4-Dimensional Imaging of Zebrafish Optic Cup Morphogenesis
In This Article
Summary
This protocol describes an approach for in toto labeling and multidimensional imaging of zebrafish early eye development. We describe labeling, embedding, and four dimensional (4D) imaging using laser scanning confocal microscopy, and considerations for optimizing acquisition of datasets for dissecting mechanisms of optic cup morphogenesis.
Abstract
Visual system function requires the establishment of precise tissue and organ structures. In the vertebrate eye, structural defects are a common cause of visual impairment, yet mechanisms of eye morphogenesis are still poorly understood. The basic organization of the embryonic eye is conserved throughout vertebrates, thus live imaging of zebrafish embryos has become a powerful approach to directly observe eye development at real time under normal and pathological conditions. Dynamic cell processes including movements, morphologies, interactions, division, and death can be visualized in the embryo. We have developed methods for uniform labeling of subcellular structures and timelapse confocal microscopy of early eye development in zebrafish. This protocol outlines the method of generating capped mRNA for injection into the 1-cell zebrafish embryo, mounting embryos at optic vesicle stage (~12 hours post fertilization, hpf), and performing multi-dimensional timelapse imaging of optic cup morphogenesis on a laser scanning confocal microscope, such that multiple datasets are acquired sequentially in the same imaging session. Such an approach yields data that can be used for a variety of purposes, including cell tracking, volume measurements, three-dimensional (3D) rendering, and visualization. Our approaches allow us to pinpoint the cellular and molecular mechanisms driving optic cup development, in both wild type and genetic mutant conditions. These methods can be employed directly by other groups or adapted to visualize many additional aspects of zebrafish eye development.
Introduction
Vertebrate eye development begins with the emergence, or evagination, of the optic vesicles from the prospective brain neuroepithelium. The optic vesicles then undergo a series of tissue shape changes, elongating and then invaginating to generate the optic cup. In the optic cup, the neural retina and retinal pigment epithelium, both derived from the neuroepithelium, enwrap the nascent lens, which is derived from the surface ectoderm. The entire process requires a complex series of cell and tissue movements and molecular signaling, coordinated between neuroepithelium, ectoderm, and mesenchymal cell populations. These initial events establish the basic structure of the eye, and later steps of eye development, including iris and cornea formation, are elaborations on early organization. Disruptions to early eye development and morphogenesis underlie numerous visual impairment conditions in humans, including anophthalmia, microphthalmia, and coloboma. Unlocking the cellular and molecular mechanisms governing optic cup morphogenesis is crucial for further understanding visual system development and the pathological conditions that result when these processes go awry.
Our understanding of vertebrate eye development and morphogenesis emerged from a tremendous amount of work spanning classic histological studies to embryological and genetic approaches in a variety of model organisms including mouse, chick, frog, and fish1,2,3,4,5. While this body of work established molecular mechanisms that regulate early eye development, historically there exists a poor understanding of the morphogenesis of the eye: the emergence of its 3D structure. The bulk of these findings have come from imaging sectioned embryos at discrete time points. While this is sufficient to provide a view of tissue morphology in 2 dimensions, morphogenesis is a dynamic, 3D process. In order to determine how the shape of the tissue changes in 3 dimensions over time, how single cells behave, and how those behaviors contribute to changes in 3D tissue shape, different approaches are necessary.
One solution to address this significant gap in knowledge is live imaging, which enables dynamic observation of cells and tissues in real time as the organ takes shape. Unfortunately, this is not readily feasible in many model organisms due to the constraints of embryonic development. For example, mouse and chick embryos (developing in utero or within an eggshell) are not easily accessed, and many live embryos are not optically transparent, causing significant light scatter and limiting the depth to which images can be acquired. Zebrafish, with external development and transparent embryos, provide a unique opportunity to carry out live imaging of eye morphogenesis6,7,8,9,10,11,12,13,14,15,16,17,18,19. Ample transgenic and mutant lines are also available, as well as tools to generate new transgenics and mutants20,21,22,23,24. Further, optic cup morphogenesis occurs rapidly in zebrafish, over a 12 h timeframe (12-24 hours post fertilization, hpf), making imaging of the entire process feasible.
Live imaging efforts have been accelerated by expansion and optimization of the family of fluorescent proteins, which allow genetically encoded vital labeling of subcellular structures, as well as improvements and innovations to microscopy methods. The protocol described here uses laser scanning confocal microscopy, rather than other current approaches to imaging zebrafish embryogenesis, including spinning disk confocal microscopy, selective plane illumination microscopy (SPIM and its variants), and other more specialized microscopy methods. For the developing zebrafish eye, we found that spinning disk confocal microscopy was not sufficient for imaging deeper in the tissue. Although SPIM boasts an extremely fast imaging time and is becoming more widely used, handling the large datasets for visualization and analysis remains a challenge. In contrast, laser scanning confocal microscopy is easily accessible, especially for individuals lacking expertise in assembling optical hardware. We hope that the wide availability of laser scanning confocal microscopy will make our protocol useful for many labs.
Here, we describe our method for capturing 4D datasets of optic cup morphogenesis, using in toto labeling of the embryo for membranes and chromatin, and a laser scanning confocal microscope for image acquisition (schematized in Figure 1). The fluorescent proteins used here (EGFP-CAAX and H2A.F/Z-mCherry) were chosen to provide tissue labeling with single cell resolution. We use datasets generated with this protocol for a variety of image analysis and visualization functions. This protocol can be easily adapted if other subcellular structures are desired. Plasma membrane labeling was selected for visualization of cell shape: we use EGFP-CAAX, in which the last 21 amino acids of H-ras, serving as a prenylation signal sequence, are fused to the C-terminus of EGFP13. Other plasma membrane targeted fluorescent proteins (e.g., transmembrane fusion or myristoylated) are likely to work just as well. To mark nuclei, we selected H2A.F/Z-mCherry, in which mCherry is fused to a histone protein13. This ensures that cell division, including mitotic spindle orientation, is easily visualized.
With any live imaging approach, one must consider trade-offs between increasing imaging speed while maximizing signal-to-noise ratio, axial resolution, and sample preservation. We have optimized our methods to maximize image quality and number of embryos that can be imaged in a single run. Often, the goal is to image optic cup morphogenesis in a homozygous mutant embryo, which may be phenotypically indistinguishable from wild type at the onset of optic cup morphogenesis and the offspring of a heterozygous incross (25% of the embryos are the desired genotype). By optimizing, and then multiplexing image acquisition, there is an increased likelihood of capturing a dataset of a homozygous mutant embryo.
Temporal resolution, or how frequently volume data (Z-stacks) are acquired, is a key aspect of timelapse imaging. Depending on the purpose for such datasets, there are different requirements for speed. Initially this protocol was developed for manual 4D cell tracking. Tracking of individual cells within a uniformly labeled tissue requires high enough temporal resolution to provide confidence that any one cell is being continuously tracked over time. We found that Z-stacks of zebrafish optic cup morphogenesis must be acquired at least every 3.1 min over 12 h; here, we have optimized our acquisition on a laser scanning confocal microscope such that we can acquire Z-stacks of 4-5 embryos every 2.5 min.
Establishing Z-step size was a crucial step in protocol optimization: for 3D rendering and visualization, isotropic data are ideal, in which the Z-step size is equal to XY pixel dimension. In reality, it is extremely difficult to acquire such timelapse data with live samples, given constraints with imaging time and photobleaching. Therefore, determining the adequate Z-step size is important for the rendering and visualization needs of the experiment, and specifically, what X:Y:Z voxel ratio is needed to maximize axial information while maintaining speed and preventing photobleaching. For this protocol, the voxel ratio established was 1:1:3.5 (0.6 µm x 0.6 µm x 2.1 µm Z-step size using a 40x long working distance water immersion objective). When acquiring a Z-depth of 130-140 µm, this yields volume data with suitable temporal resolution and little photobleaching.
As discussed above, this protocol is specific for 4D imaging of zebrafish optic cup morphogenesis, using embryos in toto labeled for plasma membrane and chromatin, and a laser scanning confocal microscope. The protocol below can be easily adapted for a variety of experiments and needs. First, with respect to subcellular structures, any structure for which a live cell marker exists can be imaged. Next, although the focus here is exclusively on optic cup morphogenesis, timelapse imaging can be adapted for other stages of eye development, for example, neurogenesis25,26,27,28,29,30,31,32,33. For imaging later development, one may need to consider embryo immobilization (as spontaneous muscle activity begins around 24 hpf), pigmentation (which begins to emerge around 24 hpf), tissue size (the eye grows significantly in volume during neurogenesis), and imaging speed (which should be adjusted depending on the speed of the process of interest). All of these considerations can be readily managed. The protocol is quite flexible; in addition to the details of the specific protocol here, there are general principles that will aid those interested in live imaging other aspects of eye development.
Protocol
All methods described here using zebrafish are covered under Dr. Kristen Kwan's animal protocol, "Cellular and Molecular Mechanisms of Visual System Development in Zebrafish", and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Utah.
1. Capped RNA Synthesis
- Generate the DNA template for in vitro transcription.
- Linearize the DNA template by digesting 10 µg of DNA in 100 µL volume of the reaction mixture. A typical reaction is assembled as follows: digest 10 µg DNA using 3 µL of enzyme and 10 µL of buffer, bring the reaction volume to 100 µL with water.
NOTE: A typical template for digest is a pCS2 vector, for example, pCS2-EGFP-CAAX or pCS2FA-H2A.F/Z-mCherry for the membrane and chromatin labeling described here. In this case, the plasmid DNAs are each digested using the enzyme NotI. Users should be cautious of star activity; in this case, a high-fidelity enzyme is recommended and commercially available. - Incubate the reaction at 37 °C overnight to ensure that the DNA is digested to completion.
- Clean up the restriction digest using a PCR purification kit following the manufacturer's protocol. Elute the DNA with 30 µL ddH2O. Store the linearized DNA at -20 °C and use as needed.
NOTE: The amount of DNA digested and the elution volume yield approximately 0.3 µg/µL; this is sufficient for ~5 rounds of in vitro transcription, each round using ~2 µg of DNA as template.
- Linearize the DNA template by digesting 10 µg of DNA in 100 µL volume of the reaction mixture. A typical reaction is assembled as follows: digest 10 µg DNA using 3 µL of enzyme and 10 µL of buffer, bring the reaction volume to 100 µL with water.
- In vitro transcribe capped RNA using an in vitro transcription kit. For pCS2 templates (as described here), use an SP6 kit.
- Assemble the in vitro transcription reaction, as follows: Digest 2 µg of DNA template or up to 6 µL with 2 µL of 10x Reaction Buffer, 10 µL of 2x Ribonucleotide Mix, and 2 µL of 10x Enzyme Mix.
- Incubate at 37 °C for 2-4 h or longer (longer times will lead to greater yield). If desired, 1 µL of Enzyme Mix can be supplemented halfway through the incubation period.
- Digest the DNA template by adding 1 µL of RNase-free DNase and incubating for 15 min at 37 °C.
- Purify the capped RNA using an RNA purification kit following manufacturer's protocol (see Table of Materials). Elute with 100 µL of RNase-free H2O.
NOTE: The addition of β-mercaptoethanol is not necessary for this application. While the method described in this protocol is straightforward, alternative reagents can also be used to purify RNA. - Precipitate the RNA.
- Add 10 µL of 3 M RNase-free NaOAc and 2.5 volumes (~275 µL) of ice-cold RNase-free 100% EtOH to the eluted RNA.
- Incubate the reaction at -20 °C for 15-30 min. Spin for 15 min at high speed at 4 °C to pellet the RNA.
NOTE: The pellet should be visible against the wall of the tube. It is helpful to note how the tube is aligned in the centrifuge at this step, in order to predict where the pellet should be at the end of the spin. - Remove EtOH carefully with a syringe, being careful to avoid heating, over-drying, or dislodging the pellet. Resuspend the pellet in 20 µL of RNase-free H2O.
- Check the RNA to ensure that the synthesis is successful. Use 1 µL to assay concentration on a spectrophotometer and run 0.5 µL on a 1% agarose gel to check for one or two discrete bands, rather than a low molecular weight smear. The yield of the in vitro transcription reaction should be ~1 µg/µL.
- Aliquot and store RNA at -20 °C or -80 °C. The RNA is not diluted until immediately prior to injections.
2. Microinjection of 1-cell zebrafish embryos
NOTE: Inject 200-300 pg per RNA to obtain ubiquitous expression of chromatin and cell membranes throughout optic cup morphogenesis. Prepare 5-10 µL of the RNA dilution, and load 2.5-5 µL/needle; plan for extra in the event the needle breaks.
- Advance preparations for injections.
- Prepare an injection mold dish to facilitate alignment and orientation of 1-cell embryos for microinjection. Melt 2% agarose in E3 (standard embryo medium) and pour into a Petri dish. Carefully float the injection mold (see Table of Materials) on the top of the hot agarose to generate the imprint of the troughs in the agarose as it solidifies. Once the agarose solidifies, remove the injection mold.
NOTE: Injection mold dishes can be used for several months; cover in E3 and store at 4 °C when not in use. - Pull microinjection needles. Pull glass capillaries to a long taper to make microinjection needles using a needle puller machine. Program the machine specific to the type of capillaries used. For 1.0 x 0.78 mm borosilicate capillaries use the following program: heat = 546 °C, pull = 130, velocity = 70, time = 90 (Figure 2F). Store the needles in a Petri dish and secure it with modeling clay.
NOTE: The pulling recipe will vary, depending on the machine and filament, and new filaments should always be calibrated. Each round of pulling produces two needles (Figure 2G); do this several times to produce enough needles in case of accidental breaks and future experiments. - Dilute the capped RNA for injection. Dilute both EGFP-CAAX and H2A-mCherry RNAs to 200-300 ng/µL concentration using RNase-free H2O and 1 µL of phenol red in a total volume of 5 µL (final concentration of phenol red is 0.1%). Flick the mixture and briefly spin down to collect the total volume. Keep the diluted RNA on ice prior to injecting.
- Prepare an injection mold dish to facilitate alignment and orientation of 1-cell embryos for microinjection. Melt 2% agarose in E3 (standard embryo medium) and pour into a Petri dish. Carefully float the injection mold (see Table of Materials) on the top of the hot agarose to generate the imprint of the troughs in the agarose as it solidifies. Once the agarose solidifies, remove the injection mold.
- Injection of 1-cell embryos
- Once the fish begin to breed, allow ~15-20 min to ensure that the eggs become fertilized. During this time, backload the needle with 2.5-5 µL of the RNA dilution using a P10 and P10 micro-loader tips.
- Using a picoinjector, calibrate the needle using an eyepiece reticle (a micrometer calibration slide is also sufficient) to measure the volume of the droplet (volume of a sphere = (4/3) * pi * radius^3). Adjust the injection time and pressure such that the injection volume is 1 nL (Figure 2I).
- With a roller/transfer pipette, carefully load eggs into the injection mold (Figure 2J). If helpful, use forceps to roll embryos such that the single cell is visible, either prior to or during injection. It is the user's preference to use a micromanipulator.
- Inject embryos at the 1-cell stage, targeting the cell and not the yolk (Figure 2M). This will ensure uniform labeling of the developing embryo.
- Raise embryos to the desired stage (prior to 12 hpf, according to standard staging34). Injected embryos will have a delayed development, so raise them at a slightly higher temperature (29.0-29.5 °C) to make up time. During the afternoon, check the embryos and remove those that are dead to preserve the health of the clutch.
3. Mounting optic vesicle stage zebrafish embryos for timelapse imaging
- Before mounting, prepare 1.6% low-melt agarose in E3. If planning multiple imaging experiments, prepare ~20 mL of low-melt agarose and store at room temperature. On the day of embryo mounting, melt this to generate a fresh aliquot (1-5 mL) in a tube that can be placed in a 42 °C heat block prior to mounting.
- Screen embryos for successful injection and overall brightness of fluorescence using a fluorescence microscope prior to mounting. An ideal sample will have strong EGFP and mCherry fluorescence and be at the correct developmental stage (Figure 3B - B'').
- Screen embryos using a fluorescence microscope. Select embryos that strongly express both EGFP and mCherry fluorescence for mounting.
- Select embryos that are 11 hpf. Count somites to properly stage the embryos34; at 11 hpf there should be 3 somites, and by 12 hpf there are 6 somites.
NOTE: By mounting embryos prior to 12 hpf (at 11 hpf), the samples will be appropriately staged at 12 hpf when the timelapse begins.
- Dechorionate embryos prior to mounting. Do this either manually with fine forceps or chemically using pronase (2 mg/mL). With embryos this young, it is essential that dechorionation is performed within an agar-coated dish and that embryos do not contact the air-water interface.
- Using a glass roller pipette, suck up an embryo and eject as much E3 as possible such that the embryo sits at the tip of the glass Pasteur pipette.
- Drop the embryo into the tube of agarose from the heat block. Let it sink into the agarose for a few seconds, then suck up some agarose first and then the embryo, making sure that the embryo remains at the tip of the pipette (Figure 3C - C').
- Place a glass-bottom dish for imaging under a dissecting scope. Eject the embryo and agarose into an agarose droplet in the glass-bottom dish. Use forceps to very quickly orient, such that the embryo is dorsal-down (top of head on the glass bottom). Allow the agarose droplet to harden (Figure 3D - D'). This step must be done quickly but carefully, as embryos at this stage are fragile. Damaged embryos will not survive the timelapse imaging process.
NOTE: It is important to orient all embryos consistently for this experiment. When performing the timelapse, the embryos must fit within the assigned field of view while maintaining additional space for the optic vesicle to grow into. An optimal orientation is to align the anterior-posterior axis of all embryos "vertically" or along 12 and 6 o'clock on a clock face (Figure 3G). - Mount between 10-12 embryos, which is more than twice as many that will be imaged, so that the best samples can be selected once evaluated on the confocal (Figure 3E - E'). Samples will be evaluated for age, uniformity of fluorescent labeling, and precision of mounting orientation.
- After mounting the embryos, pipette more agarose to completely cover the bottom of the dish, thereby encasing all of the separate agarose droplets in a single large agarose disc (Figure 3F - F'). A sufficient amount of agarose will ensure that the individual embryo droplets or the entire disc does not lift up from the bottom of the dish and float out of view.
- Once hardened, overlay the agarose with E3 to keep the samples hydrated for the duration of the timelapse imaging experiment (a concern depending on the humidity of the building and in the environment).
NOTE: Tricaine is not necessary for these specific timelapse imaging experiments, as these embryos are sufficiently young; however, if desired, and if working with older stages of embryos, tricaine can be added into the agarose itself and overlaid in the media. - On paper, draw a map of the embryos for easy reference during timelapse set up. This is crucial to later associate the position with the genotype of each sample.
4. Multiple position confocal timelapse with a laser scanning confocal microscope
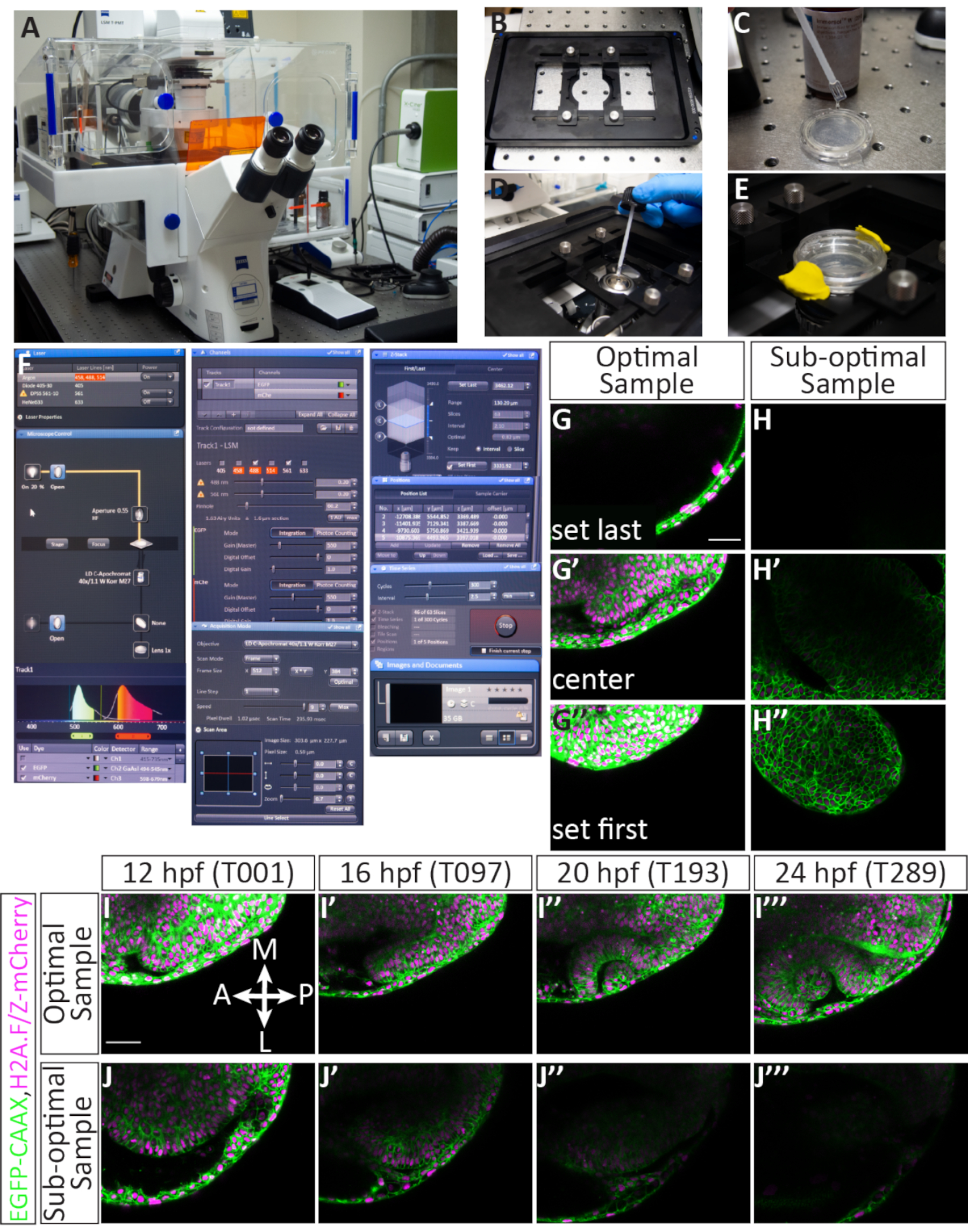
NOTE: This timelapse imaging protocol was designed to be used with a laser scanning confocal microscope equipped with manufacturer's software (see Table of Materials). This system is equipped with a piezo Z-stage device that allows rapid acquisition of Z-stacks. The laser lines used in this protocol are a 488 nm Argon ion laser and a DPSS 561 nm laser. The 561 nm laser is well-suited for imaging the mCherry fluorophore: it is close enough to the peak of mCherry excitation (587 nm) and well-powered.
- Set up the confocal.
- Power on the confocal and log into the desktop computer.
- Install the piezo Z-stage insert (Figure 4B), then start the acquisition software.
- In the software within Acquisition mode, turn on the 488 and 561 lasers using the drop-down menu (Figure 4F). The lasers take several minutes to warm up, so this step is performed early to save time.
NOTE: The acquisition parameters are as follows: Check the Z-stack, Time Series, and Position boxes. Set Frame Size to 512 (X) x 384 (Y), Scan Speed to 9, Scan Mode to bidirectional, Zoom to 0.7, Pinhole to 60.2 (1.63 Airy Units ~= 1.6 µm section), and Z-interval to 2.1 µm. This will yield an image size of 303.6 µm x 227.7 µm, and pixel size of 0.59 µm. The 488 and 561 lasers should be checked and assigned to the same track; the EGFP detection range is 494-545 nm and the mCherry detection range is 598-679 (Figure 4F). - Liberally coat the bottom of the glass dish with immersion medium matching the refractive index of water, being careful to avoid air bubbles (Figure 4C). Select the 40x water objective and apply a small drop of immersion medium to the objective (Figure 4D). Secure the glass-bottomed dish in the stage insert, apply E3 to keep the embryos moist overnight, then use modeling clay to seal the plastic lid over the dish (Figure 4E). Raise the objective to make contact with the dish.
- Screen samples and prepare for timelapse.
- Switch from the Acquisition tab to the Locate tab and click on the lightbulb symbol to turn on transmitted light. With the transmitted light, locate the first sample with the joystick: by eye, first move the sample into the beam of light, then use the eyepieces to the center and focus on an optic vesicle.
- Return to the Acquisition tab. Ensure that the Z-stack, Time Series, and Positions boxes are checked, and the lasers are warmed up for use.
- Set the Frame Size to 512 (X) x 384 (Y), Scan Speed to 9, Scan Mode to bidirectional, and set Zoom to 0.7.
- Under the Channels heading, begin by assigning the 488 and 561 lasers to Power 2.0 and Gain 550 (this can be adjusted later). Set the pinhole to 60.2 (1.63 Airy Units ~= 1.6 µm section).
- Begin scanning using the Continuous button. With the fine focus knob, locate the sample in the Z-axis and position the X-Y axis in frame. Stop scanning.
- Under the Positions heading, click on Add to save the XYZ information on the first sample (Figure 4F). Do this only for samples to be timelapsed. Select samples for their strong fluorescence and optimal mounting.
- Switch to the Locate tab, move to the next sample, and repeat steps 4.2.5-4.2.6, being selective about choosing samples.
- Finalize sample positions, assign z-range, and then start timelapse. Once all the samples are selected (it is possible to image 4-5 samples within the 2.5-minute total time interval) and positions are saved, go through each sample and adjust the XYZ position within the viewing frame.
- Highlight the first position and click on Move To. While continuously scanning, line up the optic vesicle within the frame. Leave ample space in the anterior and distal regions relative to the optic vesicle and the brain.
- Next, assign the first and last Z-slices by selecting Set First and Set Last while moving through the Z-direction with the fine focus knob. Maintain a total slice number of ~63, as this will accommodate the growth of the optic cup. Provide extra room on the ventral side of the optic vesicle to allow room for growth.
- Once first and last Z-slices have been set, click on the C button to move to the center of the Z-stack. Adjust laser power and gain for both lasers; if possible, keep the laser power below 5 and use a higher gain, if necessary. Stop Scanning. Update the position information by clicking on Update.
- Move to the next position by clicking on Position 2. Repeat steps 4.3.1-4.3.3 for each assigned position. The laser power and gain should be set such that all samples are sufficiently illuminated.
- Once both position information and laser settings are finalized, assign Time Series settings. Under the Time Series heading, assign Time Interval to 2.5 min, and Cycles to 300 (Figure 4F). The number of cycles is calculated such that the timelapse proceeds past the 24 hpf stage, when optic cup development is complete.
- To begin the timelapse, click on Start. Monitor the first complete cycle: use a timer to ensure one complete cycle (imaging all positions) is less than 2.5 min and verify that each position looks correct. The microscope runs independently overnight and does not need to be monitored.
NOTE: Although the confocal room is temperature controlled, the room temperature is 20-25 °C. With the equipment running and the lasers scanning, the temperature on the stage appears to be closer to 28.5 °C, since embryonic development proceeds at this expected rate, as assayed visually with morphological landmarks. - With these settings, the timelapse will last 12.5 h. Save the file once the timelapse is complete. It will be large (~40 GB) and can be separated into individual positions after it is saved using the acquisition software.
Representative Results
Injection of fluorescent RNAs enables subcellular labeling
RNAs that label the cell's plasma membrane and histones are injected in order to capture the individual cell morphologies and movements that underlie optic cup morphogenesis. Figure 2 demonstrates each step of the process of microinjecting zebrafish embryos at the 1-cell stage. Briefly, zebrafish are mated (Figure 2E), and embryos are collected and loaded into the injection mold (Figure 2J). A microinjection needle is backfilled (Figure 2H), and embryos are injected directly into the single cell (Figure 2K-M), which is necessary to obtain uniform labeling (Figure 2M, Figure 4I-I''', Movie 1). In addition to uniform labeling, robust fluorescence is observed and development proceeds unperturbed (Figure 3B-B'').
Effective mounting is essential for timelapse imaging OCM from 12-24 hpf
To image optic cup morphogenesis, which occurs over an extended period of time, it is critical to both select ideal embryos and properly position each sample while embedding. Figure 3 shows a detailed account for successful mounting of 11 hpf embryos. Injected embryos are first screened for sufficient fluorescence (Figure 3B',B'',F'',F'''). Individual embryos are immersed in agarose (Figure 3C,C') and mounted dorsally in an individual drop of agarose (Figure 3D,D'). All samples are mounted in a collective disc of agarose (Figure 3E-F'). When embryos are staged correctly, sufficiently fluorescent, and adequately mounted (Figure 3G), the samples will stay in the imaging frame during the timelapse allowing for the entire organ to be imaged (Figure 4G-G'', I-I''', Movie 1). Failure to accomplish any of these steps will result in a mounted embryo that is not an optimal sample for timelapse imaging. The slightest variation in the degree of rotation will have a dramatic effect on the direction of embryo growth during the timelapse. Sub-optimal rotations are shown in Figure 3, including an embryo that is over-rotated (Figure 3I), which will result in a more posterior timelapse, and an embryo that is under-rotated (Figure 3J) in which only the most anterior tissue can be observed. In addition to proper rotation during mounting, samples that are mounted with respect to the orientation of the frame are more likely to yield a successful timelapse (Figure 4G-G'', I-I''', Movie 1). Optimal samples are those that are oriented vertically on the dish, with the anterior-posterior axis in line with 12 and 6 o'clock on a clock face (Figure 3G). Sub-optimal samples are those that are not oriented on this axis, such as those that are horizontal or diagonal (Figure 3H). These samples will grow out of the imaging frame as the timelapse proceeds and cannot be used for further analysis (Figure 4H-H'', J-J''').
Acquiring a timelapse dataset that is suitable for future analysis
Embryos that are accurately injected, raised, and mounted will make for successful confocal imaged samples. Figure 4 shows an optimal sample for timelapse: there is even fluorescence, and the sample is 12 hpf. The z-stack for imaging is assigned such that the first slice is just dorsal to the optic vesicle, while the last slice leaves several slices beyond the ventral boundary of the 12 hpf optic vesicle (Figure 4G-G''). This bias toward the ventral side provides excess space for tissue growth in the ventral direction. These factors, when met, result in an optimal timelapse (Figure 4I-I''', Movie 1). A sub-optimal sample has mosaic or dim fluorescence, has already developed past 12 hpf (when optic cup morphogenesis is under way), or is positioned poorly in the Z-stack or the XY frame (Figure 4H-H''). When these criteria are not met, the timelapse will no longer capture the entire 3D volume of the tissue as it develops and cannot be used for further analysis (Figure 4J-J''').

Figure 1: Workflow of 4D timelapse imaging of zebrafish optic cup morphogenesis. (A) To fluorescently label subcellular structures of interest, synthesize the appropriate capped mRNAs. (B) Microinject mRNA(s) into zebrafish embryos at the 1-cell stage. (C) Embryos are mounted dorsally, or head-down for an inverted confocal microscope, at the optic vesicle stage, ~11 h post fertilization or 3 somite-stage. (D) Multiple embryos can be sequentially imaged during a single timelapse imaging session. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: A visual guide for microinjecting 1-cell zebrafish embryos. (A) Items to include in an injection kit (clockwise): medium sized box that can hold the entire kit with disposable nitrile gloves; a dish containing microinjection needles; forceps; an aliquot of mineral oil and cut up squares of parafilm; microloading tips; diluted RNA on ice; agarose injection mold; marker; roller/transfer pipette; and P10 pipette. (B) Microinjection set-up: a dissecting microscope next to a pico-injector that is connected to an external compressed gas source. A foot pedal and micromanipulator are attached to the pico-injector. The micromanipulator is equipped with a needle holder and positioned in place above the microscope stage. (C) An agarose injection mold containing six rows of grooves with a 90-degree angle on one side and a 45-degree angle on the other side. (D) The pico-injector front panel. The pressure display reads 7.8 PSI; this can be adjusted with the knob labeled Pinject. Beneath, there are pushbuttons to manually trigger pressure to inject, to change the injection time, or to clear, fill, or hold the liquid in the needle. The inject time is set to 08 (equivalent to 8 x 10 msec). An output hose is connected to Pout and is attached to the injection needle. The pressure meter source switch is set to Pinject while setting the injection pressure, and it can be switched to Pbalance to adjust the basal pressure applied to the needle when injections are not being made. The Pbalance regulator is next to the Pinject regulator. The power button is lit to indicate the machine is turned on. (E) Adult zebrafish are mated in small breeding tanks that contain a nested tank with a mesh bottom that separates adult fish from fertilized eggs and allows easy collection. Water level is lowered to mimic shallow water conditions and tank dividers are removed to initiate mating behavior. (F) The micropipette (needle) puller front panel: contains a display which shows the specific conditions used to pull needles. The pressure setting is P = 500, followed by the last date and time the program was edited, the program number, HEAT = 546, PULL = 130, VEL = 70, and TIME = 90. (G) One glass capillary is inserted into the micropipette (needle) puller and secured into place. Each programmed pull run results in two long tapered needles (red arrowheads). (H) The RNA dilution is backloaded into a needle; the phenol red dye allows easy visualization of the solution. (I) After breaking the tip of the needle, the bolus of RNA is measured using an eyepiece reticle on the dissecting scope. For this stereomicroscope, at 30x total magnification 6 hash marks is equal to 1 nL volume. (J) Fertilized eggs are loaded into the agarose injection mold and distributed along the troughs of the mold using a roller pipette. (K) The injection mold containing one-cell stage embryos is positioned under the microscope and embryos are injected sequentially. (L) The injection needle is inserted into each embryo targeting the single cell. (M) Once in the cell, the foot pedal is used to trigger a regulated amount of pressure and the release of 1 nL of RNA into the cell of the embryo (as visualized by phenol red). Scale bars: I = 0.1 mm; L, M = 0.5 mm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: A visual guide for mounting embryos for imaging. (A) Mounting set up (from left to right): a heat block programmed to 42 °C, heating tubes of low-melt agarose; a roller pipette; a pair of forceps; an agarose coated dish containing dechorionated embryos; and a dissecting stereomicroscope connected to a metal halide fluorescent lamp. (B,B',B'') An 11 hpf embryo injected with EGFP-CAAX mRNA and mCherry-H2A mRNA, shown under brightfield, GFP fluorescence and mCherry fluorescence, respectively. (C,C') A glass pasteur pipette containing agarose and an embryo sitting in the tip; C' is a magnified view. (D,D') One embryo mounted in a droplet of low-melt agarose on a glass bottomed dish, seen both from the stage of the microscope and through the microscope eyepieces. (E,E') 12 embryos mounted in individual droplets of low-melt agarose on a glass bottomed dish, seen both from the stage of the microscope and through the microscope eyepieces. (F,F',F'',F''') All mounted embryos are overlaid with a complete layer of agarose to fill the bottom of the dish, seen both from the stage of the microscope and from the microscope eyepieces shown in bright field; F'' GFP fluorescence; and F''' mCherry fluorescence. (G) An optimal sample at 12 hpf is mounted dorsally and oriented vertically in line with 12 and 6 o'clock with both optic vesicles in the same plane. (H) A sub-optimal sample: although mounted dorsally and optic vesicles are in plane with each other, this sample is oriented on a diagonal axis and will grow out of the frame size as development proceeds. (I) A sub-optimal sample: this sample is over-rotated and not a dorsal mount, as a result, the anterior portion of the optic vesicle will not be captured in the timelapse. (J) A sub-optimal sample: this sample is under-rotated and not a dorsal mount, as a result the posterior portion of the optic vesicle will not be captured in the timelapse. Dotted lines indicate each optic vesicle. Scale bars: B = 0.1 mm; D' = 1.5 mm; E', F' = 2.5 mm; G = 0.1 mm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Setting up a multiple position timelapse and potential results. (A) Laser scanning confocal microscope. (B) Piezo Z-stage insert. (C) The bottom of a glass bottom dish containing embryos embedded in agarose is coated in immersion medium matching the refractive index of water. (D) A droplet of immersion medium is placed on the 40x W objective (long working distance). (E) The dish is held into place with the stage insert, E3 is added on top of the agarose layer, and the lid is secured with modeling clay. (F) Acquisition software settings: The desired laser lines are turned on from the drop-down menu. For embryos injected with EGFP-CAAX RNA and H2A-mCherry RNA, the Argon and DPSS 561-10 lasers are turned on. The red highlight indicates that the Argon laser is warming up. To locate the sample over the objective, the transmitted light is turned on through the microscope control panel. Both EGFP and mCherry are assigned to Track 1 (for simultaneous imaging) and the range of each detector is set. Here, the EGFP range is set to 494-545 nm, and the mCherry range is set to 598-679 nm. Under the Channels menu, laser power can be set. After the lasers have warmed up, the power can be increased up to 5.0, and gain (master) can be adjusted. The pinhole is set at 60.2, which is equal to 1.63 Airy Units or 1.6 µm section. Under the Acquisition menu, the frame size is set to 512 x 384, scan speed is 9.0, averaging is bidirectional, and zoom is set to 0.7. Under the Z-Stack menu, the first and last position in the Z-axis are set while the confocal is scanning. The interval (step size) is set to 2.1 µm. While choosing first and last Z-position, a standard number of 63 slices is generally maintained; this accommodates the overall growth of the eye. The "use piezo" option is selected. Under the Positions menu, each selected position is listed including an assigned number and X, Y, and Z position. There are additional buttons to control the number of embryos (separate positions) to image, including add, update, or remove. Under the Time Series menu, the cycle is set to 300 (the number of Z-stacks that will be acquired) and interval (time step) is set to 2.5 min. Once the settings are finalized, timelapse acquisition is initiated by pressing start. The software will show the slice of the Z-stack, the cycle, and the position that is currently scanning. When the timelapse is complete, the file is saved by clicking on the floppy disk icon in the Images and Documents panel. (G-J) Cell membranes (green) are labeled with EGFP-CAAX and cell nuclei (chromatin, magenta) are labeled with H2A-mCherry RNAs. (G,G',G'') An example of setting the first and last Z-slice for an optimally mounted sample. The first Z-slice (at the dorsal side) comes at the expense of dorsal ectoderm, while the last Z-slice (at the ventral side) is well below the optic vesicle, to accommodate its growth in the ventral direction. (H,H',H'') An example of setting the first and last Z-slice of a sub-optimal sample. The sample has weak mCherry fluorescence and it is mounted diagonally, such that the developing optic cup is unlikely to stay in frame for the duration of the timelapse. (I,I',I'',I''') An example of a timelapse from an optimal sample. Single slice images from the middle of the Z-stack are taken from the entire 63 slice volume at 4 time points (T value). Br, brain; NR, neural retina; RPE, retinal pigmented epithelium; Le, lens. (J,J',J'',J''') An example of a timelapse from a suboptimal sample. In this case, the sample moved out of the Z-plane and only an oblique portion of the anterior optic cup was captured. Scale bars: G, I = 50 µm. Please click here to view a larger version of this figure.
{kind=link}
Movie 1: Timelapse of optic cup morphogenesis in an optimal wild type embryo. A dorsal view of a wild type embryo that is labeled with EGFP-CAAX (plasma membranes, green) and H2A.F/Z-mCherry (chromatin, magenta). The timelapse is from ~12-24 hpf and contains a single confocal section from an entire 4D dataset. Time interval is 2.5 min between z-stacks and is shown at 22.5 frames per second. Please click here to download this movie.
Discussion
Here, we describe a protocol for in toto labeling and 4D timelapse imaging of optic cup morphogenesis. We step through the process of generating capped RNA encoding fluorescent proteins to mark different subcellular compartments; injecting zebrafish 1-cell embryos; embedding 11 hpf embryos in agarose for multiplexed imaging; and acquiring 4D datasets of multiple embryos for the duration of optic cup morphogenesis (12–24 hpf).
A myriad of questions can be answered with these information-dense datasets. 4D data can be visualized and quantitatively analyzed in a variety of ways. Although outside of the scope of this protocol, we include here some of our goals and standard applications as an example of the types of things that can be accomplished. Of course, quantitative image analysis methods are constantly being developed, and both commercially available and custom-built tools can be used. If one has not used such methods before, one should be prepared to work through some optimization to ensure that the acquired datasets are adequate for the quantitative analysis approach of choice.
Visualizing and quantitatively evaluating the 4D datasets can be challenging, due to the size of the files. The acquisition software can be used to separate the datasets into individual embryos, and ImageJ/Fiji can be used to convert confocal files from commercial formats to more standard tif stacks, in which each timepoint is saved as a separate file. This will decrease the file sizes and standardize the file formats. Individual optical sections from each timepoint can be assembled as a 2D (XY) timelapse using ImageJ/Fiji, enabling rapid 2D visualization and evaluation of the data. Movie 1 is an example of precisely this: a single optical section over time assembled as a timelapse of the optimal sample shown in Figure 4I–I’’’. From there, for 3D and 4D visualization, we commonly use FluoRender35,36, which is freely available but has specific graphics card requirements. Using FluoRender, one can rotate 3D-rendered data in any axis, visualize the dataset in 4D over time, generate cutaways in any plane, and save the rotations and visualizations as a movie or series of tif images.
In terms of quantitative analysis, there are numerous questions to be answered. We have developed software in-house to aid our own specific goals of understanding the cell behaviors underlying optic cup morphogenesis. Our program, LongTracker, uses the nuclear signal as a proxy for position to track cells13. With these data, we can determine when, where, and how cells move; how fast and how far cells move; and how frequently cells divide. In addition to our own software, there are multiple commercially available and custom-built options for 4D cell tracking. We have also developed the program LongAxis to carry out cell segmentation and quantify cell shape and organization within the neural retina37. The datasets input into LongAxis, however, are single Z-stacks, taken at a high resolution. A persistent challenge has been generating timelapse datasets with high enough resolution that cells can be segmented with confidence and their morphology extrapolated. One alternative is mosaic (sparse) labeling using a photoconvertible fluorophore such as Kaede for direct visualization of cell shape, as we and others have carried out in the developing eye8,11,13. This simplifies the cell segmentation problem, and cell shape and size can be readily quantified through 3D rendering in FluoRender.
Each step of this protocol was optimized specifically for our purposes. The specificity of this protocol results in several limitations: the protocol, as written, is not optimized for imaging other aspects of zebrafish eye development (such as retinal neurogenesis), other eye structures, other developmental stages, or other subcellular compartments. The orientation of embedding, the speed of imaging, and the labels are all designed to allow us to answer our biological questions. In order to image retinal neurogenesis, for example, the dorsal orientation of the embryo as described here may not allow for visualization of specific features of interest, and the speed of imaging may not be appropriate. Many aspects of the protocol can be adapted and modified for a variety of needs, depending on one’s specific interests and goals. First, using RNA injections makes the labeling process very flexible. Fluorescent protein fusion constructs can be used to label subcellular structures of interest, and RNA injection amount can be varied to optimize labeling. Based on our work with the photoconvertible fluorophore Kaede, RNA injections appear to support a burst of translation that is over by 12 hpf11,13. High levels of fluorescent protein expression from RNA injection could combat photobleaching, but such an approach does not support sustained expression of the fluorescent label of interest. If sustained expression is necessary, such as when imaging embryos at later stages, transgenic lines are an option, and constructing new lines in zebrafish is straightforward24.
Next, the protocol can be adapted for later stages of development. Because pigment may impede imaging at later stages, embryos can be treated with phenylthiourea (PTU) to inhibit pigment formation, or genetic mutants for pigment synthesis can be used. To prevent embryo twitching, tricaine can be added to the agarose and embryo media overlay solutions, and the percent agarose can be adjusted as necessary. As the eye grows, it may be necessary to change the mounting orientation; here, we mount embryos dorsally, but at later stages, it may make more sense to mount laterally or anteriorly, depending on the structure of interest. Because different processes occur over different spatial and temporal scales, one can also optimize the Z-step and time step of image acquisition. These features can really only be determined empirically for each lab’s needs.
Finally, this protocol was developed specifically for a laser scanning confocal microscope, with embryos embedded in a relatively high percentage of agarose at an early stage of eye development. If one is imaging at different times or different locations during eye development, this protocol will need to be adapted for the process of interest. Many different imaging approaches are currently possible, with more being developed by optical engineers. Each approach brings its own set of challenges, from different ways to embed and mount embryos for imaging, to different file sizes and formats. We outline considerations in the introduction to guide the optimization process, in which maximal spatial and temporal resolution are balanced with photobleaching, phototoxicity, and immense file sizes. We hope that these general principles, in addition to the practical information described above, will aid others in establishing timelapse imaging approaches to study the many open questions in eye development.
Acknowledgements
We are grateful to past and present members of the Kwan Lab for work on timelapse approaches and discussions of this protocol. This work was supported by the NIH (R01 EY025378 and R01 EY025780 to KMK; F31 EY030758 to SL; and T32 GM007464 to MAC).
Materials
| Name | Company | Catalog Number | Comments |
| Delta TPG Dish 0.17mm clear | Bioptechs | 0420041500C | coverglass bottom for optical compatibility |
| Disposable Pasteur Pipettes | VWR | 14672-608 | overall length 14.6 cm (53/4") |
| Dumont #5, #55, or #5SF Forceps | Fine Science Tools | 11254-20 | For style #5: straight tip, 0.05 x 0.01 mm tip, inox, 11 cm long |
| Flaming/Brown Micropipette Puller | Sutter | P-97 | |
| Immersol W | Carl Zeiss Microscopy | 4449690000000 | immersion fluid for water-emission objectives, halogen-free, low fluorescence |
| Microinjection Mold | Adaptive Science Tools | TU-1 | Six ramps, one 90 degree and one 45 degree beveled side. |
| Microloader Tips | Eppendorf | 930001007 | Microloader, tip for filling glass microcapillaries, 0.5 – 20 µL |
| mMessage mMachine SP6 Transcription Kit | Invitrogen | AM1340 | contains SP6 Enzyme Mix, 10X Reaction Buffer, 2XNTP⁄CAP Solution, GTP Solution, pTRI-Xef, TURBO DNase, Ammonium Acetate Stop Solution, Lithium Chloride Precipitation Solution, and Gel Loading Buffer and are all stored at –20°C. Nuclease-free Water may be stored at any temperature. |
| Nikon SMZ645 Stereo Microscope | Nikon | SMZ645 | used for injecting embryos (see Fig. 2B) |
| NotI-HF enzyme | NEB | R3189S | comes with Gel Loading Dye, Purple (6X) |
| NuSieve GTG Agarose | Lonza | 50081 | fine resolution, low melt agarose |
| Objective LD "C-Apochromat" 40x/1.1 W Korr M27 | Carl Zeiss Microscopy | 421867-9970-000 | |
| Olympus SZX16 Stereo Microscope | Olympus | SZX2-ZB16 | used for screening, embedding embryos (see Fig. 3A) |
| Phenol red solution | Sigma-Aldrich | P0290 | 0.5% in DPBS, sterile filtered, endotoxin tested, cell culture tested |
| Pico-liter Microinjector | Harvard Apparatus | PLI-100 | Femtoliter to microliter injections; digital readouts for injection count, time, and pressure; contains 5 pressures including inject, balance, clear, fill, and hold |
| Pipette Pump Pipettor | SP Bel-Art | F37898 | fits a disposable pasteur pipette, contains thumbwheel on side for aspirating or dispensing and plunger for quick emptying |
| Premium Thin Wall Borosilicate Glass Capillaries | Warner Instruments | G100T-4 | 1.0 x 0.78 mm |
| QIAquick PCR Purification Kit | Qiagen | 28106 | contains QIAquick Spin Columns, Buffers, Collection Tubes (2 ml) |
| RNase-free H2O | Invitrogen | AM9906 | DNase-Free, Molecular Biology Grade, RNase-Free |
| RNeasy Mini Kit | Qiagen | 74104 | contains RNeasy Mini Spin Columns, Collection Tubes (1.5 ml and 2 ml), RNase-free Reagents and Buffers |
| Stage Attachment Z PIEZO WSB 500 | Carl Zeiss Microscopy | 432339-9000-000 | |
| Stage Insert Z PIEZO WSB Universal | Carl Zeiss Microscopy | 432339-9030-000 | |
| Zeiss LSM 880 with Airyscan | Carl Zeiss Microscopy | N/A | inverted Axio Observer microscope |
| Zen Black 2.3 SP1 software | Carl Zeiss Microscopy | N/A | Zeiss Efficient Navigation (ZEN) controls all light microscope systems from Zeiss |
References
- Adler, R., Canto-Soler, M. V. Molecular mechanisms of optic vesicle development: complexities, ambiguities and controversies. Developmental Biology. 305 (1), 1-13 (2007).
- Chow, R. L., Lang, R. A. Early eye development in vertebrates. Annual Review of Cell and Developmental Biology. 17, 255-296 (2001).
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Current Topics in Developmental Biology. 93, 61-84 (2010).
- Martinez-Morales, J. R., Wittbrodt, J. Shaping the vertebrate eye. Current Opinion in Genetics and Development. 19 (5), 511-517 (2009).
- Yang, X. J. Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Seminars in Cell & Developmental Biology. 15 (1), 91-103 (2004).
- Bogdanovic, O., et al. Numb/Numbl-Opo antagonism controls retinal epithelium morphogenesis by regulating integrin endocytosis. Developmental Cell. 23 (4), 782-795 (2012).
- Bryan, C. D., Casey, M. A., Pfeiffer, R. L., Jones, B. W., Kwan, K. M. Optic cup morphogenesis requires neural crest-mediated basement membrane assembly. Development. 147 (4), (2020).
- Bryan, C. D., Chien, C. B., Kwan, K. M. Loss of laminin alpha 1 results in multiple structural defects and divergent effects on adhesion during vertebrate optic cup morphogenesis. Developmental Biology. 416 (2), 324-337 (2016).
- Cavodeassi, F., Ivanovitch, K., Wilson, S. W. Eph/Ephrin signalling maintains eye field segregation from adjacent neural plate territories during forebrain morphogenesis. Development. 140 (20), 4193-4202 (2013).
- England, S. J., Blanchard, G. B., Mahadevan, L., Adams, R. J. A dynamic fate map of the forebrain shows how vertebrate eyes form and explains two causes of cyclopia. Development. 133 (23), 4613-4617 (2006).
- Gordon, H. B., et al. Hedgehog signaling regulates cell motility and optic fissure and stalk formation during vertebrate eye morphogenesis. Development. 145 (22), (2018).
- Ivanovitch, K., Cavodeassi, F., Wilson, S. W. Precocious acquisition of neuroepithelial character in the eye field underlies the onset of eye morphogenesis. Developmental Cell. 27 (3), 293-305 (2013).
- Kwan, K. M., et al. A complex choreography of cell movements shapes the vertebrate eye. Development. 139 (2), 359-372 (2012).
- Martinez-Morales, J. R., et al. Ojoplano-mediated basal constriction is essential for optic cup morphogenesis. Development. 136 (13), 2165-2175 (2009).
- Miesfeld, J. B., et al. Yap and Taz regulate retinal pigment epithelial cell fate. Development. 142 (17), 3021-3032 (2015).
- Nicolas-Perez, M., et al. Analysis of cellular behavior and cytoskeletal dynamics reveal a constriction mechanism driving optic cup morphogenesis. eLife. 5, (2016).
- Picker, A., et al. Dynamic coupling of pattern formation and morphogenesis in the developing vertebrate retina. PLoS Biology. 7 (10), e1000214 (2009).
- Rembold, M., Loosli, F., Adams, R. J., Wittbrodt, J. Individual cell migration serves as the driving force for optic vesicle evagination. Science. 313 (5790), 1130-1134 (2006).
- Sidhaye, J., Norden, C. Concerted action of neuroepithelial basal shrinkage and active epithelial migration ensures efficient optic cup morphogenesis. eLife. 6, (2017).
- Driever, W., et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development. 123, 37-46 (1996).
- Haffter, P., et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development. 123, 1-36 (1996).
- Nusslein-Volhard, C. The zebrafish issue of Development. Development. 139 (22), 4099-4103 (2012).
- Hoshijima, K., et al. Highly efficient CRISPR-Cas9-based methods for generating deletion mutations and F0 embryos that lack gene function in zebrafish. Developmental Cell. 51 (5), 645-657 (2019).
- Kwan, K. M., et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics. 236 (11), 3088-3099 (2007).
- Almeida, A. D., et al. Spectrum of Fates: a new approach to the study of the developing zebrafish retina. Development. 141 (9), 1971-1980 (2014).
- Baye, L. M., Link, B. A. Interkinetic nuclear migration and the selection of neurogenic cell divisions during vertebrate retinogenesis. The Journal of Neuroscience. 27 (38), 10143-10152 (2007).
- Clark, B. S., et al. Loss of Llgl1 in retinal neuroepithelia reveals links between apical domain size, Notch activity and neurogenesis. Development. 139 (9), 1599-1610 (2012).
- Das, T., Payer, B., Cayouette, M., Harris, W. A. In vivo time-lapse imaging of cell divisions during neurogenesis in the developing zebrafish retina. Neuron. 37 (4), 597-609 (2003).
- Kay, J. N., et al. Transient requirement for ganglion cells during assembly of retinal synaptic layers. Development. 131 (6), 1331-1342 (2004).
- Norden, C., Young, S., Link, B. A., Harris, W. A. Actomyosin is the main driver of interkinetic nuclear migration in the retina. Cell. 138 (6), 1195-1208 (2009).
- Suzuki, S. C., et al. Cone photoreceptor types in zebrafish are generated by symmetric terminal divisions of dedicated precursors. Proceedings of the National Academy of Sciences of the United States of America. 110 (37), 15109-15114 (2013).
- Wan, Y., et al. The ciliary marginal zone of the zebrafish retina: clonal and time-lapse analysis of a continuously growing tissue. Development. 143 (7), 1099-1107 (2016).
- Yoshimatsu, T., et al. Presynaptic partner selection during retinal circuit reassembly varies with timing of neuronal regeneration in vivo. Nature Communications. 7, 10590 (2016).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Wan, Y., Otsuna, H., Chien, C. B., Hansen, C. An interactive visualization tool for multi-channel confocal microscopy data in neurobiology research. IEEE Transactions on Visualization and Computer Graphics. 15 (6), 1489-1496 (2009).
- Wan, Y., Otsuna, H., Chien, C. B., Hansen, C. FluoRender: an application of 2D image space methods for 3D and 4D confocal microscopy data visualization in neurobiology research. IEEE Pacific Visualization Symposium. , 201-208 (2012).
- Carney, K. R., Bryan, C. D., Gordon, H. B., Kwan, K. M. LongAxis: a MATLAB-based program for 3D quantitative analysis of epithelial cell shape and orientation. Developmental Biology. 458 (1), 1-11 (2020).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved