A subscription to JoVE is required to view this content. Sign in or start your free trial.
Systematic Approach to Identify Novel Antimicrobial and Antibiofilm Molecules from Plants' Extracts and Fractions to Prevent Dental Caries
* These authors contributed equally
In This Article
Summary
Natural products represent promising starting points for the development of new drugs and therapeutic agents. However, due to the high chemical diversity, finding new therapeutic compounds from plants is a challenging and time-consuming task. We describe a simplified approach to identify antimicrobial and antibiofilm molecules from plant extracts and fractions.
Abstract
Natural products provide structurally different substances, with a myriad of biological activities. However, the identification and isolation of active compounds from plants are challenging because of the complex plant matrix and time-consuming isolation and identification procedures. Therefore, a stepwise approach for screening natural compounds from plants, including the isolation and identification of potentially active molecules, is presented. It includes the collection of the plant material; preparation and fractionation of crude extracts; chromatography and spectrometry (UHPLC-DAD-HRMS and NMR) approaches for analysis and compounds identification; bioassays (antimicrobial and antibiofilm activities; bacterial "adhesion strength" to the salivary pellicle and initial glucan matrix treated with selected treatments); and data analysis. The model is simple, reproducible, and allows high-throughput screening of multiple compounds, concentrations, and treatment steps can be consistently controlled. The data obtained provide the foundation for future studies, including formulations with the most active extracts and/or fractions, isolation of molecules, modeling molecules to specific targets in microbial cells and biofilms. For example, one target to control cariogenic biofilm is to inhibit the activity of Streptococcus mutans glucosyltransferases that synthesize the extracellular matrix’ glucans. The inhibition of those enzymes prevents the biofilm build-up, decreasing its virulence.
Introduction
The earliest models of medicine used in societies were based on natural products (NPs). Since then, humans have been searching for new chemicals in nature that can be transformed into drugs1. This search caused a continuous improvement of technologies and methods for ethnobotanical screening1,2,3. NPs offer a rich source of structurally diverse substances, with a wide range of biological activities useful for developing alternative or adjuvant therapies. However, the inherent complex plant matrix makes the isolation and identification of the active compounds a challenging and time-consuming task4.
NPs-based drugs or formulations can be used to prevent and/or treat several conditions affecting oral, including dental caries4. Dental caries, one of the most prevalent chronic diseases globally, derives from the interaction of sugar-rich diet and microbial biofilms (dental plaque) formed on the tooth surface that leads to demineralization caused by organic acids derived from microbial metabolism, and if not treated, leads to teeth loss5,6. Although other microorganisms may be associated7, Streptococcus mutans is a critical cariogenic bacterium because it is acidogenic, aciduric, and an extracellular matrix builder. This species encodes multiple exoenzymes (e.g., glycosyltransferases or Gtfs) that use sucrose as a substrate8 to build the extracellular matrix rich in exopolysaccharides, which are a virulence determinant9. Also, the fungus Candida albicans can drive up the production of that extracellular matrix7. Albeit fluoride, administered in various modalities, remains the basis for preventing dental caries10, new approaches are needed as adjuvants to increase its effectiveness. In addition, the available anti-plaque modalities are based on the use of broad-spectrum microbicidal agents (e.g., chlorhexidine)11. As an alternative, NPs are potential therapies for controlling biofilms and preventing dental caries12,13.
The further advance in the discovery of new bioactive compounds from plants includes necessary steps or approaches such as: (i) the use of reliable and reproducible protocols for sampling, considering that plants often show intraspecific variability; (ii) the preparation of comprehensive extracts and their respective fractions in small scale; (iii) the characterization and/or dereplication of their chemical profiles thought the acquisition of multidimensional data such as GC-MS, LC-DAD-MS, or NMR, for example; (iv) the use of viable and high-yield models to assess bioactivity; (v) the selection of potential new hits based on multivariate data analysis or other statistical tools; (vi) to perform the isolation and purification of the targeted compounds or promising candidates; and (vii) the validation of the corresponded biological activities using the isolated compounds2,14.
Dereplication is the process of rapidly identifying known compounds in crude extract and allows differentiating novel compounds from those that have already been studied. Besides, this process prevents isolation when bioactivity has already been described for certain compounds, and it is particularly helpful to detect “frequent hitters”. It has been used in different untargeted workflows ranging from major compound identification or the acceleration of activity-guided fractionation up to the chemical profiling of collections of extracts. It can be fully integrated with metabolomic studies for the untargeted chemical profiling of CE or the targeted identification of metabolites. All of this ultimately leads to prioritizing extracts before the isolation procedures1,15,16,17.
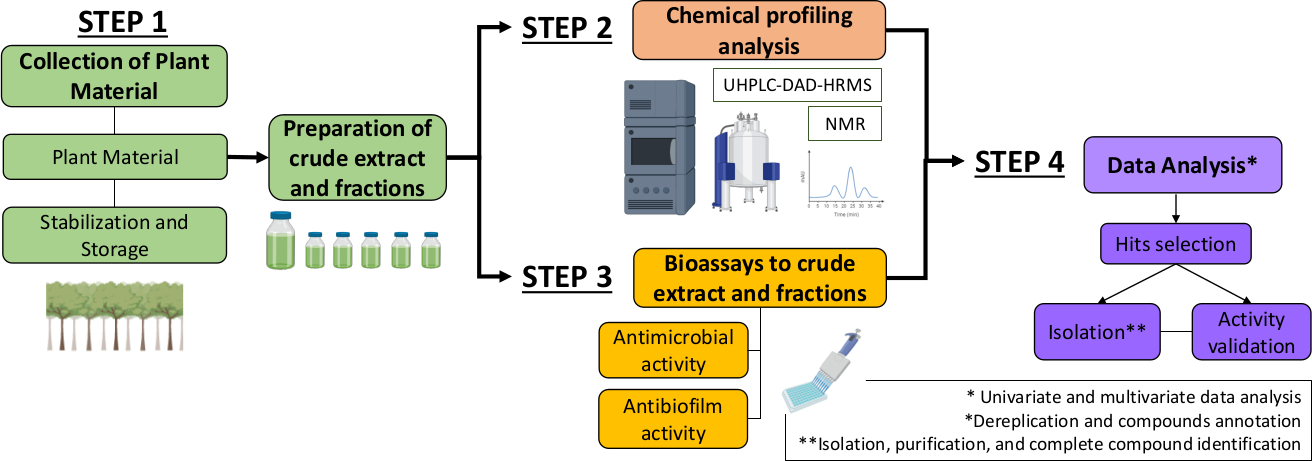
Therefore, in the present manuscript, we describe a systematic approach to identify antimicrobial and antibiofilm molecules from plant extracts and fractions. It includes four multidisciplinary steps: (1) collection of plant material; (2) preparation of crude extracts (CE) and fractions (CEF), followed by their chemical profile analysis; (3) bioassays; and (4) biological and chemical data analyses (Figure 1). Thus, we present the protocol developed to analyze of the antimicrobial and antibiofilm activities of Casearia sylvestris extracts and fractions against Streptococcus mutans and Candida albicans13, as well as the procedures for the phytochemical characterization and data analysis. For simplicity, the focus here is to demonstrate the approach for screening natural compounds using the bacterium.

Figure 1: Flow-chart of the Systematic Approach to identify active molecules from plants extracts and fractions. Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. Collection of Plant Material
- Plant material
- Record the access to the plant material on electronic platforms that regulate access to genetic heritage in the country where the collection will take place. For example, in Brazil, register with the National System for the Management of Genetic Heritage and Associated Traditional Knowledge – SisGen (website https://sisgen.gov.br/paginas/login.aspx).
- Collect samples of the plant material of interest (e.g., leaves, stems, roots, flowers, fruits). Register if the material was collected during the reproductive or the vegetative phase.
- Record the collection parameters (date, georeferencing, average annual temperature, and mean humidity percentage).
- Identify the samples precisely, and a taxonomist must confirm the authenticity.
- Plant samples stabilization and storage
- Separate plant organs in individual plastic bags or flasks immediately after collections.
- Inactivate potential enzymatic reactions by (i) freezing immediately in liquid nitrogen, (ii) dehydrating in a circulating air oven (40 °C), or (iii) freeze-drying the samples by lyophilization.
- Store the stabilized material in hermetically sealed bags at room temperature or in a freezer until use (-20 or -80 °C, depending on the storage period or intended use).
- Grind the samples in an analytical mill (knife or ball, depending on the tissue type or availability) and standardize particle size using standardized sieves.
- Weigh the samples individually for the subsequent extraction steps.
2. Preparation of Crude Extracts (CE) and Fractions (CEF) to Chemical Profile Analysis and Bioassays
- Preparation of Crude Extracts (CE)
NOTE: The steps are illustrated in the flow-chart in Figure 2A.- Prepare an extraction solvent with a hydroalcoholic mixture (e.g., ethanol (EtOH) 70% or ternary mixtures of water, EtOH, and other modifiers, defined by the experimental design of according to previous reports).
- Use the ratio sample weight (dry weight, mg)/ extraction solvent (mL) varying from 50 to 100 mg for each mL of solvent.
- For fast and reproducible extractions, use batch extractions using microtubes.
NOTE: At least three replicates should be used at this point to allow statistical analysis. - Perform ultrasound-assisted extractions (UAE) to make it quick, easy, and cheap.
- Repeat the procedure three times (15 min each) for the best efficiency.
- After each extraction step, decant the solid residue by centrifugation and remove the supernatant.
- Combine the supernatants individually, filter, and save aliquots for simultaneous chemical analysis and bioassays. If necessary, remove the extracting solvent under vacuum, nitrogen flow, or lyophilization, and register the weigh and yield.
- Store at -20 °C protected from light.
NOTE: The protocol can be paused here to screen the CE and select the ones that present the desired activity.
- Fractionation of Crude Extracts (CEF)
NOTE: The steps are illustrated in the flow-chart in Figure 2B.- Use cartridges with at least 1 g of adsorbent. If alkaloids are potentially present in the CE, use solvents containing 0.1% of formic acid (FA).
- Dilute sample in the most suitable solvent (or solvent mixture) to obtain a 100 mg/mL sample solution. Then, transfer 1 mL of the sample solution to a pre-conditioned a solid-phase extraction cartridges - SPE (1 g of adsorbent, 6 mL of capacity).
- Perform the fractionation using around three dead volumes of each extraction eluent (to a cartridge of 1 g, it will correspond to 2 mL of each solvent). If alkaloids are potentially present in the CE, use solvents containing 0.1% of FA.
- Collect one fraction by eluent composition and save aliquots for simultaneous chemical analysis and bioassays.
- Remove the solvent under vacuum, nitrogen flow, or lyophilization and register the weigh and yield.
NOTE: If the CE is difficult to dissolve in the initial elution mixture, disperse the CE in a solid phase (e.g., C18 or celite) in 1:1 (w/w) proportion before loading the material on the top of the cartridge.
CAUTION: Weigh the microtubes in advance to calculate the mass yield of extracts and fractions.
- Chemical profiling analysis
NOTE: Considering that each plant species requires optimized and specific methods for its chemical analysis, in the following sections, we describe the most common analytical approaches used to analyze plant materials. As a practical example, a reverse-phase liquid chromatography was developed and validated for the simultaneous analysis of phenolic compounds and clerodane-type diterpenes differentially biosynthesized by two varieties of Casearia sylvestris Swartz (Salicaceae). The UPLC-DAD apparatus was equipped with a degasser, a quaternary pump, an automatic sampler, a UV-Vis photodiode array detector, and an oven (see details in Bueno et al. 201518). Similar approaches described in the following examples can be optimized according to other plant species and/or plant materials.- Chromatographic analysis and hyphenation possibilities
- For separations using ultra-high-performance liquid chromatography (UPLC), use a C18 chromatographic column (e.g., 150 × 2.1 mm, 2.6 µm, 100 Å) protected by a compatible pre-column.
NOTE: Other column phases or chromatographic modes can be used depending on the plant species/material. Conventional HPLC can also be used; in that case, a suitable chromatographic column must be chosen. To achieve excellent separations, adjust the chromatographic conditions considering the flow rate (µL/min), column temperature (°C), and injection volume (µL). The mobile phase usually consists of water (A) and acetonitrile or methanol (B) using linear or multistep elution gradients or isocratic elution. Modifiers such as buffers, acids, bases, or others can also be used. - Perform the plant material analysis (CE and/or CEF) and register all related data, such as spectral data (using a UV-Vis and/or, preferentially, MS detectors), retention time (min), and others, depending on the available hyphenation.
NOTE: Liquid chromatography (LC) is usually hyphenated (coupled) with high-resolution mass spectrometry (HRMS), as LC−HRMS, and is commonly use for the rapid annotation of metabolite in CE ou CEF15. - If qualitative data is required and quantitative data is necessary, carefully prepare and inject calibration curves, following the same protocol.
NOTE: The development of the best chromatographic conditions can be performed with the assistance of the design of experiments, as described by Bueno et al. 201518, or similar literature. It is important to consider the inclusion of internal standards during the method development. They are very much appreciated since they allow correct technical deviations during sample preparation and injections, and further normalization for data analysis.
- For separations using ultra-high-performance liquid chromatography (UPLC), use a C18 chromatographic column (e.g., 150 × 2.1 mm, 2.6 µm, 100 Å) protected by a compatible pre-column.
- Chromatographic analysis and hyphenation possibilities
- Univariate and multivariate data analysis
- Export the registered chromatograms in a suitable format (e.g., ASCII, .txt. or .csv format). A single data matrix can be set by joining and aligning the chromatograms if several samples are being analyzed, and comparisons are required. The resulting matrices must be normalized the chromatograms according to the internal standard used.
- Analyze the plant metabolomics data using multivariate and univariate methods. Explore and visualize metabolomics datasets through the simultaneous analysis of multiple variables using multivariate statistical methods, including unsupervised principal component analysis (PCA) and hierarchical clustering analysis (HCA), or supervised partial least squares analysis (such as PLS, OPLS, PLS-DA). Univariate methods, such as ANOVA, Student’s, Tukey and Welch’s t-test, are especially interesting for the precise analysis of quantitative differences among samples19.
- Dereplication and compounds annotation
NOTE: The objective of this step is dedicated to rapid on-line identification of known NPs to avoid tedious isolation that can be performed simultaneously to the uni- or multivariate data analysis.- Perform the identification levels of the detected or target compounds:
- Identified compound, including full 3D structure and stereochemistry (level 0);
- Identification achieved by two orthogonal parameters, such as retention time and MS/MS spectrum (level 1);
- Putatively annotated compounds and compound classes (levels 2 and 3);
- Unidentified or unclassified metabolites that can be differentiated based on analytical data (level 4)19,20.
- Characterize the known compounds using the commercial or public databases. Among the most important databases, it can be highlighted: NIST (https://www.nist.gov), Wiley (https://www.sisweb.com/software/ms/wiley.htm), MassBank (https://massbank.eu/MassBank/), GMD (http://gmd.mpimp-golm.mpg.de/), METLIN (https://metlin.scripps.edu), and the Global Natural Products Social Molecular Networking – GNPS database (https://gnps.ucsd.edu)19.
NOTE: There are different levels of annotations and they depend on the hyphenated technique employed during the study and may include: the assistance of MS- (or NMR) based spectral databases, and in silico spectral prediction algorithms. - Isolation, purification, and complete compound identification
NOTE: If a given compound (whose identity was suspected by the statistical methods) needs complete structural identification, the first step to accomplish this task is to isolate and purify the desired compounds in a greater scale. It can be accomplished by scaling up the already developed protocols.- Perform the rapid and direct isolation of the target compound (s) by preparative chromatographic techniques well established and optimized. Semi-preparative HPLC with dry load injection can be used to avoid the compromises that generally need to be made between high loading and sample solubilization1.
- Accomplish the complete structural characterization and identification of isolated compounds. This can be made through the combination of different techniques:
- Nuclear Magnetic Resonance (NMR);
- Mass spectrometry (MS);
- Spectrometric techniques in the ultraviolet (UV) and infrared (IR) regions are also very useful for the characterization of the functional groups;
- The use of chiroptical spectroscopy such as electronic and vibrational circular dichroism (ECD and VCD, respectively), Raman optical activity (ROA), and X-ray crystallography are important techniques for absolute configuration characterization.
- Perform the identification levels of the detected or target compounds:
3. Bioassays
NOTE: Biological screening: To quickly assess CE and CEF’s potential bioactivity, the initial screening of natural substances should be organized and straightforward.
- Preparation of CE and CEF for bioassays
- Reconstitute the dry matter with the best possible solvents (which can be determined experimentally). The experimental design21,22 define the stock solution and the concentration of solvents.
- Calculate the solvent concentration of the stock solution. To do this, use the formula: C1 x V1 = C2 x V2, where C1 represents the stock solution (mg) of CE and/or CEF; V1 represents the volume of solvent; C2 is the weight of the CE and/or CEF; V2 is the final volume (mL) of the stock solution.
NOTE: For example, we selected 84.15% EtOH and 15% dimethyl sulfoxide (DMSO) as the solvent concentration of the stock solution. We prepared the stock concentration of the CE to 6 mg/mL and the CEF to 1 mg/mL13. The solvent to dilute the CE and CEF will depend on the method of evaluation of the biological activity. The solvent used as a vehicle must not interfere with biological and toxicological activity. Typically, water, DMSO, EtOH, or an aqueous solvent-based on EtOH is used to solubilize plant extracts or plant derivatives4,13.
- Preparation of test organisms
- Reactivate a microbial strain, for example S. mutans UA159 on blood agar (48 h, 37 °C, 5% CO2), and culture it in liquid culture medium (e.g., tryptone-yeast extract broth [TYE: 2.5% (w/v) tryptone with 1.5% (w/v) yeast extract] containing 1% glucose (w/v) (TYEg) for 16 h, 37 ° C, 5% CO2.
- Perform a 1:20 dilution of the initial culture of the microorganism in the same culture medium (the dilution ratio of the initial culture may change according to the experimental design).
- Incubate until it reaches the mid-log growth phase.
- Prepare the inoculum for bioassays with a defined population (e.g., 2x106 colony-forming units per milliliter - CFU/mL) in TYEg for antimicrobial assays and TYE with 1% sucrose (w/v) (TYEs) for biofilms assays.
NOTE: The growth conditions will depend on the microorganism tested.
- Antimicrobial activity
NOTE: The steps are illustrated in Figure 3.- In a 96-well plate, add an aliquot (µL) of the CE and/or CEF stock solution (treatments). The volume of the aliquot is defined by the test concentration, which must be selected based on previous studies. For example, to test CE at the 0.5 mg/mL test concentration, use a 16.67 µL aliquot of the stock solution at 6 mg/mL. For this calculation, use the formula: C1 x V1 = C2 x V2, where C1 is the stock concentration, V1 is the volume of the stock solution aliquot, C2 is the test concentration, and V2 is the volume of the 96-well plate (which corresponds to 200 µL). In this experimental condition, the test concentration of the solvents (vehicle) will be 7% EtOH and 1.25% DMSO.
- Include a set of controls for each plate: a column with treatments, without the inoculum (blank control per treatment, help to differentiate turbidity by the treatment used itself from microbial growth); a column with vehicle and inoculum (diluent of CE or CEF or 0 mg/mL control); a column with only the culture medium (culture medium control) and a column with only inoculum (microbial growth control).
- Using TYEg, adjust the volume to 100 µL. Next, incubate, for example, 24 h, 37 °C, 5% CO2 (depending on the microorganism tested).
- Inoculate 100 µL of microorganism inoculum (1x 106 CFU/mL) into the 96-well plate.
- Analyze the bacterial growth according to turbidity by visual inspection of the wells (clear or cloudy). Clear: means that there is no visual growth of the microorganism. Cloudy: means that there is visual growth of the microorganism.
- Measuring the absorbance (optical density or O.D.) of the bacterial culture in each well (ELISA reader using 540 nm). Next, transfer 100 µL of the cultures to microtubes containing 900 µL of saline solution (0.89% NaCl), mix well by vortexing. Next, continue performing a ten-fold serial dilution until the desired value.
- Inoculate an aliquot of the desired dilution in specific agar plates (in duplicate). For example, 10 µL of a specific dilution on blood agar plates.
- Incubate. Conditions can change between microorganisms, for example, S. mutans: 48 h, 37 °C, 5% CO2.
- Perform colony counts on the plates for later transformation into CFU/mL as (Anumber of colonies x 10n)/q. In this formula,n equals the absolute value of the dilution (0, 1, 2, or 3), and q equals the amount, in mL, pipetted for each dilution plated on the agar plate. Also, the CFU/mL can be converted to log values.
NOTE: When plant extracts are added to the culture medium, precipitation of particles from the extracts may occur. This fact can make it difficult to interpret the results. The same occurs when a microplate reader measures the turbidity as, in some cases, cells clump on the bottom of the microplate. In addition, depending on the extract used, the color of plant leaf extracts can make it difficult to quantify turbidity23,24. An alternative method uses dyes that reveal whether the microbial cells are metabolically active or not24.
- Antibiofilm activity
NOTE: The steps to evaluate the effect of treatments on biofilm formation are illustrated in Figure 4.- Formation and processing of biofilms
- Dilute treatments in culture medium (TYEs) in a 96-well plate as described in the steps of the Antimicrobial Activity protocol.
- Incubate the plate. In the example with S. mutans, the incubation is performed during 24 h, at 37 °C, and 5% CO2.
- After incubation, place the plates on an orbital shaker (5 min, 37 °C, 75 rpm) to loosen the cells not adhered to the biofilm. Then, discard the culture medium containing the not adhered cells, and wash the remaining biofilms three times with 0.89% NaCl to remove non-adherent cells.
- Quantification of biomass from treated biofilms
NOTE: The steps are illustrated in the flow-chart in Figure 4A.- Keep the biofilms washed on the plate and add 50 µL of 1% crystal violet aqueous solution to each well.
- Incubate the plate at room temperature for 35 min.
- Wash the stained wells with MilliQ water (three times) and then air dry them for 60-90 min.
- Elute the crystal violet from the stained wells with 200 µL of 99% EtOH by incubating the plate in an orbital shaker (5 min, 37 °C, 75 rpm).
- Transfer a 150 µL aliquot from each well with the eluted dye to another plate and quantify the sample biomass (ELISA reader 570 nm).
- Quantification of the viable microbial population (CFU/mL) of the treated biofilms
NOTE: The steps are illustrated in the flow-chart in Figure 4B.- Remove the washed biofilms from the plate with a pipette and 200 µL of NaCl 0.89% and transfer the resulting suspension individually to sterile microtubes.
- Use an additional 200 µL of NaCl 0.89% per well and transfer it to the corresponding tube, already containing 200 µL of the initial biofilm suspension. Perform this process until reaching a total suspension of 1 mL biofilm per original well.
- Use an aliquot from each tube to perform a ten-fold serial dilution.
- Inoculate an aliquot of the desired dilution in specific agar plates (in duplicate). For example, 10 µL of a specific dilution on blood agar plates.
- Incubate agar plates (e.g., 48 h, 37 °C, 5% CO2), then count the colonies to determine the CFU/mL as described above.
- Formation and processing of biofilms
- Biological Activity Validation Phase
- Salivary pellicle formation
- Use Hydroxyapatite (HA) beads (Macro-Prep Ceramic Hydroxyapatite Type I 80 μm) as a surface to form the salivary film25. These beads surface mimic dental enamel.
- Weigh the HA beads (e.g., 10 mg) in microtubes and sterilize. Then, use adsorption buffer (AB buffer: 50 mM KCl, 1 mM KPO4, 1 mM CaCl2, 1 mM MgCl2, in dd-H2O, pH 6.5]25 containing 0.1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.02% sodium azide (NaN3) to wash the beads.
- Collect and prepare human saliva26. It is necessary to have institutional ethics committee approval.
- Add 500 μL of saliva into microtubes and incubate (40 min, 37 °C, 24 rpm).
- Next, remove the saliva supernatant and wash the beads (three times with AB buffer containing PMSF and NaN3). The sHA beads (HA bead with salivary pellicle) are now ready for downstream assays.
NOTE: Saliva is collected from healthy volunteers. After collection, dilute the saliva (1: 1 v/v) with AB buffer and centrifuge (1699 x g, 20 min, 4 °C). Sterilize by filtration (polyethersulfone membrane filter with low binding to 0.22 μm proteins)26. The Institutional Ethics Committee must approve the study. In our case, the Institution's Ethics Committee approved the study (CAAE: 68161417.0.0000.5416).
- Detachment of S. mutans after adhesion to the film salivary and glucans treated with selected extracts
- Cultivate the microorganism until the mid-log growth phase, as described above.
- When cultures reached the desired O.D., centrifuge (4000 × g for 20 min), wash with 0.89% NaCl solution and resuspend the pellet with 0.89% NaCl using the same initial volume of the culture medium.
- If using a streptococci, such as S. mutans, sonicate the cultures with a probe to dechain (30 s, 7 W, three times). If using a single cell organism, this step can be skipped.
- Check the O.D. (540 nm) to adjust the concentration to 2 x 106 CFU/mL.
- Adhesion of S. mutans to the salivary pellicle (sHA) and detachment of adhered cells
NOTE: The steps are illustrated in the flow-chart in Figure 5.- Obtain the sHA samples as described above.
- Add an aliquot (in the example, we add 500 μL) of selected treatments (at the test concentration; for example, 0.5 mg/mL) or controls in microtubes containing samples of sHA.
- Incubate the sHA samples with treatments or controls (30 min, 37 °C, 24 rpm); then, wash the beads three times with AB buffer (containing PMSF and NaN3).
- Add the microorganism culture. In the example, we add 500 µL of S. mutans culture (2 x 106 CFU/mL) to each microtube.
- Incubate (1 h, 37 °C, 24 rpm) and then remove unbound cells by washing three times with AB buffer.
- Resuspend each sample with an aliquot (in the example, we add 1000 μL) of AB buffer and sonicate with a probe (30 s, 7 W).
- Use an aliquot of each suspension for a ten-fold serial dilution to determine the number of viable colonies by plating on specific agar plates (48 h, 37 °C, 5% CO2). Next, count the colonies to determine the CFU/mL as described above.
NOTE: The step of sonicate is performed to detach cells adhered to sHA.
- Adhesion of S. mutans to the initial glucan matrix (gsHA) and detachment of adhered cells
NOTE: The steps are illustrated in the flow-chart in Figure 6. The GtfB enzyme was purified from the culture supernatant Streptococcus milleri KSB8 engineered to produce GtfB. The purification was performed with a chromatography column containing hydroxyapatite beads using buffers containing two protease inhibitors (0.1 mM PMSF and 0.02% NaN3)27,28. Then, the enzyme was checked on acrylamide gel (SDS-PAGE) and stained with silver nitrate. Aliquots of the enzyme were stored at -80 °C until use.- Obtain the sHA samples as described above. Next, add an aliquot (in the example, we add 500 μL) of GtfB enzyme to each tube and incubate in a homogenizer (40 min, 37 °C, 24 rpm). Then, wash three times with AB buffer (containing PMSF and NaN3).
- Add an aliquot (e.g., 500 μL) of sucrose substrate (100 mmol of sucrose) containing the treatments (or controls-at the test concentration, e.g., 0.5 mg/mL) to each microtube.
- Incubate the samples in a homogenizer (4 h, 37 °C, 24 rpm). Then, perform three washes with AB buffer (with PMSF and NaN3) to remove the treatments and excess of sucrose not incorporated in the synthesized glucans (samples of gsHA).
- Add an aliquot (in the example, we add 500 μL) of S. mutans inoculum (2 x 106 CFU/mL) to each microtube.
- Incubate in a homogenizer (1 h, 37 °C, 24 rpm) and wash three times with AB buffer (with PMSF and NaN3) to remove unbound cells.
- Resuspend each sample with an aliquot (e.g., 1000 μL) of AB buffer (with PMSF and NaN3) and sonicate with a probe to detach cells adhered to gsHA (30 s, 7 W).
- Use an aliquot of each suspension for a ten-fold serial dilution to determine the number of viable colonies by plating on specific agar plates (48 h, 37 °C, 5% CO2). Next, count the colonies to determine the CFU/mL as described above.
- Salivary pellicle formation
4. Biological Data Analysis
- Bioassays Data
- Input raw data for the bioassays in a spreadsheet. Calculate the log of microbial growth inhibition by each treatment as (ACFU/mL of treatments + 1) x log10. Then, calculate the log percentage of microbial growth inhibition, compared to vehicle control using (Alog10 CFU/mL of treatment/mean Alog10 CFU/mL of vehicle control) x 100%.
- Correct O.D. of planktonic cultures and biomass treated by treatments (CE and CEF treated groups) and by vehicle control (negative control). For correction, subtract the absorbance of treated wells from that obtained in wells containing only culture medium (Ablank) as (Atreated groups medium /Anegative control medium) x 100%.
- After this correction, calculate the percentage of the biomass inhibition, compared to vehicle control as (Atreated biomass/mean Avehicle control) x 100%.
- Submit the raw data generated for statistical analysis of the data using specific software.
NOTE: The interpretation of the effectiveness of a given treatment is determined using breakpoints, such as the IC50/IC90. These values are defined as the minimum concentration of a treatment capable of inhibiting 50% and 90%, respectively, of bacterial growth or biofilm formation24. These parameters can help interpret the data and provide a basis for selecting compounds with better activity13,29.
Results
We provide an example of using a systematic approach to screen the biological activity of plant extracts and fractions to identify potentially active molecules for possible new anti-caries therapies: antimicrobial and antibiofilm activities of Casearia sylvestris extracts from distinct Brazilian biomes against Streptococcus mutans and Candida albicans13.
Background
Complex interactions between specific oral microorganism...
Discussion
The main challenges related to the work with natural crude extracts comprise their complex composition and the inadequacies of classic bio-guided isolation studies. Although this process is slow, it is effective and has led to major findings in NP research. To rationalize, prioritization-driven studies are needed to rationalize. Thus, the use of modern chemical profiling approaches for the analysis of CE and dereplication before isolation are important to characterize the studied material and especially useful to avoid r...
Disclosures
No conflicts of interest declared.
Acknowledgements
We express our gratitude to Núcleo de Bioensaios, Biossíntese e Ecofisiologia de Produtos Naturais (NuBBE) of the Chemistry Institute of UNESP, Araraquara/SP for providing the laboratories for preparing plant material. We also thank the Applied Microbiology Laboratory of the Department of Dental Materials and Prosthodontics, UNESP, Araraquara/SP. This research was supported by a research grant from the São Paulo Research Foundation (FAPESP #2013/07600–3 to AJC) and scholarships plus overhead funds (FAPESP #2017/07408–6 and FAPESP #2019/23175-7 to SMR; #2011/21440–3 and #2012/21921–4 to PCPB). The National Council for Scientific and Technological Development in association with FAPESP provided additional support (INCT CNPq #465637/2014–0 and FAPESP #2014/50926–0 to AJC).
Materials
| Name | Company | Catalog Number | Comments |
| 96-well microplates | Kasvi | Flat bottom | |
| Activated carbon | LABSYNTH | Clean up and/or fractionation step | |
| Analytical mill | Ika LabortechniK | Model A11 Basic | |
| Blood agar plates | Laborclin | ||
| Chromatographic column C18 | Phenomenex Kinetex | 150 × 2.1 mm, 2.6 µm, 100Â | |
| Dimethyl sulfoxide | Sigma-Aldrich | Vehicle solution | |
| ELISA plate reader | Biochrom Ez | ||
| Ethanol | J. T. Baker | For extraction and fractionation steps, and mobile phase composition | |
| Ethanol | Sigma-Aldrich | Vehicle solution | |
| Ethyl acetate | J. T. Baker | Fractionation step | |
| GraphPad Software | La Jolla | GraphPad Prism7 | |
| Hexane | J. T. Baker | Fractionation step | |
| Incubator | Thermo Scientific | ||
| Isopropanol | J. T. Baker | For extraction step | |
| Lyophilizer (a freeze dryer) | Savant | Modulyo | |
| Nylon Millipore | LAC | 0.22 µm x 13 mm | |
| Orbital shaker | Quimis | Model G816 M20 | |
| Polyamide solid phase extraction cartridge | Macherey-Nagel | Clean up and/or fractionation step | |
| Silica gel | Merck | 40–63 μm, 60 Â | |
| Sodium Chloride (NaCl) | Synth | 0,89% in water | |
| Solid phase extraction cartridges (SPE) | Macherey-Nagel | Clean up and/or fractionation step | |
| Tryptone | Difco | ||
| UHPLC-DAD | Dionex | Ultimate 3000 RS | |
| Ultrasonic bath | UNIQUE | Model USC 2800 | |
| Yeast extract | Difco |
References
- Newman, D. J., Cragg, G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. Journal of Natural Products. 83 (3), 770-803 (2020).
- Wolfender, J. L., Litaudon, M., Touboul, D., Queiroz, E. F. Innovative omics-based approaches for prioritisation and targeted isolation of natural products – new strategies for drug discovery. Natural Product Report. 36 (6), 855-868 (2019).
- Michel, T., Halabalaki, M., Skaltsounis, A. New Concepts, Experimental Approaches, and Dereplication Strategies for the Discovery of Novel Phytoestrogens from Natural Sources. Planta Medica. 79 (7), 514-532 (2013).
- Jeon, J. G., Rosalen, P. L., Falsetta, M. L., Koo, H. Natural products in caries research: current (limited) knowledge, challenges and future perspective. Caries Research. 45 (3), 243-263 (2011).
- Tonetti, M. S., Jepsen, S., Jin, L., Otomo-Corgel, J. Impact of the global burden of periodontal diseases on health, nutrition and wellbeing of mankind: A call for global action. Journal of Clinical Periodontology. 44 (5), 456-462 (2017).
- Peres, M. A., et al. Oral diseases: a global public health challenge. Lancet. 394 (10194), 249-260 (2019).
- Bowen, W. H., Burne, R. A., Wu, H., Koo, H. Oral biofilms: pathogens, matrix, and polymicrobial interactions in microenvironments. Trends Microbiology. 26 (3), 229-242 (2018).
- Paes Leme, A. F., Koo, H., Bellato, C. M., Bedi, G., Cury, J. A. The role of sucrose in cariogenic dental biofilm formation--new insight. Journal of Dental Research. 85 (10), 878-887 (2006).
- Koo, H., Falsetta, M. L., Klein, M. I. The exopolysaccharide matrix: a virulence determinant of cariogenic biofilm. Journal of Dental Research. 92 (12), 1065-1073 (2013).
- Cury, J. A., de Oliveira, B. H., dos Santos, A. P., Tenuta, L. M. Are dental fluoride releasing materials clinically effective on caries control. Dental Materials. 32 (3), 323-333 (2016).
- Mattos-Graner, R. O., Klein, M. I., Smith, D. J. Lessons Learned from Clinical Studies: Roles of Mutans Streptococci in the Pathogenesis of Dental Caries. Current Oral Health Reports. 1, 70-78 (2014).
- Rocha, G. R., Florez Salamanca, E. J., de Barros, A. L., Lobo, C. I. V., Klein, M. I. Effect of tt-farnesol and myricetin on in vitro biofilm formed by Streptococcus mutans and Candida albicans. BMC Complementary and Alternative Medicine. 18 (1), 61 (2018).
- Ribeiro, S. M., et al. Antimicrobial and antibiofilm activities of Casearia sylvestris extracts from distinct Brazilian Biomes against Streptococcus mutans and Candida albicans. BMC Complementary and Alternative Medicine. 19 (1), 308 (2019).
- Pilon, A. C., et al. Metabolômica de plantas: métodos e desafios. Quimica Nova. 43 (3), 329-354 (2020).
- Wolfender, J. L., Nuzillard, J. M., Hooft, J. J. J., Renault, J. H., Bertrand, S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography-High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Analytical Chemistry. 91 (1), 704-742 (2019).
- Allard, P. M., et al. Pharmacognosy in the digital era: shifting to contextualized metabolomics. Current opinion in biotechnology. 54, 57-64 (2018).
- Hubert, J., Nuzillard, J., Renault, J. Dereplication strategies in natural product research: How many tools and methodologies behind the same concept. Phytochemistry Reviews. 16, 55-95 (2017).
- Bueno, P. C. P., Pereira, F. M. V., Torres, R. B., Cavalheiro, A. J. Development of a comprehensive method for analysing clerodane-type diterpenes and phenolic compounds from Casearia sylvestris Swartz (Salicaceae) based on ultra-high performance liquid chromatography combined with chemometric tools. Journal of separation science. 38 (10), 1649-1656 (2015).
- Bueno, P. C. P., Lopes, N. P. Metabolomics to Characterize Adaptive and Signaling Responses in Legume Crops under Abiotic Stresses. American Chemical Society omega. 5 (4), 1752-1763 (2020).
- Blaženović, I., Kind, T., Ji, J., Fiehn, O. Software tools and approaches for compound identification of LC-MS/MS data in metabolomics. Metabolites. 8 (2), 31 (2018).
- Eloff, J. N. Quantifying the bioactivity of plant extracts during screening and bioassay-guided fractionation. Phytomedicine: International Journal Of Phytotherapy And Phytopharmacology. 11 (4), 370-371 (2004).
- Rios, J. L., Recio, M. C. Medicinal plants and antimicrobial activity. Journal of Ethnopharmacology. 100 (1-2), 80-84 (2005).
- Eloff, J. N. A sensitive and quick microplate method to determine the minimal inhibitory concentration of plant extracts for bacteria. Planta Medica. 64, 711-714 (1998).
- Eloff, J. N. Avoiding pitfalls in determining antimicrobial activity of plant extracts and publishing the results. BMC Complementary and Alternative Medicine. 19 (1), 106 (2019).
- Klein, M. I., Xiao, J., Heydorn, A., Koo, H. An analytical tool-box for comprehensive biochemical, structural and transcriptome evaluation of oral biofilms mediated by mutans streptococci. Journal of Visualized Experiments. (47), e2512 (2011).
- Lemos, J. A., Abranches, J., Koo, H., Marquis, R. E., Burne, R. A. Protocols to study the physiology of oral biofilms. Methods in molecular biology. 666, 87-102 (2010).
- Venkitaraman, A. R., Vacca-Smith, A. M., Kopec, L. K., Bowen, W. H. Characterization of glucosyltransferase B, GtfC, and GtfD in solution and on the surface of hydroxyapatite. Journal of Dental Research. 74, 1695-1701 (1995).
- Vacca-Smith, A. M., Venkitaraman, A. R., Quivey, R. G., Bowen, W. H. Interactions of streptococcal glucosyltransferases with alpha-amylase and starch on the surface of saliva-coated hydroxyapatite. Archives of Oral Biology. 41, 291-298 (1996).
- Van Dijck, P., et al. Methodologies for in vitro and in vivo evaluation of efficacy of antifungal and antibiofilm agents and surface coatings against fungal biofilms. Microbial Cell. 5 (7), 300-326 (2018).
- Marsh, P. D. Are dental diseases examples of ecological catastrophes. Microbiology. 149 (2), 279-294 (2003).
- Bowen, W. H., Koo, H. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Research. 45 (1), 69-86 (2011).
- Lobo, C. I. V., et al. Dual-species biofilms of Streptococcus mutans and Candida albicans exhibit more biomass and are mutually beneficial compared with single-species biofilms. Journal of Oral Microbioly. 11 (1), 1581520 (2019).
- Kim, D., et al. Candida albicans stimulates Streptococcus mutans microcolony development via crosskingdom biofilm-derived metabolites. Scientific reports. 7, 41332 (2017).
- Ferreira, P. M. Folk uses and pharmacological properties of Casearia sylvestris: a medicinal review. Anais da Academia Brasileira de Ciencias. 83 (4), 1373-1384 (2011).
- Xia, L., Guo, Q., Tu, P., Chai, X. The genus Casearia: a phytochemical and pharmacological overview. Phytochemistry Reviews. 14, 99-135 (2015).
- Ferreira, P. M. P., et al. Toxicological findings about an anticancer fraction with casearins described by traditional and alternative techniques as support to the Brazilian Unified Health System (SUS). Journal of Ethnopharmacol. 15, 241 (2019).
- Koo, H., Xiao, J., Klein, M. I., Jeon, J. G. Exopolysaccharides produced by Streptococcus mutans glucosyltransferases modulate the establishment of microcolonies within multispecies biofilms. Journal of Bacteriology. 192 (12), 3024-3032 (2010).
- Maske, T. T., van de Sande, F. H., Arthur, R. A., Huysmans, M. -. C. D. N. J. M., Cenci, M. S. In vitro biofilm models to study dental caries: a systematic review. Biofouling. 33 (8), 661-675 (2017).
- Fu, Y., Luo, J., Qin, J., Yang, M. Screening techniques for the identification of bioactive compounds in natural products. Journal of Pharmaceutical and Biomedical Analysis. 168, 189-200 (2019).
- Sarker, S. D., Nahar, L. An introduction to natural products isolation. Methods in molecular biology. 864, 1-25 (2012).
- Clinical Laboratory Standards Institute (CLSI). Performance standards for antimicrobial susceptibility testing; twenty-fifth informational supplement. Clinical Laboratory Standards Institute (CLSI). , (2015).
- Saputo, S., Faustoferri, R. C., Quivey, R. G. A drug repositioning approach reveals that Streptococcus mutans is susceptible to a diverse range of established antimicrobials and nonantibiotics. Antimicrobial Agents and Chemotherapy. 62 (1), 01674 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved