DNA Curtains Shed Light on Complex Molecular Systems During Homologous Recombination
* These authors contributed equally
In This Article
Summary
DNA curtains present a novel method for visualizing hundreds or even thousands of DNA-binding proteins in real-time as they interact with DNA molecules aligned on the surface of a microfluidic sample chamber.
Abstract
Homologous recombination (HR) is important for the repair of double-stranded DNA breaks (DSBs) and stalled replication forks in all organisms. Defects in HR are closely associated with a loss of genome integrity and oncogenic transformation in human cells. HR involves coordinated actions of a complex set of proteins, many of which remain poorly understood. The key aspect of the research described here is a technology called "DNA curtains", a technique which allows for the assembly of aligned DNA molecules on the surface of a microfluidic sample chamber. They can then be visualized by total internal reflection fluorescence microscopy (TIRFM). DNA curtains was pioneered by our laboratory and allows for direct access to spatiotemporal information at millisecond time scales and nanometer scale resolution, which cannot be easily revealed through other methodologies. A major advantage of DNA curtains is that it simplifies the collection of statistically relevant data from single molecule experiments. This research continues to yield new insights into how cells regulate and preserve genome integrity.
Introduction
The maintenance of genome integrity is crucial for proper functioning of all living cells1. Defects in genome integrity can lead to severe health conditions, including various types of cancer and age-related degenerative diseases2. Homologous recombination (HR) uses template-dependent DNA synthesis to repair DNA double-stranded breaks (DSB), single-stranded DNA (ssDNA) gaps, and interstrand DNA crosslinks3. HR is also necessary for the recovery of stalled and collapsed replication forks3,4. Moreover, HR is essential for the accurate chromosome segregation during meiosis5,6.
HR involves the coordinated actions of a complex set of proteins, many of which remain poorly understood1. Examples include replication protein A (RPA), Rad51, and Rad54, among many others7. HR reactions in both prokaryotic and eukaryotic cells involve an ssDNA intermediate, which is rapidly coated by ssDNA-binding proteins (SSB in prokaryotes and RPA in eukaryotes)8. These proteins protect the ssDNA from nucleases, eliminate secondary structure, and promote the recruitment of downstream factors8,9. Rad51 is a member of the highly conserved ATP-dependent Rad51/RecA family of DNA recombinases, which are present in all living organisms1. Rad51 promotes DNA strand invasion of the homologous dsDNA donor. Given its importance, Rad51 is highly regulated, and defects in these regulatory processes are commonly associated with a loss of genome integrity and oncogenic transformation7. Rad54 is a member of the Swi2/Snf2 family of dsDNA translocases and chromatin remodelers10,11. These proteins serve as essential Rad51 regulatory factors. Importantly, Rad54 removes Rad51 from the dsDNA product of strand invasion and is also necessary to prevent misaccumulation of Rad51 on chromatin11. Molecular activities for proteins involved in HR both in yeast and bacterial cells have shed light on their function in HR, but exactly how their activity contributes to HR remains poorly understood 12.
DNA curtains have emerged as a unique platform providing direct access to molecular mechanisms and macromolecular dynamics that would otherwise remain inaccessible13,14. To prepare DNA curtains, the surface of a microfluidic chamber is coated with a lipid bilayer, and DNA molecules are tethered to the bilayer through a biotin-streptavidin linkage. The bilayer renders the surface inert by mimicking natural cell membranes. Hydrodynamic force aligns the DNA along nanofabricated barriers, allowing visualization of hundreds of molecules in a single field-of-view by total internal reflection fluorescence microscopy (TIRFM). The barriers are made by electron-beam lithography, and variations in barrier design allow for precise control over the distribution and tethering geometry of the DNA. These approaches are readily applicable with either ssDNA or dsDNA13,14,15,16,17,18. Pedestals may be also nanofabricated (together with the barriers) to allow both ends of the DNA to be tethered to the sample chamber surface such that steady state experiments may be carried out in the absence of buffer flow.
Time-dependent changes in individual protein-nucleic acid complexes are revealed by the inspection of real-time videos and are represented in print using kymographs, which present the changing position of proteins on DNA over time. An important aspect of the DNA curtains approach is that it does not necessarily require a priori models or assumptions about molecular mechanisms, because the behavior of individual reaction components can be observed in real-time. This allows the direct observation of molecular behaviors. Herein, this protocol describes how to prepare DNA curtains with dsDNA substrates as well as its application to study intermediates in homologous recombination.
Protocol
1. Preparation of lipid stock
- Dissolve 1 g of 18:1 (Δ9-Cis) PC, 100 mg of 18:1 PEG2000 PE, and 5 mg of 18:1 biotinyl cap PE in 10 mL of chloroform.
NOTE: Both DOPC and DPPE are well-characterized and (along with PEG modification) selected for their ability to minimize non-specific adsorption to the surface of the flow cell. Dissolved lipid master mix can be aliquoted and stored at -20 ˚C up to 12 months. - Use chloroform to clean an organic-solvent glass syringe, then transfer 200 µL (10% of the final desired volume) of the lipid master mix to the glass vial.
- Evaporate the chloroform from the glass vial using a very gentle stream of compressed nitrogen. Slightly increase the pressure towards the end of the evaporation to ensure that all traces of chloroform are removed.
NOTE: Tilt and rotate the vial during the evaporation process so that the lipids dry as a uniform layer of white film on the wall. - Place the vial uncapped under vacuum overnight.
- Add 2 mL of lipid buffer (10 mM Tris-HCl [pH 8.0], 100 mM NaCl) to the dried lipid film and cap the vial. Incubate at room temperature (RT) for at least 1 h and vortex until dissolved.
- Transfer the solution to a 5 mL polystyrene round-bottom tube and sonicate on ice using a microtip with the following parameters: amplitude = 50, process time = 1.5 min, pulse = 15 s, time off = 2 min. The solution will be clear after sonication (total energy = 1,500-2,000 J).
- Filter the solution through a 0.22 µm PVDF syringe filter and store the lipid stock at 4 ˚C for up to 1 month.
2. Preparation of dsDNA substrate
- Warm the λ-DNA stock (500 μg/mL) to 65 ˚C.
- Prepare annealing/ligation mix containing: 1.6 pM of λ-DNA, 100x molar excess (160 pM) of each oligonucleotide (BioL: 5'Phos-AGG TCG CCG CCC-3Bio and DigR: 5'Phos-GGG CGG CGA CCT-3Dig_N) and 60 µL of 10x T4 ligase reaction buffer (in a total volume of 600 µL).
NOTE: BioL and DigR oligonucleotides are biotinylated (Bio) and modified with digoxigenin (Dig), respectively. Biotinylation allows the attachment of DNA molecules to biotinylated lipids via streptavidin linkages. The Dig modification enables double tethering through binding of anti-Dig antibodies that are coated on pedestals to Dig moieties. - Anneal the oligonucleotides to the λ-DNA cos-ends by incubating the reaction at 65 ˚C for 10 min, then allow it to slowly cool to RT.
- Add 5 µL of T4 DNA ligase and incubate at RT overnight.
NOTE: Annealing and ligation steps may also be carried out in a thermal cycler. - Precipitate the λ-DNA by adding 300 µL of PEG-8000 solution (30% w/v PEG-8000 and 10 mM MgCl2). Incubate at 4 ˚C for at least 1 h.
- Centrifuge at 14,000 x g for 5 min at RT. Carefully remove the supernatant without disturbing the pellet. Optionally, wash the pellet 1x with 500 µL of chilled 70% ethanol.
- Resuspend the pellet in 40 µL of TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]).
3. Nanofabrication of chromium patterns
- Drill two holes at the center of a 1" x 3" quartz microscope slide (~3 cm apart) using a diamond-coated 1.4 mm drill bit on a table-top drill press (see Figure 1 for geometry of the drilling position).
- Clean the drilled slides by submerging them into 200 mL of Piranha solution (75% sulfuric acid [97%] and 25% hydrogen peroxide). Incubate for 30 min.
CAUTION: Extreme care must be taken to avoid all contact with exposed skin. Prepare the solution by very slowly mixing cold hydrogen peroxide into sulfuric acid. Dispose of Piranha waste properly in a designated waste container. - Rinse slides by submerging in 200 mL of double-distilled water. Repeat 2x to eliminate any remaining Piranha. Dry the slides with a gentle stream of compressed nitrogen.
- Using a spin coater (4,000 rpm for 45 s, ramp rate of 300 rpm/s), coat the clean slides with photoresist and conductive polymer as follows:
- Coat slides with a layer of 3% (w/v) polymethylmethacrylate (PMMA; 24.3K MW; dissolved in anisole).
- Coat slides with a layer of 1.5% (w/v) polymethylmethacrylate (PMMA; 495K MW; dissolved in anisole).
- Coat slides with a layer of antistatic agent for electron beam lithography.
- Write custom patterns for barriers and pedestals on the coated slides using electron beam lithography with a scanning electron microscope equipped with nanopattern generation system software.
- Develop the slides by first rinsing off the antistatic agent for the electron beam lithography with double-distilled water. Then, place them into 50 mL tubes containing the developing solution (75% methyl isobutyl ketone [MIBK] and 25% isopropanol; keep chilled at -20 ˚C) and sonicate in an ice bath on low power for 1 min.
- Rinse off the developing solution with isopropanol and dry with a gentle stream of compressed nitrogen.
- Using an electron beam evaporator, deposit a 250 Å thick layer of chrome onto the patterned surface at 0.5 Å/s.
- Lift off the chrome and remaining PMMA using acetone from a squirt bottle, followed by sonicating the slides in an acetone bath for 10 min. Wash again with acetone from a squirt bottle.
- To prevent any deposits from the remaining acetone, rinse the slides with isopropyl alcohol and dry with a gentle stream of compressed nitrogen.
4. Assembly of flow cell
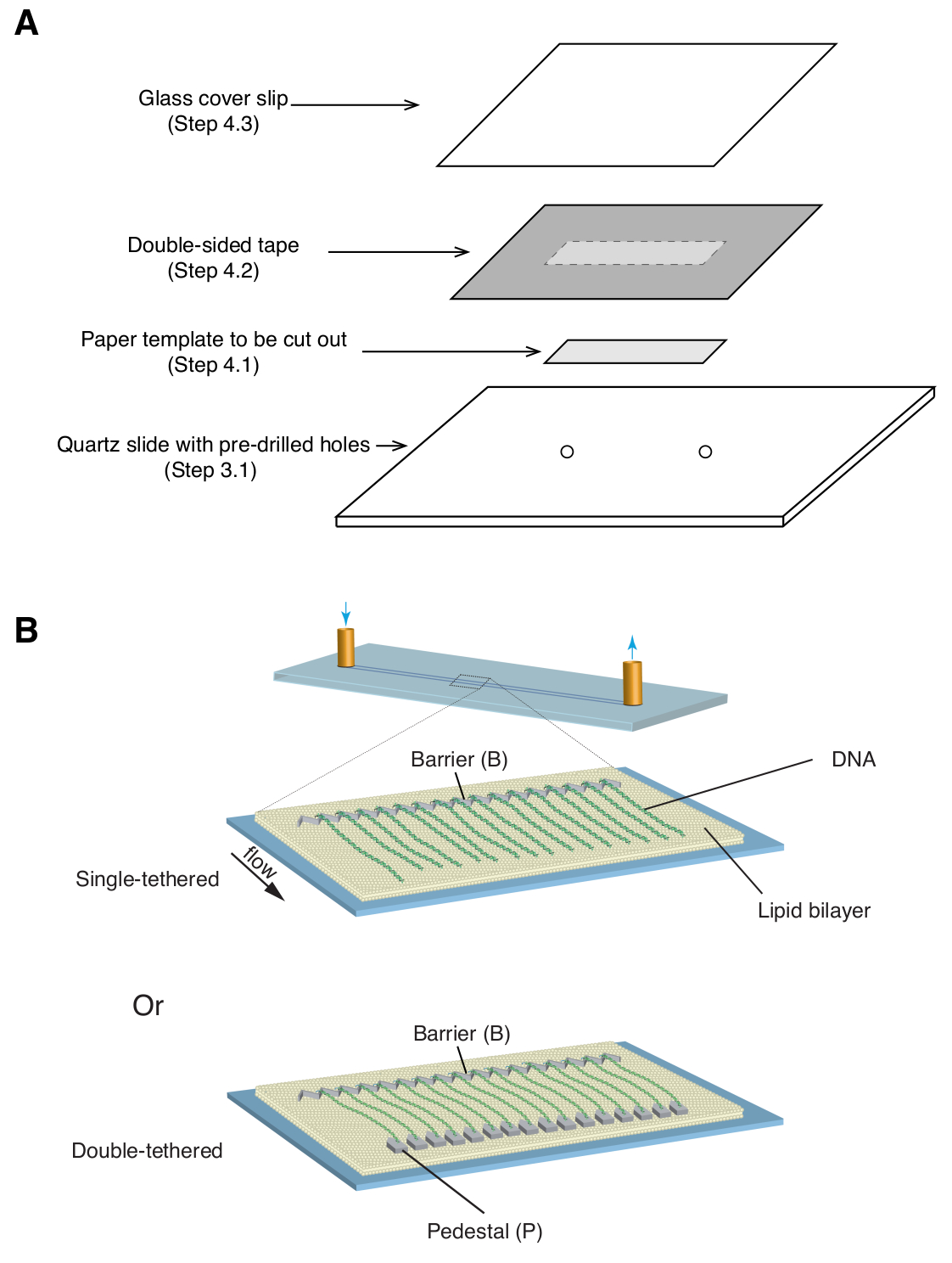
- Place a 35 mm x 5 mm rectangular paper template (covering the chrome pattern as well as the predrilled inlet/outlet holes) over the quartz slide, with the patterned side facing up ( Figure 1A).
- Apply a piece of double-sided tape (1 mm thickness) over the quartz slide, covering the paper template, then use a razor blade to cut out the rectangular paper template ( Figure 1A).
- Place a glass coverslip over the double-sided tape and apply gentle pressure to seal the coverslip to the tape ( Figure 1A). The space between the coverslip and slide, sealed by double-sided tape, forms the microfluidic chamber.
NOTE: It is crucial that the seal between the cover slip and slide is sufficiently secure to prevent leakage during the experiment. - Place the coverslip-slide sandwich in between glass slides and attach binder clips on all four sides to distribute pressure evenly. Place the assembly in a vacuum oven for 60 min at 135 ˚C to melt the double-sided tape and seal the chamber.
- Remove it from the oven, then release the binder clips and glass slides.
- Attach microfluidic ports to the predrilled inlet/outlet holes with a hot glue gun by placing the glued assembly onto a heating block (220 ˚C) for 1 min, taking it off, and carefully applying gentle pressure to properly seal the microfluidic ports. Allow glue to cure completely before proceeding.
NOTE: Assembled flow cells may be placed in 50 mL tubes and stored under vacuum.
5. Assembly of dsDNA curtain
- Attach inlet/outlet tubing to the flow cell microfluidic ports and wash the chamber with 20 mL of double-distilled water using two 10 mL syringes.
- With 10 mL syringe filled with double-distilled water connected to both sides of the flow cell, apply force to push/pull the water between the inlet and outlet syringes. This step removes all air bubbles from the flow cell.
- Wash the chamber with 3 mL of lipid buffer (10 mM Tris-HCl [pH 8.0], 100 mM NaCl).
NOTE: Make sure all connections to flow cells are drop-to-drop to avoid introducing air bubbles into the flow cell. - Mix 40 µL of liposome solution with 1 mL of lipid buffer. Inject 300 µL of the lipid solution into the flow cell and incubate for 5 min. Repeat the injection and incubation 2x.
NOTE: Alternate the inlet/outlet side for injection each time a syringe is connected to the flow cell. - Wash the chamber with 3 mL of lipid buffer and incubate for 15 min.
NOTE: This incubation period can vary from 5 min to 2 h, depending on the scheduling of experiments, without noticeable differences in final curtain assembly. - Mix 40 µL of 1 mg/mL anti-digoxigenin stock solution with 200 µL of lipid buffer. Inject 100 µL of the solution and incubate for 10 min. Repeat the injection and incubation 1x before washing with 3 mL of lipid buffer.
NOTE: This step is needed only for double-tethered dsDNA curtains. Skip this step for single-tethered dsDNA curtains. - Wash the chamber with 3 mL of BSA buffer (40 mM Tris-HCl [pH 8.0], 2 mM MgCl2, 1 mM DTT, and 0.2 mg/mL BSA).
- Mix 10 µL of 1 mg/mL streptavidin stock solution with 990 µL of BSA buffer. Inject as two separate 400 µL injections with a 10 min incubation after each injection.
- Wash the chamber with 3 mL of BSA buffer to remove any excess streptavidin.
- Dilute 25 ng of dsDNA substrate stock in 1 mL BSA buffer. Slowly inject the DNA solution 200 µL at a time, followed by a 5 min incubation after each injection. The amount of DNA needed may be optimized for specific types of experiments.
- Mount the flow cell onto the microscope stage and adjust the focus as necessary.
- Connect the input and output ports to a sample injection system comprised of a syringe pump and high-pressure switch valve.
- Double-tether the DNA to the pedestals (12 µm tether length) by continuously washing the chamber with BSA buffer at 0.8 mL/min for 5 min.
NOTE: It may be necessary to adjust flow rates and times to optimize for different double-tether lengths. This step is needed only for double-tethered DNA curtains. Skip this step for single-tethered dsDNA curtains. - The curtains are now ready for experiments.
6. Recycling of flow cells
- Submerge the slides in ethanol for at least 30 min before using a razor blade to carefully remove the microfluidic ports and coverslip. Properly dispose of the coverslip and clean any remaining glue off the microfluidic ports so they can be reused. Scrub clean any glue residue on the slide.
NOTE: Take care not to scratch the patterned area in the center of the slide. - Thoroughly rinse the slides with double-distilled water and submerge in 2% alkaline liquid quartz cleaning concentrate overnight, with stirring.
- Thoroughly rinse the slides with double-distilled water and submerge in 2% alkaline liquid quartz cleaning concentrate solution for 1 h with stirring at 80 ˚C, followed by sonicating for 15 min at 45 ˚C.
- Thoroughly rinse the slides with double-distilled water and submerge in 1 M NaOH for 30-60 min, with stirring.
- Thoroughly rinse the slides with double-distilled water, then submerge in isopropyl alcohol and sonicate for 15 min at 45 ˚C.
- Thoroughly rinse the slides with ethanol and submerge in ethanol for 1 h, with stirring.
- Place the slides in a vacuum oven for 1 h to dry at 135 ˚C. The slides are now ready to be reassembled into flow cells.
Results
Described above are the preparation, assembly, and imaging of single- and double-tethered dsDNA curtains in the context of studying protein-DNA interactions in DNA repair intermediates. Figure 1A shows all components in a flow cell, layered in the order that they are assembled. Figure 1B depicts the schematic of a single- or double-tethered DNA curtain. A lipid bilayer is used to passivate the surface of the flow cell. The DNA curtain consists of a parallel array of DNA molecules tethered at one end to the lipid and aligned at the chromium barrier, oriented perpendicular to the direction of flow. For double-tethered DNA curtains, the other end of DNA is tethered to the pedestal through Digoxigenin and anti-Digoxigenin interactions, such that imaging can be carried out in the absence of flow while DNA remains extended. dsDNA can be stained with fluorescent dye YoYo1 and visualized by TIRF microscopy. Figure 2A shows a representative wide-field image of YoYo1-stained double-tethered DNA curtain.
Time-lapse image series collected from DNA curtain experiments are typically analyzed by first generating a kymograph, which plots the position along the DNA molecule on the vertical axis vs. time on the horizontal axis for each DNA molecule of interest in ImageJ. Figure 2B are representative kymographs showing the resection of YoYo1-stained single-tethered λ-DNA (green) by the yeast resection machineries GFP-Sgs1 (not visible), Top3-Rmi1, and Dna2 in the presence of single-stranded DNA binding protein RPA-mCherry (magenta) and ATP. YoYo1 signal is lost over time as dsDNA is resected from the free end by Sgs1-Dna2. Simultaneously, mCherry signal colocalizes with DNA ends at which ssDNA is being generated as a result of the resection. Biophysically relevant characteristics of the resection, such as velocity and processivity, can be extracted from by quantifying the slopes of the YoYo1 signal loss trajectories. Figure 2C,D shows the distribution of velocities and processivity, respectively, of resection by Sgs1-Dna2.

Figure 1: Schematics of flow cell assembly and DNA curtains. (A) Stepwise illustration for flow cell assembly. Double-sided tape is placed on top of the quartz slide, and a paper template is used to cut out a rectangular channel from the center, which is sealed with a cover slip on top to form a chamber. (B) Schematics of fully assembled DNA curtains. Middle panel presents a single-tethered flow cell, while the bottom panel presents a double-tethered flow cell. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Single molecule DNA curtain visualization and analysis. (A) Representative wide-field image of a double-tethered λ-DNA curtain stained with YoYo1 (green). (B) Representative kymographs showing the resection of YoYo1-stained dsDNA (green) in the presence of pre-bound GFP-Sgs1 (not visible) when chased with Dna2, in the presence of RPA-mCherry (magenta) and 2 mM ATP. Velocity distribution (C) and processivity plot (D) of dsDNA resection by Sgs1-Dna2, obtained by quantifying the rate and extent of the YoYo1 signal loss over time. Panels (B), (C), and (D) are adapted from Xue et al.19. Please click here to view a larger version of this figure.
{kind=link}
Discussion
DNA curtains has proven to be a versatile platform for studying various DNA repair processes13,14,15,16. DNA molecules that remain extended near the surface throughout the experiment, through either continuous buffer flow or double-tethering to the pedestal, allow for TIRFM imaging of protein-DNA interactions on repair intermediates at the single molecule level. The method provides improvement in experimental throughput and predictability, since hundreds of DNA molecules are orderly aligned at nanofabricated barriers rather than non-specifically attached to the surface, which further benefits processing of large data sets.
Careful analysis of the resulting time-lapse image series provides quantitative measurements of protein-DNA interactions (i.e., binding lifetimes, positions, stoichiometry, dissociation kinetics, translocation rates, processivities of motor proteins, and colocalizations and interactions between proteins and/or DNA). Compared to other single molecule imaging techniques that either tether DNA molecules directly to the surface or combine confocal imaging with force manipulation in commercial instruments, the major trade-off of DNA curtains for higher throughput is establishing the techniques necessary for initial set-up13,14,15,16. Once established, however, the ability to collect data on hundreds of molecules simultaneously in one flow cell leads to significant savings in time spent on data collection.
As with many other single molecule imaging methods, the ability to prepare a clean surface largely dictates the user's ability to reliably assemble DNA curtains. In this case, clean surfaces require not only starting with a clean slide but also making sure not to introduce microscopic air bubbles into the chamber while making any of the fluidic connections. First, regarding cleanliness of the slides, the steps detailed above in section 6 should suffice as a routine cleaning process. Use of filtered solutions and buffers is also recommended. Second, making drop-to-drop connections during attachment of the flow cell to the buffer injection system is essential to avoid introducing bubbles. It may be helpful to degas the double-distilledwater used for making buffers. It is also recommended to carefully observe any injection process to ensure that no bubbles are present. Should a microscopic bubble be introduced before it reaches the center patterned area, it may be remedied by injecting buffer from the microfluidic port on the opposite side to push the bubble back out.
Another important point of consideration in any fluorescence-based experiments is signal-to-noise. A key to observing single fluorescent proteins (i.e., GFP) also resides in clean surfaces. After extended usage, protein aggregates typically accumulate at the chrome barriers and pedestals. Therefore, if a strong fluorescence signal is observed at the barriers prior to the addition of fluorescent proteins, it is advised to treat slides with a 10 min submersion in Piranha solution. The time period is kept short to avoid eroding the chrome features. The Piranha wash should be followed by the regular cleaning procedure.
Nanofabrication using electron beam lithography can be challenging, due to its requirements of equipment and facilities as well as existing expertise in material science. As an alternative, UV lithography-based method for fabrication of chromium features for DNA curtains has been developed20. Also provided with this protocol is a basic pattern for double-tethering lambda DNA at 12 μm (see supplemental information). Several aspects of the design of this pattern may be adjusted to suit various experimental needs. These include the distance between barriers and pedestals, spacing between adjacent wells in the barrier to control DNA density, and the shape and size of pedestals to optimize double-tethering efficiency and minimize uncertainty in double-tethering lengths. Other practical challenges may include choice of fluorescent labeling strategies for proteins, minimizing bias in manual data analysis, and development of scripts for more streamlined data processing.
While the protocol described here involves dsDNA substrates, DNA curtains based on ssDNA have also been used extensively13,14,15,16. Preparation of ssDNA substrates utilizes rolling circle replication of the M13 ssDNA plasmid with biotinylated primers13. Assembly of the ssDNA curtain also requires the use of urea and ssDNA-binding protein RPA to eliminate secondary structure and extend the ssDNA substrate13. It may also be possible to extend DNA curtains to using RNA or DNA/RNA hybrids for studies of RNA-interacting proteins. As it currently stands, the DNA curtain platform can benefit from automation in the assembly process as detailed in section 5, most of which is repetitive injections of a standard set of buffers. This may lead to improved efficiency in carrying out multiple experiments simultaneously. In addition, a unified and potentially machine learning-based suite of analysis scripts would further quicken data processing with minimal biasing.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The authors thank the past and current members of the Greene lab for developing, optimizing, and discussing the DNA curtains protocol, as well as commenting on this manuscript. This work was supported by grants from the National Science Foundation (MCB1154511 to E.C.G.) and the National Institutes of Health (R01CA217973 to E.C.G.).
Materials
| Name | Company | Catalog Number | Comments |
| nano pattern generation system software | JC Nabity Lithography Systems | N/A | NPGS |
| PFA Tubing, Natural 1/16" OD x .020" ID | IDEX | 1512L | |
| Poly(Methyl Methacrylate), Atactic (24.3K MW) | Polymer Source Inc. | P9790-MMA | |
| Quartz Microscope Slide | G. Finkenbeiner Inc. | N/A | 1" x 3" x 1 mm thick |
| Scanning Electron Microscope | FEI | N/A | Nova Nano SEM 450 |
| Scotch Double Sided Tape, 3/4 x 300 inches | 3M | N/A | |
| Sonicator | Misonix | S-4000 | For preparation of lipids |
| Sonicator | Branson | 1800 | For sonicating slides |
| Streptavidin from Streptomyces avidinii | Sigma-Aldrich | S4762 | Dissolved in 1X PBS to a final concentration of 1 mg/mL |
| Sulfuric Acid | Avantor - J.T.Baker | 9681-01 | |
| Syringe Pump | KD Scientific | 78-0200 | |
| Syringe, 500 µL, Model 750 RN SYR, Large Removable NDL, 22 ga, 2 in, point style 2 | Hamilton | 80830 | Reserve for use only with lipids |
| T4 DNA Ligase (400,000 units/ml) | NEB | M0202S |
References
- Kowalczykowski, S. C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harbor Perspectives in Biology. 7, 016410 (2015).
- Ferlay, J., et al. Estimating the global cancer incidence and mortality in 2018: globocan sources and methods. International Journal of Cancer. 144, 1941-1953 (2018).
- Li, X., Heyer, W. D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Research. 18, 99-113 (2008).
- Michel, B., Baoubakri, H., Baharoglu, Z., LeMasson, M., Lestini, R. Recombination proteins and rescue of arrested replication forks. DNA Repair. 6, 967-980 (2007).
- Paques, F., Haber, J. E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and Molecular Biology Reviews. 63, 349-404 (1999).
- Heyer, W. D., Ehmsen, K. T., Liu, J. Regulation of homologous recombination in eukaryotes. Annual Review of Genetics. 44, 113-139 (2010).
- Mason, J. M., et al. RAD54 family translocases counter genotoxic effects of RAD51 in human tumor cells. Nucleic Acids Research. 43, 3180-3196 (2015).
- Wold, M. S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annual Review of Biochemistry. 66, 61-92 (1997).
- Bianco, P. R. The tale of SSB. Progress in Biophysics and Molecular Biology. 127, 111-118 (2017).
- Hopfner, K. P., Gerhold, C. B., Lakomek, K., Wollmann, P. Swi2/Snf2 remodelers: hybrid views on hybrid molecular machines. Current Opinion in Structural Biology. 22, 225-233 (2012).
- Shah, P. P., et al. Swi2/Snf2-related translocases prevent accumulation of toxic Rad51 complexes during mitotic growth. Molecular Cell. 39, 862-872 (2010).
- Wright, W. D., Heyer, W. D. Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D loop formation. Molecular Cell. 53, 420-432 (2014).
- Ma, C. J., Steinfeld, J. B., Greene, E. C. Single-Stranded DNA Curtains for Studying Homologous Recombination. Methods in Enzymology. 582, 193-219 (2017).
- Qi, Z., et al. DNA sequence alignment by microhomology sampling during homologous recombination. Cell. 160, 856-869 (2015).
- Visnapuu, M. L., Greene, E. C. Single-molecule imaging of DNA curtains reveals intrinsic energy landscapes for nucleosome deposition. Nature Structural Molecular Biology. 16, 1056-1062 (2009).
- Kaniecki, K., De Tullio, L., Greene, E. C. A change of view: homologous recombination at single-molecule resolution. Nature Reviews Genetics. 19, 191-207 (2018).
- Redding, S., et al. Surveillance and Processing of Foreign DNA by the Escherichia coli CRISPR-Cas System. Cell. 163, 854-865 (2015).
- Lee, J. Y. Base triplet stepping by the Rad51/RecA family of recombinases. Science. 349, 977-981 (2015).
- Xue, C., et al. Regulatory control of Sgs1 and Dna2 during eukaryotic DNA end resection. Proceedings of the National Academy of Sciences USA. 116 (13), 6091-6100 (2019).
- Gallardo, I. F., Finkelstein, I. J. High-Throughput Universal DNA Curtain Arrays for Single-Molecule Fluorescence Imaging. Langmuir. 31 (37), 10310-10317 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved