Production and Characterization of Human Macrophages from Pluripotent Stem Cells
In This Article
Summary
This protocol describes the robust generation of macrophages from human induced pluripotent stem cells, and methods for their subsequent characterization. Cell surface marker expression, gene expression, and functional assays are used to assess the phenotype and function of these iPSC-derived macrophages.
Abstract
Macrophages are present in most vertebrate tissues and comprise widely dispersed and heterogeneous cell populations with different functions. They are key players in health and disease, acting as phagocytes during immune defense and mediating trophic, maintenance, and repair functions. Although it has been possible to study some of the molecular processes involved in human macrophage function, it has proved difficult to apply genetic engineering techniques to primary human macrophages. This has significantly hampered our ability to interrogate the complex genetic pathways involved in macrophage biology and to generate models for specific disease states. An off-the-shelf source of human macrophages that is amenable to the vast arsenal of genetic manipulation techniques would, therefore, provide a valuable tool in this field. We present an optimized protocol that allows for the generation of macrophages from human induced pluripotent stem cells (iPSCs) in vitro. These iPSC-derived macrophages (iPSC-DMs) express human macrophage cell surface markers, including CD45, 25F9, CD163, and CD169, and our live-cell imaging functional assay demonstrates that they exhibit robust phagocytic activity. Cultured iPSC-DMs can be activated to different macrophage states that display altered gene expression and phagocytic activity by the addition of LPS and IFNg, IL4, or IL10. Thus, this system provides a platform to generate human macrophages carrying genetic alterations that model specific human disease and a source of cells for drug screening or cell therapy to treat these diseases.
Introduction
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) represent a self-renewing cell source that can be differentiated to produce cells of all three germ layer lineages. Technologies that allow for the genetic manipulation of human pluripotent stem cells (PSCs), such as Zinc Finger Nuclease, TALENS, and CRISPR-Cas9, have revolutionised medical research1,2,3,4. Genetic manipulation of human PSCs is a particularly attractive strategy when the primary cell of interest is difficult to expand and/or to maintain in vitro, or is difficult to genetically manipulate, such as is the case for macrophages5,6,7,8,9. As human iPSCs can be derived from any somatic cell, they circumvent the ethical limitations associated with ESCs, and provide a strategy for delivering personalized medicine. This includes patient-specific disease modelling, drug testing, and autologous cell therapy with a reduced risk of immune rejection and infection6,8,10,11.
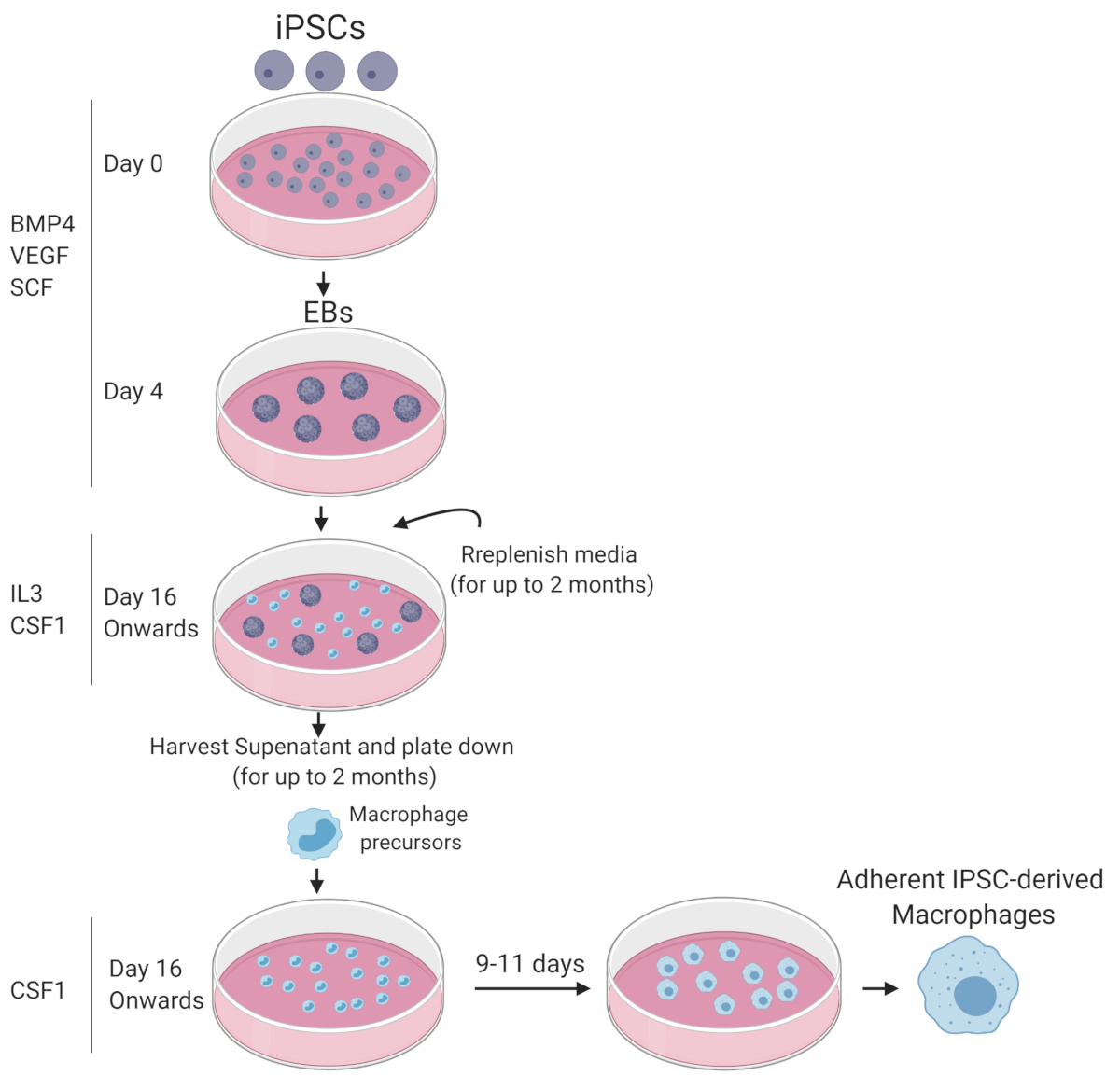
Protocols describing the generation of macrophages from iPSCs consist of a three-step process that includes: 1) Generation of embryoid bodies; 2) Emergence of hematopoietic cells in suspension; 3) Terminal macrophage maturation.
The formation of three-dimensional aggregates, known as embryoid bodies (EBs) initiates differentiation of iPSCs. Bone morphogenetic protein (BMP4), stem cell factor (SCF), and vascular endothelial growth factor (VEGF) are added to drive mesoderm specification and support emerging hematopoietic cells7,8,9,11,12. The differentiating cells within the EBs also initiate the activation of endogenous signalling pathways such as Wnt and Activin. Some differentiation protocols do not go through the stage of EB formation. In these cases, Wnt and Activin signalling regulators, such as recombinant human Activin A and/or Chiron are added to the differentiating iPSCs in a monolayer format13,14,15. Here, we focus on a protocol that uses EB formation. For the second step of differentiation, EBs are plated onto an adherent surface. These attached cells are then exposed to cytokines that promote the emergence of suspension cells that include hematopoietic and myeloid progenitors. In these in vitro culture conditions, interleukin-3 (IL3) likely supports hematopoietic stem-progenitor cell formation and proliferation16,17, as well as myeloid precursors proliferation and differentiation18. Macrophage colony stimulating factor (CSF1) supports the production of myeloid cells and their differentiation to macrophages19,20. During the third stage of differentiation, these suspension cells are cultured in the presence of CSF1 to support terminal macrophage maturation.
The differentiation of human iPSCs into macrophages in vitro mimics the early wave of macrophage production during development. Tissue-resident macrophages are established during embryogenesis and have a distinct developmental lineage from adult monocytes. Several studies have shown that iPSC-DMs have a gene signature that is more comparable to fetal liver-derived macrophages than blood-derived monocytes, suggesting that iPSC-DMs are more akin to tissue-resident macrophages. iPSC-DMs express higher levels of genes that encode for the secretion of proteins involved in tissue remodelling and angiogenesis and express lower levels of genes encoding for pro-inflammatory cytokine secretion and antigen presentation activities21,22. In addition, iPSC-DMs have an analogous transcription factor requirement to that of tissue-resident macrophages23,24. Using knockout iPSC cell lines that are deficient in the transcription factors RUNX1, SPI1 (PU.1), and MYB, Buchrieser et al. showed that the generation of iPSC-DMs is SPI1 and RUNX1 dependent, but MYB independent. This indicates that they are transcriptionally similar to yolk-sac derived macrophages that are generated during the first wave of hematopoiesis during development23. Therefore, it is widely accepted that iPSC-DMs represent a more appropriate cell model to study tissue-resident macrophages such as microglia14,25 and Kupffer cells11, and a more desirable source of cells that could potentially be used in therapies to repair tissues. For example, it has been shown that macrophages produced in vitro from mouse ESCs were effective in ameliorating fibrosis in a CCl4-induced liver injury model in vivo11. Furthermore, these ESC-derived macrophages were more efficient than bone marrow-derived macrophages at repopulating Kupffer cell compartments depleted of macrophages using liposomal clodronate11 in mice.
Here, we describe serum- and feeder-free protocols for the maintenance, freezing, and thawing of human iPSCs, and for the differentiation of these iPSCs into functional macrophages. This protocol is very similar to that described by Van Wilgenburg et al.12, with minor alterations including: 1) iPSC-maintenance media; 2) ROCK inhibitor is not used in the EB formation stage; 3) A mechanical approach rather than an enzymatic approach is used to generate uniform EBs from iPSC colonies; 4) The method for EB harvest and plating down is different; 5) Suspension cells are harvested 2x a week, rather than weekly; and 6) Harvested suspension cells are cultured under CSF1 for macrophage maturation for 9 days rather than 7 days. We also describe protocols used to characterize iPSC-derived macrophage phenotype and function, including analyses for gene expression (qRT-PCR), cell surface marker expression (flow cytometry), and functional assays to assess phagocytosis and polarization.
Protocol
NOTE: All reagents and equipment used in this protocol are listed in Table of Materials. Media should be at 37 °C for cell culture. Media and reagents used in the differentiation protocol must be sterile.
1. Human iPSC Line Thawing and Maintenance
- Prepare the cell maintenance medium, growth factors, and other reagents.
- Prepare hESC-serum free media (hESC-SFM; see Table of Materials) by supplementing Dulbecco's modified Eagle medium-F12 (DMEM/F12) with hESC supplement, 1.8% w/v bovine serum albumin (BSA), and 0.1 mM 2-mercaptoethanol.
- Prepare human basic fibroblast growth factor (bFGF) stock solution (10 μg/mL) by dissolving bFGF in a sterile 0.1% human serum albumin (HSA)-phosphate buffered saline (PBS) solution. Distribute the stock solution as 200 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 1 year. Once thawed, stock bFGF can be stored at 4 °C for 7 days.
- Prepare Rho kinase inhibitor (ROCK Inhibitor)-Y27632 stock solution (1 mg/mL) by dissolving it in sterile water. Distribute the stock solution as 50 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 1 year. Once thawed, stock ROCK Inhibitor can be stored at 4 °C for 7 days.
- Dilute stem cell substrate (see Table of Materials) 1:50 in Dulbecco’s Phosphate Buffered Saline with calcium and magnesium.
- Place the diluted stem cell substrate solution on culture plates so the final volume per surface area is 78 μL/cm2. To coat the well of a 6 well plate, add 750 μL of solution.
- Incubate the coated plate for 1 h in a humified atmosphere at 37 °C and 5% CO2.
- Aspirate the stem cell substrate coating and add 1 mL of hESC supplemented with 20 ng/mL bFGF and 10 μM ROCK Inhibitor.
- To thaw a vial of frozen human iPSC cells, incubate the vial at 37 °C until thawed and transfer the cells into 5 mL hESC-SFM media.
- Centrifuge cells at 100 x g for 3 min.
- Resuspend in 0.5 mL hESC-SFM supplemented with 20 ng/mL bFGF and 10 μM ROCK Inhibitor. Transfer cells to the coated well.
- Culture cells for 24 h.
- Change media to hESC-SFM supplemented with 20 ng/mL bFGF, but without ROCK inhibitor.
- To maintain the cells, change the medium every day until the cells reach 80% confluency. Undifferentiated iPSCs usually take 3–4 days to reach 80% confluency.
- Once the cells have reached 80% confluency, passage cells.
- Replace spent culture medium with 1.5 mL of fresh hESC-SFM supplemented with 20 ng/mL bFGF (without ROCK inhibitor).
- Hold culture vessel in one hand and roll a disposable cell passaging tool (see Table of Materials) across the plate in one direction (i.e., left to right). All blades in the roller must be touching the plate. Maintain uniform pressure while rolling.
- Repeat rolling in the same direction until the whole well has been covered.
- Rotate culture vessel 90° and repeat rolling as described in steps 1.12.2 and 1.12.3.
- Discard passaging tool after use.
- With a sterile pipette, use media in the well to dislodge cut colonies.
- Transfer cells at a 1:4 ratio onto precoated stem cell substrate wells (steps 1.2–1.5) to a final media volume (hESC -SFM supplemented with 20 ng/mL bFGF) of 1.5 mL per well.
2. Human iPSC Line Freezing
- To freeze iPSC cells, replace media of a 70%–80% confluent well of a 6 well plate with hESC-SFM supplemented with 20 ng/mL bFGF and 10 μM ROCK Inhibitor.
- Incubate well at 37 °C and 5% CO2 for 1 h.
- Cut colonies with the cell passaging tool and place dislodged colonies into a centrifuge tube.
- Centrifuge cells at 100 x g for 3 min.
- Aspirate the media and resuspend the cells in 1 mL of cell cryopreservation media (see Table of Materials).
- Divide cells equally into two cryovials and place them into a pre-chilled cell cryopreservation container at 4 °C.
- Store cells at -80 °C for 24–48 h.
- Transfer vials to either a -135 °C freezer or to a liquid nitrogen tank.
3. Human iPSC Differentiation to Macrophages
NOTE: A schematic summary of the macrophage differentiation protocol is depicted in Figure 1.
- Preparing cell differentiation growth factors and other reagents
- Prepare hESC-SFM media (see previous section).
- Prepare 0.1% w/v solution of porcine gelatin by dissolving the gelatin into sterile water. Gelatin solution can be stored at 4 °C for up to 2 years.
- Prepare human BMP4 stock solution (25 μg/mL) by dissolving BMP4 into a 4 mM hydrogen chloride (HCl)-0.2% BSA PBS solution. Distribute the stock solution as 50 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 1 year. Once thawed, stock BMP4 can be stored at 4 °C for 5 days.
- Prepare human VEGF stock solution (100 μg/mL) by dissolving VEGF into a 0.2% BSA PBS solution. Distribute the stock solution as 10 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 1 year. Once thawed, stock VEGF can be stored at 4 °C for 7 days.
- Prepare human SCF stock solution (100 μg/mL) by dissolving SCF into a 0.2% BSA PBS solution. Distribute the stock solution as 5 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 1 year. Once thawed, stock SCF can be stored at 4 °C for 10 days.
- Prepare human IL3 stock solution (10 μg/mL) by dissolving IL3 into a 0.2% BSA PBS solution. Distribute the stock solution as 500 μL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 2 years. Once thawed, stock SCF can be stored at 4 °C for 15 days.
- Prepare human CSF1 stock solution (10 μg/mL) by dissolving CSF1 into a 0.2% BSA PBS solution. Distribute the stock solution as 1 mL aliquots in cryotubes. Stock solutions can be stored at -20 °C up to 2 years. Once thawed, stock SCF can be stored at 4 °C for 15 days.
- Prepare separate 10 μg/mL stock solutions of interferon-gamma (IFNg), interleukin 4 (IL4), and interleukin 10 (IL10) by dissolving into 0.2% BSA PBS solutions. Prepare lipopolysaccharide (LPS) to a stock solution of (100 U/mL) by dissolving into a 0.2% BSA PBS solution. Distribute each stock solution as 35 μL aliquots. Store at -80 °C up to 2 years. Once thawed, stocks can be stored at 4 °C for 7 days.
- Stage 1: Generation of embryoid bodies (day 0–day 3)
- On day 0, add 2.25 mL of Stage 1 media (hESC-SFM supplemented with 50 ng/mL BMP4, 50 ng/mL VEGF, and 20 ng/mL SCF) into two wells of an ultralow attachment 6 well plate.
- Replace maintenance media of one 80% confluent well of iPSCs in a 6 well plate with 1.5 mL of Stage 1 media.
- Cut colonies using the cell passaging tool and transfer cut colonies with a pipette into the two wells of an ultralow attachment 6 well plate (see Table of Materials).
- On day 2, bring cytokines to a final concentration of 50 ng/mL BMP4, 50 ng/mL VEGF, and 20 ng/mL SCF using 0.5 mL of hESC-SFM media.
NOTE: IPSC colonies will become EBs (Figure 2A).
- Stage 2: Emergence of hematopoietic cells in suspension
- On day 4, coat 4 wells of a 6 well tissue culture plate with 0.1% w/v gelatin and incubate for at least 10 min.
- Remove gelatin and add 2.5 mL of Stage 2 media (X-VIVO15 supplemented with 100 ng/mL CSF1, 25 ng/mL IL-3, 2 mM glutamax, 1% penicillin-streptomycin, and 0.055 mM 2-mercaptoethanol).
- Collect formed EBs into a 50 mL centrifuge tube and allow them to settle at the bottom of the tube by gravity. Carefully aspirate the media.
- Resuspend EBs in 2 mL of Stage 2 media.
- Transfer 10–15 EBs (no more than 15) to a gelatin-coated well containing 2.5 mL of Stage 2 media.
- Incubate EBs at 37 °C and 5% CO2 air.
- Change media on plated EBs every 3–4 days for 2–3 weeks.
- After 2–3 weeks, the EBs start releasing nonadherent hematopoietic cells into suspension.
NOTE: This period of suspension cell release can vary and is cell line dependent. Cells in this suspension can be harvested and matured into macrophages (see Stage 3) (Figure 2A).
- Stage 3: Terminal macrophage maturation
- Collect suspension hematopoietic cells and replenish media (Stage 2 media) on EB plate.
- Centrifuge suspension cells at 200 x g for 3 min.
- Resuspend suspension cells in Stage 3 media (X-VIVO15 supplemented with 100 ng/mL CSF1, 2 mM glutamax, and 1% penicillin-streptomycin).
- Plate collected and spun cells onto untreated plastic 10 cm bacteriological-grade plates (10 mL) or uncoated 6 well tissue culture plates (3 mL) at a density of 0.2 x 106 cells/mL.
- Keep cells in Stage 3 media for 9–11 days, changing media every 5 days.
NOTE: Steps 3.4.1–3.4.5 from Stage 3 can be repeated every 3–4 days and suspension cells harvested from the original EB plate for up to 3 months.
- Macrophage polarization
- To activate macrophages to an M(LPS + IFNg) phenotype, stimulate the cells with LPS (final concentration: 100 ng/mL) and IFNg (final concentration: 10 U/mL) for 48 h. To activate cells to an M(IL4) phenotype, stimulate cells with IL4 (final concentration: 20 ng/mL). To activate to an M(IL10) phenotype, stimulate macrophages with IL10 (final concentration: 5 ng/mL).
4. iPSC-derived Macrophages Quality Control Check
- Determine the number of hematopoietic suspension cells produced per 6 well plate of EBs by counting them with a hematocytometer.
- Assess macrophage morphology as previously described (e.g., commercial kit staining)8,9.
- Detect the expression of macrophage specific markers and polarization markers using gene expression analyses and flow cytometry as previously described8,9.
- For flow cytometry experiments on one well of a 6 well plate of macrophages, harvest cells by aspirating their maturation media, wash with 2 mL of PBS, and incubate them with 2 mL of enzyme free cell dissociation buffer for 5 min at room temperature (RT). Pipette up and down repeatedly to detach and harvest macrophages.
- Count cells with a hematocytometer and resuspend them in 80 μL of a 2% BSA, 0.5 mM ethylenediaminetetraacetic acid (EDTA)-PBS solution.
- Add 20 μL of MACS human FC blocker.
- Incubate on ice for 20 min and protect from light.
- Add an appropriate volume of 2% BSA, 0.5 mM EDTA PBS solution to bring the cell concentration to 1 x 106 macrophages/mL.
- Stain 1 x 105 cells in 100 μL of 2% BSA, 0.5 mM EDTA PBS solution with corresponding antibody (see NOTE below) and incubate for 15 min at RT protected from light.
- Wash 1x with at least 100 μL of 2% BSA, 0.5 mM EDTA PBS.
- Resuspend the cells in 200 μL of 2% BSA, 0.5 mM EDTA PBS.
- Add 4',6-diamidino-2-phenylindole (DAPI, diluted 1:1,000) as a live-dead dye. Incubate 3 min.
- For flow cytometry analyses, gate on the main population, then single cells, and then live cells. On the live cell population, macrophage-related marker expression is evident (Figure 3A).
NOTE: Antibodies should be carefully titrated for each cell line used to derive macrophages. Antibodies presented in the results section are in the Table of Materials. The dilution factor for SFCi55-derived macrophages flow cytometry assays is also included.
5. High Throughput Phagocytosis Assay
- Harvest iPSC-derived macrophages (iPSC-DMs) by aspirating the media, adding ice cold enzyme free cell dissociation buffer, and incubating for 5 min. Collect macrophages by pipetting repeatedly.
- Plate 8 x 104 iPSC-DMs in a well of an imaging tissue culture grade 96 well plates (e.g., Cellcarrier Ultra, Perkin Elmer) at least 2 days before high throughput imaging in 200 μL of Stage 3 media.
- Prepare pHrodoGreen Zymosan-A Bioparticles by resuspending one vial in 2 mL of PBS (“Solution 1”). Vortex solution for 10 s.
- Dilute the 2 mL of PBS bead suspension 1:5 with more PBS (“Solution 2”).
- Sonicate Solution 2 for 8 s and vortex solution for 10 s. Keep it at 4 °C. This solution will be used in step 5.11.
- Remove the media on plated iPSC-DMs and wash with PBS.
- Stain iPSC-DMs with a PBS solution containing Hoechst 33342 diluted 1:20. Incubate for 20 min at 37 °C.
- Wash cells with PBS.
- Stain with a PBS Solution containing deep red plasma membrane stain diluted 1:1,000 (see Table of Materials). Incubate for 30 min at 37 °C.
- Wash cells with PBS.
- Add 100 μL of bead solution kept at 4 °C to each well of iPSC-DMs. The plates are now ready for imaging.
- Image the plate by using a high content imaging system and acquire three or more fields across the well to obtain a good representation of the well.
- Quantify phagocytosis by using Columbus software (High-Content Imaging analysis system software). A specific algorithm can be developed for unambiguous image batch analysis:
- Measure blue intensity and define in the software that blue signal indicates the nuclei.
- Measure red intensity and define in the software that red signal indicates the cytoplasm.
- Define that nucleus and cytoplasm together corresponds to a cell.
- Measure green intensity in the cells and establish a strict cut-off/threshold value to consider a cell as phagocytic.
- Quantify the phagocytic cell fraction and the average phagocytic index per cell. Because bead color intensity is proportional to the number of beads, phagocytic activity can be measured by the number of beads ingested.
- Apply the algorithm/pipeline to all images within every field and at all time-points acquired, allowing a robust and unbiased batch approach to determine the phagocytic capabilities of cells.
NOTE: Columbus is a high content analysis software, which offers cell segmentation analysis for cell phenotyping and functional testing.
Representative Results
Differentiation progression, macrophage number, and morphology
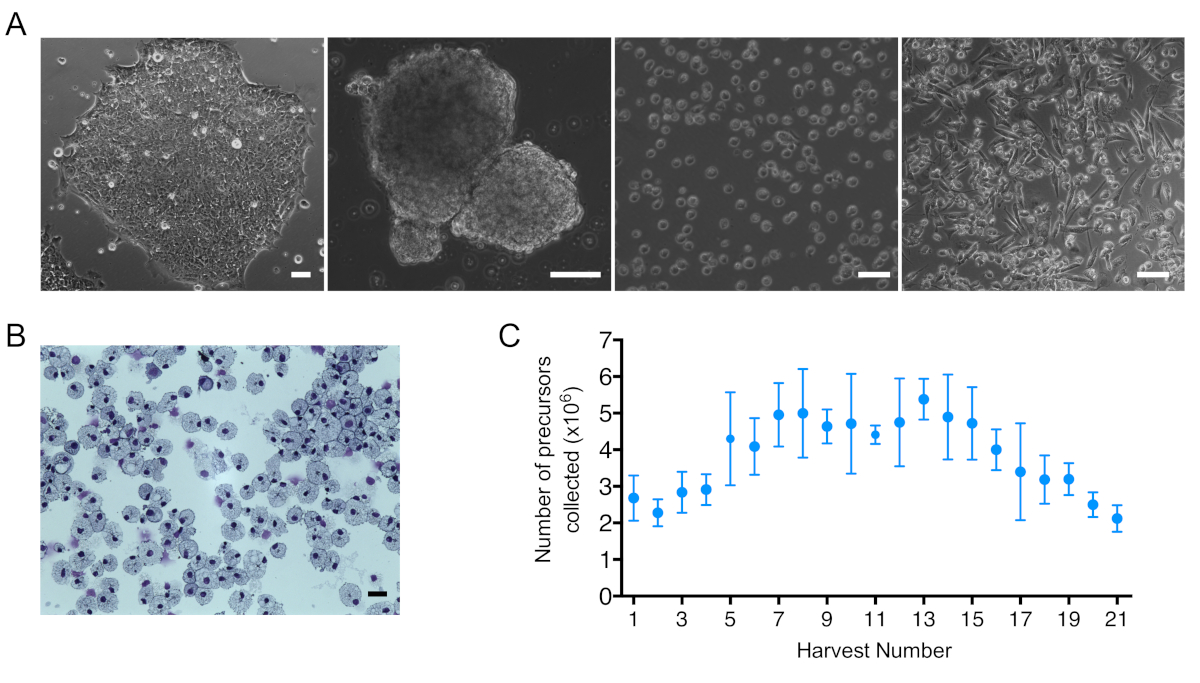
The results presented are from the differentiation of the SFCi55 human iPSC line that has been described and used in a number of studies8,9,10,26. The process of IPSC differentiation towards macrophages could be monitored by optical microscopy. iPSC colonies, embryoid bodies (EBs), hematopoietic suspension cells, and mature macrophages were morphologically distinct (Figure 2A). Mature macrophage morphology could be further validated by staining of centrifuged cytospin preparations. IPSC-derived macrophages were large, and had single small oval-shaped nuclei and abundant cytoplasm containing many vesicles (Figure 2B).
The first 2 weeks of hematopoietic suspension cells harvests (days 16–28) of one full 6 well plate of EBs contained, on average, 2.59 x 106 ± 0.54 cells. After day 28, an average of 4.64 x 106 ± 0.94 of suspension cells per 6 well plate of EBs were produced. From day 80 onwards, the number of suspension cells started to drop as the EBs become exhausted (Figure 2C). It is recommended to stop the differentiation protocol after numbers drop below 3 x 106 precursors per harvest per 6 well plate of EBs.
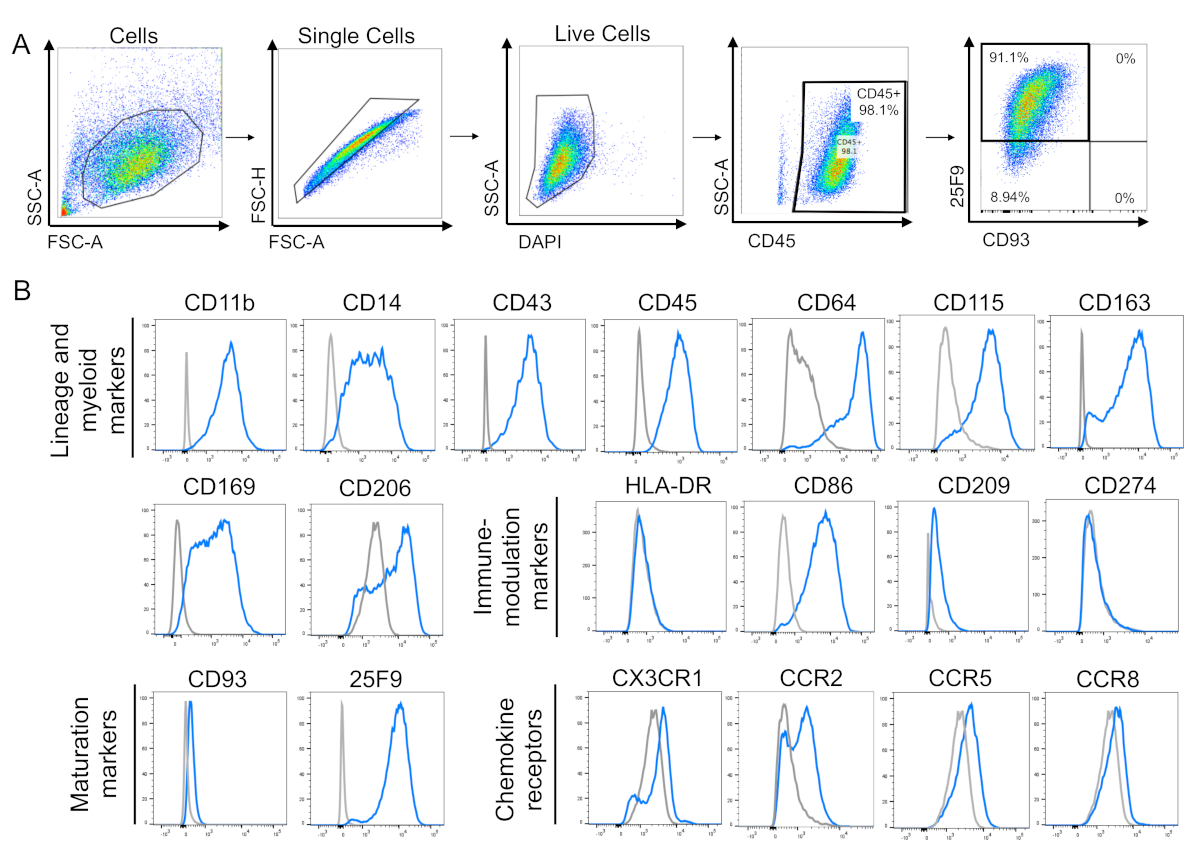
IPSC-derived macrophage cell surface markers expression
Flow cytometry remains the most common method used to assess the phenotype of human macrophages. The gating strategy to assess cell surface marker expression consists of gating the main population of cells using physical parameters like size and granularity, followed by gating single cells and then live cells (Figure 3A). Mature iPSC-derived macrophages should express the lineage marker CD45 and macrophage maturation marker 25F9, and be negative for monocyte/immature macrophage marker CD93. This is consistent with our observations (Figure 3A). IPSC-derived macrophages were also positive for lineage myeloid markers CD11b, CD14, CD43, CD64, CD115, CD163, and CD169 (Figure 3B). They were positive for immune-modulation marker CD86, and a small proportion of them expressed chemokine receptors CX3CR1, CCR2, CCR5, and CCR8 at the naive state (Figure 3B). The plots were obtained from data previously published by our laboratory8.
iPSC-derived macrophages phagocytosis and polarization
One of the key features of macrophages in host defense and tissue homeostasis is their ability to phagocytose pathogens, apoptotic cells, and debris27. The rate of phagocytosis is closely associated with specific phenotypic states28. To evaluate iPSC-DM phagocytic ability, we used a high content imaging system approach8,9,11 that makes use of the PerkinElmer Operetta Microscope and pHrodo Zymosan bioparticles (pH-sensitive dye conjugates). Bioparticles were nonfluorescent when added to the cultures (Figure 4A) but fluoresced bright green in the intracellular acidic pH (Figure 4B). The live-imaging Operetta microscope was set to image every 5 min after the addition of beads for a total time of 175 min. A high throughput and unbiased image analysis pipeline was then used in the Columbus platform to quantify activity in terms of the phagocytic fraction that represents the proportion of cells that phagocytosed beads and the phagocytic index that is a measure of the number of beads that each cell ingested.
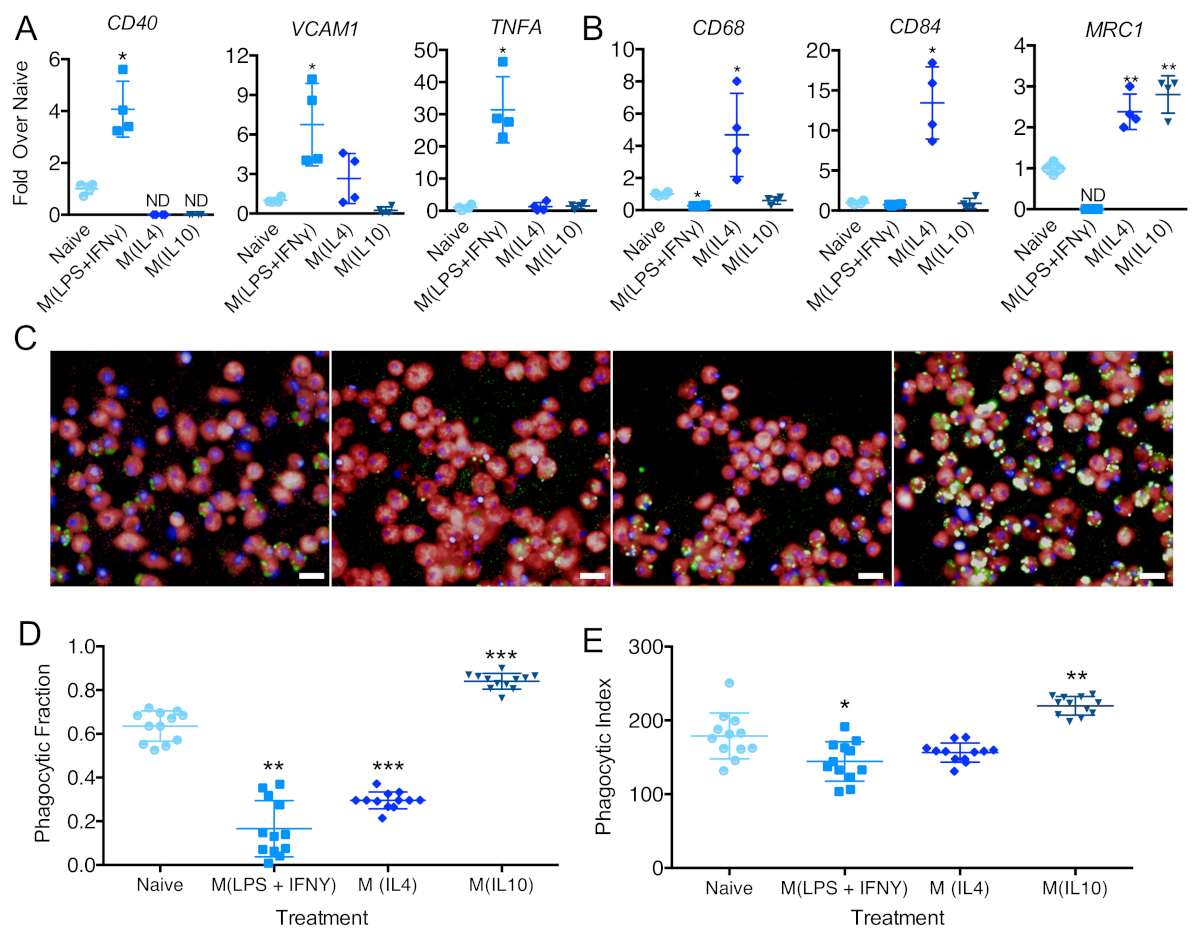
Macrophages can respond and change their phenotype depending on environmental cues. To assess their ability to react and change upon environmental stimuli, iPSC-DMs can be treated with LPS and IFNg, IL4, or IL10. After 48 h of treatment, they changed phenotype, herein referred to as M (LPS + IFNg), M (IL4), and M (IL10), respectively29. Gene expression analysis is a useful tool to test the polarization status of macrophages. Upon LPS and IFNg stimulation, macrophages upregulated mRNA expression of genes CD40, VCAM1, and TNFA (Figure 5A). Upon IL4 stimulation, cells upregulated mRNA expression of genes CD68, CD84, and MRC1 (Figure 5B). Upon IL10 stimulation, iPSC-DMs upregulated expression of MRC1 (Figure 5B). In terms of phagocytosis, macrophages treated with LPS + IFNg or IL4 showed a significantly lower percentage of phagocytic cells when compared to naive macrophages. iPSC-DMs treated with IL10 showed an increased percentage of phagocytic cells and phagocytic index (Figure 5C–E).

Figure 1: Graphic summary of iPSC differentiation to mature functional macrophages. Diagram drawn with Biorender. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: iPSC differentiation towards macrophages and iPSC-DM number and morphology. (A) Bright Field images obtained from (left to right): an IPSC colony, embryoid bodies (EBs), harvested suspension cells, and mature macrophages. Scale bar = 100 μm. (B) Image of macrophage cytospins stained with Kwik-diff kit. Scale bar = 25 μm. (C) Number of suspension cells collected per harvest per one 6 well plate of EBs. Plot shows mean + SEM; (n = 6 biologically independent experiments). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Macrophage cell surface marker phenotype. (A) Gating strategy for analysis of mature iPSC-derived macrophages. Single, live cells were gated, then analysed for the expression of cell surface markers CD45, CD93, and 25F9. Gates for the cell surface markers were drawn using fluorescence minus one (FMO) controls. (B) Representative flow cytometry histograms for single stains of iPSC-DMs (blue) and isotype controls (grey) for lineage and myeloid markers, immune-modulation markers, maturation markers, and chemokine receptors. Plots are representative of n = 5 biologically independent experiments for all lineage and myeloid markers, except CD105 and CD206 (n = 3); maturation markers (n = 5); immune-modulation markers (n = 3); and chemokine receptors (n = 3). Plots use previously published data8. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: IPSC-DM polarization and phagocytosis assays. Representative images of iPSC-DMs (A) immediately after the addition of Zymosan pHrodo green beads and (B) 175 min after the addition of beads. Blue represents the cells’ nuclei; red represents the cells’ cytoplasm. Green represents ingested beads (Scale bar = 20 μm). (C) Fraction of phagocytic macrophages and (D) phagocytic index/green intensity per phagocytic macrophage over time in the naive state (n = 6 biologically independent experiments). Plots show the mean value and the bars represent the SEM. Plots use previously published data8. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Evaluation of iPSC-DM polarization states. Relative quantification of RT-PCR analyses of naive and polarized iPSC-DMs to assess the expression of (A) M(LPS+IFNϒ); (B) M(IL4) and M (IL10)-associated genes (n = 6 biologically independent experiments; One-way ANOVA and Holm-Sidak's multiple comparisons post-test. Polarized groups were statistically compared to the naive group only). Plots show the mean value, and the error bars represent the standard deviation. ND in plots = transcript not detected. Data for these plots were previously published8. (C) Representative images of iPSC-DMs 175 min after the addition of pHrodo beads from iPSC-DMs treated with (left to right): no cytokines, LPS+IFN-Y, IL-4, and IL-10 (Scale bar = 20 μm). (D) Fraction of phagocytic macrophages and (E) phagocytic index/green intensity per phagocytic macrophage in naive and polarized macrophage treatments 175 min after the addition of beads (n = 12 biologically independent experiments, one-way ANOVA and Holm-Sidak's multiple comparisons post-test. Polarized groups were statistically compared to the naive group only). Plots show the mean value and the error bars represent the standard deviation (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Please click here to view a larger version of this figure.
{kind=link}
Discussion
The protocol for the generation of iPSC-DMs described here is robust and allows for the production of a large number of homogeneous cells from a relatively small number of iPSCs. Following the initial differentiation of approximately 1 x 106 iPSCs, the subsequent cultures can be harvested every 4 days for up to 2–3 months, resulting in the production of at least 6.5 x 107 macrophages over that time. These in vitro-generated human macrophages are morphologically similar to primary human macrophages, express the key macrophage cell surface markers, and exhibit phagocytic activity. The protocol for macrophage differentiation is reproducible and can be applied to other hiPSC and hESC cell lines, but the precise timing of the first harvest of the macrophage precursors and the absolute numbers of cells that can be generated varies between iPSC lines.
It has been demonstrated that macrophages can be generated from iPSCs that have been genetically manipulated. For example, a transgene cassette consisting of the fluorescent reporter ZsGreen under the control of the constitutive CAG promoter was inserted into the AAVS1 locus of the SFCi55 iPSC line, and it was subsequently shown that this iPSC line could be differentiated into ZsGreen-expressing macrophages8. These fluorescent macrophages could be used in the future to track the migration and stability of therapeutic macrophages in models of disease. In another study, macrophages were generated from an iPSC line that had been genetically manipulated to express a tamoxifen-induced transcription factor, KLF1. Activation of KLF1 in iPSC-derived macrophages resulted in the production of macrophages with a phenotype comparable to macrophages of the erythroid island9. Potentially, this strategy could be used to genetically program iPSC-derived macrophages into phenotypes associated with other tissue-specific macrophage populations such as Kupffer cells of the liver or Langerhans cells of the skin. This would be possible once the key transcription factors that define these cell types are identified.
In terms of the protocol, it is very important to note that the condition of the starting population of iPSCs is critical for successful differentiation. Human iPSC cultures can be overrun with karyotypically abnormal subpopulations over several passages, so robust curation of iPSC stocks and large batch master stocks subjected to genome quality control is recommended. In our hands, the maintenance protocols described here can maintain karyotypically normal iPSCs for up to 2 months in continuous culture, but this may vary for different cell lines and in different laboratories. If problems are encountered, it is advisable to use a fresh vial of undifferentiated iPSCs for each differentiation experiment. In addition, the starting culture of undifferentiated iPSCs should be no more than 80% confluent. At the EB plating stage, only 10–15 EBs should be plated per well of a 6 well tissue culture plate, and it is critical that these EBs are spread out evenly across the well. A higher number of EBs and/or clumping of EBs in the center of the well had a negative effect on the numbers of macrophages generated. Care should be taken when replenishing media and harvesting monocyte-like progenitor suspension cells from the EB cultures to avoid disturbing the adhesion of EBs to the surface of the coated culture plates. The number of hematopoietic suspension cells produced gradually increases with each harvest, with optimal production between days 40–72 of differentiation (Figure 2). Production progressively declines after day 68 and plates tend to exhaust after 2.5 months, although the precise timing can vary depending on the iPSC line.
One limitation of our protocol is that it has not been possible to cryopreserve the hematopoietic suspension cells generated at the end of Stage 2. Protocols that rely on the exogenous activation of WNT report about a 40% recovery rate after cryopreservation, but these protocols report only one cell harvest, so the absolute number of macrophages generated is low30. The protocol described here, inducing endogenous signalling via the formation of EBs, can be harvested biweekly, producing a much higher total macrophage yield.
In summary, we present a detailed protocol for the production of functional iPSC-derived macrophages. Setting up in vitro experiments with iPSC-derived macrophages to study macrophage biology in health and disease has many advantages over experiments with monocyte-derived macrophages (MDMs). These advantages include the ease of accessibility to the material (e.g., no donors are required), very large amounts of macrophages can be produced, and it is feasible and relatively straightforward to produce genetically modified macrophages. Furthermore, iPSC-derived macrophages might be better resource for the study of tissue-resident macrophage biology.
Acknowledgements
We thank Fiona Rossi and Claire Cryer for assistance with flow cytometry, Eoghan O’Duibhir and Bertrand Vernay with microscopy. This work was funded by CONACYT (M.L.-Y.), Wellcome Trust (102610) and Innovate UK (L.M.F), Wellcome Trust PhD studentship (A.M), MRC Precision Medicine Studentship (T.V). L.C. and J.W.P. were supported by Wellcome Trust (101067/Z/13/Z), Medical Research Council (MR/N022556/1), and COST Action BM1404 Mye-EUNITER (http://www.mye-euniter.eu).
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol (50 mM) | Invitrogen | 31350010 | |
| Abtibody CD64-APC -CY7 | Biolegend | 305026 | Dilution factor: 1:100 |
| Antibody 25F9-EFLUOR 660 | Ebioscience | 15599866 | Dilution factor: 1:20 |
| Antibody CCR2-PE-Cy7 | Biolegend | 357212 | Dilution factor: 1:100 |
| Antibody CCR5 PE | Biolegend | 313707 | Dilution factor: 1:100 |
| Antibody CCR8 PE | Biolegend | 360603 | Dilution factor: 1:100 |
| Antibody CD11b-PE | Biolegend | 301305 | Dilution factor: 1:50 |
| Antibody CD14-APC | Ebioscience | 10669167 | Dilution factor: 1:20 |
| Antibody CD163-PE-CY7 | BIolegend | 333614 | Dilution factor: 1:25 |
| Antibody CD169-APC | Biolegend | 346007 | Dilution factor: 1:25 |
| Antibody CD206-PE | Biolegend | 321106 | Dilution factor: 1:100 |
| Antibody CD209-PE-CY7 | Biolegend | 3310114 | Dilution factor: 1:100 |
| Antibody CD274-PE-CY7 | Biolegend | 329718 | Dilution factor: 1:100 |
| Antibody CD43-PE | Ebioscience | 10854419 | Dilution factor: 1:100 |
| Antibody CD45-APC | Ebioscience | 15577936 | Dilution factor: 1:20 |
| Antibody CD86-APC | Biolegend | 305412 | Dilution factor: 1:100 |
| Antibody CD93-PE | Ebioscience | 10804637 | Dilution factor: 1:100 |
| Antibody CX3CR1-PE | Biolegend | 307650 | Dilution factor: 1:100 |
| Antibody HLA-DR-BV650 | Biolegend | 307650 | Dilution factor: 1:100 |
| Antiboy CD115-PE | Biolegend | 347308 | Dilution factor: 1:40 |
| Cell Dissociation Buffer, enzyme free | Thermofisher | 13151014 | |
| Cell Dissociation Buffer, enzyme-free, PBS | Gibco | 13151014 | |
| CellCarrier-96 Ultra Microplates, tissue culture treated, black, 96-well | PerkinElmer | 6055302 | |
| CellMask Deep Red Plasma Membrane Stain | Thermofisher | C10046 | Cryopreservation media |
| Cryostor CS10 | Sigma | C2874 | |
| CTS CELLstart Substrate | Invitrogen | A1014201 | Stem cell substrate |
| DAPI | Merck | D9542-1MG | Final concentration 1 μg/mL |
| DPBS, calcium, magnesium (500ml) | Thermofisher | 14040091 | |
| FcR Blocking Reagent, human | MACS Miltenyi Biotec | 130-059-901 | |
| FGF-Basic (AA 10-155) Recombinant Human Protein | Thermofisher | PHG0021 | |
| GlutaMAX Supplement | Thermofisher | 35050061 | |
| Human Recombinant IFNY | Thermofisher | 14-8319-80 | |
| Human Serum Albuminum | Irvine Scientific | 9988 | |
| Lipopolysaccharide (LPS) from E. Coli | Sigma | L2630 | |
| NucBlue (Hoechst33342) | Thermofisher | R37605 | |
| pHrodo Green Zymosan Bioparticles Conjugate for Phagocytosis | Thermofisher | P35365 | |
| Porcine Skin Gelatin | Sigma | G9136 | |
| Recombinant Human BMP4 Protein | R&D | 314-BP-010 | |
| Recombinant Human IL10 | Preprotech | 200-10 | |
| Recombinant Human IL3 | Preprotech | 200-03-10 | |
| Recombinant Human IL4 | Preprotech | 200-04 | |
| Recombinant Human MCSF (carrier-free) 100ug | Biolegend | 574806 | |
| Recombinant Human VEGF Protein | R&D | 293-VE-010 | |
| Rock Inhibitor Y-27632 | Merck | SCM075 | |
| SCF (C-Kit Ligand) Recombinant Human Protein | Thermofisher | PHC2111 | |
| StemPro hESC SFM | Thermofisher | A1000701 | |
| StemPro EZPassage Disposable Stem Cell Passaging Tool | Thermofisher | 23181010 | |
| Ultralow attachment plates: Cell culture multi-well plate, 6 well, cell star cell repellent surface | Greiner | 657970 | |
| UltraPure 0.5 M EDTA, pH 8.0 | Invitrogen | 15575020 | |
| X-Vivo 15 500 mL bottle | Lonza | BE02-060F | |
References
- Takahashi, K., et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 131 (5), 861-872 (2007).
- Shi, Y., Inoue, H., Wu, J. C., Yamanaka, S. Induced pluripotent stem cell technology: a decade of progress. Nature Reviews Drug Discovery. 16 (2), 115-130 (2017).
- Focosi, D., Amabile, G. Induced Pluripotent Stem Cell-Derived Red Blood Cells and Platelet Concentrates: From Bench to Bedside. Cells. 7 (1), (2017).
- Inoue, H., Nagata, N., Kurokawa, H., Yamanaka, S. IPS cells: A game changer for future medicine. EMBO Journal. 33 (5), 409-417 (2014).
- Lachmann, N., et al. Large-Scale Hematopoietic Differentiation of Human Induced Pluripotent Stem Cells Provides Granulocytes or Macrophages for Cell Replacement Therapies. Stem Cell Reports. 4 (2), 282-296 (2015).
- Kuhn, A., et al. TALEN-mediated functional correction of human iPSC-derived macrophages in context of hereditary pulmonary alveolar proteinosis. Scientific Reports. 7 (1), 1-11 (2017).
- Ackermann, M., et al. Ex vivo Generation of Genetically Modified Macrophages from Human Induced Pluripotent Stem Cells. Transfusion Medicine and Hemotherapy. 44 (3), 135-142 (2017).
- Lopez-Yrigoyen, M., et al. A human iPSC line capable of differentiating into functional macrophages expressing ZsGreen: A tool for the study and in vivo tracking of therapeutic cells. Philosophical Transactions of the Royal Society B: Biological Sciences. 373 (1750), (2018).
- Lopez-Yrigoyen, M., et al. Genetic programming of macrophages generates an in vitro model for the human erythroid island niche. Nature Communications. 10 (1), 881 (2019).
- Cassetta, L., et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell. 35 (4), 588-602 (2019).
- Haideri, S. S., et al. Injection of embryonic stem cell derived macrophages ameliorates fibrosis in a murine model of liver injury. NPJ Regenerative Medicine. 2, 1-10 (2017).
- van Wilgenburg, B., Browne, C., Vowles, J., Cowley, S. A. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One. 8 (8), 71098 (2013).
- Pandya, H., et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nature Neuroscience. 20 (5), 753-759 (2017).
- Douvaras, P., et al. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Reports. 8 (6), 1516-1524 (2017).
- Heideveld, E., et al. Methods for macrophage differentiation and in vitro generation of human tumor associated-like macrophages. Methods in Enzymology. , 1-18 (2019).
- Leary, A. G., et al. Synergism between interleukin-6 and interleukin-3 in supporting proliferation of human hematopoietic stem cells: comparison with interleukin-1 alpha. Blood. 71 (6), 1759-1763 (1988).
- Robin, C., et al. An Unexpected Role for IL-3 in the Embryonic Development of Hematopoietic Stem Cells. Developmental Cell. 11 (2), 171-180 (2006).
- Moldenhauer, A. Hematopoietic progenitor cells and interleukin-stimulated endothelium: Expansion and differentiation of myeloid precursors. BMC Immunology. 9, 56 (2008).
- Kodama, H., Nose, M., Shumpei Niida, S., Yarnasakit, A. Essential Role of Macrophage Colony-Stimulating Factor in the Osteoclast Differentiation Supported by Stromal Cells. Brief Definitive Report. , 1291-1294 (1991).
- Bender, A. T., Ostenson, C. L., Giordano, D., Beavo, J. A. Differentiation of human monocytes in vitro with granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor produces distinct changes in cGMP phosphodiesterase expression. Cellular Signalling. 16 (3), 365-374 (2004).
- Klimchenko, O., et al. Monocytic cells derived from human embryonic stem cells and fetal liver share common differentiation pathways and homeostatic functions. Blood. 117 (11), 3065-3075 (2011).
- Alasoo, K., et al. Transcriptional profiling of macrophages derived from monocytes and iPS cells identifies a conserved response to LPS and novel alternative transcription. Scientific Reports. , 1-15 (2015).
- Buchrieser, J., et al. Human Induced Pluripotent Stem Cell-Derived Macrophages Share Ontogeny with MYB-Independent Tissue-Resident Macrophages. Stem Cell Reports. 8 (2), 334-345 (2017).
- Vanhee, S., et al. In vitro human embryonic stem cell hematopoiesis mimics MYB-independent yolk sac hematopoiesis. Haematologica. 100 (2), 157-166 (2015).
- Abud, E. M., et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 94 (2), 278-293 (2017).
- Yang, C. -. T., et al. Activation of KLF1 Enhances the Differentiation and Maturation of Red Blood Cells from Human Pluripotent Stem Cells. Stem Cells. 35 (4), 886-897 (2017).
- Karavitis, J., Kovacs, E. J. Macrophage phagocytosis: effects of environmental pollutants, alcohol, cigarette smoke, and other external factors. Journal of Leukocyte Biology. 90 (6), 1065-1078 (2011).
- Sica, A., Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. The Journal of Clinical Investigation. 122 (3), 787-795 (2012).
- Murray, P. J., et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 41 (1), 14-20 (2014).
- Cao, X., et al. Differentiation and Functional Comparison of Monocytes and Macrophages from hiPSCs with Peripheral Blood Derivatives. Stem Cell Reports. 12, 1282-1297 (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved