Live Imaging of Chemokine Receptors in Zebrafish Neutrophils During Wound Responses
In This Article
Summary
Here we describe protocols to perform live imaging and quantitative analysis of chemoattractant receptor dynamics in zebrafish neutrophils
Abstract
Leukocyte guidance by chemical gradients is essential for immune responses. Neutrophils are the first cells to be recruited to sites of tissue damage where they execute crucial antimicrobial functions. Their trafficking to these loci is orchestrated by several inflammatory chemoattractants, including chemokines. At the molecular level, chemoattractant signaling is regulated by the intracellular trafficking of the corresponding receptors. However, it remains unclear how subcellular changes in chemokine receptors affect leukocyte migration dynamics at the cell and tissue level. Here we describe a methodology for live imaging and quantitative analysis of chemokine receptor dynamics in neutrophils during inflammatory responses to tissue damage. These tools have revealed that differential chemokine receptor trafficking in zebrafish neutrophils coordinates neutrophil clustering and dispersal at sites of tissue damage. This has implications for our understanding of how inflammatory responses are self-resolved. The described tools could be used to understand neutrophil migration patterns in a variety of physiological and pathological settings and the methodology could be expanded to other signaling receptors.
Introduction
Leukocyte migration is of paramount importance for immune responses. Immune cells are prototypical migratory cells, which are remarkably capable of traversing tissues and blood vessels and sensing a range of chemical guidance cues to migrate directionally towards microbes or other host cells of importance. Correct guidance relies on the recognition of chemoattractants, among which chemokines represent the most prominent category1. Chemokines are recognized by highly specific seven-transmembrane G protein coupled receptors. Upon chemokine binding, chemokine receptors change conformation leading to the activation of associated trimeric G proteins and their dissociation into functional signaling subunits that promote cytoskeletal changes and directed migration1. Secondarily, chemokine receptors are phosphorylated, and this modification leads to desensitization to attractant, which can be followed by rapid re-sensitization/recycling or intracellular degradation and down-regulation from the cell surface2. These receptor dynamics influence the duration and dose of signaling received by the cells but how they affect leukocyte migration behavior has been difficult to elucidate in vivo.
Tracking receptor dynamics in live leukocytes in traditional mammalian systems faces several challenges. For live studies, receptor fusions with fluorescent proteins must be expressed in the cells. This is challenging in primary leukocytes, particularly in neutrophils, and studies so far have used surrogate neutrophil cell lines to express chemokine receptors3,4. Generation of transgenic mouse models, in which leukocytes express a fluorescent receptor or mutant receptors with informative trafficking defects5,6, entails considerable investment of time and resources. Even in these instances, the imaging resolution and contrast for imaging receptor dynamics in the live animal can be limited and studies have used immunohistochemistry on fixed tissue sections5. Given these technical challenges, our understanding of how chemoattractant receptors dynamics affect cell behavior in a live tissue setting is currently limited.
Here we provide a methodology to monitor receptor trafficking in zebrafish neutrophils. Zebrafish are genetically tractable, like mice, but transgenesis is relatively more straightforward through the use of efficient transposon systems and direct zygote manipulation7. The transparent larva is ideally amenable to imaging. The chemokine receptor dynamics have been visualized in primordial germ cells and the lateral line primordium by expression of corresponding fusions with fluorescent reporters8,9,10. Zebrafish larvae are equipped with mature neutrophils that have highly conserved genetic and cellular properties with respect to mammalian neutrophils. Subcellular signaling dynamics such as cytoskeletal dynamics and polarity regulators have been visualized in these cells by the generation of corresponding transgenic lines11,12,13. Recently, we visualized and functionally analyzed chemokine receptor dynamics in neutrophils during the course of inflammatory responses to tissue damage14. Here, we describe the generation of transgenic reporter lines for chemokine signaling in neutrophils, preparation of embryos for live imaging, a wound assay for studying neutrophil signaling and the protocol for acquisition and analysis of images. We also provide a side-protocol to test chemokine receptor responses to candidate ligands, which is useful when trying to establish ligand recognition patterns in uncharacterized receptors. These techniques can be used in combination with further genetic manipulations, such as inhibition of endogenous chemokine expression or generation of mutant receptors with altered ligand-induced trafficking, to interrogate how specific signaling dynamics affect leukocyte behavior in vivo. The transgenic lines expressing fluorescently tagged chemokine receptors can also be used as reporters for endogenous chemokine gradients, which are otherwise difficult to detect by direct antibody staining. The described methodology provides scope for expanding the generation of reporters to other immuno-signaling receptors.
Protocol
NOTE: All zebrafish were kept according to the ARRIVE guidelines and UK Home Office regulations, UK Animals (Scientific Procedures) Act 1986.
1. Generation of transgenic reporter zebrafish larvae for imaging receptor trafficking in leukocytes
- Generate Tol2-based construct for tissue specific expression of the fluorescently tagged receptor of interest. For neutrophils, use promoter sequences from the lysozyme C15 and myeloperoxidase gene16. The construct can be designed as a fusion with a single fluorescent protein (e.g. GFP), a tandem fluorescent timer (e.g., a fast-folding GFP and a slower maturing tagRFP8,14,17) or a bicistronic expression of reporter GFP and a control membrane marker9 (see Discussion for considerations when choosing the approach).
NOTE: This construct does not recapitulate endogenous levels of receptor expression but is useful for obtaining high level of receptor expression in the cell type of interest. Consult the literature on similar receptors3,5,6,8,9,10,14 to decide on the position of the fluorescent tag. - Set up a tank containing wild type adult males and females following standard husbandry practices18, separated by a barrier the day before egg spawning.
- On the day of egg spawning, prepare transgenesis mixture for microinjection containing 12.5 ng/μL of Tol2 DNA construct and 17.5 ng/μL of transposase mRNA7. Lift barriers from fish tanks shortly after the lights come in the morning (this may vary in different fish facilities) and collect eggs within 15 min for mRNA injection.
NOTE: Ensure the DNA solution is RNase free to avoid degradation of the transposase mRNA in the mixture. An option to circumvent this is to inject eggs with separate solutions of Tol2 construct and transposase. - Follow standard protocols for transgenesis and microinjection of zebrafish eggs19.
- Inject 1 nL of the solution into the cell of one-cell stage embryos.
NOTE: The expression results are more consistent when injecting inside the cell and discarding the injections that may not be clearly inside the cell. One-cell stage embryos are aimed for because of the variability of volume injection per cell when injecting 2-16 cell stage embryos. An option would be to separate the one-cell stage injections from batches of later injections, in case these have different efficacies. - Check the injected embryos later in the day and remove unfertilized or dead eggs to keep the clutch healthy.
- At 3 days post-fertilization (dpf), screen larvae under a fluorescent microscope. The marker will be visible in neutrophils, particularly in the caudal hematopoietic tissue (CHT), which is rich in these cells.
NOTE: The percentage of cells labeled varies with different constructs, but usually 20-60% of neutrophils are expected to express the construct. Lower percentages usually predict more screening at the adult stage. It is a good practice to also verify correct localization of the receptor at the membrane, with a higher-resolution imaging approach, in a sample of these embryos before growing the fish. - Grow positive larvae following standard husbandry practices20. These represent the F0 generation.
- At 3 months of age, screen F0 fish for founders. Cross individual fish with a non-transgenic wild type and screen their offspring at 3 dpf for the expression of the receptor by viewing under the dissecting scope. Depending on the transgenesis success, which varies with each construct, observe a percentage of positive offspring in a subset of the crosses.
NOTE: For good transgenesis, about a third to a half of adults will give positive offspring. The percentage of offspring that is positive in a clutch varies with the copy number of transgenes inserted and, can be between 10-60%. It is helpful to keep track of mendelian ratios within the clutches to identify single insertion transgenics (these are more easily identified in F2 clutches by looking for a 50% ratio of positive larvae)20. - Grow the positive offspring, which represent the F1 generation.
- At 3 months of age, screen F1 adults in the same way to establish stable F2 transgenic line.
- Perform experiments on F2 larvae after validating the neutrophil-specific expression of the transgene.
NOTE: During the transgenesis, one may observe varying levels of receptor expression and it is advisable to keep different transgenic clutches to obtain the most appropriate expression level for the biological questions. Initial results may be obtained in F0 or F1 larvae.
2. Collecting zebrafish embryos for assessing leukocyte wound responses
- After having established a stable transgenic reporter line, set up a cross between adult and female transgenic fish and collect eggs the next morning.
- Grow embryos at 28.5 °C in E3 medium (or egg water18).
- Optionally, at 24 hpf, incubate embryos in a centrifuge tube containing 50 mL of E3 medium supplemented with 0.003% bleach for 5 min. Subsequently rinse 3 times in E3 medium by letting the tube stand for a couple of minutes to allow the embryos to settle in the bottom and then decanting and replacing the medium.
NOTE: This provides a level of control on the infection exposure of the larvae, which may affect the behavior of leukocytes during wounding. - After bleaching, keep embryos in E3 medium supplemented with 0.003% of 1-phenyl-2-thiourea to prevent melanin synthesis. Methylene blue, which is often used to prevent fungal infections, is not added here, to minimize tissue autofluorescence.
- Allow larvae to hatch naturally and use at 3 dpf when neutrophils are abundant21.
3. Ventral fin wounding of larvae
- Prepare larvae for wounding. Use larvae at 2.5-3.5 dpf, when abundant neutrophils are observed. Transfer larvae to E3 medium supplemented with 160-200 mg/L tricaine MS222.
NOTE: Concentrated solutions of tricaine should be prepared and frozen in aliquots in advance and thawed on the day. - Leave the larvae in E3+tricaine medium for 15 min to ensure they are anesthetized. Check their responses by gently touching with a small paintbrush or similar tool.
- Select larvae with transgenic receptor expression under a fluorescent dissecting scope.
- Transfer larvae to a 120 mm Petri dish in E3 + tricaine for wounding. Using a sterile scalpel cut the ventral fin of the larva, while observing under the dissecting scope (Figure 1).
NOTE: The idea is to perform a deep enough cut to cause substantial neutrophil recruitment but without cutting the vessels of the CHT. The cut is made perpendicular to the CHT axis such that the incision nearly reaches the vessels of the CHT. This takes some practice and should first be performed with supervision on larvae that are not generated for this purpose (e.g., excess larvae from another experiment).
4. Preparation of larvae for live imaging
- Dissolve low melting point (LMP) agarose in E3 medium by heating to obtain a liquid 2% agarose solution.
- Allow this solution to cool down to 60 °C.
NOTE: Keep a flask or tube with agarose in an incubator at 60 °C to avoid the agarose setting between the mounting of different larvae. - Pipette 0.5 mL of the liquid LMP + E3 in a glass bottom dish for microscopy imaging.
- Pipette a wounded, anesthetized zebrafish larva along with 0.5 mL of E3 + 2x Tricaine in the glass bottom dish.
- Gently mix the two solutions to obtain a 1% LMP/1x Tricaine agarose/E3 solution, avoiding generation of bubbles. Orient the embryo laterally and gently push down so that the caudal part of the fish is as close as possible to the glass.
NOTE: The tissue to be imaged must be as close as possible to the glass bottom when imaging with an inverted microscope. The setting of orientation for the larva must be quick so that the agarose does not set before the larva is positioned. - Let the agarose solution cool down and solidify for 5-10 min. Test whether the agarose is set by gently touching the agarose gel with a small paint brush or tip.
- Once the agarose is solid, add 2 mL of E3 supplemented with 0.2 mg/mL of tricaine to the imaging plate.
5. Live confocal imaging
NOTE: Image embryos on a spinning disk confocal microscope or equivalent (Figure 2). A laser scanning microscope can also be used but the temporal resolution of the dynamics will be more limiting. Prepare the imaging settings before bringing the wounded larva, so the response can be imaged as quickly as possible after wounding. Neutrophils arrive to the wound within 5 min and receptors in the first arriving cells may internalize within this time frame. With practice it is possible to image as early as 15 min post-wounding.
- Turn on the microscope: laser, camera and computer as per manufacturer instructions.
- Use acquisition software to set up the imaging settings. Choose lasers for the appropriate fluorophores and approximate exposure times based on previous experiments (acquire GFP with 488 nm and tagRFP with 561 nm laser).
- Transfer the plate with the mounted embryo, as soon as possible after agarose sets, onto the confocal imaging spinning disc platform.
- Use the microscope eye piece to find the fish in the dish using the stage joystick.
- Focus on the wound area, using the focusing knob.
NOTE: To find the area of interest it may be easier to use a low magnification air objective (10x). - Select the field to image around the wound. Use a 30x/40x objective with high numerical aperture to obtain sufficient resolution.
- Use the software buttons to adjust the exposure time so that the fluorescent marker can be seen with good contrast but without saturating the signal.
NOTE: The exposure time must be as low as possible to minimize fluorescent exposure and maximize temporal resolution in the time-lapse. The laser power depends on the condition of the laser but needs to be adjusted to a level that permits low enough exposure time for dynamic imaging. - Use the software buttons to select the volume to image as a z-stack
- Set up a time lapse every 30 s for the desired duration.
NOTE: For sterile ventral fin wounds, the maximum recruitment is observed by 2-3 h. - Before launching the time-lapse, take a bright field snapshot to document the field of view. If possible, acquire bright field within the time-lapse movie.
6. Quantification of receptor internalization in zebrafish neutrophils
- Record the time-interval of image acquisition and the pixel size of the image. Keep a record of how many minutes post-wounding the imaging started.
- Open the image datasets using Fiji by dragging the image onto the software interface, select a representative frame of interest for each dataset using the time slider, e.g., at 1-1.5 h post-wounding, and save it.
- Proceed with MATLAB to process the image dataset.
- Create a new script and include functions for image reading (line 6 in script called ‘select_neutrophils.m’ for centroid definition in Supplementary File 1), opening (line 11 in Supplementary File 1) and manual selection of points on the image (line 12 in Supplementary File 1).
- Open the frame of interest by running this script (Supplementary file 1), identify the neutrophils to analyze by visual inspection, click on them and record an estimation of their centroids, both in the ventral fin wound and in the CHT.
NOTE: The non-mobilized neutrophils in the CHT serve as an internal reference for neutrophils whose receptor distribution remains constant. This allows normalization of contrast values of cells at the wound to an internal reference. - Proceed with segmentation of the neutrophils in each frame using active contours technique22 as described in steps below.
- Create a function to include the metadata required for segmentation by active contours (i.e., number of iterations, bias of contour, estimated centroid etc.)22 (see ‘wound data.m’ in Supplementary File 2).
- Create a script that calls the function ‘wound_data.m’ to input the necessary information for segmentation of each neutrophil (line 28 in script called ‘calc_contrast.m’ in Supplementary File 3).
- Include in the script commands for image reading (line 32 in Supplementary File 3).
- Add the generation of a black image (i.e. image where pixel values are zeros) with an equal size to the input image (line 44 in Supplementary File 3) and the definition of a square of 10 × 10 pixels around the centroid of each neutrophil (line 45 in Supplementary File 3).
- Include the neutrophil segmentation using active contours (lines 48-49 in Supplementary file 3) and the removal of small false detected objects (line 52 in Supplementary File 3).
NOTE: The initial segmentation contour is the square around the centroid, which evolves by active contour technique based on the pixel intensities, the number of iterations and the bias of contour. The result of segmentation is a binary mask where all pixels have value 0 apart from the neutrophil area whose pixels have value 1. - Include the multiplication of the segmented binary image with the original one to get the pixel intensities of the neutrophil only, with the rest of the image being not-a-number, so it does not contribute to calculations (lines 56-57 in Supplementary File 3).
- Add the calculation of the gray-level co-occurrence matrix for each neutrophil (GLCM)14,23 (line 61 in Supplementary File 3). GLCM is another representation of the image showing relative position of pixels in terms of pixel intensity.
- Include the calculation of contrast of the neutrophil based on the GLCM (lines 62,65 in Supplementary File 3). The contrast metric measures differences in intensity between neighboring pixels. Pixels are compared with pixels certain distance apart, which can be adjusted empirically based on the size of local peaks in intensity. As an indication, for the images, with a pixel size of 0.389 µm, when the receptor showed vesicular distribution each bright dot was in the range of 5 pixels. Therefore, intensities were compared in pixels spaced 5 pixels apart.
- Add commands to save the values separately for individual neutrophils in the ventral fin (line 68 in Supplementary File 3).
- Include the calculation of the mean neutrophil contrast value from all CHT neutrophils the same way as for neutrophils at the wound (lines 72-119 in Supplementary file 3). For CHT neutrophils, call the function ‘cht_data.m’ (line 77 in Supplementary File 4).
- Include the normalization of the contrast value of individual neutrophils at the wound to the mean contrast of CHT neutrophils calculated above in step 6.16 (i.e., division) (line 122 in Supplementary File 3). This gives a normalized contrast that reflects how ‘dotty’ the appearance of receptor is in individual responding cells relative to control non-responding cells (Figure 3 and Figure 4).
- Run the script (Supplementary File 3) by clicking the run symbol in the software.
- Repeat all steps for different conditions.
- Use statistical software (see Table of Materials) to import the results for the different conditions by creating a column table, plot the results and perform statistical test to check significance of difference between the mean values.
NOTE: The codes for the analysis can also be found in GitHub at https://github.com/LeukocyteMotionAndDynamics/ReceptorTraffic
7. Chemokine response assays in early embryos
NOTE: This is an optional side experiment that allows testing of receptor distribution changes in response to a candidate chemokine and is independent from the experiments described above concerning neutrophil expression of the receptor constructs. Differences in ligand-induced trafficking between receptors are difficult to establish with this technique as the ligand levels are saturating14. However, if one sees ligand-internalization of a receptor in this system, this can be an indication that the ligand is recognized by the receptor in instances where the ligand identity is unclear. This is useful, because expression of chemokine receptors in established cell lines such as HEK293T cells14 can be cumbersome.
- Set up a cross of wild type fish (e.g., AB) and collect eggs the next morning shortly after lifting the separators (as described above).
- Inject 100 pg of fluorescently tagged receptor mRNA (e.g., Cxcr1-FT), together with 100 pg of mRNA for a membrane marker (e.g., membrane CFP). Include in the mixture varying doses of mRNA for chemokine ligand.
NOTE: As an indication 150 pg Cxcl8a mRNA gave prominent internalization of fluorescent Cxcr1-FT (see Figure 5). - Rinse embryos with E3 medium and incubate at 28°C.
- At about 7 hpf, test expression of the mRNA on a fluorescent dissecting scope and select the embryos to image.
- Prepare 0.8 % LMP agarose in advance and keep in glass tube in a heatblock at 60 ˚C. Use a glass pipette to manipulate the embryos. Gently dechorionate embryos using a pair of forceps in each hand.
- Aspirate an individual dechorionated embryo with the glass pipette ensuring no bubbles are at the tip. Gently release the embryo into the tube of agarose allowing it to sink into the tube.
- Aspirate the embryo from the agarose tube, collecting some liquid agarose along the way. Gently release embryo onto the center of a glass-bottomed imaging dish. Quickly rotate embryo so that the animal pole is facing the bottom of the dish (this side must be closest to the objective when using an inverted microscope).
NOTE: One may need to readjust the orientation of embryos while the agarose is still setting. - After the agarose is set, supplement with 2 mL of E3 medium.
NOTE: The embryos at this stage are very fragile in comparison to larvae and it takes some practice to dechorionate and mount. It is important to aspirate and release as gently as possible, to avoid embryo rupture. - Repeat the process aiming to load 3-5 embryos per dish.
- Image embryos on an inverted confocal microscope (see Table of Materials). Use a 40x/1.3 NA oil objective to obtain high enough resolution. Visualize mCFP, sfGFP, and tagRFP with 405, 488, and 552 nm, respectively, on the scope. Adjust filters and settings to have high contrast while avoiding saturation and minimize leak through between the channels.
- Repeat the mounting and imaging for different conditions.
Representative Results
Ventral fin wounding is followed by rapid neutrophil mobilization from the CHT into the ventral fin and clustering at the wound margin, within 30-60 min (Figure 1). We visualized the distribution of two chemokine receptors, Cxcr1 and Cxcr2, which are expressed by zebrafish neutrophils24 and recognize Cxcl8a and Cxcl8b14, using spinning-disk confocal microscopy. We generated two corresponding transgenic lines, Tg(lyz:Cxcr1-FT) and Tg(lyz:Cxcr2-FT), in which neutrophils express a fluorescent timer (FT) construct of the receptor, i.e. a fusion with a tandem of sfGFP and tagRFP (Figure 2 and reference14). The use of the two fluorophores was intended to allow monitoring of a broad range of receptor fates and provide estimates of protein turnover time at the plasma membrane, as newly synthesized receptors would fluoresce in green and progressively become red as they age8,14. However, these receptors were found to have fast constitutive turnover at the neutrophil plasma membrane and that the residence time was shorter than the maturation time of tagRFP, with sfGFP showing membrane localisation and tagRFP showing vesicular localization at steady state (Supplementary Video 1 and ref14). Therefore, we focused on the distribution of sfGFP to monitor ligand-induced internalization at sites of tissue damage. The pattern of receptor distribution was quantified using the contrast metric, which reports differences in intensity between neighboring pixels. The rationale is that when the receptor distribution is membranous and smooth, the contrast value is low. When the receptor distribution is vesicular and more punctate, then the contrast value is high (Figure 3).
An alternative method is to quantify the ratio of receptor levels (sfGFP intensity) over the levels of a control membrane marker e.g. membrane CFP (mCFP) (Figure 3). Both methods could detect receptor internalization, as indicated by more vesicular receptor distribution pattern globally in the cell (higher contrast value) or lower receptor levels at the membrane (lower sfGFP/mCFP ratio). However, the contrast metric could also detect receptor internalization in neutrophil clusters at the wound, in which membrane segmentation was less accurate and not applicable (Figure 3). Using this metric, we were able to quantify visible differences between Cxcr1 and Cxcr2 trafficking in neutrophils at wounds (Figure 4 and Supplementary Video 2). Cxcr1-FT internalized in cells located at the wound whereas Cxcr2-FT remained membranous in neutrophils at the wound (Figure 4A-C, Supplementary Video 2 and Supplementary Video 3). Suppression of Cxcl8a and Cxcl8b, through morpholino treatment, differentially affected Cxcr1-FT internalization at wounds (Figure 4C,D). To further validate that Cxcr1-FT responds to Cxcl8a, we performed chemokine response assays in early embryos. We found that Cxcr1-FT markedly internalized in embryos in which Cxcl8a was co-expressed (Figure 5). Altogether these results indicate that the described methods can be deployed to measure chemokine-induced receptor internalization in neutrophils and establish the identity of the ligand mediating these effects.

Figure 1: Neutrophil migration to ventral fin wounds. (A) (Left) Cartoon of 3 dpf larva showing the location of the caudal hematopoietic tissue (CHT), the venus circulation (VC, blue), the ventral fin (VF) and the wound site. (Right) Cartoon depicting the area of the wound (W) with neutrophils getting mobilized from the CHT and clustering at the wound. The caudal vein plexus (CVP) of the CHT tissue is drawn in blue. (B) Bright field image (left) and confocal projection (right) showing the ventral fin wound and the distribution of neutrophils in Tg(mpx:GFP) larvae at 2 h post-wounding. Dashed lines show VF and CHT outlines. Scale bar = 25 µm. Cartoon and fluorescent image modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
{kind=link}

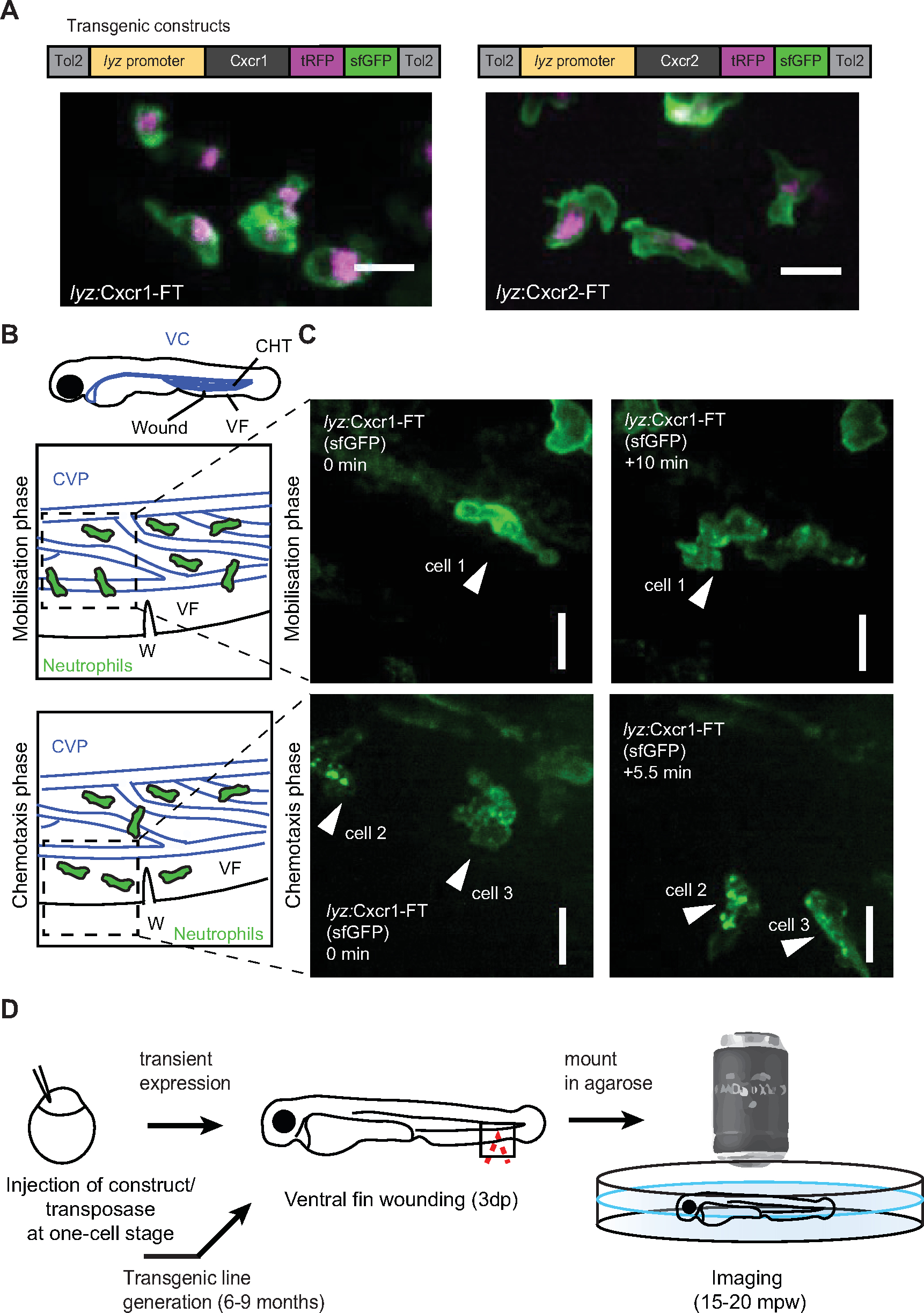
Figure 2: Live imaging of chemokine receptor trafficking in neutrophils. (A) Constructs used for neutrophil-specific transgenic expression of Cxcr1-FT (Fluorescent Timer) and Cxcr2-FT. Confocal projections of neutrophils in the head of a 3 dpf transgenic larva (Tg(lyz:Cxcr1-FT), top; Tg(lyz:Cxcr2-FT), bottom) showing tRFP (magenta) and sfGFP (green) channels. Scale bar = 20 µm. (B) Anatomical scheme of 3 dpf larva as in Figure 1A. Below the larva are schemes depicting the area of the wound (W) with neutrophils getting mobilized from the CHT (top) or performing chemotaxis upon entering the ventral fin (bottom). Dashed square indicates area imaged in snapshots on the right. (C) Neutrophils in Tg(lyz:Cxcr1-FT) larvae (sfGFP is shown) upon mobilization from the CHT (top panels) or chemotaxis towards the wound (bottom panels). Arrows show the same cells over time. Time points on the right image are minutes elapsed after image on the left. Scale bar = 10 µm. (D) Schematic representation of experimental approach for live imaging of chemokine receptor trafficking. Panels A-C modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Quantification examples of receptor dynamics. Single (blue) or clustered neutrophils (green) at wounds or non-mobilized neutrophils in the CHT (red, orange) were segmented and analyzed by different methods to compare results. The same example cells shown were analyzed with two methods to relate what is seen in the image with the range of values extracted. (A) The surface of the selected, example cells were segmented based on contour definition in the sfGFP channel. (B) Contrast was computed from the example cells shown in A. (C) The membrane of the selected, example cells were segmented based on contour definition in the CFP channel. Ratiometric analysis of sfGFP/CFP followed. (D) The ratio of sfGFP/CFP was computed on the example cells shown in C. Error bars represent S.E.M. from individual cells, in cases of n>1, values here were not used for statistical analysis but merely to exemplify measurements obtained with the different quantification methods. Scale bar = 10 µm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
{kind=link}

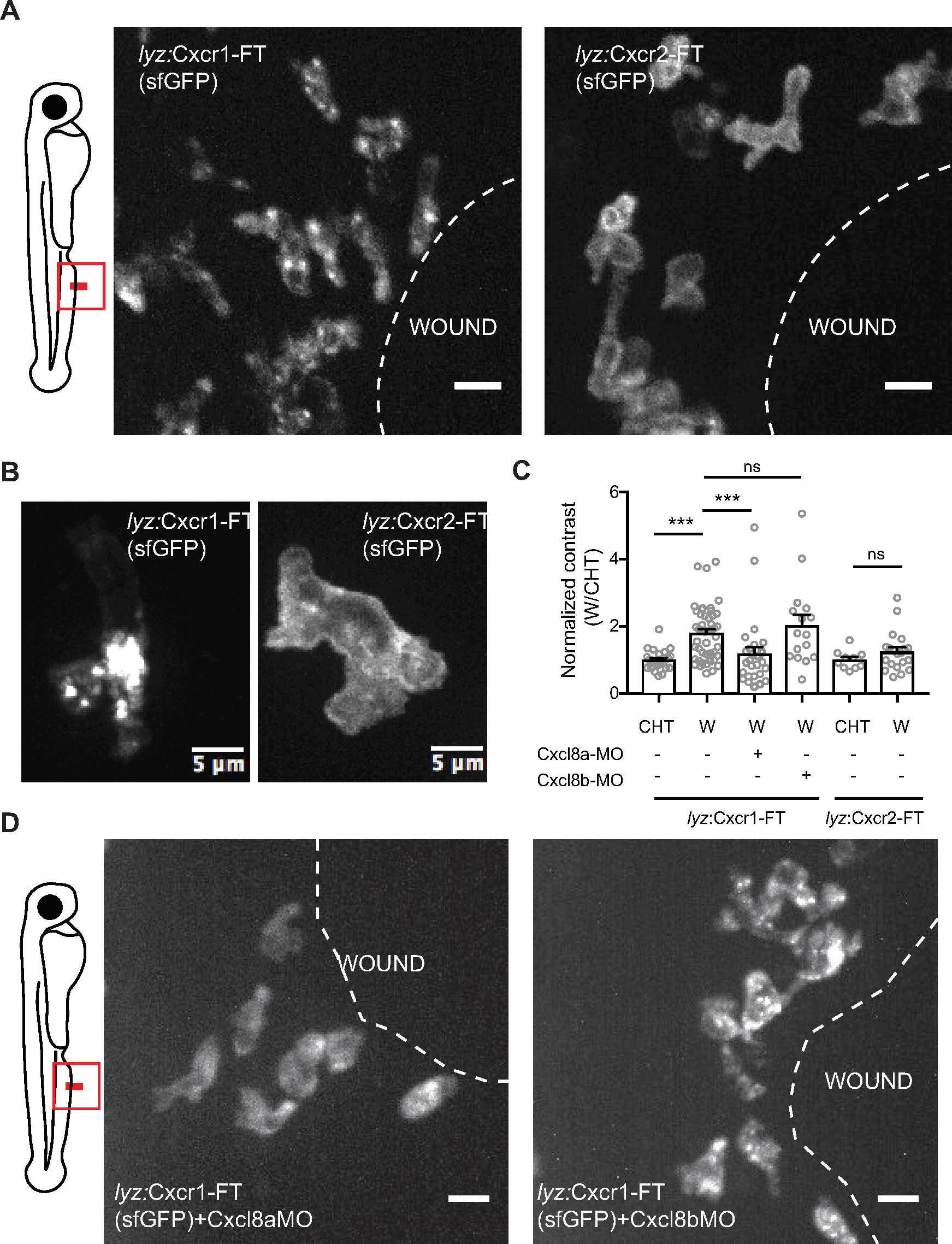
Figure 4: Differential dynamics of Cxcr1 and Cxcr2 in response to wounding. (A) Confocal projection of neutrophils in Tg(lyz:Cxcr1-FT) or Tg(lyz:Cxcr2-FT) larvae at the wound at 80 min post-wounding (sfGFP channel shown). Scale bar = 10 µm. (B) Magnified Cxcr1-FT neutrophil (left) and Cxcr2-FT (right) at the wound. Green receptor is shown in gray. Scale bar = 5 µm. (C) Normalized contrast (contrast per individual neutrophil normalized to the mean contrast of non-mobilized cells in the CHT). cxcl8a refers to injection of a splice-blocking together with a translation-blocking morpholino for cxcl8a. cxcl8b refers to injection with a splice-blocking morpholino for cxcl8b. For Tg(lyz:Cxcr1-FT): n=24 cells (CHT), n=47 cells (wound) from 8 larvae. For Tg(lyz:Cxcr1-FT) with morpholinos: n=28 cells (Cxcl8a-MO) from 5 larvae, n=16 cells (Cxcl8b-MO) from 5 larvae. For Tg(lyz:Cxcr2-FT): n=10 cells (CHT) and n=20 cells (wound) from 3 larvae. Data were pooled from independent larvae acquired in 1-5 imaging sessions. Kruskal-Wallis test with Dunn’s multiple comparisons test for Tg(lyz:Cxcr1-FT), two-tailed unpaired Mann-Whitney test for Tg(lyz:Cxcr2-FT). (D) Confocal projection of neutrophils in Tg(lyz:Cxcr1-FT) transgenic larvae treated with cxcl8a morpholino (MO) (left) and cxcl8b MO (right) responding to fin wounds (sfGFP channel shown in green). Snapshot taken at timepoints of equivalent neutrophil accumulation (85 min post-wounding in left image and 45 min post-wounding in right image). Scale bar = 10 µm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
{kind=link}

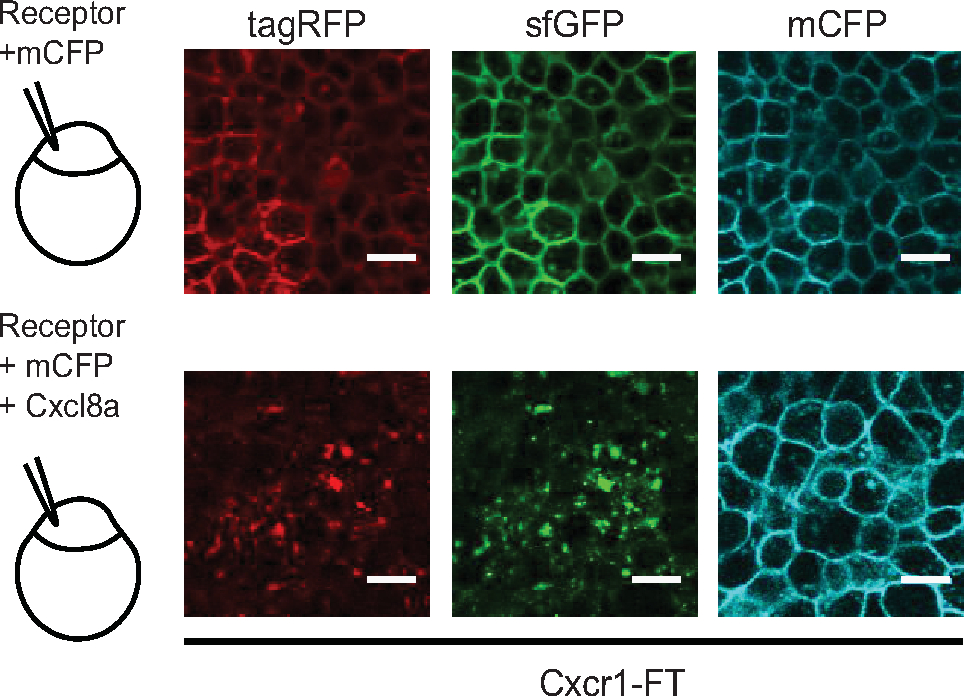
Figure 5: Chemokine response assay in early embryos. Laser-scanning confocal slices of gastrulating embryos showing expression and distribution of Cxcr1-FT. 100 pg of Cxcr1-FT mRNA was injected into one cell-stage eggs with or without 150 pg Cxcl8a mRNA. Green and red receptors are shown in separate channels. Control membrane CFP marker (mCFP) is shown in the cyan channel. Scale bar = 20 μm. Figure modified from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to view a larger version of this figure.
{kind=link}
Supplementary Movie 1: Transgenic neutrophils in the head of a Tg(lyz:Cxcr1-FT) (left) and Tg(lyz:Cxcr2-FT) (right) larva at 3 dpf. sfGFP(green), tagRFP (magenta). Frame interval is 30 sec and frame rate is 5 fps. Scale bar = 20 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary Movie 2: Neutrophils in Tg(lyz:Cxcr1-FT) (left) and Tg(lyz:Cxcr2-FT) (right) transgenic larvae responding to fin wounds. Movie starts within 10 min post-wounding and lasts 60 min. sfGFP (green), tagRFP (magenta). Frame interval is 30 sec and frame rate is 10 fps. CHT = caudal hematopoietic tissue. VF = ventral fin. Scale bar = 25 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary Movie 3. Additional examples of neutrophils from a wounded Tg(lyz:Cxcr1- FT) transgenic larva (different larva to that shown in Video 2), acquired at higher resolution, showing receptor internalization (sfGFP channel shown in green) upon mobilization in the CHT or upon entry and chemotaxis in the ventral fin. Frame interval is 30 sec and frame rate is 2 fps. Scale bar = 10 μm. Video originates from ref.14 (http://creativecommons.org/licenses/by/4.0/). Please click here to download this video.
Supplementary File 1: Please click here to download this file.
Supplementary File 2: Please click here to download this file.
Supplementary File 3: Please click here to download this file.
Supplementary File 4: Please click here to download this file.
Discussion
The method described allows live imaging of receptor dynamics in response to endogenous ligands in situ during an inflammatory response to tissue damage. The use of Cxcr1/Cxcr2 neutrophil reporters could be expanded to other physiological settings, such as infection, tumor models or other types of tissue damage14,25,26,27. In addition, transgenic rescue lines, in which the endogenous receptor is suppressed and rescued by an exogenous mutant receptor, could provide useful tools to dissect the importance of specific neutrophil migration patterns in immune responses. For example, Cxcr1 receptor mutants that have impaired desensitization cause more prominent neutrophil clustering at inflammatory sites14. This gain of function phenotype could be used to understand the role of neutrophil congregation in different physiological processes, e.g., wound repair, infectious disease, or tumor evolution, and complement receptor knockdown/knockout experiments. The methodology also provides a basis to expand the range of available reporters. The choice of fluorescent reporter is important to consider and depends on the biological question. We found that the constitutive turnover of these chemokine receptors in neutrophils was high, in comparison to epithelial cells, and that reporters with fast maturation (e.g., sfGFP) were required to report membrane levels at steady state and resolve differences upon neutrophil stimulation8,14. Thus, membrane ratios of sfGFP/tagRFP are not applicable for measuring ligand-induced internalization in this cell type, but the pattern of tagRFP allows tracking of the intracellular fates of the receptor, which could be useful in some studies. We also found that the more concentrated intracellular signal of tagRFP is useful for screening individual larvae. An alternative approach for measuring receptor levels at the plasma membrane would be to co-express a fluorescent membrane marker in neutrophils either in the same transgene9 or in an independent transgene14. In the former scenario the transgene would provide an additional means for screening the fish and expression levels would be comparable between the marker and the receptor. The latter approach would be more modular, in that a zebrafish line with a receptor reporter could be combined with different reporter lines. In either case, it is worth noting that membrane quantification of the receptor levels is challenging in clustered neutrophils (see below). Finally, we note that a possible extension of this protocol would be to follow up the live imaging by immunohistochemistry for more detailed localization analyses.
The Tol2 transgenesis system is well established7 and the lysozyme C promoter has been used extensively for neutrophil expression11,15. The transgenesis approach is, therefore, relatively straightforward and the expression level achieved with this promoter is high enough to provide sufficient contrast for analysis of receptor dynamics. A possible limitation is that the expression level does not recapitulate endogenous receptor expression levels. New CRISPR technologies could be deployed to establish knock-in lines for receptors of particular interest28. These technologies are still cumbersome and may not guarantee the required expression levels for subcellular imaging, but their successful development would be an important breakthrough for understanding endogenous signaling dynamics. Functional validations are important for interpreting data with transgenic receptor constructs. For example, ligand recognition assays can be used to establish that the fluorescent fusion protein is functional and rescue of knockout phenotypes could be used to establish that the transgenic neutrophil expression levels are compatible with functionality14. Finally, a more direct way to validate the receptor fusion would be to utilize an in vitro functional assay with labelled receptor alongside non-labeled versions14.
The quantification approach addresses specific difficulties in accurate membrane segmentation in neutrophils in vivo. In cells of epithelial nature, quantification of receptor levels can be executed automatically by normalizing membrane receptor levels to a control marker, which can be expressed in tandem or separately9. Indeed, we have applied such an approach, when using the ligand-recognition assay in gastrulating embryos14. However, neutrophils undergo complex, rapid changes in cell shape in vivo, making the membrane segmentation difficult both in 2D and 3D14. This is even more challenging when neutrophils cluster, which occurs in many physiological settings29. The contrast metric overcomes this limitation as it does not require membrane segmentation but instead reflects the overall state of receptor distribution in the cell (membranous/smooth vs vesicular/dotty). It is important to note that contrast metric can be affected by the overall contrast of the image, so normalization of individual cell values to an internal reference is required to account for variability of signal in different embryos/samples. For example, we used the mean cell contrast value of non-responsive neutrophils in the CHT (i.e., neutrophils that remain stationary and do not migrate into the ventral fin)14. An additional possibility would be to normalize with contrast values of a control marker in the same cell. This would provide a solution when an internal reference of non-responding cells is not available and may likely resolve finer quantitative differences in receptor dynamics between different conditions.
The location of imaging is another variable to consider. The reason for choosing the ventral fin wound here, as opposed to the more commonly used tail fin wound model16,30, is because the site of wounding is nearby the site of neutrophil residence/migratory origin. This accelerates the timeline of the assay, as it takes relatively little time for neutrophils to arrive. Additionally, it provides the opportunity to capture cell behavior both at the migration origin (CHT) and the target location of interest (wound). This is relevant here, because the spatial and temporal resolution required for subcellular imaging is difficult to combine with a large field of view or multi-position scanning. Thus, the ventral fin wound assay permits tracking of the evolution of the migratory response from the migration origin and simultaneous capturing of unspecific receptor fluctuations in cells that do not respond. As mentioned above, the latter is useful for quantification purposes as it provides an internal reference for unspecific dynamics. In other systems, it may not be possible to have such an internal reference, in which case the contrast values of a co-expressed membrane marker would provide an alternative control.
In summary, we anticipate that the methodology is applicable to other systems and can be deployed for a variety of purposes. For example, the same reporters could be utilized in other inflammatory settings, such as infection settings or other disease models. The repertoire of zebrafish receptor reporter lines could be expanded to other signaling receptors, to understand signaling mechanisms or report ligand dynamics in vivo. The approach can be combined with knockdown/knockout techniques to interrogate the mechanistic basis of observed dynamics. For example, perturbation of ligand expression can indicate the ligand dependency for observed receptor dynamics. In the future, we envisage that the system could be further refined to incorporate knock-in insertion of reporters. Ultimately, findings using this methodology would provide novel insights valuable beyond the zebrafish community, given the conservation of these signaling receptors in mammals and the relative challenge of conducting these studies in larger organisms.
Acknowledgements
We thank Christine Holt and Bill Harris for help with confocal microscopy. We thank Darren Gilmour for the fluorescent timer backbone constructs and Anna Huttenlocher for the Tol2-Lyz backbone vector. We thank Steve Renshaw for the Tg(mpx:GFP)i114 line. This work was supported by the MRC (MR/L019523/1), the Wellcome Trust [204845/Z/16/Z]; Isaac Newton Trust [12.21(a)i] and an Isaac Newton Trust grant 19.23(n). C.C. was supported by an MRC DTP studentship and partly by the Wellcome Trust [204845/Z/16/Z]; Isaac Newton Trust [12.21(a)i] grant. H.W. was supported by an MRC DTP Studentship. H.P. was supported by a Wellcome Trust PhD grant (105391/Z/14/Z) and partly by a Wellcome Trust [204845/Z/16/Z]; Isaac Newton Trust [12.21(a)i] grant and the MRC (MR/L019523/1).
Materials
| Name | Company | Catalog Number | Comments |
| 1-phenyl-2-thiourea | Sigma-Aldrich | P7629-25G | |
| 50mL CellStar Centrifuge Tube | Grenier Bio-One Limited | 210270 | |
| Clark capillary glass | Harvard Apparatus | 30-0020 | |
| Cleanline Thin Bleach | Scientific Laboratory Supplies | CLE0301 | |
| Dissecting scope | Nikon | SMZ645 | |
| Dri Block heater | Techne | FDB02AD | |
| Fiji | National Institutes of Health, Bethesda, MD | ||
| Forceps, Jewelers Dumont No. 5 | Sigma-Aldrich | F6521 | |
| Glass bottom microwell dishes | MatTek | P35G-1.5-14-C | |
| Glass pasteur pipette | Fisherbrand | 11795098 | |
| Invitrogen Ambion mMESSAGE mMACHINE SP6 Transcription Kit | Invitrogen | 10391175 | |
| Leica SP8 confocal microscope | Leica | N/A | |
| Low melting point agarose | Invitrogen | 16520-100 | |
| Magnetic stand | Narishige | GJ-1 | |
| MATLAB | The MathWorks, Inc., Natick, MA | ||
| Methylene blue | Sigma-Aldrich | M9140-25G | |
| MgSO4 | Sigma-Aldrich | M-2643 | |
| Microinjection system | Parker | Picospritzer III | |
| Microloader pipette tips | Eppendorf | 5242956003 | |
| Micromanipulator | Narishige | M-152 | |
| Micropipette Puller | Sutter Instrument Company | Flaming/Brown P-97 | |
| Microscope slide | Thermo-Scientific | J1800AMNZ | |
| PerkinElmer Spinning Disk UltraVIEW ERS, Olympus IX81 spinnng disk confocal microscope | Olympus | N/A | |
| Petri dish (120mm) | Grenier Bio-One Limited | 632180 | |
| Phenol red | Sigma-Aldrich | P0290-100ML | |
| Poly(A) Tailing Kit | Invitrogen | AM1350 | |
| Prism | GraphPad Software | ||
| Small paintbrush | N/A | ||
| Spectral Applied Research LMM5 laser module | N/A | ||
| Surgical Scalpel Blade No. 23 | Swann-Morton | 233-5528 | |
| Tricaine MS222 | Sigma-Aldrich | E10521-50G | |
| Volocity software | N/A | ||
| Yokogawa CSU10 spinning disc confocal scanner | Yokogawa | N/A |
References
- Rot, A., von Andrian, U. H. Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annual Review of Immunology. 22, 891-928 (2004).
- Cotton, M., Claing, A. G protein-coupled receptors stimulation and the control of cell migration. Cellular Signalling. 21 (7), 1045-1053 (2009).
- Liu, X., et al. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nature Immunology. 13 (5), 457-464 (2012).
- Wen, X., Jin, T., Xu, X. Imaging G Protein-coupled Receptor-mediated Chemotaxis and its Signaling Events in Neutrophil-like HL60 Cells. Journal of Visualized Experiments. (115), e54511 (2016).
- Arnon, T. I., et al. GRK2-dependent S1PR1 desensitization is required for lymphocytes to overcome their attraction to blood. Science. 333 (6051), 1898-1903 (2011).
- Jung, H., Mithal, D. S., Park, J. E., Miller, R. J. Localized CCR2 Activation in the Bone Marrow Niche Mobilizes Monocytes by Desensitizing CXCR4. PloS One. 10 (6), 0128387 (2015).
- Kwan, K. M., et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics: An Official Publication of the American Association of Anatomists. 236 (11), 3088-3099 (2007).
- Donà, E., et al. Directional tissue migration through a self-generated chemokine gradient. Nature. 503 (7475), 285-289 (2013).
- Venkiteswaran, G., et al. Generation and dynamics of an endogenous, self-generated signaling gradient across a migrating tissue. Cell. 155 (3), 674-687 (2013).
- Minina, S., Reichman-Fried, M., Raz, E. Control of receptor internalization, signaling level, and precise arrival at the target in guided cell migration. Current Biology. 17 (13), 1164-1172 (2007).
- Yoo, S. K., et al. The role of microtubules in neutrophil polarity and migration in live zebrafish. Journal of Cell Science. 125, 5702-5710 (2012).
- Yoo, S. K., et al. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Developmental Cell. 18 (2), 226-236 (2010).
- Buchan, K. D., et al. A transgenic zebrafish line for in vivo visualisation of neutrophil myeloperoxidase. PLoS One. 14 (4), 0215592 (2019).
- Coombs, C., et al. Chemokine receptor trafficking coordinates neutrophil clustering and dispersal at wounds in zebrafish. Nature Communications. 10 (1), 5166 (2019).
- Hall, C., Flores, M. V., Storm, T., Crosier, K., Crosier, P. The zebrafish lysozyme C promoter drives myeloid-specific expression in transgenic fish. BMC Developmental Biology. 7, 42 (2007).
- Renshaw, S. A., et al. A transgenic zebrafish model of neutrophilic inflammation. Blood. 108 (13), 3976-3978 (2006).
- Khmelinskii, A., et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nature Biotechnology. 30 (7), 708-714 (2012).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Brachydanio rerio). , (2007).
- Rosen, J. N., Sweeney, M. F., Mably, J. D. Microinjection of Zebrafish Embryos to Analyze Gene Function. Journal of Visualized Experiments. (25), 1115 (2009).
- Detrich, H. W., Zon, L. I., Westerfield, M. . The Zebrafish: Genetics, Genomics and Informatics. , (2004).
- Le Guyader, D., et al. Origins and unconventional behavior of neutrophils in developing zebrafish. Blood. 111 (1), 132-141 (2008).
- Chan, T. F., Vese, L. A. Active contours without edges. IEEE Transactions on Image Processing. 10 (2), 266-277 (2001).
- Haralick, R. M., Shanmugam, K., Dinstein, I. Textural features for image Classification. IEEE Transactions on Systems, Man, and Cybernetics. SMC-3 (6), 610-621 (1973).
- Deng, Q., et al. Localized bacterial infection induces systemic activation of neutrophils through Cxcr2 signaling in zebrafish. Journal of Leukocyte Biology. 93 (5), 761-769 (2013).
- Torraca, V., Mostowy, S. Zebrafish infection: From pathogenesis to cell biology. Trends in Cell Biology. 28 (2), 143-156 (2018).
- Feng, Y., Santoriello, C., Mione, M., Hurlstone, A., Martin, P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biology. 8 (12), 1000562 (2010).
- Poplimont, H., et al. Neutrophil swarming in damaged tissue is orchestrated by connexins and cooperative calcium alarm signals. Current Biology. 30 (14), 2761-2776.e7 (2020).
- Cornet, C., Di Donato, V., Terriente, J. Combining Zebrafish and CRISPR/Cas9: Toward a more efficient drug discovery pipeline. Frontiers in Pharmacology. 9, 703 (2018).
- Kienle, K., Lämmermann, T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunological Reviews. 273 (1), 76-93 (2016).
- Lisse, T. S., Brochu, E. A., Rieger, S. Capturing tissue repair in zebrafish larvae with time-lapse brightfield stereomicroscopy. Journal of Visualized Experiments. (95), e52654 (2015).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved