Site-Directed φC31-Mediated Integration and Cassette Exchange in Anopheles Vectors of Malaria
In This Article
Summary
The protocol describes how to achieve site-directed modifications in the genome of Anopheles malaria mosquitoes using the φC31 system. Modifications described include both the integration and the exchange of transgenic cassettes in the genome of attP-bearing docking lines.
Abstract
Functional genomic analysis and related strategies for genetic control of malaria rely on validated and reproducible methods to accurately modify the genome of Anopheles mosquitoes. Amongst these methods, the φC31 system allows precise and stable site-directed integration of transgenes, or the substitution of integrated transgenic cassettes via recombinase-mediated cassette exchange (RMCE). This method relies on the action of the Streptomyces φC31 bacteriophage integrase to catalyze recombination between two specific attachment sites designated attP (derived from the phage) and attB (derived from the host bacterium). The system uses one or two attP sites that have been integrated previously into the mosquito genome and attB site(s) in the donor template DNA. Here we illustrate how to stably modify the genome of attP-bearing Anopheles docking lines using two plasmids: an attB-tagged donor carrying the integration or exchange template and a helper plasmid encoding the φC31 integrase. We report two representative results of φC31-mediated site-directed modification: the single integration of a transgenic cassette in An. stephensi and RMCE in An. gambiae mosquitoes. φC31-mediated genome manipulation offers the advantage of reproducible transgene expression from validated, fitness neutral genomic sites, allowing comparative qualitative and quantitative analyses of phenotypes. The site-directed nature of the integration also substantially simplifies the validation of the single insertion site and the mating scheme to obtain a stable transgenic line. These and other characteristics make the φC31 system an essential component of the genetic toolkit for the transgenic manipulation of malaria mosquitoes and other insect vectors.
Introduction
The ability to modify the genome of mosquito vectors of diseases reliably and reproducibly has bolstered in vivo functional validation of genes and opened the doors to realizable genetic vector control strategies, such as those targeting Anopheles mosquitoes that transmit malaria1.
Early mosquito genome editing relied solely on transposable element (TE)-mediated transformation, with piggyBac being the most commonly used transposon in Anopheles2,3,4. However, the random nature of TE integration can lead to undesirable modifications such as gene knockouts (insertional mutagenesis) and significant position effects on transgene expression5,6,7,8. Multiple insertions are also a common occurrence when using piggyBac5,9, which makes the validation and the isolation of transgenic lines with single insertions laborious. Other drawbacks include their potential remobilization, as observed in the germline of Anopheles stephensi when providing a source of piggyBac transposase10,11,12, and their limited size of DNA cargo (10-15 kb in length) with transformation efficiency declining with increasing size of the donor plasmid13,14.
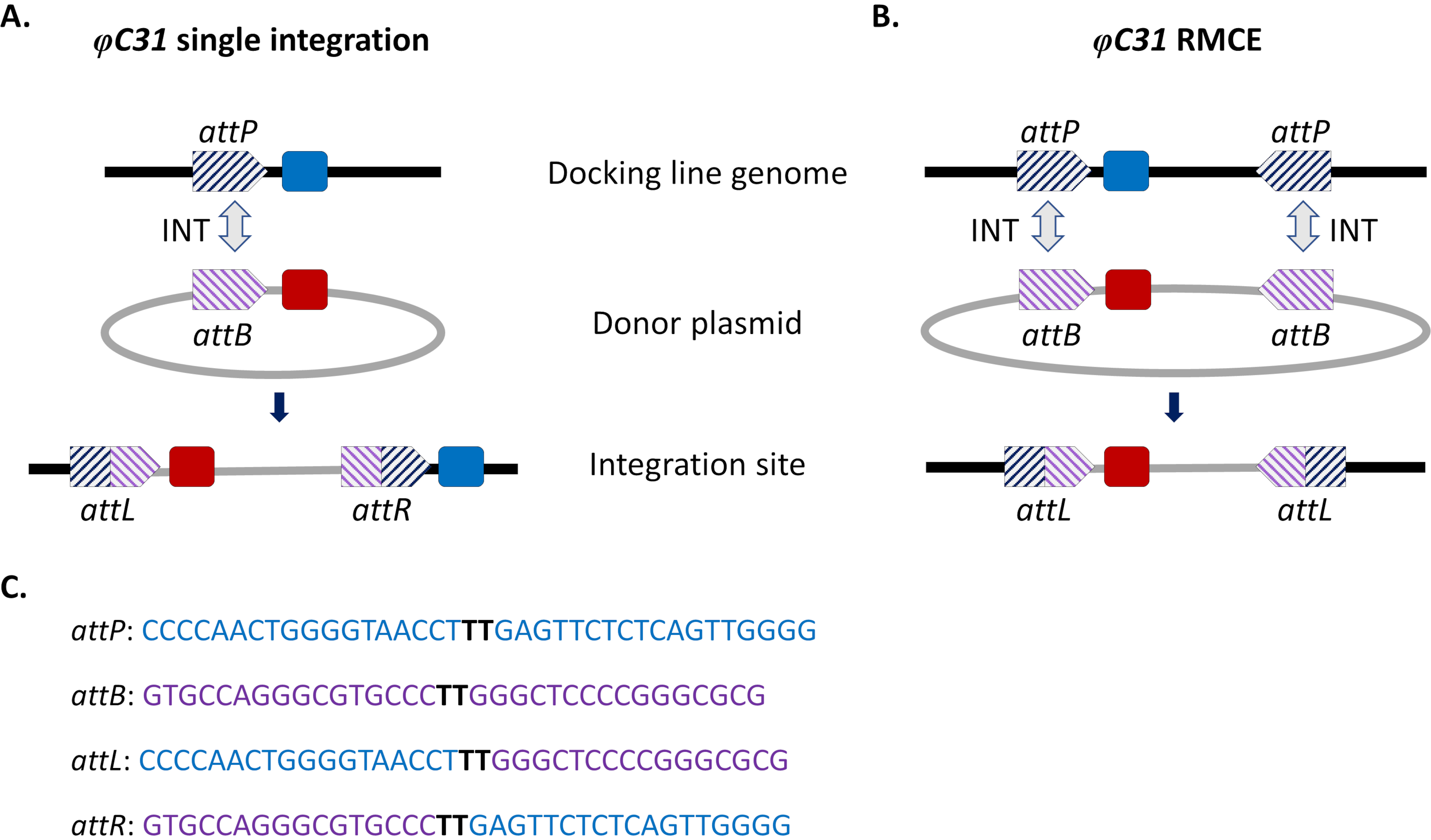
Site-directed integration approaches were introduced to circumvent these issues. The most common site-directed genome modification in mosquitoes is that mediated by the φC31 system (Figure 1a). This is driven by a viral integrase that catalyzes the recombination between two heterospecific attachment (att) sites occurring naturally in the genome of the bacteriophage φC31 (attP) and in the Streptomyces bacterium host (attB)15. Recombination of the two sites is unidirectional and results in the formation of hybrid sites (attL and attR). The recombination of such hybrid sites (leading to DNA excision) would require not only the presence of an active viral integrase but also another phage-encoded recombination factor16,17. A stable integration site is thus generated that relieves the issue of potential undesired remobilization15. Moreover, the system allows the integration of large cargoes (e.g., integration of >100 kb constructs was reported in D. melanogaster18), significantly increasing carrying capacities. Integration occurs in a single predefined genomic locus which greatly simplifies the validation of insertion and the mating scheme to obtain a stable transgenic line. Finally, the site-directed nature of the integration allows normalization of expression as alternative transgenes are located in the same locus and therefore are regulated within the same neighboring genomic context. Indeed, one of the main applications of the technique is the direct comparison of phenotypes conferred by different transgenes following insertion into an identical locus.
Achieving φC31-mediated integration involves two phases: phase I is the creation of transgenic docking lines carrying attP site(s), and phase II is the site-directed integration of an attB-flanked cargo in the genome of the docking line19. The creation of phase I docking lines has relied on the TE-mediated random integration of attP-tagged constructs and thus involved an initial laborious process (including southern blot and inverse PCR analyses on single-female progeny) to isolate and validate transgenic lines carrying a single integration event in unique, transcriptionally active, and fitness neutral genomic locations. Nevertheless, several docking lines for φC31-mediated single integration have been developed and validated in An. gambiae19,20,21,22 and in An. stephensi23,24,25 (Table 1). Each of these lines varies in terms of the genomic location of the docking site and the strain-specific genetic background and from them a great variety of new transgenic lines can be created. The complex validation of TE-mediated integrations for producing docking lines can now be circumvented by the CRISPR/Cas9 technology26; however this relies on the a priori knowledge of neutral loci to be targeted and their surrounding sequences.
φC31-mediated integration has been applied extensively to insect genome editing from the model organism D. melanogaster27, to the mosquitoes Aedes aegypti13,28, Ae. albopictus29, An. gambiae19, and An. stephensi24, as well as other insects including Ceratitis capitata30 and Bombyx mori31.
A limitation of φC31-mediated integration, especially in view of potential field releases for vector control, is the integration in the mosquito genome of the entire attB-bearing donor plasmid, including undesirable sequences such as antibiotic-resistance gene markers and plasmid backbone components of bacterial origin. To address this, a modification of the standard system, recombinase-mediated cassette exchange (RMCE), was implemented that allows the precise replacement of a previously integrated transgenic cassette with a new donor DNA (Figure 1b). This is achieved by using two inverted att sites flanking the donor and recipient cassettes at each end, which drives two independent recombination events to take place simultaneously resulting in cassette exchange without integration of the plasmid backbone. This improved design circumvents the integration of undesired sequences and expands the application of φC31 systems to include for example the integration of unmarked DNA cargos by screening for the loss of a previously integrated fluorescent marker32.
RMCE was achieved first with D. melanogaster32 and later applied successfully to non-model insects including An. gambiae9,26,33, Ae. aegypti34, Plutella xylostella34, and B. mori35. Several docking lines for RMCE have been developed and validated in An. gambiae5,9,26 (Table 1). To our knowledge, RMCE is yet to be explored in other Anopheles vectors species.
To date, the φC31 system has been used widely in Anopheles mosquitoes to introduce and study a variety of molecules including antimalaria effectors19,24,36, components of the GAL4/UAS system to overexpress and knockdown genes for insecticide resistance studies9,33, regulatory elements, reporter genes5,21,37, and gene-drive elements26,38.
This protocol describes how to perform 1) site-directed integration of an attB-flanked cargo and 2) RMCE of a construct flanked by inverted attB sites into the genome of Anopheles docking lines. This is achieved by using two plasmids: a donor attB-tagged plasmid carrying the transgene of interest, and a helper plasmid expressing the φC31 integrase. The major malaria vectors An. gambiae and An. stephensi are used as specific examples, however these protocols are applicable to other Anopheles species.

Figure 1. Site-directed genome modifications, single integration and recombinase-mediated cassette exchange (RMCE) , using the φC31 system. The φC31 integrase (INT, grey double arrow) catalyzes the recombination between the attB site(s) (purple striped) present in a donor plasmid and the attP site(s) (blue striped) present in a receiving docking line, which results in the formation of hybrid sites attL and attR. A) Integration is achieved when single attB and attP sites recombine and results in the presence of two integrated markers (blue and red). B) RMCE occurs when two attB/P sites recombine simultaneously and results in the replacement of the cassette between the att sites of the docking line (blue marker) with that carried by the donor plasmid (red marker). C) Partial nucleotide sequences of attP (blue) and attB (purple) and the hybrid sites attL/R. Recombination occurs between the 'TT' core sequences highlighted in bold black. Please click here to view a larger version of this figure.
{kind=link}
Protocol
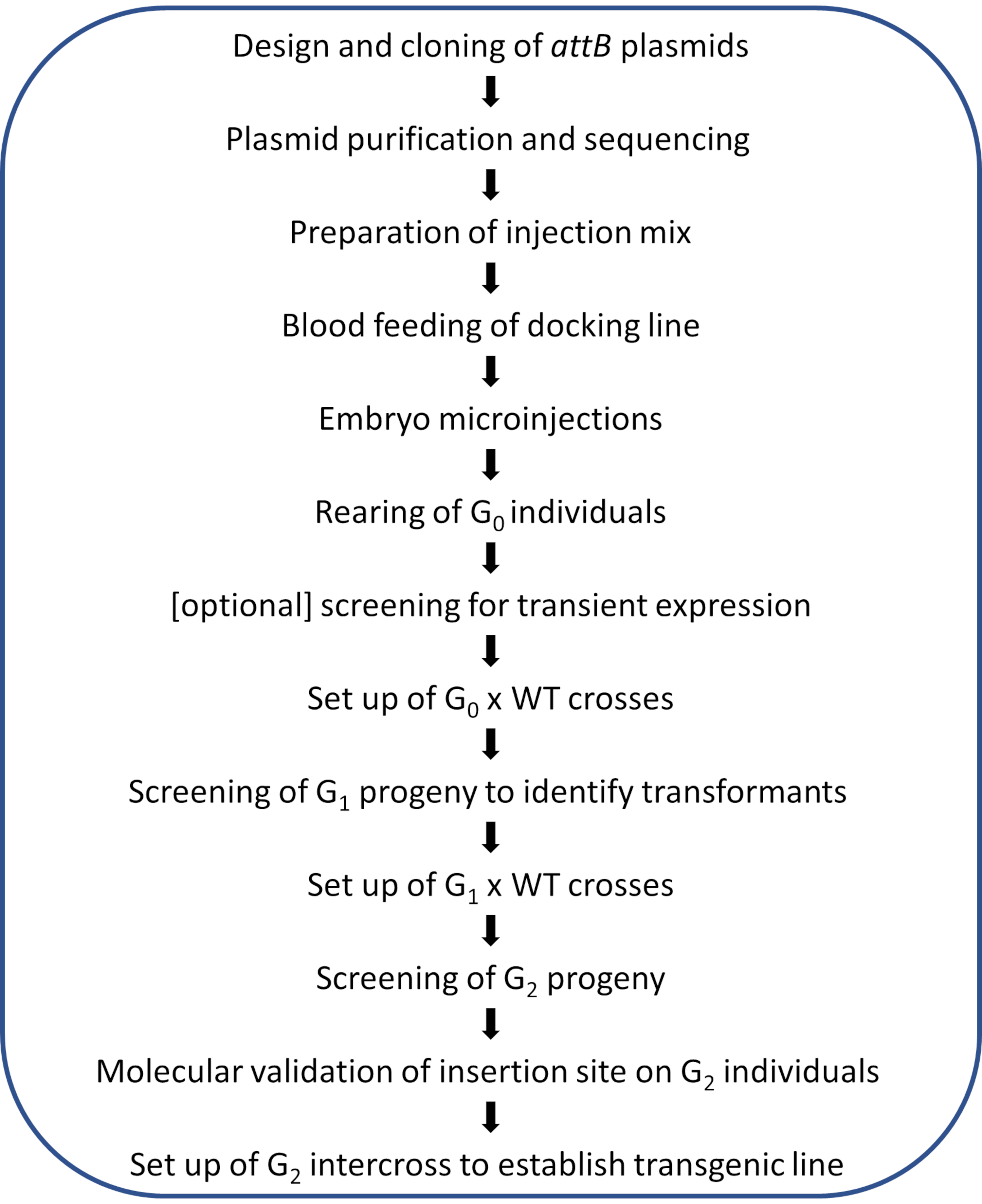
NOTE: A schematic workflow of the illustrated protocol is shown in Figure 2.
1. Design of φC31 attB -tagged plasmids (Figure 3)
- Create attB donor plasmids carrying the following essential components
- Dominant fluorescent marker
- Choose a promoter to drive the expression of the fluorescent marker.
NOTE: For Anopheles transgenesis, fluorescent markers are usually under the regulation of the 3xP3 promoter39, which drives expression in the eyes and nerve cord. Alternatively, the PUBc promoter5 can be used when expression in multiple tissues is desired. Donor plasmids and docking lines used as examples in this protocol are marked using the 3xP3 promoter. - Choose a fluorescent protein (FP) that is compatible with that of the receiving docking line so that they are readily distinguishable.
NOTE: Do not use the same marker that is already present in the docking line and avoid the simultaneous use of GFP (green)/YFP (yellow) and GFP (green)/CFP (cyan) as they are very difficult to reliably differentiate. Donor plasmids used as examples in this protocol are marked with either DsRed or YFP as they are to be integrated in a docking line marked with CFP.
- Choose a promoter to drive the expression of the fluorescent marker.
- attB recombination site(s)
- Use a single attB site for integration of a transgenic cassette (single-attB design) (Figure 3A).
- Use two inverted attB sites for RMCE (double-attB design) where the sites lay inverted in respect to one another and enclose the donor DNA template (Figure 3B).
NOTE: The orientation of the attB site(s) must be compatible with that of the attP site(s) present in the docking line.
- Desired transgene cargo
- Use any other desired features to be integrated in the mosquito genome based on the specific purpose of the experiment. Here, we describe the integration of an antimalarial effector molecule into the genome of An. stephensi and the integration of the components of the GAL4/UAS system into An. gambiae mosquitoes.
- Plasmid backbone components
- Include, amongst other essential components for plasmid replication in bacteria, a marker for plasmid selection in vitro (i.e., an antibiotic resistance gene).
NOTE: The plasmid backbone will be integrated in the mosquito genome in the single-attB design for integration (Figure 3A), while it will not be inserted in the double-attB design for RMCE (Figure 3B).
- Include, amongst other essential components for plasmid replication in bacteria, a marker for plasmid selection in vitro (i.e., an antibiotic resistance gene).
- Dominant fluorescent marker
2. Preparation of plasmids for the microinjection mix
NOTE: The protocol illustrated here involves the use of two plasmids: an attB-tagged donor plasmid carrying the transgene of interest, and a helper plasmid that expresses the φC31 integrase under the regulation of the Drosophila Hsp70 promoter40.
- Purify donor and helper plasmids using an endotoxin-free plasmid purification kit.
NOTE: Sequence the final plasmid preparation used for injection to verify the integrity of all components. - Combine appropriate amounts of the two plasmids to obtain a mix with a final concentration of 350 ng/µL of the donor plasmid and 150 ng/µL of the helper plasmid when resuspended in injection buffer.
NOTE: When calculating the necessary volume of mix, consider that 10-15 µL are sufficient for each day of planned injections and DNA can be prepared in advance and stored at -20 °C. Integrase helper plasmid concentrations of 60-500 ng/µL and donor plasmid concentrations of 85-200 ng/µL have also been reported21,22,26,41. - Precipitate the DNA by adding 0.1 volumes of 3 M sodium acetate (pH 5.2) and 2.5 volumes of ice-cold 100% EtOH and vortex. A white precipitate should be immediately visible. Having highly concentrated initial plasmid preparations (i.e., ~1 µg/µL) improves precipitation efficiency.
NOTE: Stopping point - The precipitate can be stored at -20 °C overnight. - Centrifuge at 15,000 x g for 20 min at 4 °C, discard the supernatant, and wash the pellet with 1 mL of ice-cold 70% EtOH.
- Wash the pellet with 1 mL of ice-cold 70% EtOH and centrifuge at 15,000 x g for 5 min at room temperature.
- Discard the supernatant without disturbing the pellet and air dry.
- Resuspend the pellet in 1x injection buffer (0.1 mM Na3PO4, 5 mM KCl, pH 7.2, 0.22 µm filter sterilized) to reach a total final concentration of 500 ng/µL.
NOTE: Assume that some DNA will be lost during the precipitation process; therefore, add a smaller volume of injection buffer first, check the concentration on a spectrophotometer (e.g., Nanodrop), and then add an appropriate remaining volume to reach 500 ng/µL. - Ensure that the DNA is thoroughly resuspended, prepare aliquots of 10-15 µL each and store them at -20 °C.
- On the day of injection, thaw one aliquot and centrifuge at 15,000 x g for 5 min to remove any particulate residues.
NOTE: An alternative method for particulate removal is to filter the solution through a 0.22 µm filter. Avoid the presence of particulate residues in the injection mix as they lead to needle blockage during embryo microinjection.
3. Microinjection of embryos from an Anopheles docking line
- Blood feed 4-7-day-old mosquitoes from the desired docking line 72 h prior to microinjection (i.e., for injection on Monday and Tuesday feed females on the previous Friday; for injection on Thursday and Friday feed females on Monday of the same week).
- Blood feed wild-type (WT) mosquitoes (i.e., mosquitoes with the same genomic background of the docking line) on the same day; these will be needed for outcrossing.
NOTE: The size and quality of the blood meal affect egg quality, so it is recommended to always use fresh blood (i.e., blood drawn within the previous 7 days). Arm feeding or feeding on mice may increase the quality and quantity of eggs, however these methods are not encouraged. Specific approved protocols will be necessary for human and animal use. - Perform embryo microinjections
- Perform An. gambiae embryo microinjections in 25 mM NaCl42 by targeting the posterior pole of the embryo at a 45-degree angle. For a detailed protocol for embryo collection, alignment, and microinjection refer to Pondeville et al.43 and Lobo et al.44.
- Perform An. stephensi embryo microinjections in halocarbon oil 700:27 (2:1) by targeting the posterior pole of the embryo at a 30-degree angle. A detailed protocol for embryo collection, alignment, and microinjection can be found in Terenius et al.45 and Lobo et al.44.
- Transfer eggs immediately after injection in a Petri dish filled with sterile distilled water (pH 7.2) and return them to insectary conditions.
- Upon hatching, transfer G0 larvae into a tray with salted distilled water (0.1% tonic salt) daily and rear to pupae.
- Record hatching rate (i.e., number of larvae hatched/number of embryos injected).
NOTE: Embryo movement aids hatching, so gentle swirling is desirable. Hatching should start ~48 h after injection. Since injection may cause a slight developmental delay it is advisable to keep monitoring for late-hatching larvae for 3-4 days.
4. Crossing and screening of transformed individuals
- [OPTIONAL STEP] Screen G0 (injected) 1st or 2nd instar larvae (L1-L2) for transient expression of the fluorescent marker.
- Use a fine-tip glass pipette to transfer G0 L1-L2 larvae to a microscope slides with wells. Place one larva in each well.
- Use a fluorescence stereoscope with the appropriate filter to screen for the presence of transient expression of the fluorescent marker.
NOTE: The pattern of transient expression is dictated by the promoter used. When using the 3xP3 promoter, transient expression of the fluorescent marker is visible in the anal papillae (see Figure 6 in Pondeville et al.43) - Rear G0 positive individuals separately.
- Sort G0 pupae by sex under a stereoscope52.
- Let males emerge in separate cages in groups of 3-5 (founder families) and add a 10-fold excess of age-matched WT females.
NOTE: Since males mate multiple times, it is important to provide an excess of WT females to maximize the mating chances of each male. - Let females emerge in separate cages in groups of 10-15 (founder families) and add an equal number of age-matched WT males.
NOTE: If there is limited space in the insectary, females can emerge all together in a single cage. The female to male ratio can be as low as 1 male to 3 females. - Allow adults to mate for 4-5 days and provide females with a blood meal.
NOTE: Blood feed and collect eggs from G0 females multiple times to maximize the chances of getting transformants from multiple gonotrophic cycles. - Blood feed WT individuals at the same time for outcrossing.
- Collect eggs and rear emerging next generation G1s.
- Screen G1 L3-L4 larvae for appropriate fluorescence to identify transformants.
- Collect larvae in a Petri dish lined with filter paper or on a microscope slide and screen using a fluorescent stereoscope with appropriate filters for the presence of the marker introduced with the attB-tagged cargo.
NOTE: Fluorescence driven by the 3xP3 promoter is visible in all postembryonic stages and the screening may be performed on younger larvae, however these are more fragile and must be handled relatively carefully. Pupae can also be screened.- For single-attB designs for integration screen for the presence of the new and pre-existing marker; they should both be present since the new cassette is inserted next to the original one (Figure 3A, Figure 4).
NOTE: Screening exception for single attB designs: When using marker-less docking lines22, screen for the presence of the new marker only. When using docking lines where integration results in the inactivation of the pre-existing marker21, screen for the presence of the new marker and the loss of the pre-existing one. - For double-attB designs for RMCE, screen for the presence of the new marker and the loss of the pre-existing one, only the newly introduced marker should be present since the new cassette replaces the original one (Figure 3B, Figure 5).
NOTE: Occasional integration events can be recovered in RMCE experiments where only a single attP recombined and thus both markers will be present. The screening of G1 individuals can be carried out also at the pupa stage following the same procedure52.
- For single-attB designs for integration screen for the presence of the new and pre-existing marker; they should both be present since the new cassette is inserted next to the original one (Figure 3A, Figure 4).
- Collect larvae in a Petri dish lined with filter paper or on a microscope slide and screen using a fluorescent stereoscope with appropriate filters for the presence of the marker introduced with the attB-tagged cargo.
- Transfer transformed G1 individuals into a larval tray and rear to pupae. Discard non-fluorescent individuals and individuals with an unexpected marker expression pattern.
- Sort transformed G1 pupae by sex and cross them en masse with opposite-sex age-matched WT individuals.
- Allow adults to mate for 4-5 days, provide a blood meal, collect the eggs, and rear the next generation G2 progeny.
- For single integration experiments, collect eggs directly from the en masse cross as the integration site is identical in all individuals.
- For RMCE experiments, collect eggs from single females and maintain progeny separate until molecular assessment is complete due to the potential presence of two alternative cassette orientations (Figure 3B).
- Screen the G2 progeny (at either the larva or pupa stage) for the presence of the fluorescent marker (50% of the individuals are expected to be positive), discard non-fluorescent progeny.
- Set aside a subset of G2 positive individuals for molecular analysis, rear the rest to adulthood.
NOTE: If all G2 individuals must be kept alive, molecular analysis can be conducted on single adult's legs46 or pupal case DNA extractions (L. Grigoraki personal communication). Alternatively, molecular analysis can be performed after all the G2 individuals have oviposited and eggs have hatched. - Allow adult males and females to intercross in the same cage to establish the new transgenic line.
NOTE: For RMCE experiments, adult intercross must occur between siblings deriving from a single female until orientation of insertion is determined via molecular analysis.
5. Molecular validation of the insertion site by DNA amplification (PCR)
- Prepare a map of the predicted insertion site in the genome of the docking line after transformation.
- Single integration: Ensure that the predicted insertion site carries the original docking construct plus the whole sequence of the donor plasmid between the two hybrid sites attL and attR (Figure 3A).
- RMCE: Ensure that the predicted insertion site is identical to that of the docking line where hybrid inverted attL sites replace the original inverted attP sites and the exchange template replaces the cassette originally present between them (Figure 3B).
- Design oligonucleotide primers to amplify the insertional junction at either side of the integration locus.
- Single integration: Design oligonucleotide primer pairs that span across the attR and/or attL sites. One primer must bind to the previously integrated docking construct and the other to the newly integrated transgene (Figure 3A).
- RMCE: Cassette replacement can occur in two different orientations with respect to the chromosome (designated A and B). Design alternative combinations of 4 oligonucleotide primers to give a discrete product in only one of the orientations, with one pair being diagnostic for orientation A, and the other for orientation B (Figure 3B, Figure 6).
- Extract genomic DNA from G2 positive individuals and perform the diagnostic PCR and gel electrophoresis to visualize the presence of expected diagnostic amplicons from the predicted integration site maps.
NOTE: DNA may alternatively be extracted from single adult's legs46 or pupal cases (L. Grigoraki personal communication). - Sequence PCR products to confirm expected sequences.

Figure 2. Workflow diagram for site-directed φC31 genome modification in Anopheles mosquitoes. Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Molecular basis of φC31-mediated single integration (A) and RMCE (B). A) Schematic maps of the genomic insertion in an An. stephensi docking line (80.9, Table 1) carrying a single attP site and marked with CFP (top), a single-attB design donor plasmid marked with DsRed (middle), and the expected insertion site resulting after successful integration (bottom). B) Schematic maps of the genomic insertion in an An. gambiae docking line (A11, Table 1) carrying two inverted attP sites and marked with CFP (top), a double-attB design donor plasmid marked with YFP (middle), and the expected insertion site resulting after successful RMCE (bottom). Wavy line: mosquito genome; Striped arrows: piggyBac transposon arms; 3xP3: promoter of the fluorescent marker; SV40: viral terminator; Ori: origin of replication; AmpR: ampicillin resistance gene. Crossing lines represent the site(s) of recombination between attP and attB sites. Numbered black arrows represent primer binding sites for the molecular validation of the insertion locus (step 5 of the protocol). Fully annotated single and double attB-tagged plasmids are available from the authors upon request. Please click here to view a larger version of this figure.
{kind=link}
Representative Results
The protocol illustrated here enables to generate a stable Anopheles transgenic line in ~10 weeks (assuming a 21-day mosquito life cycle).
Post-injection larval hatching rates in An. gambiae are expected to be generally lower than An. stephensi, however hatching rates between 10-50% have been reported9,20,24,26,33,43,47. Given appropriate injection technique, hatching rates of ≥20% are generally sufficient to yield transformants. DNA uptake by the embryos can be assessed by screening young larvae for transient expression of the fluorescent marker. In successful RMCE experiments in An. gambiae using the 3xP3 promoter up to 50% of the surviving G0 larvae showed episomal expression of the marker in the anal papillae48.
Generalized estimates of transformation efficiency are difficult to evaluate among laboratories and even among experiments as transformation depends on a complex interplay of variables including purity, concentration, size, and potential toxicity of the injected DNA, quality of eggs, pre- and post-injection handling of eggs, mosquito rearing, and most importantly the experience of the operator. Transformation rates up to 7% have been obtained for RMCE in An. gambiae (calculated as the number of independent transformation events in the total G0 individuals)9,26,33, and up to 2.2% transformation rate for integration in An. stephensi. We suggest injecting at least 500 embryos, which should lead to the hatching of at least 100 G0 larvae and to 2-7 G0 adult founders from which stably transformed progeny can be obtained. If screening for transient expression in G0 larvae, up to 40 positive larvae can be expected.
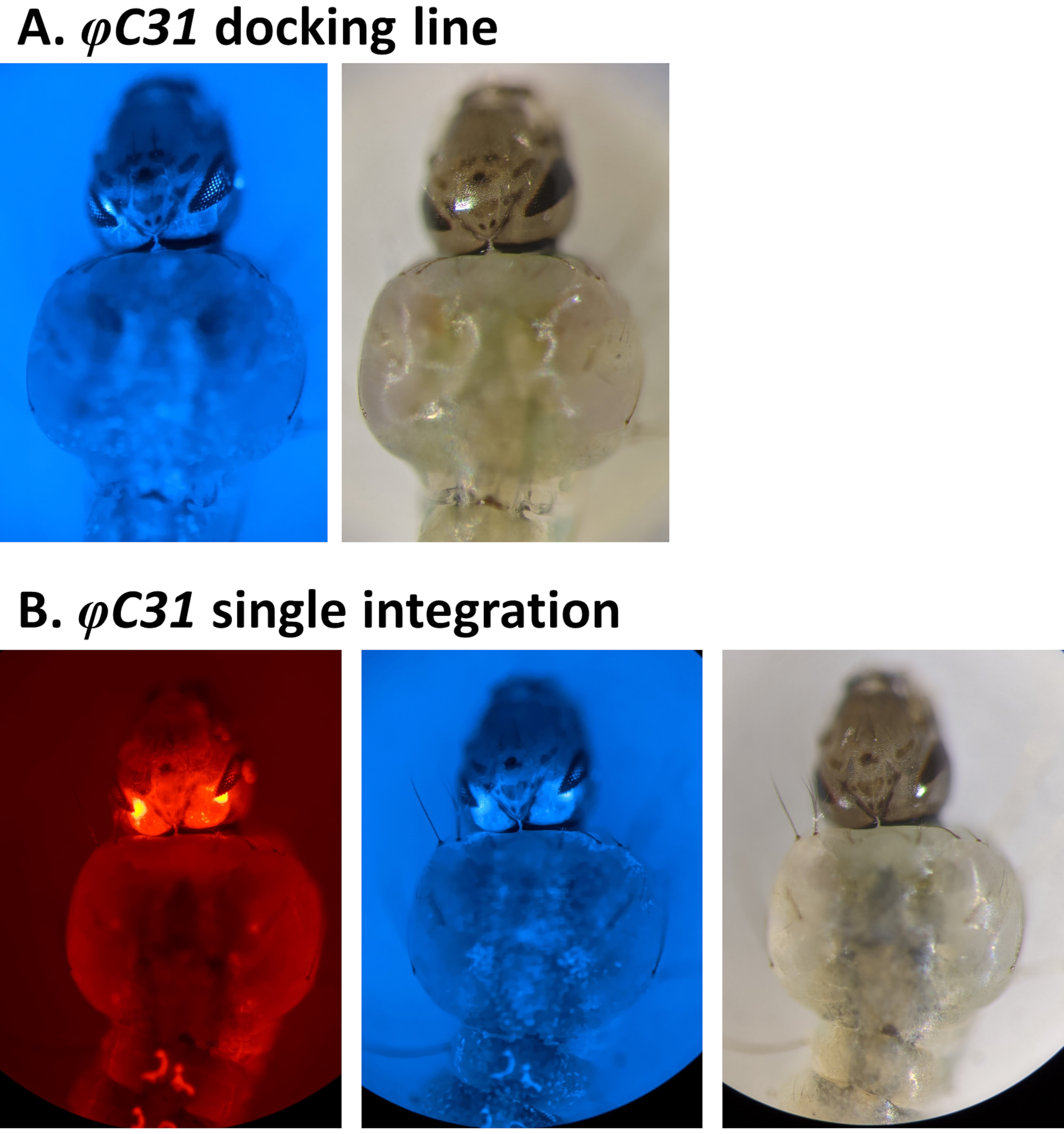
Examples of phenotypic validation of transformation via the screening of fluorescent markers regulated by the 3xP3 promoter are reported in Figure 4 and Figure 5. Figure 4 shows a new An. stephensi line obtained by insertion of a DsRed-marked cassette into a docking line marked with CFP (80.9, Table 1), resulting in G1 progeny expressing both markers as indicated by the red and blue fluorescence detected in the eyes.
RMCE designs are instead expected to result in the replacement of the marker originally inserted into the docking line with that of the donor plasmid. Figure 5A and Figure B illustrate this marker exchange in an An. gambiae docking line marked with CFP (A11, Table 1) where after successful RMCE the CFP marker is lost and the YFP marker is acquired resulting in yellow (but not blue) eye and nerve cord fluorescence33. Occasionally, RMCE can result in a single integration event instead of the exchange of the desired transgenic cassette as illustrated in Figure 5C, where a larva marked with both the original CFP and the new YFP markers is shown. It is reported that up to 50% of the total number of transformation events are single integrations9, 33.
When screening for the presence of a fluorescent marker it is crucial to distinguish its signal from possible background autofluorescence. This is particularly important when using CFP as Anopheles larvae display natural blue autofluorescence (Figure 6A). Increasing the magnification and focusing on the tissues and organs where fluorescence is expected to be driven by the promoter is necessary to identify true CFP-positive individuals as illustrated in Figure 6B using the 3xP3-CFP marker.
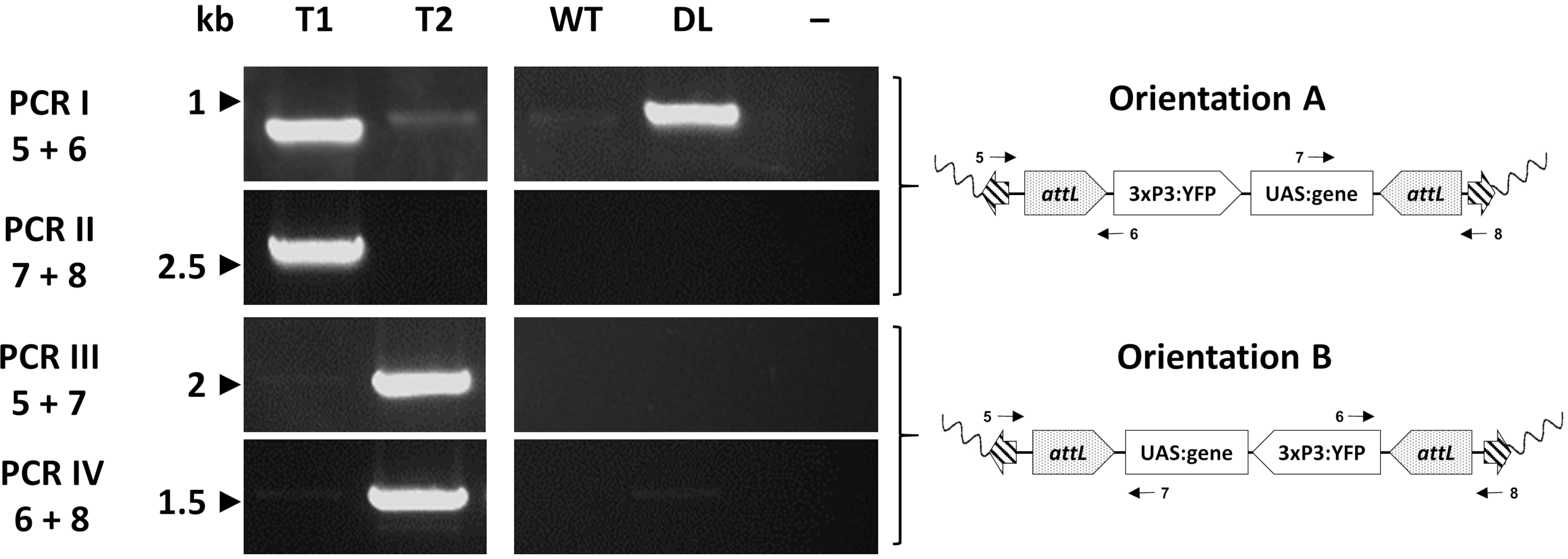
Individual transformants are finally assessed molecularly via PCR to confirm the expected insertion site. Figure 7 reports the PCR validation in individuals from an exchange An. gambiae line showing the two potential orientations of insertion in the mosquito genome33.

Figure 4. Validation of φC31 single integration in An. stephensi larvae (dorsal view). A) The docking line (80.9, Table 1) expresses CFP in the eyes under the regulation of the 3xP3 promoter. B) Successful integration results in the expression of the newly acquired DsRed as well as the original CFP marker in the eyes. Please click here to view a larger version of this figure.
{kind=link}

Figure 5. Validation of φC31 RMCE in An. gambiae larvae (ventral view). A) The docking line (A10, Table 1) expresses CFP under the regulation of the 3xP3 promoter in the eyes (e) and the nerve cord (nc)5. B) Successful RMCE results in the swap of fluorescent marker from CFP to YFP33. C) Single integration event occurred during RMCE experiment where the transformant larva expresses both the CFP and YFP markers. This larva carries GAL4/UAS components that cause a broad expression pattern of YFP, particularly strong in the abdominal muscles (am). Please click here to view a larger version of this figure.
{kind=link}

Figure 6. CFP autofluorescence in An. gambiae larvae (dorsal view). A) Side-by-side image of a positive (CFP+) and a negative (CFP-) L4 larva using the CFP filter. B) Close-up image of the larval eyes that reveals a CFP+ vs CFP- individual. Please click here to view a larger version of this figure.
{kind=link}

Figure 7. Molecular validation of the orientation of cassette insertion in representative transgenic An. gambiae created by φC31 RMCE. The transgenic cassette can be inserted in one of two alternative orientations (A or B) in respect to the insertion site. Each PCR reaction (I - IV) uses a combination of primers (5-8)33 designed to give a diagnostic amplification fragment for each orientation as indicated in the schematic plasmid maps. T1: representative transgenic individual carrying orientation of insertion A; T2: representative transgenic individual carrying orientation of insertion B; WT: wild type; DL: docking line; -: reaction negative control where water was used as template.This figure has been modified from Adolfi et al. (2019)33. Please click here to view a larger version of this figure.
{kind=link}
| Species | Strain | Name | attP(s) | Chromo-some | Promoter-marker | Institution of origin | Reference |
| An. stephensi | Indiana | 26.10b | Single | 2R | 3xP3-eCFP | Univ. of California Irvine | 25 |
| An. stephensi | Indiana | 44Cb | Single | X | 3xP3-eCFP | Univ. of California Irvine | 23, 24 |
| An. stephensi | Indiana | 80.9b | Single | 2L | 3xP3-eCFP | Univ. of California Irvine | This study |
| An. gambiae | G3 | 113 | Single | 2R | 3xP3-eCFP | Univ. of California Irvine | This study |
| An. gambiae | KIL | Ec | Single | 3R | 3xP3-eCFP | Keele Univ. | 19, 43 |
| An. gambiae | G3 | X1 | Single | 2L | No marker | Univ. of Strasbourg | 22 |
| An. gambiae | G3 | YAttP | Single | Y | 3xP3-RFP | Imperial College London | 21 |
| An. gambiae | G3 | A10b | Double | 2R | 3xP3-eCFP | Liverpool School Trop. Med. | 5 |
| An. gambiae | G3 | A11b | Double | 2R | 3xP3-eCFP | Liverpool School Trop. Med. | 9 |
| a. Strain from Johns Hopkins University (gift of M. Jacobs-Lorena) and in culture at the Univ. of California Irvine for >20 years. | |||||||
| b. These lines are available from the authors upon reasonable request. | |||||||
| c. This line is available at the BEI repository www.beiresources.org as MRA-1163. | |||||||
Table 1. Anopheles attP docking lines.
Discussion
The accurate design of attB-tagged plasmids that are compatible with the docking line of choice is paramount for the success of the experiment. Careful consideration must be given to the choice of the marker used for the screening of transformants, including the fluorescence color and its pattern of expression, which will be subject to the pattern already present in the docking line. It is necessary to use fluorescent markers that are easily distinguishable: good marker combinations include RFP (red)/CFP (cyan), RFP (red)/GFP (green), RFP (red)/YFP (yellow), and YFP (yellow)/CFP (cyan), while combinations to avoid are YFP (yellow)/GFP (green) and CFP (cyan)/GFP (green). The 3xP3 promoter39, specific to the eyes and nerve cord, is the most frequently used to drive the expression of fluorescent markers for mosquito transgenesis. Indeed, all the Anopheles docking lines currently available utilize this promoter. Alternative regulatory regions are that of the An. gambiae polyubiquitin gene (PUBc)5 or the viral promoter IE120, which drive expression in multiple tissues. When used along with 3xP3, these promoters would expand the possible color combinations and even the use of the same fluorophore. The indicated promoters are active throughout the mosquito life cycle allowing screening and fluorescence monitoring at all life stages. An additional consideration during plasmid design is the size of the cargo to be integrated or exchanged. While the φC31 system has remarkable carrying capacities18, it should be considered that the size of the donor plasmid generally correlates negatively with transformation efficiency22.
In the described protocol the source of integrase is a helper plasmid expressing the enzyme ubiquitously40. The ubiquitous presence of the integrase may lead to the transformation of somatic cells if microinjections are not precisely directed to the area where the germline forms. While such transformation events will be lost as they are not heritable, somatic effects can decrease the fitness of injected individuals. To avoid this and increase transformation efficiency, integrase expression can be restricted to the germline, for example by using the vasa promoter22,26. Other protocols describe the use of in vitro transcribed messenger RNA (mRNA) as source of φC31 integrase19,24,43. However, this involves the laborious preparation of mRNA and requires careful handling of the injection mix and the use of RNase free reagents to avoid degradation. Plasmid sources of integrase have been demonstrated in both An. gambiae9,21,22,26,33,37 and An. stephensi (A.A. personal communication) to be reliable and lead to efficient transformation, and are thus our preferred option. A further option for integrase delivery is its in vivo production in self-docking helper lines. Such lines were created in An. gambiae that express the φC31 integrase under the regulation of the germline-specific promoter nanos and were found to lead to an improved survival and transformation efficiency20. However, potential fitness loads imposed by the in vivo production of the integrase enzyme on the helper line must be considered.
As with other transgenic techniques, special care must be reserved to the rearing and crossing of individuals deriving from injected embryos to maximize the chances to recover transformants. Individuals that have stably inherited the transgene can be firstly recovered at the G1 progeny. However, early signs of potential transformation can be evaluated by the presence of transient episomal expression of the fluorescent marker in the anal papillae and/or nerve cord of G0 first and second instar larvae when using the 3xP3 promoter43. While the presence of transient fluorescence suggests successful plasmid delivery, it does not guarantee heritable germline transformation. Similarly, the lack of transient expression does not exclude successful transformation. Nevertheless, it has been observed that transiently positive individuals are more likely to yield transgenic progeny compared to transiently negative ones43,48. In expert hands, rearing and crossing of only positive individuals may be an option to reduce mosquito numbers. However, given the importance and fragility of small G0 larvae, the least amount of manipulation is still advisable and the rearing of all G0 individuals is always recommended.
The mating scheme reported in this protocol is designed to maximize the chance of mating and to isolate independent transformation events. However, if insectary space or personnel availability is an issue, G0 adults can be pooled by sex in single cages if enough opposite-sex individuals are provided. Such a setup will not allow discrimination between multiple transformation events occurring in individuals from the same cage. Depending on the experimental setup, the presence of a double (single integration) or single (RMCE) marker is expected during the screening process. In single integration experiments it is important to verify the presence of the original marker from the docking line, while in RMCE is important to verify the loss of the previously integrated marker. Indeed, it is not uncommon in RMCE designs to recover transformants in which single integration instead of exchange occurred due to the recombination of a single attP site9,33. In such individuals both fluorescent markers are present as well as the whole donor plasmid backbone highlighting the importance of conducting a thorough screening for both fluorescent markers.
While the presence of expected fluorescence patterns indicates successful transformation, molecular characterization of the insertion site must be undertaken. To do so, the preparation of accurate maps of the predicted insertion locus, including the flanking genomic regions of the docking line, is crucial for the design of adequate diagnostic oligonucleotide primers for gene amplification analyses. Single integration events result in the formation of attR and attL hybrid sites at the junction between the newly integrated DNA and the previously inserted cassette. These sites can be targeted for insertion site validation. In RMCE designs, the insertion of the donor cassette can occur in two alternative orientations in respect to the genomic locus, thus four primers can be used in alternative PCR combinations to detect which orientation the line carries. As the orientation of cassette insertion may affect transgene expression, in comparative gene expression analysis it is important to use lines carrying the same orientation of insertion.
When working with low numbers of transformants it may not be desirable to sacrifice whole individuals for molecular analysis. An option to this is conducting molecular analysis on DNA extracted from single adult's legs46 as leg loss does not affect an adult female ability to mate and oviposit49. However, there is a risk of damaging the individual in the process of leg removal. Success has been obtained using discarded pupal cases (L. Grigoraki personal communication), however the safest approach is to perform molecular analysis on G2 parents after obtaining viable G3 progeny.
In recent years, CRISPR/Cas9 has revolutionized the way of performing site-specific genome editing26,41,50,51. Unlike site-directed RMCE, CRISPR/Cas9-mediated gene integrations (knock-ins) are independent of the presence of pre-inserted recombination sites with only a one-step transformation event needed. Nevertheless, the CRISPR/Cas9 system relies on the presence of large known genomic sequences flanking the desired insertion site for successful homology directed repair as well as on the efficient site recognition mediated by guide RNAs. These conditions cannot always be met or may be laborious to troubleshoot and, given the availability of multiple docking lines in An. gambiae and An. stephensi and lines derived from them, the φC31 system remains a very valuable tool to perform direct phenotypic comparisons between transgenes at the same genomic locations.
Acknowledgements
We are grateful to Kiona Parker (UCI) for providing images of transgenic An. stephensi larvae, and to Fraser Colman (LSTM) and Beth Poulton (LSTM) for providing transgenic An. gambiae larvae. Beth Poulton (LSTM) also provided precious assistance during the imaging of An. gambiae larvae. This work was funded by the Tata Institute for Genetics and Society (TIGS) and the LSTM's Director Catalyst Fund awarded to A.A. (DCF2014AA). A.A.J. is a Donald Bren Professor at the University of California,Irvine.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL eppendorf tubes | |||
| 8-well microslides | VWR | MARI1216690 | |
| DNeasy Blood & Tissue Kit | Qiagen | 69504 | |
| EndoFree Plasmid Maxi Kit (10) | Qiagen | 12362 | |
| Ethanol, Absolute, Molecular Biology Grade | |||
| Filter set CFP for Leica MZ FLIII Excitation 436/20 nm, extinction 480/40 nm | Leica | 10446363 | |
| Filter set dsRED for Leica MZ FLIII Excitation 545/30 nm, extinction 620/60 nm | Leica | 10447079 | |
| Filter set YFP customised for Leica MZ FLIII | Omega Optical | 500QM25, 500QM35 | |
| Halocarbon oil 27 | Sigma | H8773 | |
| Halocarbon oil 700 | Sigma | H8898 | |
| Petri dishes | |||
| Potassium chloride | |||
| Sodium Chloride | |||
| Sodium phosphate dibasic | |||
| Sodium Acetate Solution (3 M), pH 5.2 | Thermo Fisher Scientific (Life Technologies) | R1181 | |
| Stable brush Size 0 |
References
- Adolfi, A., Lycett, G. J. Opening the toolkit for genetic analysis and control of Anopheles mosquito vectors. Current Opinion in Insect Science. 30, 8-18 (2018).
- Grossman, G. L., Rafferty, C. S., Clayton, J. R., Stevens, T. K., Mukabayire, O., Benedict, M. Q. Germline transformation of the malaria vector, Anopheles gambiae, with the piggyBac transposable element. Insect Molecular Biology. 10 (6), 597-604 (2001).
- Nolan, T., Bower, T. M., Brown, A. E., Crisanti, A., Catteruccia, F. piggyBac-mediated germline transformation of the malaria mosquito Anopheles stephensi using the red fluorescent protein dsRED as a selectable marker. Journal of Biological Chemistry. 277 (11), 8759-8762 (2002).
- Perera, O. P., Harrell, R. A., Handler, A. M. Germ-line transformation of the South American malaria vector, Anopheles albimanus, with a piggyBac/EGFP transposon vector is routine and highly efficient. Insect Molecular Biology. 11 (4), 291-297 (2002).
- Adolfi, A., Pondeville, E., Lynd, A., Bourgouin, C., Lycett, G. J. Multi-tissue GAL4-mediated gene expression in all Anopheles gambiae life stages using an endogenous polyubiquitin promoter. Insect Biochemistry and Molecular Biology. 96, 1-9 (2018).
- Carballar-Lejarazú, R., Jasinskiene, N., James, A. Exogenous gypsy insulator sequences modulate transgene expression in the malaria vector mosquito, Anopheles stephensi. Proceedings of the National Academy of Sciences of the United States of America. 110 (18), 7176-7181 (2013).
- Galizi, R., et al. A synthetic sex ratio distortion system for the control of the human malaria mosquito. Nature Communications. 5, 3977 (2014).
- Nolan, T., Petris, E., Müller, H. M., Cronin, A., Catteruccia, F., Crisanti, A. Analysis of two novel midgut-specific promoters driving transgene expression in Anopheles stephensi mosquitoes. PLoS ONE. 6 (2), 16471 (2011).
- Lynd, A., Balabanidou, V., Vontas, J., Lycett, G. J. Development of a functional genetic tool for Anopheles gambiae oenocyte characterisation: appliction to cuticular hydrocarbon synthesis. BioRxiv. , (2019).
- O'Brochta, D. A., Alford, R. T., Pilitt, K. L., Aluvihare, C. U., Harrell, R. A., Harrell, R. A. piggyBac transposon remobilization and enhancer detection in Anopheles mosquitoes. Proceedings of the National Academy of Sciences of the United States of America. 108 (39), 16339-16344 (2011).
- O'Brochta, D. A., Pilitt, K. L., Harrell, R. A., Aluvihare, C., Alford, R. T. Gal4-based enhancer-trapping in the malaria mosquito Anopheles stephensi. G3. 2 (11), 1305-1315 (2012).
- Macias, V. M., et al. nanos-Driven expression of piggyBac transposase induces mobilization of a synthetic autonomous transposon in the malaria vector mosquito, Anopheles stephensi. Insect Biochemistry and Molecular Biology. 87, 81-89 (2017).
- Nimmo, D. D., Alphey, L., Meredith, J. M., Eggleston, P. High efficiency site-specific genetic engineering of the mosquito genome. Insect Molecular Biology. 15 (2), 129-136 (2006).
- Kim, A., Pyykko, I. Size matters: Versatile use of PiggyBac transposons as a genetic manipulation tool. Molecular and Cellular Biochemistry. 354, 301-309 (2011).
- Thorpe, H. M., Smith, M. C. M. In vitro site-specific integration of bacteriophage DNA catalyzed by a recombinase of the resolvase/invertase family. Proceedings of the National Academy of Sciences of the United States of America. 95 (10), 5505-5510 (1998).
- Khaleel, T., Younger, E., Mcewan, A. R., Varghese, A. S., Smith, M. C. M. A phage protein that binds φC31 integrase to switch its directionality. Molecular Microbiology. 80 (6), 1450-1463 (2011).
- Farruggio, A. P., Chavez, C. L., Mikell, C. L., Calos, M. P. Efficient reversal of phiC31 integrase recombination in mammalian cells. Biotechnology Journal. 7 (11), 1332-1336 (2012).
- Venken, K. J. T., He, Y., Hoskins, R. A., Bellen, H. J. P[acman]: A BAC Transgenic Platform for Targeted Insertion of Large DNA Fragments in D. melanogaster. Science. 314 (5806), 1747-1751 (2006).
- Meredith, J. M., et al. Site-specific integration and expression of an anti-malarial gene in transgenic Anopheles gambiae significantly reduces Plasmodium infections. PLoS ONE. 6 (1), 14587 (2011).
- Meredith, J. M., Underhill, A., McArthur, C. C., Eggleston, P. Next-Generation Site-Directed Transgenesis in the Malaria Vector Mosquito Anopheles gambiae: Self-Docking Strains Expressing Germline-Specific phiC31 Integrase. PLoS ONE. 8 (3), 59264 (2013).
- Bernardini, F., et al. Site-specific genetic engineering of the Anopheles gambiae Y chromosome. Proceedings of the National Academy of Sciences of the United States of America. 111 (21), 7600-7605 (2014).
- Volohonsky, G., et al. Tools for Anopheles gambiae Transgenesis. G3. 5 (6), 1151-1163 (2015).
- Amenya, D. A., et al. Comparative fitness assessment of Anopheles stephensi transgenic lines receptive to site-specific integration. Insect Molecular Biology. 19 (2), 263-269 (2010).
- Isaacs, A. T., et al. Transgenic Anopheles stephensi coexpressing single-chain antibodies resist Plasmodium falciparum development. Proceedings of the National Academy of Sciences of the United States of America. 109 (28), 1922-1930 (2012).
- Pham, T. B., et al. Experimental population modification of the malaria vector mosquito, Anopheles stephensi. PLoS Genetics. 15 (12), 1008440 (2019).
- Hammond, A., et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nature Biotechnology. 34 (1), 78-83 (2015).
- Groth, A. C., Fish, M., Nusse, R., Calos, M. P. Construction of Transgenic Drosophila by Using the Site-Specific Integrase from Phage phiC31. Genetics. 166 (4), 1775-1782 (2004).
- Franz, A. W. E., et al. Comparison of transgene expression in Aedes aegypti generated by mariner Mos1 transposition and site-directed recombination. Insect Molecular Biology. 20 (5), 587-598 (2011).
- Labbé, G., Nimmo, D., Alphey, L. piggybac-and PhiC31-Mediated Genetic Transformation of the Asian Tiger Mosquito, Aedes albopictus (Skuse). PLoS Neglected Tropical Diseases. 4 (8), 788 (2010).
- Schetelig, M. F., Scolaric, F., Handler, A. M., Kittelmann, S., Gasperi, G., Wimmer, E. A. Site-specific recombination for the modification of transgenic strains of the Mediterranean fruit fly Ceratitis capitata. Proceedings of the National Academy of Sciences of the United States of America. 106 (43), 18171-18176 (2009).
- Yonemura, N., et al. phiC31-integrase-mediated, site-specific integration of transgenes in the silkworm, Bombyx mori (Lepidoptera: Bombycidae). Applied Entomology and Zoology. 43 (11), 997-1008 (2013).
- Bateman, J. R., Lee, A. M., Wu, C. T. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics. 173 (2), 769-777 (2006).
- Adolfi, A., Poulton, B., Anthousi, A., Macilwee, S., Ranson, H., Lycett, G. J. Functional genetic validation of key genes conferring insecticide resistance in the major African malaria vector, Anopheles gambiae. Proceedings of the National Academy of Sciences of the United States of America. 116 (51), 25764-25772 (2019).
- Haghighat-Khah, R. E., et al. Site-specific cassette exchange systems in the aedes aegypti mosquito and the Plutella xylostella moth. PLoS ONE. 10 (4), 0121097 (2015).
- Long, D., Lu, W., Zhang, Y., Bi, L., Xiang, Z., Zhao, A. An efficient strategy for producing a stable, replaceable, highly efficient transgene expression system in silkworm, Bombyx mori. Scientific Reports. 5 (1), 8802 (2015).
- Volohonsky, G., et al. Transgenic Expression of the Anti-parasitic Factor TEP1 in the Malaria Mosquito Anopheles gambiae. PLoS Pathogens. 13 (1), 1006113 (2017).
- Grigoraki, L., Grau-Bové, X., Yates, H. C., Lycett, G. J., Ranson, H. Isolation and transcriptomic analysis of anopheles gambiae oenocytes enables the delineation of hydrocarbon biosynthesis. eLife. 9, 58019 (2020).
- Kyrou, K., et al. A CRISPR-Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nature Biotechnology. 36 (11), 1062-1066 (2018).
- Berghammer, A. J., Klingler, M., Wimmer, E. A. A universal marker for transgenic insects. Nature. 402 (6760), 370-371 (1999).
- Ringrose, L. Transgenesis in Drosophila melanogaster. Methods in Molecular Biology. 561, 3-19 (2009).
- Dong, Y., Simões, M. L., Marois, E., Dimopoulos, G. CRISPR/Cas9 -mediated gene knockout of Anopheles gambiae FREP1 suppresses malaria parasite infection. PLoS Pathogens. 14 (3), 1006898 (2018).
- Lombardo, F., Lycett, G. J., Lanfrancotti, A., Coluzzi, M., Arcà, B. Analysis of apyrase 5' upstream region validates improved Anopheles gambiae transformation technique. BMC research notes. 2, 24 (2009).
- Pondeville, E., et al. Efficient ΦC31 integrase-mediated site-specific germline transformation of Anopheles gambiae. Nature Protocols. 9 (7), 1698-1712 (2014).
- Lobo, N. F., Clayton, J. R., Fraser, M. J., Kafatos, F. C., Collins, F. H. High efficiency germ-line transformation of mosquitoes. Nature protocols. 1 (3), 1312-1317 (2006).
- Terenius, O., Juhn, J., James, A. A. Injection of An. stephensi embryos to generate malaria-resistant mosquitoes. Journal of Visualized Experiments. 5, 216 (2007).
- Lynd, A., et al. Insecticide resistance in Anopheles gambiae from the northern Democratic Republic of Congo, with extreme knockdown resistance (kdr) mutation frequencies revealed by a new diagnostic assay. Malaria Journal. 17 (1), 412 (2018).
- Marinotti, O., et al. Development of a population suppression strain of the human malaria vector mosquito, Anopheles stephensi. Malaria Journal. 12 (1), 142 (2013).
- Adolfi, A. In vivo functional genetic analysis of insecticide resistance in the malaria mosquito Anopheles gambiae. University of Liverpool. , (2017).
- Isaacs, A. T., Lynd, A., Donnelly, M. J. Insecticide-induced leg loss does not eliminate biting and reproduction in Anopheles gambiae mosquitoes. Scientific Reports. 7, 46674 (2017).
- Gantz, V. M., et al. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proceedings of the National Academy of Sciences of the United States of America. 112 (49), 6736-6743 (2015).
- Li, M., Akbari, O. S., White, B. J. Highly efficient site-specific mutagenesis in malaria mosquitoes using CRISPR. G3: Genes, Genomes, Genetics. 8 (2), 653-658 (2018).
- Poulton, B. C., et al. Using the GAL4-UAS System for Functional Genetics in Anopheles gambiae. J. Vis. Exp. , (2021).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved