルギーはハーツリーの単位で、他のすべての量はkcal mol-1の単位である。PW91/6-311++G**レベルの理論と振動周波数を計算した場合、水とグリシンを最適化しました。1気圧の熱力学補正と298.15Kの温度をthermo.plスクリプトを用いて計算した。
| E[PW91/6-311++G**] | 0 K | 216.65 K | 273.15 K | 298.15 K | ||||||||
| N | 名前 | LB-UF | ZPVE | Δ H | S | Δ G | Δ H | S | Δ G | Δ H | S | Δ G |
| 1 | グリ-h2o-1 | -360.88481 | 63.96 | 3.61 | 80.12 | 50.22 | 5.12 | 86.27 | 45.52 | 5.85 | 88.83 | 43.33 |
| 2 | グリ-h2o-2 | -437.33763 | 79.33 | 4.53 | 90.86 | 64.17 | 6.46 | 98.78 | 58.81 | 7.40 | 102.06 | 56.30 |
| 3 | グリ-h2o-3 | -513.78620 | 94.52 | 5.67 | 105.08 | 77.42 | 8.08 | 114.94 | 71.19 | 9.23 | 119.00 | 68.27 |
| 4 | グリ-h2o-4 | -590.23667 | 109.80 | 6.03 | 104.98 | 91.30 | 8.78 | 116.21 | 84.40 | 10.11 | 120.87 | 81.14 |
| 5 | グリ-h2o-5 | -666.68845 | 125.80 | 7.26 | 121.70 | 106.69 | 10.47 | 134.83 | 99.44 | 12.01 | 140.24 | 96.00 |
表2:クラスターエネルギー図 1に示す手順を使用して見つかった最低エネルギー Gly(H2O)n=1-5構造体のエネルギー 。電子エネルギーはハーツリーの単位で、他のすべての量はkcal mol-1の単位である。
| 総水分補給: Gly + nH2O <> Gly(H2O)n | シーケンシャルハイドレーション: Gly(H2O)n-1 + H2O <-> Gly(H2O)n | ||||||||||||||||
| E[PW91/6-311++G**] | 216.65 | 273.15 | 298.15 | 216.65 | 273.15 | 298.15 | |||||||||||
| N | システム名 | LB-UF | Δ E(0) | Δ H(T) | Δ G(T) | Δ H(T) | Δ G(T) | Δ H(T) | Δ G(T) | LB-UF | Δ E(0) | Δ H(T) | Δ G(T) | H(T) | Δ G(T) | Δ H(T) | Δ G(T) |
| 1 | グリ-h2o-1 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 | -12.22 | -9.85 | -10.61 | -3.68 | -10.61 | -1.87 | -10.59 | -1.07 |
| 2 | グリ-h2o-2 | -26.22 | -21.53 | -23.10 | -9.27 | -23.11 | -5.66 | -23.09 | -4.06 | -14.00 | -11.68 | -12.49 | -5.59 | -12.50 | -3.79 | -12.50 | -2.99 |
| 3 | グリ-h2o-3 | -37.56 | -30.72 | -32.88 | -12.90 | -32.87 | -7.69 | -32.82 | -5.38 | -11.34 | -9.19 | -9.78 | -3.63 | -9.76 | -2.03 | -9.73 | -1.32 |
| 4 | グリ-h2o-4 | -50.10 | -40.34 | -43.48 | -15.87 | -43.54 | -8.71 | -43.51 | -5.55 | -12.54 | -9.62 | -10.60 | -2.97 | -10.67 | -1.02 | -10.69 | -0.17 |
| 5 | グリ-h2o-5 | -63.45 | -51.41 | -55.42 | -20.58 | -55.51 | -11.48 | -55.48 | -7.45 | -13.35 | -11.07 | -11.94 | -4.71 | -11.97 | -2.77 | -11.97 | -1.90 |
表3:水和エネルギーGly(H2 O)n=1-5の水2和と順次水和の総エネルギー (kcal mol-1の単位) 。ここで、E[PW91/6-311++G**]は電子エネルギーの変化であり、δ E(0)はゼロ点振動エネルギー(ZPVE)補正されたエネルギー変化、ΔH(T)は温度Tでのエンタルピー変化、およびδG(T)は各Gly(H2O)n=1-5クラスターのギブス自由2エネルギー変化であるn=1-5。
| 温度及び相対湿度の関数としての平衡水和物分布 | |||||||||
| T=298.15K | T=273.15K | T=216.65K | |||||||
| グリ(H2O)n | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% | RH=100% | RH=50% | RH=20% |
| 0 | 1.3E+06 | 2.2E+06 | 2.7E+06 | 1.1E+06 | 2.0E+06 | 2.7E+06 | 6.1E+05 | 1.5E+06 | 2.5E+06 |
| 1 | 2.3E+05 | 1.9E+05 | 9.5E+04 | 2.0E+05 | 1.9E+05 | 9.9E+04 | 1.2E+05 | 1.5E+05 | 9.5E+04 |
| 2 | 1.0E+06 | 4.3E+05 | 8.4E+04 | 1.3E+06 | 6.1E+05 | 1.3E+05 | 1.8E+06 | 1.1E+06 | 3.0E+05 |
| 3 | 2.8E+05 | 5.8E+04 | 4.5E+03 | 3.2E+05 | 7.4E+04 | 6.3E+03 | 3.1E+05 | 9.6E+04 | 1.0E+04 |
| 4 | 1.1E+04 | 1.1E+03 | 3.4E+01 | 1.3E+04 | 1.5E+03 | 5.0E+01 | 1.1E+04 | 1.8E+03 | 7.5E+01 |
| 5 | 7.5E+03 | 3.9E+02 | 4.9E+00 | 1.2E+04 | 7.2E+02 | 9.7E+00 | 2.4E+04 | 1.9E+03 | 3.1E+01 |
表4:Gly(H2O)n=0-5の平2衡水和n=0-5物濃度を機能温度として(T=298.15K,273.15K,216.65K)及び相対湿度(RH=100%,50%,20%)。水和物の濃度は、実験値,-356、57、58、056,57,58= 2.9 x 106 cm-3および[H2O]=7.7 x 1017 cm-3、1.6x 10 1014 cm -3を100%の湿度で100%の温度と290%の温度で与えるcm-3の単位で与えられる。 273.15 K、および 216.65 K、それぞれ59.
ディスカッション
このプロトコルによって生成されるデータの精度は、主に、(i)ステップ2でサンプリングされた構成の多様性、(ii)システムの電子構造の精度、(iii)および熱力学的補正の精度の3つの要素に依存する。これらの各要因は、含まれているスクリプトを編集してメソッドを変更することで対処できます。最初の要因は、ランダムに生成された構造のより大きな初期プール、GAのより多くの反復、およびGAに関与する基準のより緩い定義を使用して簡単に克服されます。また、異なる物理的記述の影響を探るために、自己一貫した電荷密度機能密着性の密結合(SCC-DFTB)6262モデルおよび有効断片電位(EFP)6363モデルのような異なる半経験的方法を用いてもよい。ここでの主な制限は、共有結合を形成または切断する方法の不能であり、単量体が凍結していることを意味する。GA手順は、半経験的記述に従って、これらの凍結モノマーの最も安定した相対位置のみを見つける。
システムの電子構造の精度は、その計算コストを持つ様々な方法で改善することができます。M06-2X 64やwB97X-V65などのより優れた密度機能、65またはMøller-Plesset69 66、67、68(MPn)66摂動理論などの量子機械(QM)法を選択して、67,68システムの物理的記述を改善することができます。機能の階層では、一般的に、PW91のような一般化された勾配近似(GGA)機能から、M06-2XのようなwB97X-DおよびメタGGAハイブリッド機能のような範囲分離されたハイブリッド機能に至ると、性能が向上する。
DFT法の欠点は、正確な値に向けた体系的な収束が不可能であるということです。しかし、DFT法は計算上安価であり、多種多様なアプリケーションに対して多種多様な機能があります。
増加する枢機卿数の相関一貫基準セットと組み合わせてMP2やCCSD(T)のような波関数法を使用して計算されたエネルギー([aug-]cc-pV[D,T,Q,...]Z) は、完全な基準セットの制限に向かって体系的に収束しますが、システムサイズが大きくなるにつれて、各計算の計算コストは非常に大きくなります。電子構造のさらなる改良は、明示的に相関するベーシスセット70を用いて、完全な基準セット(CBS)71制限に外挿することによって達成71することができる。我々の最近の研究は、密度が明示的に相関する第2次Møller-Plesset(DF-MP2-F12)摂動アプローチがMP2/CBS計算32のエネルギーに近づくエネルギーをもたらすことを示唆している。現在のプロトコルを変更して異なる電子構造メソッドを使用するには、(i)ソフトウェアの構文に従ってテンプレート入力ファイルを作成し、(ii)run-pw91-sb.csh、run-pw91-lb.csh、およびrun-pw91-lb-ultrafine.cshスクリプトを使用して正しい入力ファイル構文を生成する2つの手順を実行します。 run-pw91-lb.csh
最後に、熱力学による補正の精度は、電子構造法と、グローバル最小の周りのPESの記述に依存します。PESの正確な記述では、非常にコストのかかる作業である、四次力場72,73(QFF),73のような核自由度の変位に関するPESの3次および高次微分の計算が必要である。現在のプロトコルは振動周波数に対して高調波発振器近似を使用するため、PESの二次導関数までしか計算する必要が出ない。このアプローチは、真のPESと高調波PESの大きな違いのために、非常にフロッピー分子や対称的なダブルウェル電位など、高い不均調性を持つシステムでは問題になります。さらに、計算負荷の高い電子構造法から高品質PESを有するコストは、振動周波数計算のコストの問題を複合化するのみである。これを克服する1つのアプローチは、高品質の電子構造計算から電子エネルギーを使用し、低品質のPESで計算された振動周波数と共に、コストと精度のバランスを取ることです。現在のプロトコルは、前の段落で説明したように、異なるPESの記述を使用するように変更することができます。ただし、スクリプトやテンプレートの振動周波数キーワードを編集して、アンコン波振動周波数を計算することもできます。
構成サンプリング プロトコルの 2 つの重要な問題は、潜在的なエネルギー サーフェスをサンプリングするための最初の方法と、各クラスターを識別するために使用される基準です。前作では様々な方法を幅広く活用してきました。最初の問題として、潜在的なエネルギー表面をサンプリングするための最初の方法は、これらの要因に基づいて半経験的な方法でGAを使用するという選択をしました。化学直感26、ランダムサンプリング、分子動力学(MD)29,30を用いた構成サンプリングは、水29,30クラスター18の研究で観察したように、10個のモノマーを超えるクラスターに対して定期的に推定グローバルミニマを見つけることができません。我々は、(H2O)111174の複雑なPESを研究するために2盆地ホッピング(BH)を使用することに成功したが、それはBHアルゴリズムが見つけ出さなかったいくつかの潜在的な低エネルギー異性体の手動包含を必要とした。水クラスターの世界的な最小値を見つける場合のBHとGAのパフォーマンスの比較, (H2O)n=10-20 GAは一貫してBH75よりも速いグローバル最小値を発見したことを示しました.OGOLEM および CLUSTER に実装されている GA は、任意の分子クラスターに適用でき、古典的な力場、半経験的、密度機能、および ab initio機能を持つ膨大な数のパッケージとのインターフェースが可能であるため、非常に汎用性があります。PM7の選択は速度および合理的な正確さによって動かされる。他のセミ経験的手法は、計算コストが大幅に高くなります。
2つ目の課題としては、電子エネルギー、双極子モーメント、オーバーラップRMSD、回転定数に至るまで、異なる基準を用いて、独自の構造を特定しました。双極子モーメントの両方が分子の向きに依存し、ダイポールモーメントの総部分が、構造が同じまたはユニークであると判断する閾値を設定することが困難であったような形状の違いに非常に敏感であったため、双極子モーメントを使用することは困難であることが判明した。電子エネルギーと回転定数の組み合わせが最も有用であることが判明しました。
2つの構造を一意と見なす現在の基準は、エネルギー差閾値0.10kcal mol-1と回転定数差1%に基づいています。したがって、エネルギーが0.10kcalモル-1(〜0.00015-1 a.u.)以上異なる場合、2つの構造が異なると考えられます。そして、その3つの回転定数(A、B、C)のいずれかが1%以上異なります。長年にわたる実質的な内部ベンチマークは、これらのしきい値が合理的な選択であることを発見しました。我々の構成サンプリング手法及びスクリーニング方法論は、水76,77,77と、アンモニア及びアミン類を含む強結合した三硫酸水和物と同様に、水と結合した多芳香族炭化水素のような非常に弱く結合したクラスターに適用された。考慮するプロネーション状態が異なるクラスターの場合、各々が異なる原始状態のモノマーから始まるさまざまな GA 計算を実行するのが最善の方法です。これにより、異なる原始状態の構造を慎重に考慮することが保証されます。ただし、低レベル DFT 計算では、多くの場合、ジオメトリ最適化の過程でプロトネーション状態が変化するため、開始ジオメトリに関係なく最も安定したプロトネーション状態が得られます。
GA コードが一般的な非パラメータ化メソッドとインターフェイスされている限り、GA の GA 構成サンプリング方法は、GA の実行中にモノマーが異なる構成を採用できる限り、うまく機能するはずです。例えば、GAとPM7をインターフェースするとモノマーの構造が変わることになりますが、原始状態が変化したときに結合が壊れた場合、構造は受け入れられない候補として廃棄される可能性があります。
高調波近似の欠点、特に低振動周波数に起因する欠点を修正するさまざまな方法を検討してきました。準高調波近似を現在の方法論に組み込むことは難しくない。しかし、準高調波法については、特に適用されるカットオフ周波数に関しては疑問が残っています。また、従来の知恵がRRHO近似値よりも改善すべきであることを示唆しているにもかかわらず、準RRHO近似の信頼性を調べる厳格なベンチマーク作業はありません。
このように提示されるプロトコルは、非共有結合ガス相分子クラスターの任意のシステムに一般化され得る。また、スクリプトやテンプレートを編集することにより、セミ経験的方法、電子構造法およびソフトウェア、および振動解析方法およびソフトウェアを使用するように一般化してもよい。これは、ユーザーが Linux コマンドライン インターフェイス、Python スクリプト、およびハイパフォーマンス コンピューティングに慣れかけられていることを前提としています。Linux オペレーティングシステムの不慣れな構文と見た目とスクリプトの経験の欠如は、このプロトコルの最大の落とし穴であり、新入生が最も苦労している場所です。このプロトコルは、主に硫酸とアンモニアがエアロゾル形成に及ぼす影響に焦点を当てて、私たちのグループで長年にわたって様々な実装で成功してきました。このプロトコルのさらなる改善には、より多くの電子構造ソフトウェアへのより堅牢なインターフェース、遺伝的アルゴリズムの代替実装、そしておそらく電子および振動エネルギーのより速い計算のための新しい方法の使用が含まれる。このプロトコルの現在のアプリケーションは、現在の大気中でのエアロゾル形成の初期段階とプレバイオティクス環境におけるより大きな生体分子の形成におけるアミノ酸の重要性を探っています。
開示事項
なし。
謝辞
このプロジェクトは、国立科学財団(GCS)、アーノルドとメイベルベックマン財団ベックマン奨学生賞(AGG)、バリーM.ゴールドウォーター奨学金(AGG)からの助成金CHE-1229354、CHE-1662030、CHE-1721511、CHE-1903871によって支援されました。水銀コンソーシアム(http://www.mercuryconsortium.org)の高性能コンピューティング資源を使用した。
資料
| Name | Company | Catalog Number | Comments |
| Avogadro | https://avogadro.cc | Open-source molecular visualization program | |
| Gaussian [09/16] Software | http://www.gaussian.com/ | Commercial ab initio electronic structure program | |
| MOPAC 2016 | http://openmopac.net/MOPAC2016.html | Open-source semi-empirical program | |
| OGOLEM Software | https://www.ogolem.org | Genetic algorithm-based global optimization program | |
| OpenBabel | http://openbabel.org/wiki/Main_Page | Open-source cheminformatics library | |
| calcRotConsts.py | Shields Group, Department of Chemistry, Furman University | Python script to compute rotational constants | |
| calcSymmetry.csh | Shields Group, Department of Chemistry, Furman University | Shell script to calculate symmetry number of a molecule given Cartesian coordinates | |
| combine-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different GA directories | |
| combine-QM.csh | Shields Group, Department of Chemistry, Furman University | Shell script to combine energy and rotational constants from different QM directories | |
| gaussianE.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 energies | |
| gaussianFreqs.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract Gaussian 09 vibrational frequencies | |
| getrotconsts | Shields Group, Department of Chemistry, Furman University | Executable to calculate rotational constants given a molecule's Cartesian coordinates | |
| getRotConsts-dft-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of large basis DFT optimized structures | |
| getRotConsts-dft-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of ultrafine DFT optimized structures | |
| getRotConsts-dft-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of small basis DFT optimized structures | |
| getRotConsts-GA.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute rotational constants for a batch of genetic algorithm optimized structures | |
| global-minimum-coords.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of global minimum structures of gly-(h2o)n, where n=0-5 | |
| make-thermo-gaussian.csh | Shields Group, Department of Chemistry, Furman University | Shell script to extract data from Gaussian output files and make input files for the thermo.pl script | |
| ogolem-input-file.ogo | Shields Group, Department of Chemistry, Furman University | Ogolem sample input file | |
| ogolem-submit-script.pbs | Shields Group, Department of Chemistry, Furman University | PBS batch submission file for Ogolem calculations | |
| README.docx | Shields Group, Department of Chemistry, Furman University | Clarifications to help readers use the scripts effectively | |
| runogolem.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run OGOLEM | |
| run-pw91-lb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of large basis DFT optimization calculations | |
| run-pw91-lb-ultrafine.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of ultrafine DFT optimization calculations | |
| run-pw91-sb.csh | Shields Group, Department of Chemistry, Furman University | Shell script to run a batch of small basis DFT optimization calculations | |
| run-thermo-pw91.csh | Shields Group, Department of Chemistry, Furman University | Shell script to compute the thermodynamic corrections for a batch of DFT optimized structures | |
| similarityAnalysis.py | Shields Group, Department of Chemistry, Furman University | Python script to determine unique structures based on rotational constants and energies | |
| symmetry | Shields Group, Department of Chemistry, Furman University | Executable to calculate molecular symmetry given Cartesian coordinates | |
| symmetry.c | (C) 1996, 2003 S. Patchkovskii, Serguei.Patchkovskii@sympatico.ca | C code to determine the molecular symmstry of a molecule given Cartesian coordinates | |
| template-marcy.pbs | Shields Group, Department of Chemistry, Furman University | Template for a PBS submit script which uses OGOLEM | |
| template-pw91.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input | |
| template-pw91-HL.com | Shields Group, Department of Chemistry, Furman University | Template Gaussian 09 input for ultrafine DFT optimization | |
| thermo.pl | https://www.nist.gov/mml/csd/chemical-informatics-research-group/products-and-services/program-computing-ideal-gas | Perl open-source script to compute ideal gas thermodynamic corrections | |
| gly-h2o-n.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet for the complete protocol | |
| table-1.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-2.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-3.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| table-4.xlsx | Shields Group, Department of Chemistry, Furman University | Excel spreadsheet | |
| water.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of water | |
| glycine.xyz | Shields Group, Department of Chemistry, Furman University | Cartesian coordinates of glycine |
参考文献
- Foster, P., Ramaswamy, V., Solomon, S., Qin, D., Manning, M., Chen, Z., Marquis, M., Averyt, K. B., Tignor, M., Miller, H. L. . Climate Change 2007 The Scientific Basis. , (2007).
- Kulmala, M., et al. Toward direct measurement of atmospheric nucleation. Science. 318 (5847), 89-92 (2007).
- Sipila, M., et al. The role of sulfuric acid in atmospheric nucleation. Science. 327 (5970), 1243-1246 (2010).
- Jiang, J., et al. First measurement of neutral atmospheric cluster and 1 - 2 nm particle number size distributions during nucleation events. Aerosol Science and Technology. 45 (4), (2011).
- Dunn, M. E., Pokon, E. K., Shields, G. C. Thermodynamics of forming water clusters at various Temperatures and Pressures by Gaussian-2, Gaussian-3, Complete Basis Set-QB3, and Complete Basis Set-APNO model chemistries; implications for atmospheric chemistry. Journal of the American Chemical Society. 126 (8), 2647-2653 (2004).
- Pickard, F. C., Pokon, E. K., Liptak, M. D., Shields, G. C. Comparison of CBSQB3, CBSAPNO, G2, and G3 thermochemical predictions with experiment for formation of ionic clusters of hydronium and hydroxide ions complexed with water. Journal of Chemical Physics. 122, 024302 (2005).
- Pickard, F. C., Dunn, M. E., Shields, G. C. Comparison of Model Chemistry and Density Functional Theory Thermochemical Predictions with Experiment for Formation of Ionic Clusters of the Ammonium Cation Complexed with Water and Ammonia; Atmospheric Implications. Journal of Physical Chemistry A. 109 (22), 4905-4910 (2005).
- Alongi, K. S., Dibble, T. S., Shields, G. C., Kirschner, K. N. Exploration of the Potential Energy Surfaces, Prediction of Atmospheric Concentrations, and Vibrational Spectra of the HO2•••(H2O)n (n=1-2) Hydrogen Bonded Complexes. Journal of Physical Chemistry A. 110 (10), 3686-3691 (2006).
- Allodi, M. A., Dunn, M. E., Livada, J., Kirschner, K. N. Do Hydroxyl Radical-Water Clusters, OH(H2O)n, n=1-5, Exist in the Atmosphere. Journal of Physical Chemistry A. 110 (49), 13283-13289 (2006).
- Kirschner, K. N., Hartt, G. M., Evans, T. M., Shields, G. C. In Search of CS2(H2O)n=1-4 Clusters. Journal of Chemical Physics. 126, 154320 (2007).
- Hartt, G. M., Kirschner, K. N., Shields, G. C. Hydration of OCS with One to Four Water Molecules in Atmospheric and Laboratory Conditions. Journal of Physical Chemistry A. 112 (19), 4490-4495 (2008).
- Morrell, T. E., Shields, G. C. Atmospheric Implications for Formation of Clusters of Ammonium and 110 Water Molecules. Journal of Physical Chemistry A. 114 (12), 4266-4271 (2010).
- Temelso, B., et al. Quantum Mechanical Study of Sulfuric Acid Hydration: Atmospheric Implications. Journal of Physical Chemistry A. 116 (9), 2209 (2012).
- Husar, D. E., Temelso, B., Ashworth, A. L., Shields, G. C. Hydration of the Bisulfate Ion: Atmospheric Implications. Journal of Physical Chemistry A. 116 (21), 5151-5163 (2012).
- Bustos, D. J., Temelso, B., Shields, G. C. Hydration of the Sulfuric Acid – Methylamine Complex and Implications for Aerosol Formation. Journal of Physical Chemistry A. 118 (35), 7430-7441 (2014).
- Wales, D. J., Scheraga, H. A. Global optimization of clusters, crystals, and biomolecules. Science. 27 (5432), 1368-1372 (1999).
- Day, M. B., Kirschner, K. N., Shields, G. C. Global search for minimum energy (H2O)n clusters, n = 3 - 5. The Journal of Physical Chemistry A. 109 (30), 6773-6778 (2005).
- Shields, R. M., Temelso, B., Archer, K. A., Morrell, T. E., Shields, G. C. Accurate predictions of water cluster formation, (H2O)n=2-10. The Journal of Physical Chemistry A. 114 (43), 11725-11737 (2010).
- Temelso, B., Archer, K. A., Shields, G. C. Benchmark structures and binding energies of small water clusters with anharmonicity corrections. The Journal of Physical Chemistry A. 115 (43), 12034-12046 (2011).
- Temelso, B., Shields, G. C. The role of anharmonicity in hydrogen-bonded systems: The case of water clusters. The Journal of Chemical Theory and Computation. 7 (9), 2804-2817 (2011).
- Von Freyberg, B., Braun, W. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. The Journal of Computational Chemistry. 12 (9), 1065-1076 (1991).
- Rakshit, A., Yamaguchi, T., Asada, T., Bandyopadhyay, P. Understanding the structure and hydrogen bonding network of (H2O)32 and (H2O)33: An improved Monte Carlo temperature basin paving (MCTBP) method of quantum theory of atoms in molecules (QTAIM) analysis. RSC Advances. 7 (30), 18401-18417 (2017).
- Deaven, D. M., Ho, K. M. Molecular geometry optimization with a genetic algorithm. Physical Review Letters. 75, 288-291 (1995).
- Hartke, B. Application of evolutionary algorithms to global cluster geometry optimization. Applications of Evolutionary Computation in Chemistry. , (2004).
- Dieterich, J. M., Hartke, B. OGOLEM: Global cluster structure optimization for arbitrary mixtures of flexible molecules. A multiscaling, object-oriented approach. Molecular Physics. 108 (3-4), 279-291 (2010).
- Herb, J., Nadykto, A. B., Yu, F. Large ternary hydrogen-bonded pre-nucleation clusters in the Earth's atmosphere. Chemical Physics Letters. 518, 7-14 (2011).
- Ortega, I. K., et al. From quantum chemical formation free energies to evaporation rates. Atmospheric Chemistry and Physics. 12 (1), 225-235 (2012).
- Elm, J., Bilde, M., Mikkelsen, K. V. Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical. Journal of Physical Chemistry A. 117 (30), 6695-6701 (2013).
- Loukonen, V., et al. Enhancing effect of dimethylamine in sulfuric acid nucleation in the presence of water - a computational study. Atmospheric Chemistry and Physics. 10 (10), 4961-4974 (2010).
- Temelso, B., Phan, T. N., Shields, G. C. Computational study of the hydration of sulfuric acid dimers: implications for acid dissociation and aerosol formation. Journal of Physical Chemistry A. 116 (39), 9745-9758 (2012).
- Jiang, S., et al. Study of Cl-(H2O)n (n = 1-4) using basin-hopping method coupled with density functional theory. Journal of Computational Chemistry. 35 (2), 159-165 (2014).
- Temelso, B., et al. Effect of mixing ammonia and alkylamines on sulfate aerosol formation. Journal of Physical Chemistry A. 122 (6), 1612-1622 (2018).
- Perdew, J. P., Ruzsinszky, A., Tao, J. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. Journal of Chemical Physics. 123, 062201 (2005).
- Riplinger, C., Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. Journal of Chemical Physics. 138, 034106 (2013).
- Riplinger, C., Pinski, P., Becker, U., Valeev, E. F., Neese, F. Sparse maps--A systematic infrastructure for reduced-scaling electronic structure methods. II. Linear scaling domain based pair natural orbital coupled cluster theory. Journal of Chemical Physics. 144 (2), 024109 (2016).
- Kildgaard, J. V., Mikkelsen, K. V., Bilde, M., Elm, J. Hydration of atmospheric molecular clusters: a new method for systematic configurational sampling. Journal of Physical Chemistry A. 122 (22), 5026-5036 (2018).
- González, &. #. 1. 9. 3. ;. Measurement of areas on a sphere Using Fibonacci and latitude-longitude lattices. Mathematical Geosciences. 42, 49-64 (2010).
- Karaboga, D., Basturk, B. On the performance of artificial bee colony (ABC) algorithm. Applied Soft Computing. 8 (1), 687-697 (2008).
- Zhang, J., Doig, M. Global optimization of rigid molecules using the artificial bee colony algorithm. Physical Chemistry Chemical Physics. 18 (4), 3003-3010 (2016).
- Kubecka, J., Besel, V., Kurten, T., Myllys, N., Vehkamaki, H. Configurational sampling of noncovalent (atmospheric) molecular clusters: sulfuric acid and guanidine. Journal of Physical Chemistry A. 123 (28), 6022-6033 (2019).
- Grimme, S., Bannwarth, C., Shushkov, P. A Robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent Interactions of large molecular systems parametrized for all spd-block elements (Z = 1-86). Journal of Chemical Theory and Computation. 13 (5), 1989-2009 (2017).
- Buck, U., Pradzynski, C. C., Zeuch, T., Dieterich, J. M., Hartke, B. A size resolved investigation of large water clusters. Physical Chemistry Chemical Physics. 16 (15), 6859 (2014).
- Forck, R. M., et al. Structural diversity in sodium doped water trimers. Physical Chemistry Chemical Physics. 14 (25), 9054-9057 (2012).
- Witt, C., Dieterich, J. M., Hartke, B. Cluster structures influenced by interaction with a surface. Physical Chemistry Chemical Physics. 20 (23), 15661-15670 (2018).
- Freitbert, A., Dieterich, J. M., Hartke, B. Exploring self-organization of molecular tether molecules on a gold surface by global structure optimization. The Journal of Computational Chemistry. 40 (22), 1978-1989 (2019).
- Stewart, J. J. P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. The Journal of Molecular Modeling. 19 (1), 1-32 (2013).
- Burke, K., Perdew, J. P., Wang, Y. Derivation of a generalized gradient approximation: The PW91 density functional. Electronic Density Functional Theory. , 81-111 (1998).
- Frisch, M. J., et al. . Gaussian 09, Revision A.02. , (2016).
- Ditchfield, R., Hehre, W. J., Pople, J. A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. The Journal of Chemical Physics. 54 (2), 724 (1971).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters. The Journal of Chemical Theory and Computation. 8 (6), 2071-2077 (2012).
- Elm, J., Mikkelsen, K. V. Computational approaches for efficiently modelling of small atmospheric clusters. Chemical Physics Letters. 615, 26-29 (2014).
- Bayucan, A., et al. . PBS Portable Batch System. , (1999).
- O'Boyle, N. M., et al. Open Babel: An open chemical toolbox. Journal of Cheminformatics. 3, 33 (2011).
- Csaszar, A. G. Conformers of gaseous glycine. Journal of the American Chemical Society. 114 (24), 9568-9575 (1992).
- Zhang, Q., Anastasio, C. Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from Northern California. Atmospheric Environment. 37 (16), 2247-2258 (2003).
- Matsumoto, K., Uematsu, M. Free amino acids in marine aerosols over the western North Pacific Ocean. Atmospheric Environment. 39 (11), 2163-2170 (2005).
- Mandalakis, M., Apostolaki, M., Stephanou, E. G. Trace analysis of free and combined amino acids in atmospheric aerosols by gas chromatography-mass spectrometry. Journal of Chromatography A. 1217 (1), 143-150 (2010).
- Seinfeld, J. H., Pandis, S. N. . Atmospheric Chemistry and Physics, 3rd Ed. , (2016).
- Myllys, N., Elm, J., Halonen, R., Kurten, T., Vehkamaki, H. Coupled cluster evaluation of atmospheric acid-base clusters with up to 10 molecules. The Journal of Physical Chemistry A. 120 (4), 621-630 (2016).
- Elm, J., Bilde, M., Mikkelsen, K. V. Assessment of binding energies of atmospherically relevant clusters. Physical Chemistry Chemical Physics. 15 (39), (2013).
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theoretical Chemistry Accounts. 116 (1-3), 316-325 (2006).
- Kaliman, I. A., Slipchenko, L. V. LIBEFP: A new parallel implementation of the effective fragment potential method as a portable software library. The Journal of Computational Chemistry. 34 (26), 2284-2292 (2013).
- Zhao, Y., Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and trasition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theoretical Chemistry Accounts. 120 (1-3), 215-241 (2008).
- Mardirossian, N., Head-Gordon, M. wB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Physical Chemistry Chemical Physics. 16 (21), 9904-9924 (2014).
- Head-Gordon, M., Pople, J. A., Frisch, M. J. MP2 energy evaluation by direct methods. Chemical Physics Letters. 153 (6), 503-506 (1988).
- Pople, J. A., Seeger, R., Krishnan, R. Variational configuration interaction methods and comparison with perturbation theory. The International Journal of Quantum Chemistry. 12, 149-163 (1977).
- Pople, J. A., Binkley, J. S., Seeger, R. Theoretical models incorporating electron correlation. The International Journal of Quantum Chemistry. 10 (10), 1-19 (1976).
- Monkhorst, H. J. Calculation of properties with the coupled-cluster method. The International Journal of Quantum Chemistry. 12 (11), 421-432 (1977).
- Klopper, W., Manby, F. R., Ten-No, S., Valeev, E. F. R12 methods in explicitly correlated molecular electronic structure theory. International Reviews in Physical Chemistry. 25, 427-468 (2006).
- Hattig, C. Optimization of auxiliary basis sets for RI-MP2 and RI-CC2 calculations: Core-valence and quintuple-z basis sets for H to Ar and QZVPP basis sets for Li to Kr. Physical Chemistry Chemical Physics. 7 (1), 59-66 (2005).
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. The Journal of Chemical Physics. 122, 014108 (2005).
- Barone, V. Vibrational zero-point energies and thermodynamic functions beyond the harmonic approximation. The Journal of Chemical Physics. 120 (7), 3059-3065 (2004).
- Temelso, B., et al. Exploring the Rich Potential Energy Surface of (H2O)11 and Its Physical Implications. Journal of Chemical Theory and Computation. 14 (2), 1141-1153 (2018).
- Kabrede, H., Hentschke, R. Global minima of water clusters (H2O)N, N≤25, described by three empirical potentials. Journal of Physical Chemistry B. 107 (16), (2003).
- Steber, A. L., et al. Capturing the Elusive Water Trimer from the Stepwise Growth of Water on the Surface of a Polycyclic Aromatic Hydrocarbon Acenaphthene. Journal of Physical Chemistry Letters. 8 (23), 5744-5750 (2017).
- Perez, C., et al. Corrannulene and its complex with water: A tiny cup of water. Physical Chemistry Chemical Physics. 19 (22), 14214-14223 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved