Automated Production of Human Induced Pluripotent Stem Cell-Derived Cortical and Dopaminergic Neurons with Integrated Live-Cell Monitoring
* These authors contributed equally
In This Article
Summary
We show the automation of human induced pluripotent stem cell (hiPSC) cultures and neuronal differentiations compatible with automated imaging and analysis.
Abstract
Manual culture and differentiation protocols for human induced pluripotent stem cells (hiPSC) are difficult to standardize, show high variability and are prone to spontaneous differentiation into unwanted cell types. The methods are labor-intensive and are not easily amenable to large-scale experiments. To overcome these limitations, we developed an automated cell culture system coupled to a high-throughput imaging system and implemented protocols for maintaining multiple hiPSC lines in parallel and neuronal differentiation. We describe the automation of a short-term differentiation protocol using Neurogenin-2 (NGN2) over-expression to produce hiPSC-derived cortical neurons within 6‒8 days, and the implementation of a long-term differentiation protocol to generate hiPSC-derived midbrain dopaminergic (mDA) neurons within 65 days. Also, we applied the NGN2 approach to a small molecule-derived neural precursor cells (smNPC) transduced with GFP lentivirus and established a live-cell automated neurite outgrowth assay. We present an automated system with protocols suitable for routine hiPSC culture and differentiation into cortical and dopaminergic neurons. Our platform is suitable for long term hands-free culture and high-content/high-throughput hiPSC-based compound, RNAi and CRISPR/Cas9 screenings to identify novel disease mechanisms and drug targets.
Introduction
Human induced pluripotent stem cells (hiPSC) are self-renewing and can differentiate in almost any adult cell type. These characteristics make hiPSC a useful tool for disease modeling in basic research and drug discovery1. Human iPSC retains the donor genetic background which allows deriving disease-relevant cell types that are most affected/involved in the disease course, for example, different neuronal subtypes for neurodegenerative diseases2,3. Also, hiPSC overcomes some of the limitations of animal and cellular over-expression models by modeling diseases in a human context and physiological protein expression levels, and have proven to be a valuable asset in modeling diseases ranging from monogenic, complex and epigenetic disorders as well as late-onset diseases4.
Despite these benefits and opportunities, several limitations of hiPSC still need to be addressed. Current hiPSC culture and differentiation protocols are not cost-effective, difficult to standardize and are labor-intensive. Manual culture steps can result in high variability in the yields and phenotypes due to differences in growth and spontaneous differentiation of hiPSC. Therefore, experimenter-dependent variation needs to be reduced by implementing more standardized handling techniques and simplifying protocols which can be achieved using automation5. The establishment of automated hiPSC culture and differentiation protocols will set common standards for both academic and industrial research projects, and allow the generation of biologically relevant disease models and more reproducible results.
Previous work has attempted automation of hiPSC cultures6,7,8 but their protocols have been restricted to specific cell culture plate formats dependent on the system and lacking adaptability to different assay formats. Such systems are useful in the bulking of cells but may not be suitable for automated differentiation into desired cell types, disease phenotyping, and screening purposes. Additionally, a large-scale automated platform for fibroblast derivation, hiPSC generation and differentiation has been described9 but on a scale that can only be achieved by high-throughput laboratories dedicated to the production of lines which seems attractive but can be unaffordable for many academic laboratories.
We developed a fully automated cell culture system based on a liquid handling station in a High-Efficiency Particulate Air (HEPA)-filtered environment in conjunction with a large-capacity CO2 incubator, a brightfield imaging cytometer and a robotic arm for plate transport. These components provide the basis for stable and reproducible hiPSC culture and differentiation. We complemented the system with an automated -20 °C storage system for compound or virus storage and a high-speed spinning disk confocal live-cell imager. Custom-made protocols were generated allowing automated cell seeding, media changes, confluency checks, cell expansion and assay plate generation with sample treatment and plate imaging, making the system compatible with high-content/high-throughput screenings. The automated cell culture and imaging system are operated using the controlling software and the custom-made graphical user interface (GUI). The GUI allows users to import CSV files containing cell line-specific parameters needed for method execution. Additionally, the GUI enables to schedule numerous experiments in any sequence using the built-in calendar view thus allowing full control of the time when each method starts.
Our automated cell culture system uses standardized pipetting speeds, passaging times, confluency thresholds, seeding densities, and medium volumes with the flexibility to culture cells in a variety of plate formats (96-, 48-, 24-, 12-, 6- or 1-well plate format). We adapted a recently published short-term differentiation protocol for converting hiPSC into neurons that can yield TUBB3 positive neurons in 6 days10,11. We also established the automated differentiation and imaging of small molecule neural precursor cells (smNPC) into neurons constitutively expressing GFP under EF1a promoter12 and iPSC into midbrain dopaminergic (mDA) neurons, adapting a previously published dual-SMAD inhibition protocol13 that yields mDA neurons within 65 days.
Protocol
1. Basic procedures for automating cell cultures and imaging
- Load new culture plates and tips
- Open the GUI and click on the Resource/Instrument process view button. To run any resource/instrument process, select the resource/instrument process and click on the Run Instrument process button.
- Run the resource process RunHepaHood and Reloading (see step 1.1.1). Open the door, load the cell culture plates and disposable tips in the corresponding position as indicated in the pop-up image on the controlling PC.
- Wipe the door with 70% ethanol for decontamination and close it. Run the instrument process Decontamination from the GUI (see step 1.1.1). The system gets sterilized by UV radiation for 30 min.
- Refill culture media and/or dissociation reagent
- Execute the instrument step RunHepahood (see step 1.1.1). Open the front door of the liquid handling station. Decontaminate reservoirs (containing media, PBS and/or dissociation reagent) with 70% ethanol and place them on the deck to the assigned positon (as defined in cfg.csv file). De-lid the reservoirs.
- Decontaminate the door with 70% ethanol and close it. Power down the HEPA hood by running the resource/instrument process “InitHepaHood” from the GUI.
- Create a new “Cell line” and project in the GUI
- Create/modify the user-defined “Cell line”-files consisting of the Config (*.cfg.csv), the import (*.imp.csv), the resource (*.rsv.csv) and the workflow (*.wfl.csv) files.
- Open the GUI and create a new “Cell line” by clicking on the Add cell line button in the “Cell line editor” view. A wizard opens where the Config file needs to be selected and a project name with a short description needs to be entered.
- Navigate through this wizard using the green arrowhead and confirm the settings by clicking on the green checkmark. Create a new project for the generated “Cell line” by clicking on the Add project button.
- Enter a project name and description and define a project color that will be visible in the calendar view of the GUI. Navigate to the next page of the wizard by clicking on the arrowhead.
- Create a new batch by clicking on the Add batch button and giving a short name and description in the pop-up window. Select all process steps and schedule the start time for each of the process step. Close the window by clicking on OK.
- Execute an automated method using the GUI
- Go to the calendar view of the GUI and click on the Add process step button.
- Select the Cell line and after navigating to the next page of the wizard choose the project. Mark the batch to be used and click on the arrowhead pointing to the right. Depending on the method used, batches have to be either empty (for receiving new plates), or contain culture plates or assay plates (all other processes).
- Navigate to the next page of the wizard and select the process step which shall be executed. Go to the last page of this wizard and schedule the experiment. In the section “Parameter Details” variables that are needed to run the method can be modified. Confirm by clicking OK.
- Loading of culture and assay plates into the CO2 incubator
NOTE: 1-well plates are defined as culture plates since they are commonly used to bulk cells. All multi-well plates are defined as assay plates.- Execute the instrument process “RunHepaHood” from the GUI as described in step 1.1.1. and open the door in front of the robotic arm.
- Wipe the bottom of the assay plates with 70% ethanol and a lint-free tissue if plates are loaded for automated imaging. Place culture or assay plates on the left shelf. Make sure that the orientation of the plate is correct. For assay plates, well A1 has to point towards the robotic arm while the edges of culture plates have to point to the right.
- Wipe the door with 70% ethanol for decontamination and close it.
- Execute the method “Loading Of Culture Plates” for importing 1-well plates or “Loading Of Assay Plates” for importing multi-well plates as described in step 1.4. Use an empty batch to run both methods. If plate barcodes are already present in this batch, deselect them by clicking on the checkboxes.
- Transport individual plates from the shelf to the CO2 incubator platform using the robotic arm. The built-in plate shuttle station retrieves the plate and stores them in one of the racks of the CO2 incubator. The cells are maintained at 37 °C and 5% CO2.
- Unloading of plates from the CO2 incubator
- Execute the method “Unload plates” (see step 1.4). Select the batch containing the plates which need to be exported. Individual plates can be selected by their barcodes.
- Transport the plates out from the CO2 incubator to the left shelf using the robotic arm.
- Start the HEPA hood by using the instrument process “RunHepaHood” as described in step 1.1.1. and open the door in front of the robotic arm. Remove the plates, decontaminate the door with 70% ethanol before closing it and power down the HEPA hood.
2. Automation protocols
- Seeding of plates from tubes
- Start the HEPA hood by executing the instrument process RunHepaHood (see step 1.1.1). Open the door in front of the liquid handling station. Input the 50 mL tube containing the cell suspension and de-lid the tube.
- Open the door in front of the robotic arm and load coated culture plates for receiving cells on the shelf. Decontaminate and close both doors. Execute the method “Seeding of plates from tubes” (see step 1.4).
- Count cells at the brightfield imaging cytometer using direct cell counting function. For cell counting, use 16 wells as replicates in a 384-well plate format.
- Transport the 384-well counting plate from the deck to the brightfield imaging cytometer via the turntable using the robotic arm and start the imaging process. The cytometer automatically determines the cell number per milliliter. Bring the counting plate back to its original position using the robotic arm.
- Transport a coated culture or assay plate from the shelf to the pipetting deck using the robotic arm. Remove coating and seed cells in the user-defined number, and volume suitable for the plate format (see Table 1). Move the plate to the on-deck shaker for 10 s at 500 rpm for cell distribution and transfer to the CO2 incubator.
- Automated confluence assessment
- Execute the method “Check Confluency” as described in step 1.4. Select a batch that contains at least one culture plate and no assay plates. In the section “Parameter Details” input “iPSCf_2020” for image acquisition and imaging analysis settings.
- Transport the first plate from the CO2 incubator to the brightfield imaging cytometer via the turntable using the robotic arm.
- Perform imaging of cells in the brightfield imaging cytometer for confluence check of hiPSC colonies. Use “confluence” application and image 13 fields of the 1-well pate and automatically calculate the average area occupied by cells.
- Transport the plate back to the CO2 incubator using the robotic arm. Repeat steps 2.2.2. to 2.2.4. for the remaining plates.
- Media change of culture plates or assay plates
- Execute the method “Media Change Of Culture Plates” as described in step 1.4. and select a batch containing only culture plates. Set the variable indicating whether tips or needles are used for the pipetting steps in the section “Parameter Details”. For media change of any multi-well plate, execute the method “Media Change Of Assay Plates” and select a batch containing assay plates.
- Transport the individual plates from the CO2 incubator to the deck using the robotic arm and de-lid the plates. Automatically tilt the plates and aspirate old media and discard it into the waste collection module. Add 12 mL of fresh media. Re-lid the plate and transport the plates back to the CO2 incubator using the robotic arm.
- Subcultivation
NOTE: Execute the instrument process “RunHepaHood” from the GUI as described in 1.1.1. and open the front door of the liquid handling station. Load media and dissociation reagent at the required positions (see step 1.2). If tips need to be refilled, use the “Reloading” as described in step 1.1. Input a 50 mL tube for receiving the cell suspension and de-lid the tube.- Execute the method “Subcultivation Of Adherent Cells” (see step 1.4). Select the batch containing culture plates that need subcultivation.
- Transport the plates from the CO2 incubator to the deck using the robotic arm and de-lid the plates. Automatically tilt the plates and remove old media and discard it into the waste collection module. Wash cells once with 8 mL of PBS. Add 8 mL of 0.5 mM EDTA for clump based passaging. Incubate on the deck for 8 min.
- Remove EDTA solution from the plates and discard it into the waste collection module. Add 12 mL of fresh medium and transport the plates to the shaker. Shake at 2000 rpm for 1 min to dislodge the colonies.
- Triturate the cell suspension for five cycles of pipetting to break the iPSC colonies into a smaller size (~50‒80 µm). Transfer the cell suspension into a 50 mL tube on the deck. Seed cells automatically as described in step 2.1.5 using a split ratio of 1 in 7.
NOTE: Optional, single cell passaging. Generate a single cell suspension using single cell dissociation reagent (see Table of Materials). Instead of 0.5 mM EDTA in step 2.4.2., use 8 mL of single cell dissociation reagent and incubate for 20 min at 37 °C. Collect the cell suspension into a 50 mL tube and add equal volume of media. Dissociation using the single cell dissociation reagent requires a manual step to pellet the cells. Centrifuge cells at 300 x g for 3 min. Remove the supernatant and resuspend the cell pellet in 12 mL of the required media and proceed with “Seeding of plates from tubes” (see step 2.1). Supplement the media with 2 µM thiazovivin on the day of seeding when using single cell suspension of hiPSCs.
- Automated high-content, high throughput imaging
NOTE: The measurement setting has to be available (see Supplementary File 1: step 1.1). The assay plates to be imaged are available in the incubator.- Execute the method “Imaging” (see step 1.4). Select a batch that contains at least one assay plate and no culture plate. In the section “Parameter Details”, enter the plate type, the plate definitions (for standard 96-well imaging plates: “Plate-96-20170918113842”) and the name of the measurement setting.
- Transport the assay plate via the turntable to the automated confocal microscope using the robotic arm to start the imaging process. Recover the plate at the end of the imaging from the microscope and transport it back to the CO2 incubator. Repeat the process of the remaining plates in the batch.
3. Automated maintenance and expansion of hiPSC
- The sequence of automated confluency checks, media changes and subcultivation
- Seed cells on 1-well plates using “Seeding of plates from tubes” method (see step 2.1). Alternatively, import manually seeded 1-well plates using “Loading of culture plate” method (see step 1.5).
- Schedule automated confluency checks daily to monitor colony growth from day 1 to day 6 of the culture for iPSC (see step 2.2).
- Refresh media every second day using the “Media change of culture plates” method (see step 2.3). 12 mL of medium is used per plate.
- On day 6, start the “Subcultivation” process (see step 2.4.).
4. Automated differentiation
- Human iPSC to cortical NGN2 neurons
NOTE: Manual hiPSC preparation. The protocol involves over-expression of NGN2 using lentiviral vectors for delivery. Transduce hiPSC with NGN2 to rTTA3 lentivirus in a 1:2 ratio (> ~107 TU/mL). Cells are then selected for one passage with 0.5 µg/mL puromycin. A detailed protocol for generating stable hiPSC-NGN2 lines is in the Supplementary File 1: step 2.- Proceed with the “Subcultivation” process as described using the single cell dissociation reagent described in note of step 2.4.4. when hiPSC reach 70‒80% confluency
- Remove the supernatant and resuspend the cell pellet in 12 mL of NGN2 media (Table of Materials) containing 2.5 µg/mL doxycycline (dox) and 2 µM thiazovivin.
- Start differentiation using the “Seeding of plates from tubes” method (see step 2.1). Set the desired seeding density of iPSC to 30,000/cm2 (see table 1). The iPSC are plated onto pre-coated plates with 0.1 mg/mL poly-L-ornithine (PLO) and 5 µg/mL laminin.
NOTE: 1-well plates are preferred for RNA and proteomics-based studies while the 96-well plate format is preferred for imaging experiments. Other assay plate formats such as 48-, 24-, 12- or 6-well plates may also be used. - On day 1 of differentiation, Refresh media using the “Media change of assay plates (see step 2.3) using NGN2 medium, supplemented with 2.5 µg/mL dox and 10 µM N-[2S-(3,5-difluorophenyl)acetyl]-L-alanyl-2-phenyl-glycine, 1,1-dimethylethyl ester (DAPT). Continue with media changes every 2‒3 days until the desired day of differentiation (see step 2.3).
- On day 4 of differentiation, carry out another media change (see step 2.3.) using NGN2 medium supplemented with 10 ng/mL brain-derived neurotrophic factor (BDNF), 10 ng/mL glial cell-derived neurotrophic factor (GDNF) and 10 ng/mL neurotrophic factor 3 (NT-3) to enhance the maturation. At the end of the experiment, export the culture plates from the system for downstream experiments (see step 1.6).
- Small molecule neural precursor cells (smNPC) to NGN2 neurons
NOTE: Derive smNPC following the adapted version of published protocol12 (see Supplementary File 1: step 5). Modify the smNPC with NGN2 and rTTA3 virus (see Supplementary File 1: step 2). One round of NGN2 and rTTA transduction is sufficient to generate a stable population. Further modify smNPC with pLVX-EF1a-AcGFP1-N1 lentivirus allowing to perform live-cell monitoring. A multiplicity of infection (MOI) of 10 is used for GFP lentiviral transduction.- Passage smNPC on reaching confluency using single cell dissociation reagent as described in step 2.4. Note.
- Seed cells using the automated cell culture system at a cell density of 50,000/cm2 using “Seeding of plates from tubes” method (see step 2.1.) onto pre-coated plates with 0.1 mg/mL PLO, and 5 µg/mL laminin in NGN2 medium supplemented with 2.5 µg/mL dox.
- On day 3, perform a media change (see step 1.3.) using fresh NGN2 medium containing 2.5 µg/mL dox and 10 µM DAPT. After day 3, perform media changes every third day with fresh NGN2 medium containing 2.5 µg/mL dox, BDNF, GDNF and NT-3 at 10 ng/mL each.
- Image cells daily using the “Automated high content, high throughput imaging” method (see step 2.5) for monitoring differentiation status using GFP as readout for neurite outgrowth. Set the laser power to 80% and use an exposure time of 30 ms. Acquire a large number of fields (e.g., 25 fields) using 20x objective.
- Batch image analysis
- Perform image analysis with image analysis software 1, using the neurite outgrowth template.
- Open the software. Select the “Data” option and browse to the imaging data folder, click ok to load data to be analyzed. Click open and from the top menu bar, select protocol and from the application menu select neurite outgrowth. Go to “algorithms” and use the “488” channel to define the nucleus, cell body and neurite. Adjust the threshold parameters for each.
- Select the wells to be used for image analysis. On the bottom right click link and select features to be analyzed such as cell count average, total skeleton length total. Proceed with “pre-analyze” followed by “preview”.
- Check if the preview settings are acceptable and start the analysis in the batch mode. The results are available in the parent folder under “reports” in .csv file format that can be opened in excel.
NOTE: Image analysis script is available upon request. - Plot average neurite length obtained by total neurite length normalized to cell number.
5. Automated differentiation of hiPSC into midbrain dopaminergic (mDA) neurons
- Manual hiPSC preparation
- Dissociate 70‒80% confluent hiPSC into single cells using the single cell dissociation reagent. Briefly, incubate cells with single cell dissociation reagent (100 µL/cm2) for 30 min at 37 °C, collect cell suspension into a conical tube, centrifuge at 200 x g for 5 min and resuspend cell pellet in iPSC culture medium.
- Seed 200,000 cells/cm2 on extracellular matrix-coated 1-well plates and iPSC culture medium supplemented with 10 µM Y-27632. Culture cells overnight at 37 °C and 5% CO2.
- Automated differentiation: Phase 1
- Prepare the automated culture system as described in step 1.1‒1.2.
- Load culture plates containing cells into the CO2 incubator of the automated culture system (see step 1.5).
- Prepare KSR medium (see Table of Materials) and supplement with small molecules (see Table 2) required for starting day 0 of differentiation. Use only freshly prepared media with small molecules and growth factors.
- Perform media changes of the culture plates as described in step 2.3 on days 0 and 1 of differentiation and then every second day until day 25.
- From day 5, shift media formulation gradually as described in detail in Table 3.
- On day 11, add mDA neuron differentiation medium supplemented with CHIR (until day 13), BDNF, AA1, GDNF, db-cAMP, TGFß3 and DAPT (see the Table of Materials).
- On day 25, unload plates (see step 1.6.)
- Manual replating 1
- Dissociate day 25 mDA precursors into single cells using the single cell dissociation reagent. Briefly, incubate cells with single cell dissociation reagent (100 µL/cm2) for 40 min at 37 °C, collect cell suspension into a conical tube, centrifuge at 200 x g for 5 min and resuspend cell pellet in mDA neuron differentiation medium.
- Seed 400,000 cells/cm2 in 1-well culture plates pre-coated with 0.1 mg/mL PLO, 10 µg/mL laminin and 2 μg/mL fibronectin in mDA neuron differentiation medium supplemented with 10 µM Y-27632 (until day 26) and small molecules and growth factors described in Table 2. Culture cells overnight at 37 °C and 5% CO2.
- Automated differentiation: Phase 2
- Load culture plates containing mDA neurons into the CO2 incubator of the automated culture system as described in step 1.5
- Prepare mDA neuron differentiation medium (see Table of Materials) and supplement with small molecules and growth factors required for the final differentiation from day 26 onwards (see Table 2).
- Perform media changes of culture plates as described in step 2.3 on day 26 of differentiation and then every 3‒4 days until day 65.
NOTE: For high-throughput imaging purposes, it is recommended to replate the mDA neurons in 96-well plates at low density on day 32 of differentiation, as described in step 5.5.
- Manual replating 2
- Unload culture plates as described in step 1.6. Dissociate day 32 mDA neurons into single cells as described in step 5.3.1.
- Seed 100,000 cells/cm2 in 96-well plates pre-coated with 0.1 mg/mL PLO, 10 µg/mL laminin and 2 μg/mL fibronectin in mDA neuron differentiation medium supplemented with 10 µM Y-27632 (until day 33) and small molecules and growth factors (see Table 2). Culture cells overnight at 37 °C and 5% CO2.
- On day 33, replace medium by freshly prepared DA neurons differentiation medium supplemented with small molecules and growth factors (without Y-27632). Change medium manually every 3‒4 days until day 65.
6. Immunostaining, automated high-throughput image acquisition and analysis
- Fluorescence staining
- Perform fluorescent immunostaining as described in the Supplementary File 1: step 4.
- Image cells as described in the Supplementary File 1: step 1.2.
- Upload imaging files generated by the imaging system to the image analysis software 2 (see Table of Materials) for further image analysis, as described in step 6.2.
- Image analysis using the image analysis software 2
- Once imaging files are uploaded in image analysis software 2, go to the “image analysis” function and build the analysis algorithm by using the drop down menu.
- Select nuclei using the task “find nuclei”, which detects regions on the image belonging to cell nuclei (here stained with Hoechst). Exclude nuclei from dead cells (pyknotic nuclei) by setting a threshold for nuclear area or diameter.
- Select the cell cytoplasm using the task “find cytoplasm”. This task detects regions around nuclei belonging to cell cytoplasm. Include only well-segmented cells. Exclude mitotic, apoptotic and badly segmented cells. Remove cells touching the border of the image.
- Add the tasks “calculate morphology properties” and “calculate intensity properties”. The morphology parameters include the calculation of morphology properties such as area for a region of interest. The intensity parameters include the calculation of intensity properties such as mean intensity for a region of interest (e.g. cytoplasm of neurons).
- Select a subpopulation (e.g., TH positive neurons) of the input population (all cytoplasms selected) using one or more conditions, morphology and/or intensity.
NOTE: Usually, intensity is chosen for neurons. Based on negative controls, it is possible to define above which threshold of intensity the neurons are considered positive for TH. - Define the output results. This is the last building block of each analysis. It defines the assay readout values for each well of a culture plate (results per well).
- Run the batch analysis and export the results. Normalize the number of positive cells to the total number of nuclei and represent data as the percentage of positive cells.
7. Quantitative real-time polymerase chain reaction (qRT-PCR)
- Perform the qRT-PCR protocol as described in the Supplementary File 1: step 3.
Representative Results
Our automated cell culture and imaging system was designed to minimize human intervention allowing us to standardize the cultivation of hiPSC and differentiation into different cell types such as cortical or midbrain dopaminergic (mDA) neurons. A schematic overview of our automated cell culture system with integrated imaging devices is depicted in Figure 1. The initial introduction of cell cultures to this automated cell culture system can either be done by automatically seeding cells from a 50 mL tube or by using the “Loading Of Culture Plates” or “Loading Of Assay Plates” method for import of culture or assay plates. A central component of our system is the liquid handling station where all liquid transfer steps such as media changes or subcultivations are carried out. The custom-made deck layout of the liquid handler is represented in Figure 2. The liquid handling station is equipped with four positions. Up to four plates can be transferred from the incubator to the deck, allowing parallel media changes. Since in the subcultivation method, both the parent and daughter culture plates have to be accommodated on the deck, the maximum number of culture plates processed in parallel is limited to two. An important feature of the liquid handling station is the possibility to tilt the plates during media change for complete removal of cell culture supernatant. Also, the liquid handling station is equipped with shakers for favoring enzymatic dissociation of cells during the execution of the subcultivation protocol. Our automated culture system is also equipped with two imaging systems: a brightfield imaging cytometer for performing cell counting and confluency checks and, therefore, monitoring the cell growth over time, and a dual spinning disk confocal microscope for rapid, high content and high-resolution imaging of cells.
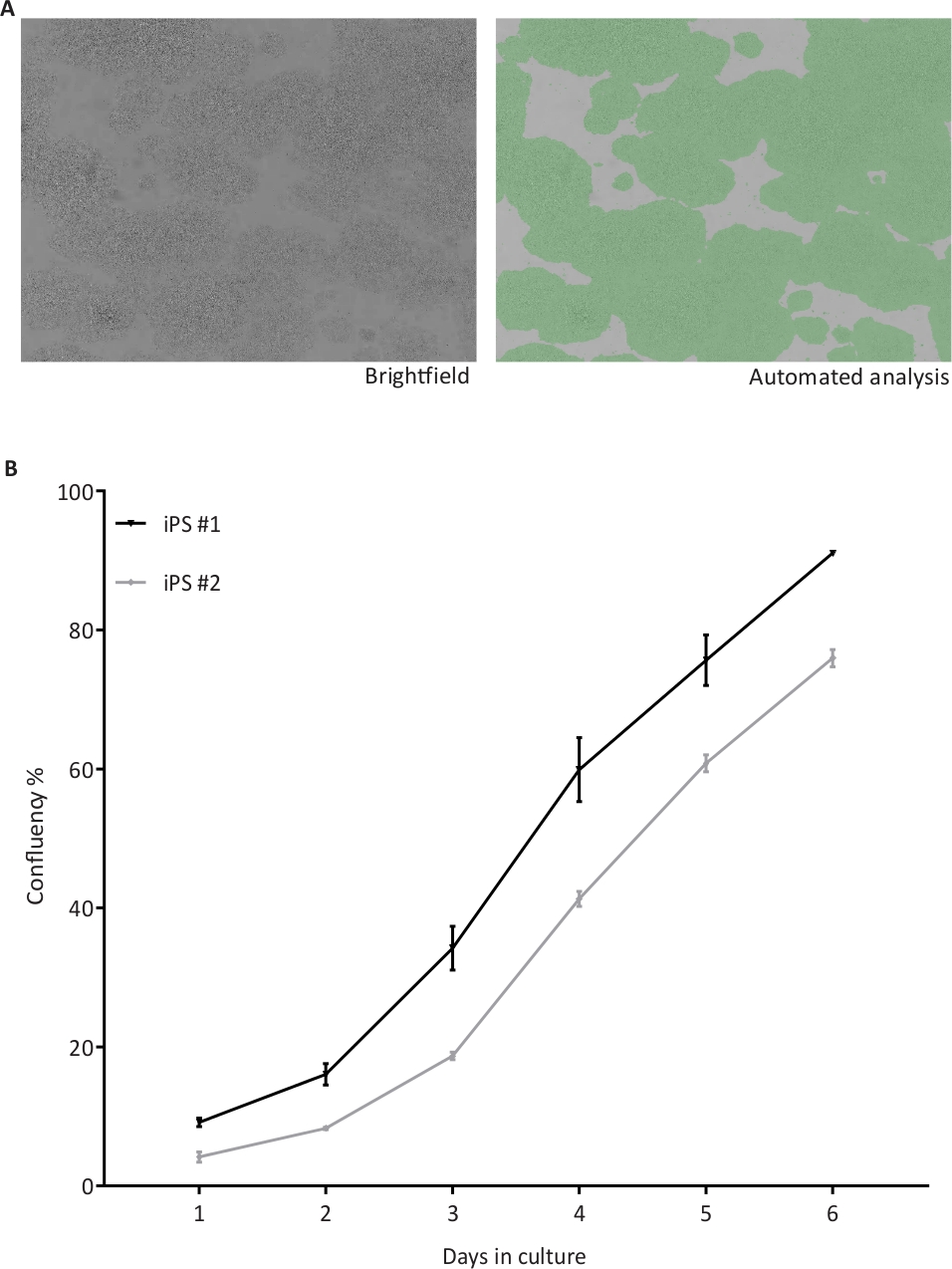
The hiPSC cultures are monitored daily for growth at the brightfield imaging cytometer and analyzed for percentage of confluency. The brightfield image in the left panel and in the right panel a green mask from the analysis of the brightfield image obtained with the cytometer (Figure 3A). A homogeneous hiPSC growth is observed over the time, as shown by the confluence percentages of two hiPSC lines (n = 4 plates) grown in parallel and subjected to confluency checks from day 1 to day 6 (Figure 3B). Upon reaching the set threshold, the hiPSC are passaged. The cell lines were cultured manually (m) or by the automation (a) system and observed for maintenance of typical stem cell morphology for at least two passages, representative brightfield images (Figure 4A). The hiPSC cultured manually (not shown) or in the automated system exhibited the typical stem cell marker OCT4 (red) and SSEA4 (green), as shown in the immunofluorescence assay (Figure 4B). The expression of the pluripotency markers OCT4, NANOG and REX1 were also assessed at mRNA level by qRT-PCR (Figure 4C). Relative quantifications were performed with samples collected from one cell line grown manually (m) and in the automated culture system (a) in duplicates (replicates 1 and 2). The expression levels of all three pluripotency markers in the replicates cultivated in the automated culture system are similar to marker expression after manual culture. On day 8 (D8), the expression of pluripotency markers was absent in cortical neurons differentiated (Diff) from hiPSC.
One important application of the automated culture system is the differentiation of hiPSC into different cell types including neurons. Here we show the differentiation of hiPSC into neurons using the NGN2 strategy, which produces a pure cortical neuron culture in a very short time (approximately 6 days). Neurons differentiated in the automated culture system (a) presented similar morphology and neuronal network organization as the neurons cultivated manually (m) (Figure 5A). Automated differentiated cortical neurons were positive for TUBB3 (neuron-specific Class III β-tubulin, red) and BRN2 (upper cortical layer marker, green) (Figure 5B), comparable to manually differentiated neurons (data not shown). The expression of neuronal markers including the microtubule-associated protein 2 (MAP2), the neural cell adhesion molecule (NCAM1) and Synapsin-1 (SYN1), as well as the cortical neuron markers BRN2 and CUX1 (upper cortical layer) were enriched in neurons at day 8 (D8) of differentiation (Figure 5C). Very low or no expression of these markers was observed in hiPSC. Relative quantifications were performed with samples collected from one cell line grown manually (m) and in the automated culture system (a) in duplicates (replicates 1 and 2). The expression levels in replicates show similar variations between manually and automated differentiations.
The integrated imaging capability of the automated culture system allows hands-free data collection for the health of cultures thus enabling long-term automated acquisition of phenotypic readouts. Using the NGN2 approach to a small molecule derived neural precursor (smNPC) line transduced with GFP lentivirus, we established a live-cell automated neurite outgrowth assay in which neurite length was measured over 11 days of differentiation without any manual intervention. The neurite complexity increased over time, as demonstrated by the area occupied with neurites on day 1, 3 and 11, GFP expression and masked images from analysis (Figure 6A, B). The increase in neurite length from day 1 to 11 of differentiation was quantified and showed a similar development across different wells. For the sake of simplicity, data from only 3 columns with 6 wells each from a 96-well plate is depicted in the representative graph although all inner 60 wells were analyzed (Figure 6C).
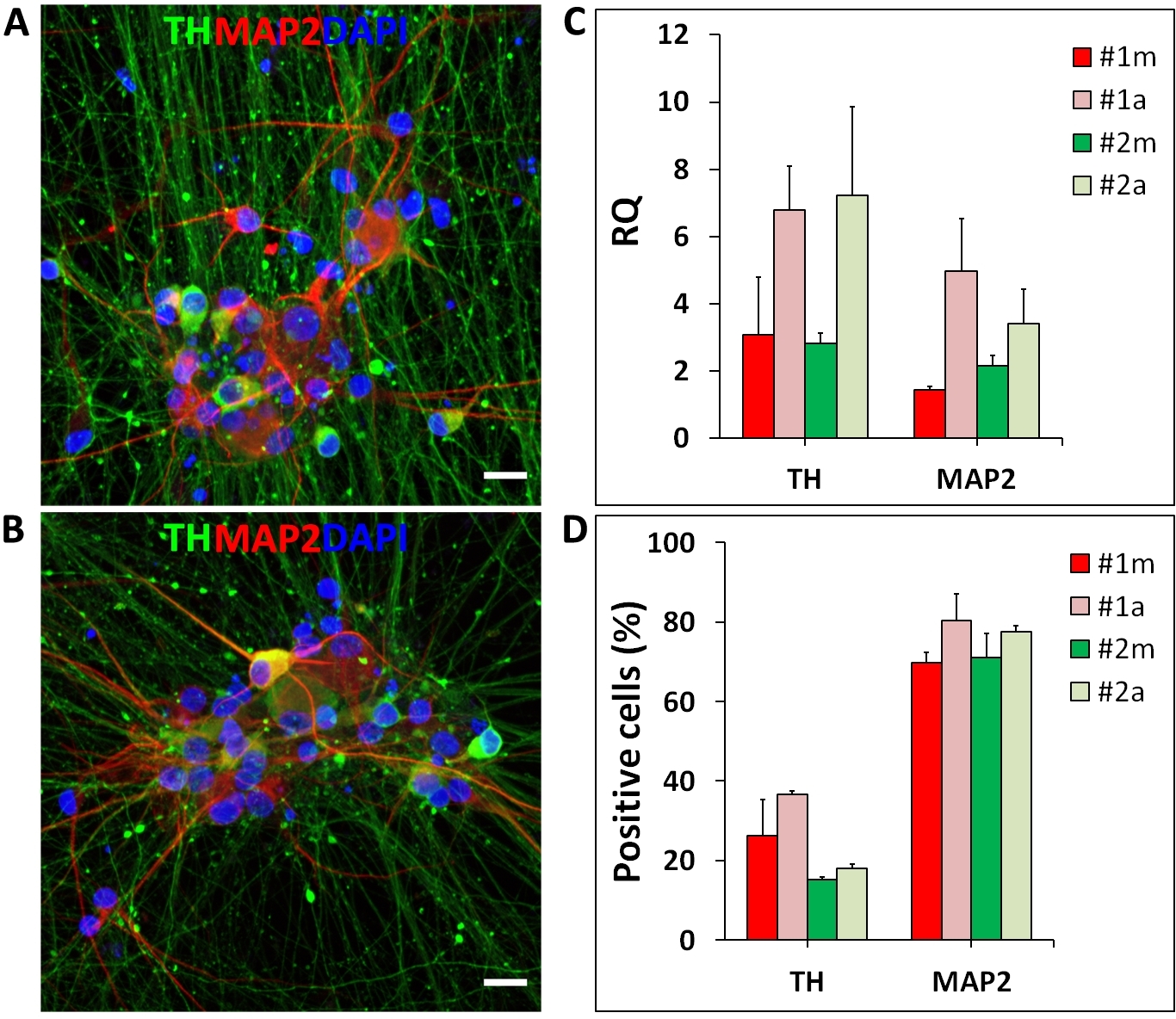
Another application of the automated culture system shown here is the differentiation of hiPSC into mDA neurons. The differentiation is based on media changes following a pre-established protocol and was performed on the automated culture system from days 0 to 65. Automated media changes did not cause cell detachment or any other visually detectable changes in the differentiation. At the end of the differentiation, on day 65, mDA neurons show cellular organization and morphology (spheric soma, long and spiny dendrites) comparable to manual differentiation (Figure 7A, B). At the mRNA level, mDA neurons differentiated in the automated culture system show the expression of neuronal and mDA markers, MAP2 and TH (tyrosine hydroxylase), respectively (Figure 7C). Both differentiations generated substantial amounts of TH and MAP2 positive neurons (Figure 7D).

Figure 1: Schematic overview of the automated cell culture and imaging platform.
The system was designed with a polycarbonate housing and two HEPA hoods (A and B) equipped with four UV lamps ensuring a sterile environment for cell culture applications. Cell culture plates are loaded on shelves in front of the robotic arm which can be accessed via the front door (C). The plates are loaded into the CO2 incubator (D) with a capacity of 456 plates. A brightfield cell cytometer (E) is used for confluency checks and cell counting during subcultivation routines. The liquid handling station is below one of the HEPA hoods (B). The deck layout of the liquid handler is described in Figure 2. The pipetting arm of the liquid handling station carries a 96 channel pipetting head, eight 1 mL pipetting channels and four 5 mL pipetting channels. In the case of the 1 mL pipetting channels, tips or needles can be used for liquid transfers. For screening purposes, cells seeded in assay plates can be treated with samples stored at -20 °C in the automated -20 °C storage system (F) after thawing these samples in a second incubator (G). High throughput imaging is performed in the automated confocal microscope (H) offering to acquire images in confocal mode using two spinning discs or in epifluorescence mode. A live-cell chamber integrated into the microscope allows performing long-term imaging of cultured cells. Please click here to view a larger version of this figure.
{kind=link}

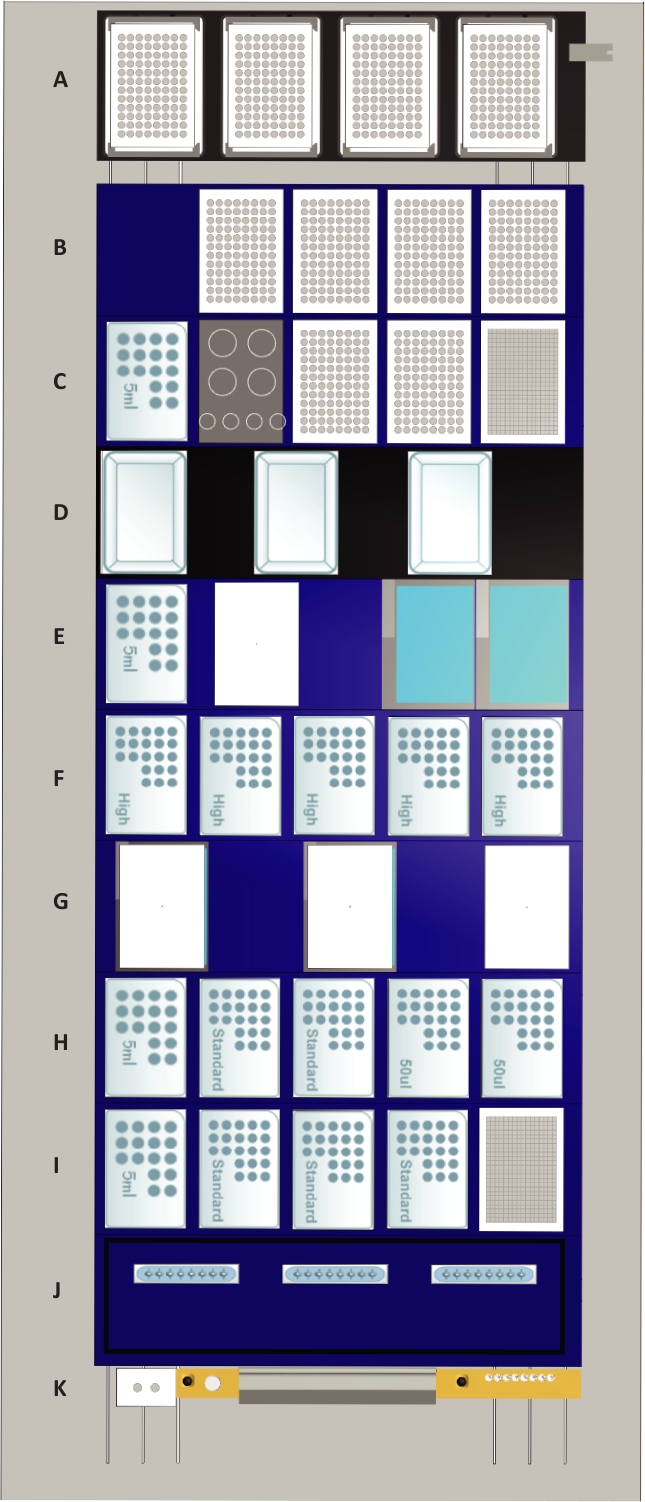
Figure 2: The deck layout of the liquid handling station.
Tip positions are indicated by “50 µL” for 50 µL tips, by “standard” for 300 µL tips, by “high” for 1 mL tips and by “5 mL” for 5 mL tips. Deck components: (A) Four heated shaker positions (max speed: 2500 rpm) which can be used for any culture plate or assay plate format. The shaker positions are equipped with clampable grippers which are also used for plate alignment following transports to the deck. Furthermore, the shaker positions function as a lid parking position for plates during liquid transfer steps. For representative purposes, all shaker positions are occupied by 96 well assay plates. (B) Four tilt modules for processing four plates of any format simultaneously are positioned on the top. The lowest position marks the waste collection chamber for culture and assay plates and the 96 channel pipetting head. For representative purposes, all tilt modules are occupied by 96-well assay plates. (C) On the top position, the 384-well plate for cell counting is located. Below are two positions for plates that are occupied by 96-well plates for representative purposes and a rack for four 50 mL and four 15 mL tubes. The lowest position is occupied by 5 mL tips. (D) Three media lines with positions for media reservoirs. The media lines possess liquid level sensors that allow to automatically fill the media reservoir with up to 250 mL of media. (E) Two liquid waste modules with active drain are based on the top, below a temperature-controlled module with a position for one container (white) and a 5 mL tip rack are located. (F) Five positions for 1 mL tips. (G) Two temperature-controlled modules are positioned at the bottom and a position for parking one of their lids on the top. (H) Positions for two 50 µL nested tip racks (NTR) in the top followed by two positions for single-channel and 96-channel pick up of 300 µL tips and a 5 mL tip rack at the bottom. (I) A stacker for 384-well counting plates at the top followed by three 300 µL NTR and a 5 mL tip rack at the bottom. (J) Storage and wash station for three sets of eight reusable metal 1 mL needles. (K) Waste position for 1 mL and 5 mL pipetting channels and empty NTR (grey) as well as a gripper block for 1 and 5 mL channels (white) used for on-deck transport steps. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Automated confluency check of hiPSC.
(A) Representative brightfield (BF) images of hiPSC taken by the cell cytometer (left) and after automated confluency analysis (right) indicating the proportional area occupied by cells in green; (B) Confluency percentages recorded from two hiPSC lines (iPS #1 and #2) from day 1 to 6 of culture, n = 4 1-well plates per cell line. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Passaging of hiPSC.
(A) BF image of one hiPSC line grown manually (left) and using the automated culture system (right). Images were taken 6 days after the second passaging; (B) Representative images of hiPSC stained for the pluripotency markers OCT4 and SSEA4, and counterstained with Hoechst 33342 (Nuclei); (C) Results of qRT-PCR for the pluripotency markers OCT4, NANOG and REX1 in one hiPSC line cultivated in duplicates (1 and 2) manually (m) and in the automated culture system (a), and the respective hiPSC-derived cortical neurons (Diff) at day 8 (D8) of differentiation. The data is represented as the relative quantity (RQ) using iPS_a_1 as the reference sample. Error bars represent standard deviation (SD) from 3 technical replicates of qRT-PCR reaction. GAPDH, RPL13A1 and RPLPO were used as housekeeping genes. Scale bar: 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Human iPSC-derived cortical neurons.
(A) Brightfield images of day 6, manually (m) and automated (a) differentiated cortical neurons showing similar neuronal networks; (B) Representative images of cells stained for TUBB3 (pan neuronal), BRN2 (cortical neurons) and Hoechst 33342 (nuclei) on day 8 of differentiation; (C) Results of qRT-PCR for marker genes of cortical neurons (MAP2, BRN2, CUX2, NCAM1 and SYN1) enriched in day 8 (D8) of differentiation. The data is represented as the relative quantity (RQ) using iPS_a_1 as the reference sample. Error bars represent the SD from 3 technical replicates of qRT-PCR reaction. GAPDH, RPL13A1 and RPLPO were used as housekeeping genes. Scale bar: 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: A high-throughput assay for neurite outgrowth.
(A) Representative images of GFP expressing cells on days 1, 3 and 11 of differentiation; (B) Representative binary images of neurites on days 1, 3 and 11 of differentiation. The neurite outgrowth was quantified using the high content image analysis software 1 and is represented as neurite length; (B) The graphic displays the increase in neurite length in NPC-derived NGN2 neurons and formation of a dense network. Three 96-well plates with cells in the inner 60 wells are imaged. For simplicity, only three columns of wells per 96-well plate are shown as an example with n = 6 wells per column. An average of 1308 cells were analyzed per well. Error bars represent the standard error of the mean (S.E.M.). Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Human iPSC-derived mDA neurons.
Midbrain DA neurons differentiated manually (m) and in the automated (a) culture system. (A, B) Representative fluorescent images of mDA neurons stained for tyrosine hydroxylase (TH, mDA neuron marker; green), MAP2 (neuronal marker; red) and Hoechst 33342 (nuclei; blue); (C) Representative qRT-PCR results for marker genes of mDA neurons differentiated manually and in the automated culture system. TH and MAP2 expression levels are represented as relative quantity (RQ) normalized to housekeeping genes (OAZ1 and GAPDH); (D) Percentages of TH and MAP2 positive neurons generated by manual and automated differentiation. Error bars represent the SD of two independent differentiations performed with two distinct iPSC lines (#1 and #2). Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}
| Cell type | Purpose | Protocol step | Cell density | Plate format | Cell number/well |
| NGN2 differentiation | |||||
| iPSC | NGN2 stable line generation | S.2.3. | 30,000 cells/cm2 | 12-well | 1,17,000 |
| iPSC | NGN2 neuron differentiation | 3.1.3. | 30,000 cells/cm2 | 1-well | 25,20,000 |
| iPSC | NGN2 neuron differentiation | 3.1.3. | 30,000 cells/cm2 | 96-well | 9,600 |
| smNPC generation | |||||
| smNPC | Replating day 12 and 16 | S5.9. and S5.11. | 70,000 cells/cm2 | 6-well | 6,72,000 |
| smNPC | Replating from passage 5 | 5.11. | 50,000 cells/cm2 | 6-well | 4,80,000 |
| smNPC | smNPC to NGN2 neurons | 3.2.2 | 50,000 cells/cm2 | 96-well | 16,000 |
| mDA differentiation | |||||
| iPSC | mDA neuron differentiation | 4.1.2. | 200,000 cells/cm2 | 1-well | 1,68,00,000 |
| DA neurons | Day 25 replating | 4.3.2. | 400,000 cells/cm2 | 1-well | 3,36,00,000 |
| DA neurons | Day 25 replating | 4.5.2. | 100,000 cells/cm2 | 96-well | 32,000 |
| Coating | Purpose | Protocol step | Concentration/ Dilution | Plate format | Details of coating |
| iPS culture | |||||
| Extracellular matrix | iPSC, NGN2 line, mDA neurons | 1.5.7. and S2.3. | 1 aliquot*; 25 mL DMEM/F-12 | 1-/12-well | 8/0.5 mL/well; 1 h at RT |
| NGN2 differentiation | |||||
| Poly-L-Ornithine | NGN2 neuron differentiation | 3.1.3. and 3.2.2. | 0.1 mg/mL; PBS | 1-/96-well | 8/0.1 mL/well; 12 h at 37°C; 3x PBS wash |
| Laminin | NGN2 neuron differentiation | 3.1.3. and 3.2.2. | 5 μg/mL; PBS | 1-/96-well | 8/0.1 mL/well; 4 h at 37°C |
| smNPC generation | |||||
| Extracellular matrix | smNPC generation and culture | S5.6., S5.9., S5.11. | 1 aliquot*; 25 mL DMEM/F-12 | 6-well | 1 mL; 2 h at RT |
| mDA differentiation | |||||
| Extracellular matrix | mDA differentiation | 4.1.2. | 1 aliquot*; 25 mL DMEM/F-12 | 1-well | 12 mL; 12 h at 37°C |
| Poly-L-Ornithine | mDA differentiation | 4.3.2. and 4.5.2. | 0.1 mg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C; 3x PBS wash |
| Laminin | mDA differentiation | 4.3.2. and 4.5.2. | 10 μg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C |
| Fibronectin | mDA differentiation | 4.3.2. and 4.5.2. | 2 μg/mL; PBS | 1-/96-well | 12/0.1 mL/well; 12 h at 37°C |
| *A extracellular matrix aliquot is defined as the dilution factor (in µL) present in the Certificate of Analysis of this product. | |||||
Table 1: Seeding cell density and coating respective to the plate format.
| Day | Reagent | ||
| Day 0 - 1 | 100 nM LDN193189, 10 µM SB431542 | ||
| Day 1 - 3 | 100 nM LDN193189, 10 µM SB431542, 1 mM SHH, 2 mM Purmorphamine, 100ng/mL FGF-8b | ||
| Day 3 - 5 | 100 nM LDN193189, 10 µM SB431542, 1 mM SHH, 2 mM Purmorphamine, 100ng/mL FGF-8b, 3 µM CHIR99021 | ||
| Day 5 - 7 | 100 nM LDN193189, 1 mM SHH, 2 mM Purmorphamine, 100ng/mLFGF-8b, 3 µM CHIR99021 | ||
| Day 7 - 9 | 100 nM LDN193189, 1 mM SHH, 3 µM CHIR99021 | ||
| Day 9 - 11 | 100 nM LDN193189, 1 mM SHH, 3 µM CHIR99021 | ||
| Day 11 - 13 | 3 µM CHIR99021, 20 ng/mL BDNF, 0.2 mM L-ascorbic acid (AA1), 20 ng/mL GDNF, 1 mM db-cAMP, 1 ng/mL TGFß3, 10 µM DAPT | ||
| Day 13 - 65 | 20 ng/mL BDNF, 0.2 mM L-ascorbic acid (AA1), 20 ng/mL GDNF, 1 mM db-cAMP, 1 ng/mL TGFß3, 10 µM DAPT | ||
Table 2: Small molecule addition for dopaminergic neuron differentiation.
| Day | KSR medium | N2 medium | Differentiation medium |
| Day 0 - 1 | 100% | 0 | 0 |
| Day 1 - 3 | 100% | 0 | 0 |
| Day 3 - 5 | 100% | 0 | 0 |
| Day 5 - 7 | 75% | 25% | 0 |
| Day 7 - 9 | 50% | 50% | 0 |
| Day 9 - 11 | 25% | 75% | 0 |
| Day 11 - 13 | 0 | 0 | 100% |
| Day 13 - 65 | 0 | 0 | 100% |
Table 3: Media gradient for dopaminergic neuron differentiation.
Supplementary File 1. Please click here to download this file.
Discussion
We introduce an automated cell culture system with integrated imaging capabilities for the standardization of hiPSC culture and neuronal differentiation. Due to minimal user intervention, experimental variation is low ensuring reproducibility of cellular phenotypes during differentiation. The calendar-based scheduler supports the organization and parallelization of experiments and allows a high degree of flexibility at which time the experiments are carried out. Existing methods can be easily adapted and the spectrum of available methods can be increased. Additionally, a large number of assay plate formats can be used adding to the flexibility of this system. The minimal system consisting of a CO2 incubator, a robotic arm, a brightfield cell cytometer, and a liquid handling station forms the basic unit needed for hiPSC culture and differentiation, with affordable costs to academic research laboratories. The combination of the automated cell culture system with an automated -20 °C storage system for storage of compounds, RNAi libraries or CRISPR/Cas9 libraries, and the integration of a high-content/high-throughput microscope enable the execution of phenotypic screenings.
In the current study, the automated cell culture system used disposable tips and the culture media was refilled manually into the reservoir, thus limiting the use of the liquid handling station for media changes and other culture processes especially overnight. To circumvent this limitation, the methods can be adjusted to needle usage instead of disposable tips and, after installing tube connections between media lines and media bags stored in a fridge, media reservoirs can be automatically refilled with fresh media pre-warmed by heater elements. This would reduce user interferences caused by manual refilling of tips, culture media and reservoir exchanges.
Our automated cell culture system offers several advantages. One is the barcode tracking system. The plates loaded in the system are identified by a unique barcode which is read and saved by the system allowing tracking of samples during and after method execution. Another advantage is the possibility to create user specific projects. Here, culture plates loaded in the system can be assigned to a specific project and grouped in batches. The structuring in batches simplifies the execution of the same procedure to all plates of a certain batch since no individual plates need to be selected. Additionally, a liquid class editor allows to adjust the pipetting speed and height as well as the aspiration and dispensing parameters for each liquid transfer step. Every process is documented in log files allowing to retrace which tasks have been performed for a given culture or assay plate.
Neurons and other cell types derived from human induced pluripotent stem cells (hiPSC) are useful in vitro tools for studying the mechanisms of neurodegenerative diseases in specific patient populations (e.g. dopaminergic neurons for Parkinson’s disease) offering the possibility for personalized drug screenings. Culturing hiPSC is very time intensive and demands trained people to execute complex differentiation protocols, usually limited to low scale production. We adapted the feeder-free culture of hiPSC to an automated culture and implemented two neuronal differentiation protocols, a rapid cortical neuron differentiation protocol based on NGN2 over-expression under a tet-on promoter10,11, and a long-term small molecule-based protocol for generation of midbrain dopaminergic (mDA) neurons13. The straightforward transfer and reproducibility of manual culture and differentiation protocols makes the automated culture system very useful. Human iPSC cultured in the automated cell culture system showed consistent stem cell morphology and expressed important pluripotency markers, reproducible between independent experiments. In addition, the automation of the hiPSC culture protocol favored the culture and expansion of a larger number of cell lines in parallel. Automated confluency checks scheduled to be performed overnight saved time leaving the system free during the day for downstream process steps carried out when the user was in the laboratory (e.g., harvesting of cells or manual replating for differentiations). On reaching the user-defined confluence threshold, cells are passaged and replated into extracellular matrix-coated plates available on the stacker of the automated cell culture system. Each passage round takes about 70 min and generates four 1-well plates from one parent plate, which translates to a capacity of 20 passages in a day.
The automation of the NGN2 differentiation protocol was done successfully and allowed the generation of a homogeneous population of neuronal cells across different passages and comparable to manual differentiations. Moreover, the experimental costs for large-scale screening studies involving multiple cell lines or screening experiments with thousands of test conditions/compounds would be reduced due to rapid differentiations. Cost-effective and high-throughput readouts including live-cell neurite outgrowth measurements can be easily developed, implemented and used as phenotypic readouts for disease modeling, as shown previously14,15,16. Thus, we further adapted the NGN2 protocol using small molecule derived neural precursor (smNPC) cells that constitutively over-express GFP. The smNPC cells offer further advantages including reduced costs with culture media (one third of the cost with iPSC culture) and time required to scale up experiments. The cell yields from smNPC are 7 to 10 times higher than that obtained with iPSC. The differentiating neurons were successfully monitored and imaged for several days using a fully automated imaging process without the need of manual antibody stainings or chemical labeling, saving costs and time required for manual procedures including the imaging by itself. The current imaging of inner 60 wells of a 96-well plate takes around 16 min per plate when 25 fields per well are imaged, which means that the data for an imaging-based screening for 1000 compounds, could be acquired and analyzed in a day. In the future, this readout could be used in compound screening studies for the rescue of neurite outgrowth defects.
Further, we also demonstrate the transfer of a manual differentiation protocol for generating midbrain dopaminergic (mDA) neurons from iPSC. This small molecule-based differentiation protocol takes 65 days and is labor intensive because of the multiple replating steps and frequent media changes, mostly every 2 days, which limits the production of mDA neurons to few iPSC lines at the same time. The automated mDA differentiation protocol has the great advantage of scaling up the differentiation to dozens of iPSC lines. Up to 30 cell lines could be differentiated in parallel. Since the differentiation is mostly based on media changes, almost the whole differentiation process can be conducted without human interference. Using the calendar-based scheduler of the automated system, we could plan the media changes according to the differentiation steps. One limitation of working with such a large number of cell lines and culture plates was the impossibility to perform overnight media changes. The main reason is the fact that our system is set up for using disposable tips and manual refilling of culture media requiring an user in the laboratory to execute this manual step. To facilitate the media change process, plates loaded in the system were assigned to a project and grouped in batches. The batch size was then adapted to the number of disposable tips and volume of culture media available. As discussed above, this limitation can be easily overcome by implementation of reusable/washable needles and automated refilling of media. Automated passaging/replating of cells, as shown for iPSC, is one of the conveniences offered by our automated cell culture system. We have tested the automated replating of mDA neurons on day 25 of differentiation. However, the dissociation of mDA neurons requires longer (40 min) incubation with the dissociation enzyme than iPSC (8 min) extending the automated replating process to more than 1 h per cell lines. As a consequence, the automated replating of 30 cell lines in the same day became impossible. Speeding up other steps during automated replating (transport of plates, pipetting) and adapting the system to the use of needles and media line that makes an overnight work possible would resolve this limitation. Despite the drawbacks, we could successfully transfer the manual protocol to an automated differentiation of mDA neurons producing cultures with substantial amounts of MAP2 (neuron) and TH (mDA neurons) positive cells.
Differentiating dozens of iPSC cell lines in parallel is of great interest in projects that investigate the molecular mechanisms of neurodegenerative diseases, including Parkinson’s disease. However, to complete tasks faster with fewer errors and at reduced costs is a big challenge. Due to the automation of the protocols presented here (iPSC, smNPC and mDA neuron), we could speed up, reduce the costs and increase reproducibility in our projects. The development of projects like FOUNDIN-PD (https://www.foundinpd.org/wp/) involving hundreds of patient cell lines shows need for automated culture and differentiation protocols. Our future perspectives include the transfer of manual 3 dimensional (3D) cell culture models to the automated system. Minor adaptations in the plate definition settings and the use of adaptors will allow the use of commercial or custom-made plates and microfluidic chambers required for the 3D cultures. Moreover, the implementation of an automated label-free imaging model will allow us to track the neuronal growth in real time and translate changes in neurite outgrowth, neuron organization and cell death into better understanding of the disease mechanisms.
Acknowledgements
The authors gratefully acknowledge the patients and their families who contributed biomaterial for this study. Cells lines used in the study were from NINDS collection with Rutgers (ND41865 as iPS#1) and the lab of Dr. Tilo Kunath (iPS#2). This work is supported in part by the NOMIS Foundation (PH), RiMod-FTD, an EU Joint Programme - Neurodegenerative Disease Research (JPND) (PH); The DZNE I2A initiative (AD); PD-Strat, an ERA-Net ERACoSysMed funded project (PH) and the Foundational Data Initiative for Parkinson's Disease (FOUNDIN-PD) (PH, EB). FOUNDIN-PD is part of The Michael J. Fox Foundation’s PATH to PD program. The authors thank Steven Finkbeiner and Melanie Cobb (Gladstone Institutes) for contributing to the establishment of the manual mDA neuron differentiation protocol and Mahomi Suzuki (Yokogawa Electric Corporation) for assistance in neurite outgrowth analysis setup.
Materials
| Name | Company | Catalog Number | Comments |
| Antibodies | Distributor | Catalog Number | Dilution |
| iPSC pluripotency marker | |||
| Mouse anti-SSEA4 | Abcam | ab16287 | 1 to 33 |
| Rabbit anti-Oct3/4 | Abcam | ab19857 | 1 to 200 |
| NGN2 neuron markers | |||
| Mouse anti- TUBB3 | R & D | MAB1195 | 1 to 500 |
| Rabbit anti-BRN2 | NEB | 12137 | 1 to 1,000 |
| mDA neuron markers | |||
| Chicken anti-TH | Pel-Freez Biologicals | 12137 | 1 to 750 |
| Mouse anti-MAP2 | Santa Cruz | sc-74421 | 1 to 750 |
| Secondary antibodies | |||
| Goat anti-chicken IgY, Alexa Fluor 488 | Invitrogen | A11039 | 1 to 2,000 |
| Goat anti-mouse IgG, Alexa Fluor 488 | Invitrogen | A11029 | 1 to 2,000 |
| Goat anti-mouse IgG, Alexa Fluor 594 | Invitrogen | A11032 | 1 to 2,000 |
| Goat anti-rabbit IgG, Alexa Fluor 488 | Invitrogen | A11008 | 1 to 2,000 |
| Goat anti-rabbit IgG, Alexa Fluor 488 | Invitrogen | A11008 | 1 to 2,000 |
| Goat anti-rabbit IgG, Alexa Fluor 594 | Invitrogen | A11012 | 1 to 2,000 |
| Nuclei counterstaining | |||
| Hoechest 33342 | Invitrogen | H3570 | 1 to 8,000 |
| Instruments | Distributor | Catalog Number | Description/Application |
| Agilent TapeStation system | Agilent technologies | 4200 | Automated electrophoresis for DNA and RNA samples |
| Automated -20 °C storage system | Hamilton Storage Technologies | Sample Access Manager (SAM -20C, 3200 series) | Storage of reagents |
| Barcode reader | Honeywell | Barcode Reader Orbit | Barcode scanner |
| Brightfield cell cytomat | Nexcelom | Celigo | Confluence check and cell counting |
| CellVoyager 7000 | Yokogawa | CellVoyager 7000 | Automated confocal microscope |
| Cytomat for cell cultures | Thermo Fisher Scientific | Cytomat 24 C, CU | 12 stackers pitch 28 mm, 12 stackers pitch 23 mm (total of 456 plates) |
| Cytomat for thawing samples | Thermo Fisher Scientific | Cytomat 2-LIN, 60 DU (Drying Unit) | 2 stackers pitch 28 mm (total of 42 plates) |
| HEPA filters | Hamilton Robotics | Hood Flow Star UV | Modified by Hamilton Robotics |
| Liquid handling station | Hamilton Robotics | Microlab Star | Channels: 8x 1-ml, 4x 5 ml and 96 Channel MPH |
| Media reservoir | Hamilton Robotics | 188211APE | Media/reagents reservoir |
| Pure water system | Veolia Water | ELGA PURELAB Classic | Provides pure water for needle wash station |
| QuantStudio 12K Flex Real-Time PCR System | Thermo Fisher Scientific | QSTUDIO12KOA | Real-time PCR machine |
| Robotic arm | Hamilton Robotics | Rackrunner | Transport of plates |
| Turn table | Hamilton Robotics | Turn Table | Adjust plate orientiation |
| Uninterruptible power supply | APC | Smart UPS RT Unit, 10000 VA Power supply | Backup power supply |
| VIAFLO-pipettes | Integra | 4500 | Electronic pipette |
| ViiA 7 Real-Time PCR System | Applied Biosystems | 4453545 | Real-time PCR machine |
| Materials | Distributor | Catalog Number | Notes |
| 1-well culture plate (84 cm2) | Thermo Fischer Scientific | 165218 | Nunc OmniTray |
| 6-well culture plates (9.6 cm2) | Greiner Bio-One | 657160 | TC treated with lid |
| 12-well culture plate (3.9 cm2) | Greiner Bio-One | 665180 | TC treated with lid |
| 96-well culture plate (0.32 cm2) | Perkin Elmer | 6005558 | CellCarrier-96 Black plate |
| Tips, 50-µL | Hamilton Robotics | 235987 | 50-uL tips |
| Tips, 300-µL | Hamilton Robotics | 235985 | 300-µL tips |
| Tips, 1000-µL | Hamilton Robotics | 235939 | 1000-µL tips |
| Tips, 5-mL | Hamilton Robotics | 184022 | 5-mL tips |
| Tubes, 15-mL | Greiner Bio-One | 188271 | 15-mL tubes |
| Tubes, 50-mL | Greiner Bio-One | 227261 | 50-mL tubes |
| Nalgene cryogenic 2.0 mL vials | Sigma Aldrich | V5007 | Cryovials |
| Plasmids | Distributor | Catalog Number | Notes |
| pLV_hEF1a_rtTA3 | Addgene | 61472 | Kind gift from Ron Weiss |
| pLV_TRET_hNgn2_UBC_Puro | Addgene | 61474 | Kind gift from Ron Weiss |
| pLVX-EF1a-AcGFP1-N1 lentivirus | Takara Bio | 631983 | |
| Primers | Sequence (forward) | Sequence (reverse) | Source |
| iPSC pluripotency | |||
| OCT4 (ID 4505967a1) | CTTGAATCCCGAATGGAAAGGG | GTGTATATCCCAGGGTGATCCTC | PrimerBank |
| NANOG (ID 153945815c3) | CCCCAGCCTTTACTCTTCCTA | CCAGGTTGAATTGTTCCAGGTC | PrimerBank |
| REX1 (ID 89179322c1) | AGAAACGGGCAAAGACAAGAC | GCTGACAGGTTCTATTTCCGC | PrimerBank |
| NGN2 neurons | |||
| MAP2 (ID 87578393c1) | CTCAGCACCGCTAACAGAGG | CATTGGCGCTTCGGACAAG | PrimerBank |
| BRN2 (ID 380254475c1) | CGGCGGATCAAACTGGGATTT | TTGCGCTGCGATCTTGTCTAT | PrimerBank |
| CUX2 (ID 291045458c2) | CGAGACCTCCACACTTCGTG | TGTTTTTCCGCCTCATTTCTCTG | PrimerBank |
| NCAM1 (ID 336285437c3) | TGTCCGATTCATAGTCCTGTCC | CTCACAGCGATAAGTGCCCTC | PrimerBank |
| SYNAPSIN1 (ID 91984783c3) | TGCTCAGCAGTACAACGTACC | GACACTTGCGATGTCCTGGAA | PrimerBank |
| mDA neurons | |||
| TH | CGGGCTTCTCGGACCAGGTGTA | CTCCTCGGCGGTGTACTCCACA | NCBI primer-BLAST |
| MAP2 | GGATCAACGGAGAGCTGAC | TCAGGACTGCTACAGCCTCA | NCBI primer-BLAST |
| Housekeeping genes | |||
| GAPDH | GAAATCCCATCACCATCTTCCAGG | GAGCCCCAGCCTTCTCCATG | NCBI primer-BLAST |
| OAZ1 | AGCAAGGACAGCTTTGCAGTT | ATGAAGACATGGTCGGCTCG | NCBI primer-BLAST |
| RPLPO | CCTCATATCCGGGGGAATGTG | GCAGCAGCTGGCACCTTATTG | NCBI primer-BLAST |
| RPL13A | GCCTACAAGAAAGTTTGCCTATC | TGGCTTTCTCTTTCCTCTTCTC | NCBI primer-BLAST |
| Media and Reagents | Distributor | Catalog Number | Use concentration |
| Coating matrix | |||
| Extracellular matrix (Matrigel) | Corning | 354277 | 10 μg/mL |
| Fibronectin | Corning | 356008 | 2 μg/mL |
| Laminin | Sigma | L2020 | 5 - 10 μg/mL |
| Poly-L-Ornithine (PLO) | Sigma | P3655 | 0.1 mg/mL |
| Culture media | |||
| iPSC culture medium (Essential 8 Flex medium) | Gibco | A2858501 | |
| NGN2 neurons - NGN2 medium | |||
| 2-mercaptoethanol | Gibco | 21985023 | 0.909 mL (50 µM) |
| B27 supplement | Gibco | 12587010 | 10 mL (1%) |
| DMEM/F-12, GlutaMAX | Gibco | 31331093 | 484.75 mL |
| GlutaMAX | Gibco | 35050038 | 5 mL ( 2 mM) |
| Insulin | Sigma | I9278 | 0.25 mL (2.5 µg/mL) |
| MEM Non-Essential Amino Acids | Gibco | 11140050 | 5 mL (0.5%) |
| N2 supplement | Gibco | 17502048 | 5 mL (0.5%) |
| Neurobasal medium | Gibco | 21103049 | 485 mL |
| mDA neurons - SRM medium | |||
| 2-mercaptoethanol | Gibco | 21985023 | 0.5 mL (55 µM) |
| GlutaMAX | Gibco | 35050038 | 5 mL (2 mM) |
| Knockout DMEM/F-12 | Gibco | 12660012 | 409.5 mL |
| Knockout serum replacement (serum replacement) | Gibco | 10828028 | 75 mL (15%) |
| MEM Non-Essential Amino Acids | Gibco | 11140050 | 5 mL (1%) |
| Penicillin-Streptomycin | Gibco | 15140122 | 5 mL (1%) |
| mDA neurons - N2 medium | |||
| B27 supplement | Gibco | 12587010 | 10 mL (2%) |
| GlutaMAX | Gibco | 35050038 | 5 mL (2 mM) |
| N2 supplement | Gibco | 17502048 | 5 mL (1%) |
| Neurobasal medium | Gibco | 21103049 | 475 mL |
| Penicillin-Streptomycin | Gibco | 15140122 | 5 mL(1%) |
| mDA neurons - Differentiation medium | |||
| B27 supplement | Gibco | 12587010 | 10 mL (2%) |
| Neurobasal medium | Gibco | 21103049 | 485 mL |
| Penicillin-Streptomycin | Gibco | 15140122 | 5 mL (1%) |
| NGN2 neurons - Supplements | |||
| Brain-Derived Neurotrophic Factor (BDNF) | Peprotech | 450-02 | 10 ng/mL |
| CHIR99021 (CHIR) | R&D | 4423/10 | 2 μM |
| Doxycyline (dox) | Sigma | D9891 | 2.5 μg/mL |
| Glial-Derived Neurotrophic Factor (GDNF) | Peprotech | 450-10 | 10 ng/mL |
| L-ascorbic acid 2-phosphate magnesium (AA2) | Sigma | A8960 | 64 mg/L |
| Neurotrophic factor-3 (NT-3) | Peprotech | 450-10 | 10 ng/mL |
| Purmorphamine (PMA) | Cayman | 10009634 | 0.5 μM |
| Puromycin | Sigma | P8833-10MG | 0.5 μg/mL |
| Thiazovivin | Merk Millipore | 420220-10MG | 2 μM |
| mDA neurons - Supplements | |||
| Brain-Derived Neurotrophic Factor (BDNF) | Peprotech | 450-02 | 20 ng/mL |
| CHIR99021 (CHIR) | R&D | 4423/10 | 3 μM |
| DAPT | Cayman | 13197 | 10 µM |
| Dibutyryl-cAMP (db-cAMP) | Sigma | D0627 | 1 mM |
| Fibroblast Growth Factor 8B (FGF-8b) | Peprotech | 100-25 | 100 ng/mL |
| Glial-Derived Neurotrophic Factor (GDNF) | Peprotech | 450-10 | 20 ng/mL |
| L-ascorbic acid (AA1) | Sigma | A4403 | 0.2 mM |
| LDN193189 (LDN) | Cayman | 11802 | 100 nM |
| Purmorphamine (Purm) | Cayman | 10009634 | 2 μM |
| Sonic Hedgehog/Shh (C24II) N-Terminus (SHH) | R&D | 1845-SH | 100 ng/mL |
| SB431542 (SB) | Cayman | 13031 | 10 μM |
| Transforming Growth Factor type ß3 (TGFß3) | R&D | 243-B3 | 1 ng/mL |
| Y-27632 | Cayman | 10005583 | 10 µM |
| Dissociation reagents | |||
| Single cell dissociation reagent (StemPro Accutase) | Gibco | A1110501 | 1x |
| UltraPure 0.5M EDTA, pH 8.0 | Gibco | 11575020 | 0.5 mM |
| RNA isolation and cDNA kit | |||
| RNA isolation kit | Qiagen | 74106 | Rneasy MiniKit |
| RNA lysis buffer | Thermo Fisher Scientific | 15596018 | Trizol lysis buffer (RNA lysis buffer) |
| Reverse transcriptase (kit) | Thermo Fisher Scientific | 18080-085 | SuperScript III Reverse Transcriptase |
| Software | Company | Catalog Number | Description/Application |
| Cell Culture Framework (CCF) (Graphical user interface, GUI) | Hamilton Robotics | Custom-made | User interface for the automated system |
| CellPathFinder software (image analysis software 1) | Yokogawa | CellPathfinder HCS Software | Image analysis tool |
| CellVoyager Measurement System | Yokogawa | Included with CellVoyager 7000 | Microscope controlling software |
| Columbus software (image analysis software 2) | Perkin Elmer | Columbus | Image analysis tool |
| Cloud based qPCR app | Thermo Fisher Scientific | Themo Fisher cloud | Analysis software for qRT-PCR data |
| Venus software | Hamilton Robotics | VENUS Two Dynamic Schedular 5.1 Software | Controlling software for the automated system |
References
- Takahashi, K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131 (5), 861-872 (2007).
- Payne, N. L., Sylvain, A., O'Brien, C., Herszfeld, D., Sun, G., Bernard, C. C. A. Application of human induced pluripotent stem cells for modeling and treating neurodegenerative diseases. New biotechnology. 32 (1), 212-228 (2015).
- Wu, Y. Y., Chiu, F. L., Yeh, C. S., Kuo, H. C. Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. Open Biology. 9 (1), 180177 (2019).
- Avior, Y., Sagi, I., Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nature Reviews Molecular cell biology. 17 (3), 170-182 (2016).
- Jain, S., Heutink, P. From single genes to gene networks: high-throughput-high-content screening for neurological disease. Neuron. 68 (2), 207-217 (2010).
- Soares, F. A. C., Chandra, A., Thomas, R. J., Pedersen, R. A., Vallier, L., Williams, D. J. Investigating the feasibility of scale up and automation of human induced pluripotent stem cells cultured in aggregates in feeder free conditions. Journal of biotechnology. 173, 53-58 (2014).
- Konagaya, S., Ando, T., Yamauchi, T., Suemori, H., Iwata, H. Long-term maintenance of human induced pluripotent stem cells by automated cell culture system. Scientific reports. 5, 16647 (2015).
- Conway, M. K., et al. Scalable 96-well Plate Based iPSC Culture and Production Using a Robotic Liquid Handling System. Journal of visualized experiments: JoVE. (99), e52755 (2015).
- Paull, D., et al. high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nature methods. 12 (9), 885-892 (2015).
- Busskamp, V., et al. Rapid neurogenesis through transcriptional activation in human stem cells. Molecular systems biology. 10, 760 (2014).
- Zhang, Y., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 78 (5), 785-798 (2013).
- Reinhardt, P., et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PloS one. 8 (3), 59252 (2013).
- Kriks, S., et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 480 (7378), 547-551 (2011).
- Koch, J. C., et al. Alpha-Synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell death & disease. 6, 1811 (2015).
- Korecka, J. A., et al. Neurite Collapse and Altered ER Ca2+ Control in Human Parkinson Disease Patient iPSC-Derived Neurons with LRRK2 G2019S Mutation. Stem cell reports. 12 (1), 29-41 (2019).
- Mehta, S. R., et al. Human Huntington's Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell reports. 25 (4), 1081-1096 (2018).
- Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., Madden, T. L. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC bioinformatics. 13, 134 (2012).
- Spandidos, A., Wang, X., Wang, H., Seed, B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic acids research. 38, 792 (2010).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved