Mapping the Structure-Function Relationships of Disordered Oncogenic Transcription Factors Using Transcriptomic Analysis
In This Article
Summary
Intrinsically disordered domains are important for oncogenic fusion transcription factor function. To therapeutically target these proteins, a more detailed understanding of the regulatory mechanisms employed by these domains is required. Here, we use transcriptomics to map important structural features of the intrinsically disordered EWS domain in Ewing sarcoma.
Abstract
Many cancers are characterized by chromosomal translocations which result in the expression of oncogenic fusion transcription factors. Typically, these proteins contain an intrinsically disordered domain (IDD) fused with the DNA-binding domain (DBD) of another protein and orchestrate widespread transcriptional changes to promote malignancy. These fusions are often the sole recurring genomic aberration in the cancers they cause, making them attractive therapeutic targets. However, targeting oncogenic transcription factors requires a better understanding of the mechanistic role that low-complexity, IDDs play in their function. The N-terminal domain of EWSR1 is an IDD involved in a variety of oncogenic fusion transcription factors, including EWS/FLI, EWS/ATF, and EWS/WT1. Here, we use RNA-sequencing to investigate the structural features of the EWS domain important for transcriptional function of EWS/FLI in Ewing sarcoma. First shRNA-mediated depletion of the endogenous fusion from Ewing sarcoma cells paired with ectopic expression of a variety of EWS-mutant constructs is performed. Then RNA-sequencing is used to analyze the transcriptomes of cells expressing these constructs to characterize the functional deficits associated with mutations in the EWS domain. By integrating the transcriptomic analyses with previously published information about EWS/FLI DNA binding motifs, and genomic localization, as well as functional assays for transforming ability, we were able to identify structural features of EWS/FLI important for oncogenesis and define a novel set of EWS/FLI target genes critical for Ewing sarcoma. This paper demonstrates the use of RNA-sequencing as a method to map the structure-function relationship of the intrinsically disordered domain of oncogenic transcription factors.
Introduction
A subset of cancers, including many malignancies of childhood and adolescence, are characterized by chromosomal translocations which generate novel fusion oncogenes1,2,3,4,5,6. The resulting fusion proteins frequently function as oncogenic transcription factors, orchestrating widespread changes in transcriptional regulation to promote tumorigenesis7,8. Cancers with these translocations commonly possess an otherwise quiet mutational landscape, with few recurring genomic aberrations aside from the pathognomonic fusion4,9. As such, directly targeting the fusion protein is an attractive therapeutic strategy in these diseases. However, these oncogenic transcription factors commonly consist of a low-complexity, intrinsically disordered, transcriptionally activating domain fused with a DNA-binding domain (DBD)10,11,12,13,14. Both the intrinsically disordered domains (IDDs) and DBDs of these proteins have proven difficult to target with conventional pharmacological approaches. Development of novel therapeutic approaches, therefore, requires a more detailed molecular understanding of the mechanisms employed by these fusions to aberrantly regulate gene expression.
The N-terminal IDD portion of EWSR1 is commonly fused to a DBD in cancer, including EWS/FLI in Ewing sarcoma, EWS/WT1 in diffuse small round cell tumor, and EWS/ATF1 in clear cell sarcoma of soft parts10. The mechanistic role of the EWS IDD in each of these fusions is incompletely understood. The EWS/ETS family of fusions, specifically EWS/FLI, is the most functionally characterized to date. EWS/FLI coordinates genome-wide epigenetic and transcriptional changes leading to the activation and repression of thousands of genes7,11,15,16. Studies have shown that the IDD is important for the recruitment of both transcriptional co-activators (such as p300, WDR5, and the BAF complex), as well as co-repressors (such as the NuRD complex)11,15,17. The fusion of the EWS IDD to the C-terminal portion of FLI1 confers novel DNA-binding specificity to the ETS DBD of FLI1, such that the fusion oncoprotein (EWS/FLI) binds to repetitive GGAA-microsatellite regions of the genome in addition to the consensus ETS motif18,19,20. Combined with the co-activator recruitment function, this emergent DNA-binding activity of EWS/FLI promotes de novo enhancer formation at GGAA-microsatellites distal to transcription start sites (TSS) (“enhancer-like” microsatellites) and recruits RNA polymerase II to promote transcription at GGAA-microsatellites proximal to TSS (“promoter-like” microsatellites)11,15,16,21.
Taken together, these data led us to hypothesize that discrete elements within the EWS domain contribute to the recruitment of distinct co-regulators to different types of EWS/FLI binding sites. However, discerning these elements within the EWS portion of EWS/FLI, and how they function, has been hindered by the highly repetitive and disordered nature of the domain. Here we utilize a previously published knockdown-rescue system in Ewing sarcoma cells to functionally map these elements in the EWS IDD. In this system EWS/FLI is depleted using an shRNA targeting the 3’UTR of the FLI1 gene, and expression is rescued with varying EWS/FLI mutant cDNA constructs lacking the 3’UTR7,17,22. These experiments focused on constructs with various deletions to map the structure-function relationship between the EWS IDD and important oncogenic phenotypes, including activation of a GGAA-microsatellite reporter construct, colony formation assays, and targeted validation of EWS/FLI-activated and -repressed genes7,17,22. However, these studies failed to find discrete sub-domains within the EWS IDD in EWS/FLI that are uniquely important for either activation or repression. All tested constructs were either able to both activate and repress specific target genes, leading to efficient colony formation, or unable to regulate any of the EWS/FLI target genes, leading to loss of colony formation7,17,22.
Transcriptomic analyses enabled by the widespread adoption of the next generation sequencing are commonly used to compare gene expression signatures in two conditions, frequently in the context of screening or descriptive studies. We instead wanted to leverage the ability to capture genome-wide expression data using RNA-sequencing (RNA-seq) to characterize the contributions of IDDs to transcription factor function. In this case RNA-seq is paired with the knockdown-rescue system to explore the structure-function relationship of the EWS domain. This approach is applicable to other fusion transcription factors, including other EWS fusions or wildtype transcription factors with poorly understood function, and has multiple advantages over the other assays used for functional mapping studies, such as reporter assays or targeted qRT-PCR. These include testing structural determinants of function in the relevant chromatin context, the ability to test multiple types of response elements in one assay (i.e., activated and repressed, GGAA-microsatellite and non-microsatellite, etc.), and the resulting ability to better detect partial function.
Successful implementation of this approach depends on a cell-based system that captures the phenotypes of interest (in this case A673 cells with shRNA-mediated EWS/FLI depletion), and a panel of mutant constructs in an expression vector appropriate for the cell-based system (in this case, pMSCV-hygro with various 3x-FLAG-tagged EWS/FLI mutants to be delivered by retroviral transduction). Viral transduction of either CRISPR-based depletion constructs, shRNA-based depletion constructs, and cDNA expression constructs with appropriate selection to generate stable cell lines is recommended over transient transfection. The downstream interpretation of results is strengthened when the transcriptomic data can be paired with other data related to localization of the transcription factor and other phenotypic readouts where available.
In this paper, we apply this approach to characterize the activity of the DAF mutant of EWS/FLI14. The DAF mutant has 17 tyrosine to alanine mutations in the repetitive regions of the EWS IDD of EWS/FLI14. This particular EWS mutant had been previously reported and is unable to activate reporter gene expression when fused to the ATF1 DBD14. However, preliminary qRT-PCR data suggested that this mutant was able to activate transcription of the EWS/FLI target NR0B123. The transcriptomic approach described here enabled successful detection of partial function of the DAF mutant. By pairing these transcriptomic data with information about EWS/FLI binding and recognition motifs we further show that the DAF mutant retains function at GGAA-microsatellite repeats. These results identify DAF as the first partially functional EWS/FLI mutant and highlight function at non-microsatellite genes as important for oncogenesis (as reported23). This demonstrates the power of this transcriptomic structure-function mapping approach to provide insight into the function of oncogenic transcription factors.
Protocol
1. Set up in vitro panel of constructs
NOTE: This step will vary depending on the specific protein to be analyzed.
- Prepare aliquots of virus for depletion and expression constructs as necessary.
- Seed a 10 cm tissue culture dish with 3-5 x 106 HEK293-EBNA or HEK293T cells for each construct needed for viral transduction. Let cells adhere overnight in Dulbecco’s Modified Eagle Media (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin/glutamine (P/S/Q), and 0.3 mg/mL G418.
NOTE: HEK293-EBNA and HEK293T cells are recommended for viral production because they are easy to grow, have high transfection efficiency, and efficiently express recombinant proteins from episomal plasmids. The cells should be between 50-70% confluent the day of transfection. - Prepare a transfection mix for each viral transduction construct. Combine 2 mL of reduced serum media with 90 µL of transfection reagent.
NOTE: Pre-warming reduced serum media is recommended. - Add 10 µg each of a viral packaging plasmid (e.g., gag-pol), viral envelope plasmid (e.g., VSV-G), and one of either CRISPR-based depletion, shRNA-based depletion, or cDNA expression construct (e.g., pMKO or pMSCV) to the transfection mix. Mix well by gentle pipetting.
- Let the transfection mix sit for 20 min at room temperature. Remove HEK293-EBNA growth media from tissue culture dishes and add 3 mL DMEM supplemented with 10% FBS, P/S/Q, and 10 mM sodium pyruvate. To each dish, add 2 mL of transfection mix dropwise. Let cells sit in transfection media overnight in an incubator at 37 ˚C and 5% CO2.
- The following morning add 20 mL of DMEM media with 10% FBS, P/S/Q supplementation, and 10 mM sodium pyruvate. Incubate the cells in it at 37 ˚C and 5% CO2 for overnight.
- The next morning, replace media with 5 mL viral collection media (VCM) (DMEM supplemented with 10% heat inactivated FBS, P/S/Q, and 20 mM HEPES).
- After 4 h, collect VCM from plates and store in a 50 mL conical tube on ice at 4 °C. Replace with 5 mL of fresh VCM.
- After 4 h, collect VCM from plates in same 50 mL conical tube and store on ice at 4 °C. Replace with 8 mL of fresh VCM for overnight collection.
- In the morning collect VCM from plates and store in the 50 mL conical tube on ice at 4 °C. Replace with 5 mL of fresh VCM.
- After 4 h, collect VCM from plates and store in the 50 mL conical tube on ice at 4°C. Replace with 5 mL of fresh VCM. After 4 h, collect VCM from plates and add to the 50 mL conical tube.
- Aliquot collections from 50 mL tube into cryotubes (2 mL per aliquot) after filtration through a 0.45 µm filter. Store viral aliquots at -80 °C until use.
NOTE: The protocol can be paused here, and the viral aliquots can be stored until ready for use.
- Seed a 10 cm tissue culture dish with 3-5 x 106 HEK293-EBNA or HEK293T cells for each construct needed for viral transduction. Let cells adhere overnight in Dulbecco’s Modified Eagle Media (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin/streptomycin/glutamine (P/S/Q), and 0.3 mg/mL G418.
- Seed cells at the appropriate density in a 10 cm tissue culture dish. Target 50% confluence. Let cells adhere overnight by placing in the incubator at 37 ˚C containing 5% CO2.
NOTE: For A673 cells this is 5 x 106 cells in 10 mL of DMEM media with 10% FBS, P/S/Q supplementation, and 10 mM sodium pyruvate. These conditions may vary depending on the growth rate of the cells used. - Deplete endogenous factor of interest. If cells do not need to have the endogenous protein of interest depleted, skip ahead to step 1.4.
- Thaw viral aliquot for transduction of shRNA or CRISPR construct targeting the protein of interest. Thaw frozen aliquots quickly in a 37 °C water bath.
- Add 2.5 µL of 8 mg/mL polybrene to each viral aliquot and mix by gentle pipetting. Remove media from plates of cells and gently add viral aliquot to 10 cm plate by pipetting along the side of the plate. Rock the plate to spread the 2 mL of viral aliquot.
- Incubate at 37 °C in the tissue culture incubator for 2 h. Rock the plate every 30 min to prevent any areas of the plate drying out.
- Add 5 mL of DMEM media with 10% FBS, P/S/Q supplementation, and 10 mM sodium pyruvate, with 5 µL of 8 mg/mL polybrene. Let cells incubate overnight.
- In the morning remove media from cells and passage cells into media supplemented with a selection reagent. When passaging cells, seed them in a manner to allow them to grow for 48-72 h and reach 50% confluency.
NOTE: For A673 cells with pSRP-iEF-2, cells are seeded in a 1:5 split and selected for 72 h with 2 µg/mL puromycin.
- Transduce cDNA expression constructs.
- Check cells to confirm 50-70% confluency.
- Thaw viral aliquot(s) for transduction of cDNA construct(s) of interest. Thaw frozen aliquots quickly in a 37 °C water bath. Add 2.5 µL of 8 mg/mL polybrene to each viral aliquot and mix by gently pipetting.
- Remove media from plated cells and gently add viral aliquot to 10 cm plate by pipetting along the side of the plate. Rock the plate to spread the 2 mL of viral aliquot.
- Incubate at 37 °C in the tissue culture incubator for 2 h. Rock the plate every 30 min to prevent any areas of the plate drying out.
- Add 5 mL of DMEM media with 10% FBS, P/S/Q supplementation, and 10 mM sodium pyruvate, with 5 µL of 8 mg/mL polybrene. Let cells incubate overnight.
- In the morning remove media from cells and passage cells into double selection media. Grow and passage cells as needed for 7-10 days to allow for double selection and expression of the cDNA construct.
NOTE: This split of this passage may require optimization for different cell lines. For A673 cells with pSRP-iEF-2 and a pMSCV-hygro construct, cells are passed without splitting into 2 µg/mL puromycin and 100 µg/mL hygromycin.
2. Collect cells, validate expression of constructs, and set up correlative phenotypic assays
- After 7-10 days of double selection collect cells in a 15 mL conical tube. Count collected cells with a hemocytometer. Aliquot collected cells for RNA-sequencing and to validate expression of cDNA constructs.
NOTE: Set up any correlative phenotypic assays required by the research question under investigation. Colony forming assays are an example of a correlative phenotypic assay that are used here.- Collect between 5 x 105 and 1 x 106 cells for RNA-sequencing and 2 x 106 cells for protein extraction. Pellet cells by centrifugation at 1,000 x g at 4 °C for 5 min and remove the supernatant.
- Wash the pellet with 1 mL cold PBS. Pellet by centrifugation at 1,000 x g at 4 °C for 5 min and remove supernatant. Flash freeze pellets in liquid nitrogen and store at -80 °C.
- Set up any correlative assays with the remaining cells.
NOTE: The protocol can be paused here with collected samples stored in the -80 °C freezer.
- Validate the knockdown of protein of interest (if used) and expression of the panel of constructs.
- Thaw cell pellets for protein extraction on ice. Resuspend cells in ice cold 500 µL nuclear extraction buffer (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1% IGEPAL) with protease inhibitor. Let it sit for 5 min on ice.
- Pellet nuclei by centrifugation at 1,000 x g at 4 °C for 5 min and remove supernatant. Wash nuclei in 500 µL ice cold nuclear extraction buffer (20 mM HEPES pH 7.9, 140 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1% IGEPAL) with protease inhibitor.
- Pellet nuclei by centrifugation at 1,000 x g at 4 °C for 5 min and remove the supernatant. Resuspend nuclei in 200 µL cold RIPA buffer with protease inhibitor (adjust the volume of RIPA buffer according to pellet size.) Let it sit on ice for 45-60 min with vigorous vortexing every 15 min.
- Pellet cell debris by centrifugation at 16,000 x g at 4 °C for 45-60 min. Keep the supernatant and transfer to a fresh cold tube
- Prepare samples for SDS-PAGE electrophoresis by boiling 5-10 µg of protein with 1x loading buffer for 5 min. Run an SDS-PAGE gel as required for the protein of interest.
- Transfer to a nitrocellulose or PVDF membrane as needed for the protein of interest. Block, and blot with the appropriate primary and secondary antibodies to confirm the knockdown of the endogenous protein (if used) and ectopic expression of the cDNA construct.
NOTE: The protocol can be paused here.
- Extract RNA. Assess RNA quality and quantity.
- Thaw cell pellets on ice. Extract total RNA using a silica spin-column based extraction kit according to the manufacturer’s instructions.
- Briefly, lyse the cells using the lysis buffer from the kit. Either apply the lysate to a silica spin-column with a brief spin at >13000 rpm for 30-60 seconds or remove gDNA by applying the lysate to a gDNA removal column with a brief spin at >13000 rpm for 30-60 seconds.
- Perform an on-column DNA digestion if lysate was directly applied to a silica spin-column. If using a gDNA removal column, apply the eluate to a silica-spin column with a brief spin at >13000 rpm for 30-60 s.
- Wash RNA on the column per the manufacturer’s instructions. Elute RNA in 30 µL of elution buffer.
- Assess RNA quality and quantity using a fluorometer, or any other comparable instrument. Make sure the 260/280 ratio is close to 2 and that there are at least 2.5 µg of RNA to submit for sequencing.
NOTE: As replicates are gathered, each replicate must be processed with the same RNA extraction protocol. - Use a small aliquot of RNA to confirm the stable knockdown of the protein of interest, if required, by qRT-PCR. Store the remaining RNA sample at -80 °C.
- Collect biological replicates by repeating steps 1-2 until 3-4 complete sets of RNA have been collected. Ensure that each replicate displays adequate expression of cDNA constructs and stable knockdown of the endogenous protein (if used).
3. Next-Generation Sequencing
- Submit extracted RNA to be sequenced using a next generation sequencing platform with a target of 50 million 150 base pair (bp) paired end reads. Follow the instructions of the facility processing the samples. Select for poly-adenylated RNAs and strand-specific sequencing.
4. Alignment and Transcript Counting Pipeline
NOTE: This protocol assumes that following sample submission and processing, a set of paired FASTQ files are returned for each sample. These files are frequently compressed with a suffix of “fastq.gz”. Further analysis of these FASTQ files will require access to a high-performance computing (HPC) facility running a Linux operating system.
- Transfer files
- Open a terminal to the HPC environment with PuTTY. Make a directory for the analysis called “project”.
- Navigate to the “path_to/project” directory and make a new directory for the compressed raw fastq.gz files called “fastq”. Also make a directory called “trimmed”. This is shown in Figure S1A-C.
- Transfer the compressed raw fastq.gz files from local storage to the “path_to/project/fastq/” directory using WinSCP or a similar program. Check that there is a “R1” and an “R2” file for each sample as shown in Figure S1B.
- Optional: If required, install TrimGalore. Set the directory containing the trim_galore executable file in the PATH environment variable in Linux.
NOTE: Low quality reads and adapters are trimmed with TrimGalore. TrimGalore is available at https://github.com/FelixKrueger/TrimGalore. - Optional: Navigate to the directory for downloaded software packages (i.e “path_to/software”). Download the latest TrimGalore package using the command “curl -fsSL https://github.com/FelixKrueger/TrimGalore/archive/[version].tar.gz -o trim_galore-[version].tar.gz.”
- Optional: Unpack the tar.gz file. Use the command “tar -xvzf trim_galore-[version_number].tar.gz”.
- Optional: Make TrimGalore executable. Use the command “chmod a+x path_to/software/TrimGalore-[version]/trim_galore”. Make sure this new directory is in the PATH. Use the command “export PATH=path_to/software/TrimGalore-[version]:$PATH”.
- Navigate to path_to/project/fastq/. Use TrimGalore to trim the low quality reads from the fastq.gz files using the command shown in Figure S1C.
NOTE: Additional flags for this command may be relevant and can be found here: https://github.com/FelixKrueger/TrimGalore/blob/master/Docs/
Trim_Galore_User_Guide.md - Check for the trimmed fastq.gz files in the path_to/project/trimmed directory. Ensure they are called sample1_R1_val_1.fq.gz and sample1_R2_val_2.fq.gz
- Align trimmed FASTQ files with STAR and generate transcript counts.
NOTE: STAR is available at https://github.com/alexdobin/STAR)- Optional: Install STAR version 2.6 or later. Set the STAR executable in the path.
- Optional: Navigate to the directory for downloaded software packages (i.e “path_to/software”).
- Optional: Download the STAR package using the command “curl -SLO https://github.com/alexdobin/STAR/archive/[version].tar.gz”. Unpack the tar.gz file.
- Optional: Use the command “tar -xzf [version].tar.gz”. Make STAR executable. Use the command “chmod a+x path_to/software/STAR-[version]/bin”.
- Optional: Make sure this new directory is in the path. Use the command “export PATH=path_to/software/STAR-[version_number]/bin/linux_x86_64_static:$PATH”.
NOTE: The STAR manual is available at: (https://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf). - Ensure there is genome index to use with STAR. Place this in a directory separate from the path_to/project/ directory. If an index was previously generated for prior experiments use that. Alternatively use an appropriate pre-generated index if available here: http://refgenomes.databio.org/. Otherwise, construct a new index using the “STAR--runMode genomeGenerate” command using the instructions in the STAR manual.
NOTE: For the remainder of this protocol the path to the STAR index will be referred to as “path_to/STAR_index”. - Navigate to the path_to/project/ directory. Make a new directory called “STAR_output” as shown in Figure S1D.
- Navigate to the path_to/project/trimmed/ directory. Use the command shown in Figure S1D to run STAR to align the trimmed fastq.gz files.
NOTE: This step is the most computationally intensive and it is recommended to perform this on a HPC cluster with multiple threads (i.e. >16) designated for the task of alignment. Depending on the number of samples and available computational resources this step may take many hours to days. - Find the required output for the next steps which contain the counts per transcript at the following location: path_to/project/STAR_output/sampleN_ReadsPerGene.out.tab.
NOTE: In the ReadsPerGene.out.tab file column 1 holds information about the feature being counted. Column 2 holds the unstranded read counts, column 3 holds the forward stranded read counts, and column 4 holds the reverse stranded read counts. The first four rows of this file will have information about the aligned reads that did not align to a single gene. This protocol requires the unstranded read counts. - Use RStudio (preferable) or R in the HPC environment to compile the data from row 5 and below for columns 1 and 2 for each sample. Set the working directory to “project” in R.
- Read in each ReadsPerGene.out.tab file using the command in Figure S2A. For the first column, take only the characters before the “.” in the “Ensembl gene ID” column for the ease of downstream processing.
- Compile counts from all samples into a dataframe called “totcts” using the commands in Figure S2B. Save this new table of raw count data as a tab delimited .txt file, i.e. sample_counts.txt, if desired, using the “write.table” command.
NOTE: The order of the Ensembl gene ID is the same for every ReadsPerGene.out.tab file across samples.
5. Differential expression and downstream analysis
- Normalize for batch effects between samples with ComBat.
NOTE: There are two possible variables that explain changes in gene expression, the first being the construct used (i.e. the sample) and the second being external factors associated with the passage of cells through time (i.e. the batch). A step to normalize samples for batch-to-batch variation with the R-package ComBat is recommended.- Install if needed and load the libraries for sva, DESeq2, AnnotationDBI, org.Hs.eg.db, pheatmap, RColorBrewer, genefilter, Cairo, ggplot2, ggbiplot, rgl, and reshape2 as shown in Figure S2C. For installation, use the “install.packages” command or Bioconductor per the documentation for each package.
- First filter the data to only those genes that have at least one count per read. Save this new table to denote filtering as seen in Figure S2D.
NOTE: Frequently, many genes will have very low or no read counts. - Prepare a second table for batch normalization called “vars” as shown in Figure S2E. Set the row names to the unique names of each sample. Set the column names to “sample”, “batch”, and “construct”.
- Assign all samples a unique number in the “sample” column from 1 to n, with n being the number of samples. Assign batch numbers to all samples in the “batch” column such that condition-a_1 and condition-b_1 are both assigned 1, and condition-a_2 and condition-b_2 are both assigned 2. Assign all condition designations to all samples in the “construct” column such that condition-a samples are all “A” and condition-b samples are all “B”.
- Define the batch variable as well, and a specific null model matrix for ComBat as shown in Figure S2F. Run ComBat with the command defined in Figure S2F.
- Further curate the data by rounding to the nearest integer. Also remove genes with a negative value. Use the commands shown in Figure S3A.
NOTE: The output of batch normalization will have non-integer read counts and some genes with negative values. This step is required because the downstream differential expression analysis doesn’t support negative read counts. - Define the differential expression profile for each construct using DESeq2.
- Input the experiment design for DESeq2 as shown in Figure S3B. Construct a DESeqDataSet (dds) using the DESeqDataSetFromMatrix function, estimate the size factors, and run DESEq2, as shown in Figure S3B.
NOTE: It is imperative that the column data entered for “condition” is in the same order as the column in the count matrix. - In order to evaluate the quality of the analysis, extract the rlog-normalized counts used by DESeq2 as shown in Figure S3B.
NOTE: During analysis, DESeq2 transforms counts with a “regularized log,” rlog, transformation to shrink the sample-to-sample differences for genes with low counts (low information) in order to preserve differences in genes with higher counts across samples (high information). - When extracting the results for each transcriptional profile from the results of DESeq2, perform pairwise comparisons in reference to either the knockdown condition or baseline empty vector as shown in Figure S3C. Further amend these results with the HGNC gene symbols as shown in Figure S3D.
- As seen in Figure S3E, extract data from DESeq2 results. Export as a single file with the Ensembl gene ID, HGNC symbol, base mean expression, and differential expression data for all constructs with log2FoldChange and raw and adjusted p-values.
NOTE: Using an adjusted p-value < 0.05 is the recommended cutoff for differential expression. - Assess successful batch normalization and intra-sample similarity. Check sample clustering with PCA and sample-to-sample distance plots using the rlog normalized counts using athe code shown in Figures S4A-B.
- Input the experiment design for DESeq2 as shown in Figure S3B. Construct a DESeqDataSet (dds) using the DESeqDataSetFromMatrix function, estimate the size factors, and run DESEq2, as shown in Figure S3B.
- Use the differential expression profiles to generate volcano plots using the code in Figure S4C. Evaluate changes in gene expression across constructs.
- Use the rlog normalized counts and hierarchical clustering to identify gene signatures unique to the different constructs. Use the code shown in Figure S4D.
- Extract the 1000 most variable genes across all constructs in a matrix. Use pheatmap to perform unsupervised hierarchical clustering of your samples based on these genes.
- Extract the clusters of interest from the dendrogram by deciding at what level of the dendrogram clusters of interest appear. Set “k” equal to the number of clusters at that level. Replot the heatmap ordered by cluster to determine which clusters are of interest as shown in Figure S5.
- Export the list of genes associated with each cluster as demonstrated in Table S1. Use this information to determine the genes in clusters of interest.
- Identify the biological roles for different clusters of genes identified and compare between the classes. This can be performed using a variety of bioinformatics tools. ToppGene24 is used here and is freely available online.
NOTE: There are many free tools which require just a list of genes to copy and paste into a field on a website. Choose the analytical tools most appropriate for the research questions under investigation. - Optionally, if there are data available about genomic binding that drives transcriptional output for transcription factor of interest, compare the transcriptional response at genes associated with different binding elements to further evaluate mutant function.
6. Comparison with Relevant Phenotypes
- Compare the correlative phenotypes with the transcriptomic profile data generated and interpret as appropriate.
Representative Results
Preliminary qRT-PCR data suggested that an EWS/FLI mutant called DAF, with specific tyrosine to alanine mutations in the repetitive and disordered region of EWS, maintained the ability to activate EWS/FLI target genes, but failed to repress critical target genes23. In order to better understand the relationship between these residues in the EWS domain and EWS/FLI function, the protocol described above and outlined in Figure 1 was used. A673 Ewing sarcoma cells were virally transduced with an shRNA targeting the 3’UTR of FLI1, resulting in the depletion of endogenous EWS/FLI. After four days of selection, EWS/FLI function was rescued with viral transduction of different 3XFLAG-tagged EWS/FLI mutant constructs, with empty vector as a control for no rescue. A non-functional mutant lacking the EWS domain, called Δ22, was used as a negative control and wild-type EWS/FLI, called wtEF, was used as a positive control (Figure 2A). DAF was used as the test construct, though more than one test construct can be used if desired. Cells were selected for an additional 10 days to allow construct expression to stabilize and then collected for RNA (with a gDNA removal step), protein and colony forming assays. Four replicates were collected and representative qRT-PCR and western blots showing effective knockdown and rescue are shown in Figure 2B-D. It should be noted that DAF-rescued cells failed to form colonies as shown in Figure 2E, suggesting impaired oncogenic transformation.
Following completion of the replicate validation and phenotypic assays, RNA was submitted to the Institute for Genomic Medicine at Nationwide Children’s Hospital for library preparation and next generation sequencing with ~50 million 150-bp paired-end reads collected. The data was returned as fastq.gz files. Low-quality reads were trimmed from these files with TrimGalore and STAR was used to align reads to the human genome hg19 and count the reads per gene. hg19 was used for purposes of compatibility with the other curated datasets for EWS/FLI used in downstream analysis. These read counts were combined into a single count matrix for all samples, the first 6 rows of which are shown in Figure 3.
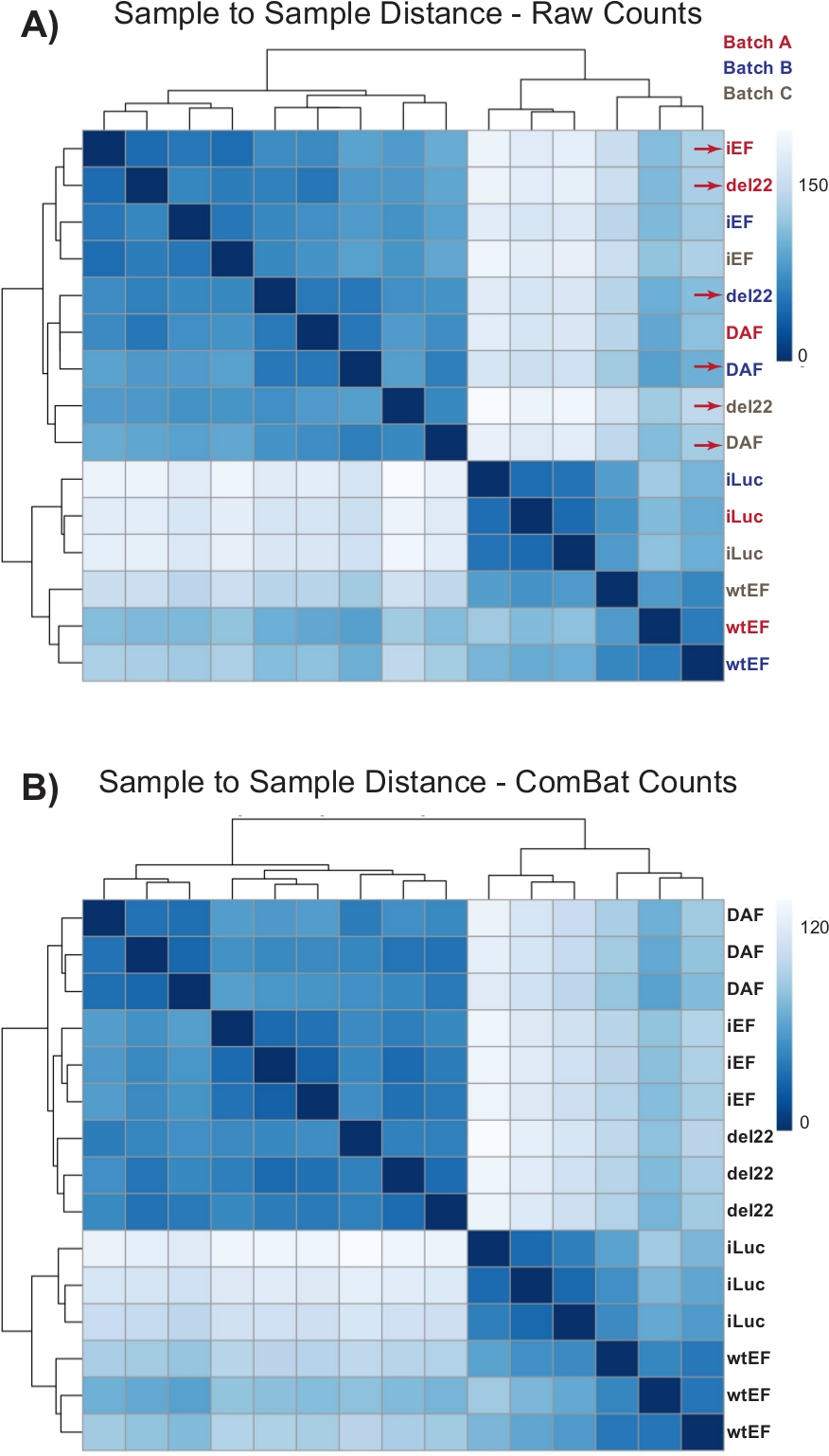
Counts were initially run through DESeq2 without batch normalization, however, visual inspection of the sample-to-sample distance showed potential confounding batch effects as shown highlighted with red arrows in Figure 4A. This likely arose due to biological variability introduced by the passage of cells in culture and differences in the processing of each batch. Normalization for batch effects was performed with ComBat and is generally recommended. The sample-to-sample distances of the batch-normalized data are shown in Figure 4B. Following batch normalization, DESeq2 was used to generate transcriptional profiles for the three constructs (wtEF, Δ22, and DAF) relative to the baseline. Note that while “parental” A673 cells (mock knockdown and mock rescue, called “iLuc” here) were included in the differential analysis, the reference for this experiment are the cells with EWS/FLI-depleted, called iEF cells. The transcriptional profile can be generated for the endogenous protein here by comparing the iLuc sample to iEF, and this may be of interest in understanding how the rescue system works, but that is not the goal of this particular analysis. The transcriptional profiles generated for the mutants include positive (wtEF) and negative (Δ22) controls, with respect to iEF, such that these should function as the benchmarks for other mutants. This is important, as the positive control in this example did not completely recapitulate the function of endogenous EWS/FLI as discussed elsewhere7,23.
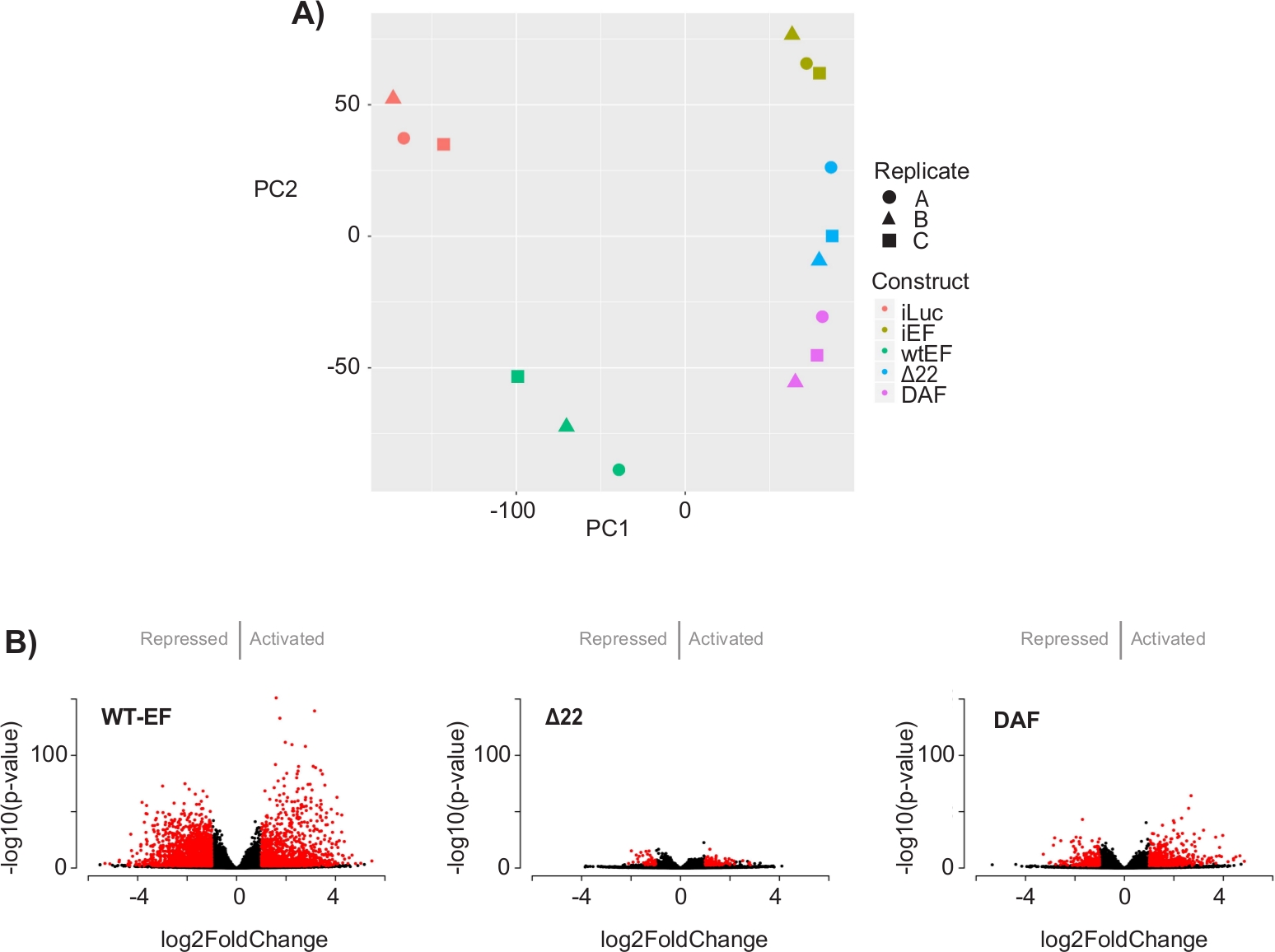
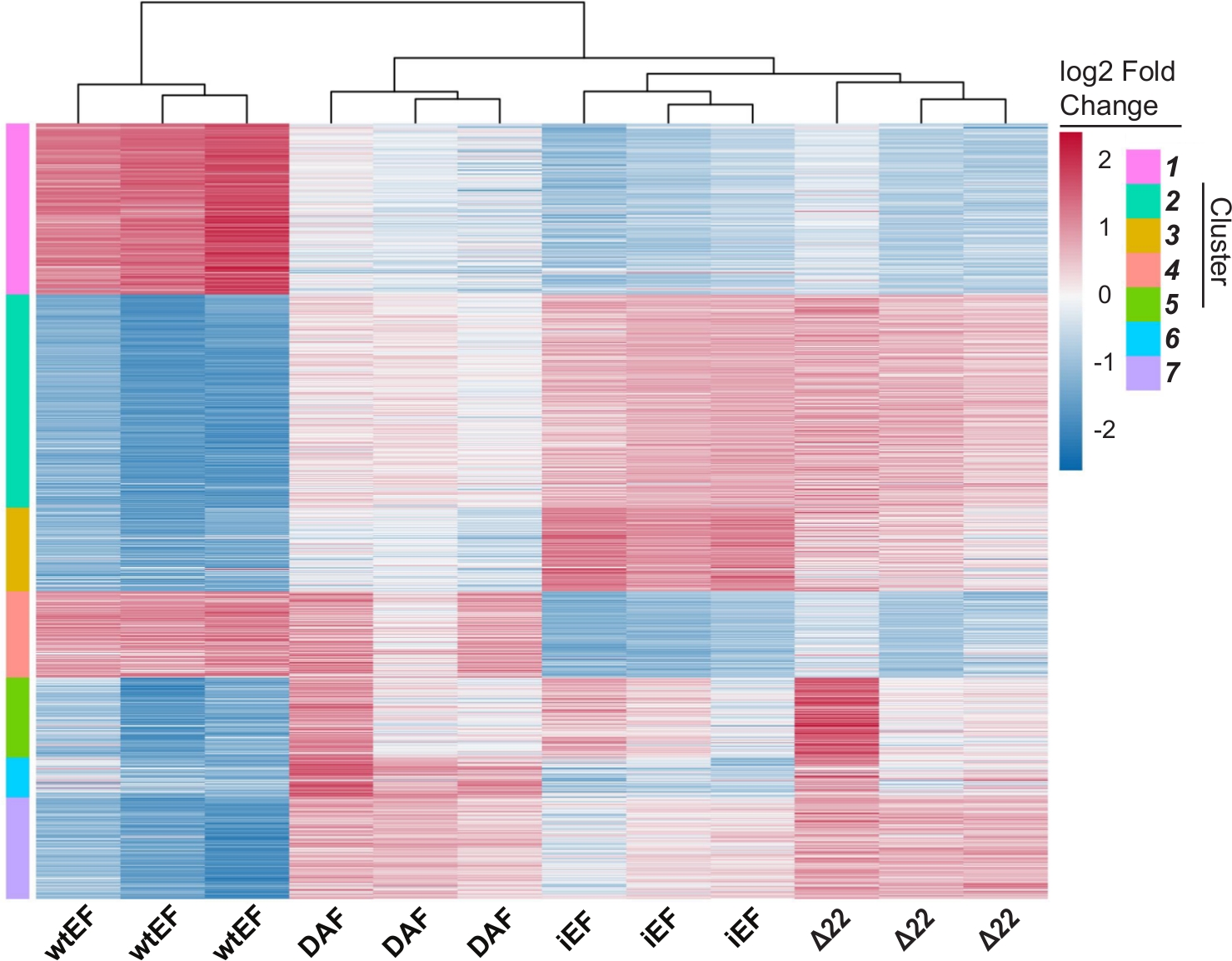
The principal component analysis (PCA) in Figure 5 suggests that the transcriptional profile of DAF is intermediate between wtEF and Δ22, confirming partial function. Moreover, hierarchical clustering of the 1000 most variable genes across samples showed that DAF failed to repress EWS/FLI target genes, and only partially retained gene activation activity as shown in Figure 6A and Figure S5. ToppGene analysis suggested that the classes of genes that DAF activates are functionally distinct from those EWS/FLI-activated targets where DAF is non-functional (Figure 6B). Interestingly, the function of activated genes rescued by wtEF, but not by DAF, appear to be related to transcriptional control and chromatin regulation. Based on the results of the colony formation assays, the genes from this core gene signature should be further analyzed for their role in EWS/FLI-mediated oncogenesis. The importance of EWS/FLI-mediated gene repression has been previously described17.
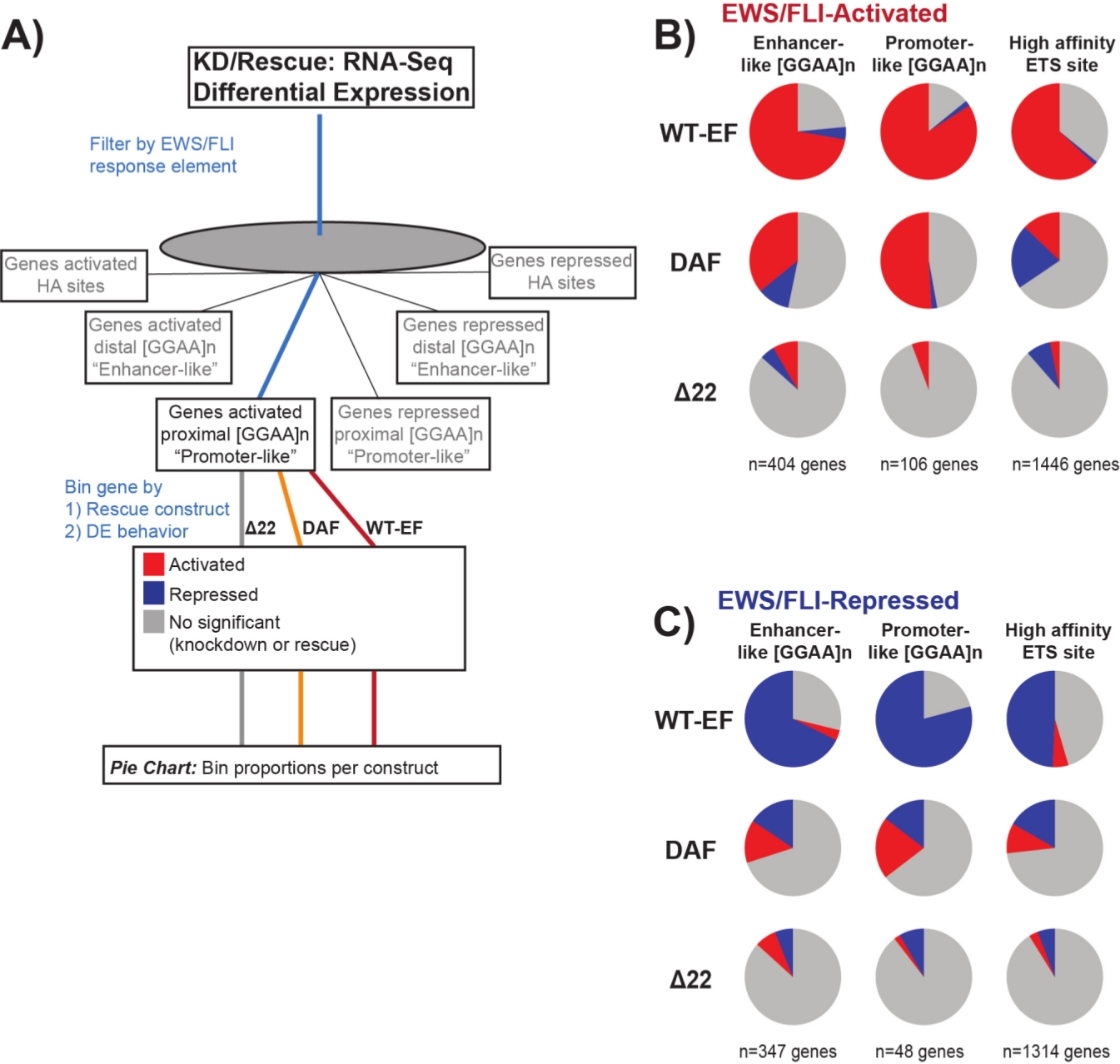
It is known that EWS/FLI possesses a unique binding affinity for GGAA-microsatellite repeat elements19,22, and that binding at these elements drives downstream gene regulation11,15,18,20,22. These microsatellites have been characterized as either associated with activation or repression, and either proximal to (< 5 kb) TSS or distal to (> 5 kb) TSS25. In addition, there are EWS/FLI-regulated genes with high affinity (HA) ETS motifs proximal to TSS23. In order to further analyze the characteristics of DAF function and what types of EWS/FLI-activated genes DAF was able to rescue, differential expression of genes associated with these different classes was analyzed. Interestingly, DAF was most able to rescue GGAA-microsatellite activated genes, but unable to rescue activated genes near an HA site as seen in Figure 7. As seen with hierarchical clustering, DAF fails to rescue EWS/FLI-mediated repression across motif classes. These data suggest that DAF retains sufficient structural features of EWS to bind to and activate from GGAA-microsatellites, both proximal and distal to TSS. This likely arises from the intact SYGQ domain thought to be important for EWS/FLI activity at GGAA repeats11. These data also suggest that the specific tyrosines mutated in DAF play important, but poorly understood, roles in EWS/FLI-mediated gene regulation from HA sites, as well as in gene repression, highlighting an important area of further investigation.

Figure 1: Workflow. Depiction of the step-by-step procedure to perform structure-function mapping by transcriptomics. Cells were first prepared to express the suite of constructs required for structure-function mapping. Following expression, cells were harvested for RNA and protein and assayed for correlative phenotypes. Expression of the constructs was validated, and this process was repeated 3-4 times to gather independent biological replicates. RNA was then submitted for next-generation sequencing (NGS). When data was received, data was trimmed for quality, aligned, and counts per transcript were calculated. Batch effects were controlled for and transcriptomic signatures and differential expression were determined using DESeq2. Hierarchical clustering and downstream analysis integrating other -omics datasets and different pathway or functional analysis can be incorporated. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Validation of construct expression and correlative assays. (A) Schematic depicting the constructs tested in this example. (B) Validation of knockdown of endogenous EWS/FLI and expression of 3X-FLAG-tagged constructs by immunoblot. (C,D) Validation of construct activity at an EWS/FLI (C) activated target gene, NR0B1, and (D) repressed target gene, TGFBR2, by qRT-PCR. Data are presented as mean +/- standard deviation. P-values were calculated with a Tukey’s honest significance test. * p < 0.05, ** p < 0.01, *** p < 0.005 (E) Colony counts from soft-agar assays performed to assess transforming activity of constructs. P-values were calculated with a Tukey’s honest significance test. * p < 0.05, ** p < 0.01, *** p < 0.005. This figure is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Final collated count data for analysis. Screenshot of the first 6 rows of the count file with gene counts for all the samples to be batch normalized and analyzed. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Sample-to-sample distance heatmaps. (A) Sample-to-sample distance plot showing the sample clustering of the raw count data. Samples which are clustering both by batch and by sample are denoted with red arrows. (B) Sample-to-sample distance plot following batch normalization with ComBat. Here, samples from all replicates cluster together, independent of batch. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Results of differential expression analysis. (A) Principle component analysis (PCA) plot of the transcriptomic signatures generated for all the samples show strong intra-sample clustering and demonstrate that DAF is intermediated between the positive (wtEF) and negative (Δ22) controls. (B) Volcano plots showing the -log(p-value) plotted against the log2FoldChange for genes in each construct. Genes with an adjusted p-value < 0.05 and a |log2(FoldChange)| > 1 are considered significant and are shown in red. Panel 5B is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Hierarchical clustering to identify gene classes. (A) Hierarchical clustering of the top 1000 most variable genes across all constructs and the baseline, iEF, shows DAF partially rescues EWS/FLI-mediated gene activation. (B) Gene ontology (molecular function) results from ToppGene showing the functional enrichment of EWS/FLI-activated genes that are either rescued or not rescued by DAF. Panel 6B is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Detailed analysis of different transcription factor response elements to different constructs: (A) Schematic depicting the data processing used to generate panels (B) and (C) by incorporating other available datasets with the transcriptomic profiles here. (B,C) Compilation showing the rescue of different classes of direct EWS/FLI- (B) activated and (C) repressed targets. Genes included were only those genes with detectable differential expression by endogenous EWS/FLI. In each pie chart, gray depicts the portion of genes which are not rescued by the construct. Red depicts the portion of genes that are differentially activated, and blue depicts the portion of genes that are differentially repressed. This figure is adapted from Theisen, et al.23 Please click here to view a larger version of this figure.
{kind=link}
Figure S1: Loading the fastq.gz files to the HPC environment, trimming and alignment. Please click here to download this figure.
{kind=link}
Figure S2: Collating read counts across samples and running batch normalization with ComBat. Please click here to download this figure.
{kind=link}
Figure S3: Running DESeq2 and extracting results of differential expression analysis. Please click here to download this figure.
{kind=link}
Figure S4: Analyzing output. Please click here to download this figure.
{kind=link}
Figure S5: Hierarchical clustering to identify gene classes: Hierarchical clustering of the top 1000 most variable genes across all constructs and the baseline, iEF, sorted into k clusters. In this instance k=7, but this parameter is set by the user as shown in Figure S4D. Please click here to download this figure.
{kind=link}
Table S1: List of genes (Ensembl gene ID) with cluster annotation. Please click here to download this table.
Discussion
Studying the biochemical mechanisms of oncogenic transcription factors is critically important to understand the diseases they cause and to design new therapeutic strategies. This is especially true in malignancies characterized by chromosomal translocations resulting in fusion transcription factors. The domains included in these chimeric proteins may lack meaningful interactions with regulatory domains present in the wild-type proteins, complicating the ability to interpret structure-function information in the context of the fusion26,27,28. Moreover, many of these oncogenic fusions are characterized by low-complexity intrinsically disordered domains10,13,29,30.
The EWS domain is an example of such an intrinsically disordered domain that is involved in a variety of oncogenic fusions10. The intrinsically disordered and repetitive nature has hindered efforts to understand the molecular mechanisms employed by the EWS domain. Prior efforts to investigate the structure-function have largely resorted to using different mutants in the context of reporter gene assays or in cell backgrounds that fail to recapitulate the relevant cellular context, or lack any structural variations which produce meaningful partial function11,17,25. The method presented here addresses these issues. Structure-function mapping is performed in a disease-relevant cell context and next generation sequencing enables transcriptomic profiling to evaluate transcription factor function in the setting of native chromatin. In the specific case of the DAF mutant of EWS/FLI, DAF was reported to show little activity in reporter assays using isolated response elements, but to show activity in the context of the full gene promoter, either in a reporter assay or in native chromatin, suggesting an interesting phenotype23. Using the method described here more directly resolves the question of which type of regulatory elements across the genome are most responsive in the disease setting. By testing all candidate target genes in their native chromatin context simultaneously, a transcriptomic approach is more likely to identify constructs with partial function.
The inherent strength of using a disease-relevant cell background is perhaps the biggest limitation of this technique. One of the most important factors is choosing the appropriate cell system for these experiments. Many cell lines derived from malignancies with pathognomonic transcription factors do not readily tolerate knockdown of that transcription factor, and in many instances, particularly for pediatric cancers, the true cell of origin remains controversial and the expression of the oncogene in other cell backgrounds is prohibitively toxic31,32. In these cases, it may be helpful to perform experiments in a different cell background, so long as the researcher exercises caution in the interpretation of results and appropriately validates any relevant findings in a more disease-relevant cell type.
It is critically important to carefully validate the stability and phenotypic consequences of expression of the oncogene and to only submit samples for sequencing that meet strict criteria. Here, this included western blot to confirm knockdown and rescue, and qRT-PCR of a small number of known target genes to validate the positive control (Figure 2). It is likewise crucial to decrease as much batch variability as possible by carefully performing the cell and RNA preparations as similarly as possible through each batch.
The method described here becomes especially powerful when paired with other types of genomic data that speak to the genome-wide function of the transcription factor under study. Future directions for this type of structure-function analysis would expand to include ChIP-seq and ATAC-seq to determine the binding of the transcription factor and any induced changes in chromatin accessibility. As a suite, this type of data can point to where different structural components of an oncogenic transcription factor contribute to different aspects of function (i.e. DNA binding vs. chromatin modification vs. co-regulator recruitment). Overall, using NGS-based approaches to map the structure-function relationships of fusion transcription factors can reveal new insights in the biochemical determinants of the oncogenic function of these proteins. This is important to further our understanding of the diseases they cause and in enabling the development of new therapeutic strategies.
Acknowledgements
This research was supported by the High Performance Computing Facility at the Abigail Wexner Research Institute at Nationwide Children’s Hospital. This work was supported by the National Institutes of Health National Cancer Institute [U54 CA231641 to SLL, R01 CA183776 to SLL]; Alex’s Lemonade Stand Foundation [Young Investigator Award to ERT]; Pelotonia [Fellowship to ERT]; and the National Health and Medical Research Council CJ Martin Overseas Biomedical Fellowship [APP1111032 to KIP].
Materials
| Name | Company | Catalog Number | Comments |
| Wet Lab Reagents | |||
| anti-FLI rabbit pAb | Abcam | ab15289 | 1:500 |
| anti-lamin B1 rabbit pAb | Abcam | ab16048 | 1:2000 |
| Cell-based system for introduction of mutant constructs | Determined by cell system used | ||
| Cryotubes | For viral aliquots | ||
| DMEM | Corning Cellgro | 10-013-CV | For viral production |
| Fetal bovine serum | Gibco | 16000-044 | For viral production |
| G418 | ThermoFisher | 10131027 | For viral production |
| HEK293-EBNAs | ATCC | CRL-10852 | For viral production |
| HEPES | Gibco | 15630106 | |
| Hygromycin B | ThermoFisher | 10687010 | |
| M2 anti-FLAG mouse mAb | Sigma | F3165 | 1:2000 |
| Near IR-secondary antibodies | Li-Cor | ||
| Optimem | Gibco | 31985062 | For viral production |
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | For viral production |
| Polybrene | Sigma | TR-1003-G | For viral transduction |
| Puromycin | Sigma | P8833 | Stored at 2 mg/mL stock |
| RNeasy Plus kit | Qiagen | 74136 | Has gDNA removal columns |
| Selection reagents | As dictated by cell system used | ||
| Sodium Pyruvate | Gibco | 11360-070 | For viral production |
| Tissue culture media | Determined by cell system used | ||
| TransIT-LT1 | Mirus | MIR 2304 | For viral production |
| Software | |||
| Access to HPC environment | |||
| AnnotationDbi | 1.38.2 | ||
| Cairo | 1.5-10 | ||
| DESeq2 | 1.16.1 | ||
| genefilter | 1.58.1 | ||
| ggbiplot | 0.55 | ||
| ggplot2 | 3.1.1 | ||
| org.Hs.eg.db | 3.4.1 | ||
| pheatmap | 1.0.12 | ||
| PuTTY | |||
| R | 3.4.0 | ||
| RColorBrewer | 1.1-2 | ||
| reshape2 | 1.4.3 | ||
| rgl | 0.100.19 | ||
| R-studio | |||
| STAR | Version 2.6 or later | ||
| sva | 3.24.4 | ||
| TrimGalore! | |||
| WinSCP |
References
- Miettinen, M., et al. New fusion sarcomas: histopathology and clinical significance of selected entities. Human Pathology. 86, 57-65 (2019).
- Knott, M. M. L., et al. Targeting the undruggable: exploiting neomorphic features of fusion oncoproteins in childhood sarcomas for innovative therapies. Cancer and Metastasis Reviews. 38, 625-642 (2019).
- Yoshihara, K., et al. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 34, 4845-4854 (2015).
- Duesberg, P. H. Cancer genes generated by rare chromosomal rearrangements rather than activation of oncogenes. Medical Oncology and Tumor Pharmacotherapy. 4, 163-175 (1987).
- Dupain, C., Harttrampf, A. C., Urbinati, G., Geoerger, B., Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Molecular Therapy - Nucleic Acids. 6, 315-326 (2017).
- Mitelman, F., Johansson, B., Mertens, F. The impact of translocations and gene fusions on cancer causation. Nature Reviews Cancer. 7, 233-245 (2007).
- Smith, R., et al. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing's sarcoma. Cancer Cell. 9, 405-416 (2006).
- Davicioni, E., et al. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Research. 66, 6936-6946 (2006).
- Gröbner, S. N., et al. The landscape of genomic alterations across childhood cancers. Nature. 555, 321-327 (2018).
- Kim, J., Pelletier, J. Molecular genetics of chromosome translocations involving EWS and related family members. Physiological Genomics. 1, 127-138 (1999).
- Boulay, G., et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 171, 163-178 (2017).
- Lessnick, S. L., Braun, B. S., Denny, C. T., May, W. A. Multiple domains mediate transformation by the Ewing’s sarcoma EWS/FLI-1 fusion gene. Oncogene. 10, 423-431 (1995).
- Leach, B. I., et al. Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure. 21, 176-183 (2013).
- Ng, K. P., et al. Multiple aromatic side chains within a disordered structure are critical for transcription and transforming activity of EWS family oncoproteins. Proceedings of the National Academy of Sciences U.S.A. 104, 479-484 (2007).
- Riggi, N., et al. EWS-FLI1 Divergent Chromatin Remodeling Mechanisms to Directly Activate or Repress Enhancer Elements in Ewing Sarcoma. Cancer Cell. 26, 668-681 (2014).
- Tomazou, E. M., et al. Epigenome Mapping Reveals Distinct Modes of Gene Regulation and Widespread Enhancer Reprogramming by the Oncogenic Fusion Protein EWS-FLI1. Cell Reports. 10, 1082-1095 (2015).
- Sankar, S., et al. Mechanism and relevance of EWS/FLI-mediated transcriptional repression in Ewing sarcoma. Oncogene. 32, 5089-5100 (2013).
- Gangwal, K., et al. Microsatellites as EWS/FLI response elements in Ewing's sarcoma. Proceedings of the National Academy of Sciences U.S.A. 105, 10149-10154 (2008).
- Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., Lessnick, S. L. Emergent Properties of EWS/FLI Regulation via GGAA Microsatellites in Ewing's Sarcoma. Genes & Cancer. 1, 177-187 (2010).
- Guillon, N., et al. The Oncogenic EWS-FLI1 Protein Binds In Vivo GGAA Microsatellite Sequences with Potential Transcriptional Activation Function. PLoS One. 4, 4932 (2009).
- Chong, S., et al. Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science. 361, (2018).
- Johnson, K. M., et al. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proceedings of the National Academy of Sciences U.S.A. 114, 9870-9875 (2017).
- Theisen, E. R., et al. Transcriptomic analysis functionally maps the intrinsically disordered domain of EWS/FLI and reveals novel transcriptional dependencies for oncogenesis. Genes & Cancer. 10, 21-38 (2019).
- Chen, J., Bardes, E. E., Aronow, B. J., Jegga, A. G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Research. 37, 305-311 (2009).
- Johnson, K. M., Taslim, C., Saund, R. S., Lessnick, S. L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLOS One. 12, 0186275 (2017).
- Kim, P., Ballester, L. Y., Zhao, Z. Domain retention in transcription factor fusion genes and its biological and clinical implications: a pan-cancer study. Oncotarget. 8, 110103-110117 (2017).
- de Mendíbil, I. O., Vizmanos, J. L., Novo, F. J. Signatures of Selection in Fusion Transcripts Resulting from Chromosomal Translocations in Human Cancer. PLOS One. 4, 4805 (2009).
- Frenkel-Morgenstern, M., Valencia, A. Novel domain combinations in proteins encoded by chimeric transcripts. Bioinformatics. 28, 67-74 (2012).
- Hegyi, H., Buday, L., Tompa, P. Intrinsic Structural Disorder Confers Cellular Viability on Oncogenic Fusion Proteins. PLoS Computational Biology. 5, 1000552 (2009).
- Latysheva, N. S., Babu, M. M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Research. 44, 4487-4503 (2016).
- Deneen, B., Denny, C. T. Loss of p16 pathways stabilizes EWS/FLI1 expression and complements EWS/FLI1 mediated transformation. Oncogene. 20, 6731-6741 (2001).
- Kendall, G. C., et al. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. eLife. 7, 33800 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved