Workflow and Tools for Crystallographic Fragment Screening at the Helmholtz-Zentrum Berlin
In This Article
Summary
Crystallographic fragment screening at the Helmholtz-Zentrum Berlin is performed using a workflow with dedicated compound libraries, crystal handling tools, fast data collection facilities and largely automated data analysis. The presented protocol intends to maximize the output of such experiments to provide promising starting points for downstream structure-based ligand design.
Abstract
Fragment screening is a technique that helps to identify promising starting points for ligand design. Given that crystals of the target protein are available and display reproducibly high-resolution X-ray diffraction properties, crystallography is among the most preferred methods for fragment screening because of its sensitivity. Additionally, it is the only method providing detailed 3D information of the binding mode of the fragment, which is vital for subsequent rational compound evolution. The routine use of the method depends on the availability of suitable fragment libraries, dedicated means to handle large numbers of samples, state-of-the-art synchrotron beamlines for fast diffraction measurements and largely automated solutions for the analysis of the results.
Here, the complete practical workflow and the included tools on how to conduct crystallographic fragment screening (CFS) at the Helmholtz-Zentrum Berlin (HZB) are presented. Preceding this workflow, crystal soaking conditions as well as data collection strategies are optimized for reproducible crystallographic experiments. Then, typically in a one to two-day procedure, a 96-membered CFS-focused library provided as dried ready-to-use plates is employed to soak 192 crystals, which are then flash-cooled individually. The final diffraction experiments can be performed within one day at the robot-mounting supported beamlines BL14.1 and BL14.2 at the BESSY II electron storage ring operated by the HZB in Berlin-Adlershof (Germany). Processing of the crystallographic data, refinement of the protein structures, and hit identification is fast and largely automated using specialized software pipelines on dedicated servers, requiring little user input.
Using the CFS workflow at the HZB enables routine screening experiments. It increases the chances for successful identification of fragment hits as starting points to develop more potent binders, useful for pharmacological or biochemical applications.
Introduction
The first step in drug development is the screening of compounds against a target of interest. Traditionally, large compound libraries in the order of 100,000-1,000,000 entries are used in high-throughput biochemical assays in the pharmaceutical industry. This strategy was complemented by fragment-based drug design (FBDD), a newer method that took a steep rise during the last 20 years and became a mainstream strategy to generate high-quality lead candidates due to several inherent advantages of the method1. The term "fragment" refers to a small organic molecule containing typically less than 20 non-hydrogen or heavy atoms (HAs). Thus, a fragment is significantly smaller than the drug- or lead-like molecules (usually less than 30 HAs) explored in conventional high-throughput screening. Fragments are weak-affinity binders. However, compared to larger molecules, fragments are more versatile, since even a small collection of them can better represent the respective chemical space of molecules of the same size2. Also, evolving fragment screening hits into lead molecules is considerably more effective than optimizing already larger molecules2,3,4,5. That means, pending sufficient sensitivity of the detection, screening of fragments can be employed efficiently and yields high-quality starting points for further compound evolution. Several biophysical methods may be applied for fragment screening, the most popular being nuclear magnetic resonance, X-ray crystallography, surface plasmon resonance and thermal shift assays. These methods are used either in a parallel or in a sequential way, with the aim to increase the confidence in the hits and reduce the numbers of false positives or false negatives, respectively. However, a recently conducted comparative study6 suggested that sequential screening cascades are to be avoided due to the low overlap between the different methods.
X-ray crystallography is a well established method for structure determination at atomic detail but has recently also been developed as a tool for screening purposes7,8. As protein crystals tolerate high fragment concentrations (e.g., 100 mM), crystallographic fragment screening (CFS) can compete with other biophysical methods for screening fragments or even outperform them as a first-step screening method6,9. However, a vital pre-requisite for CFS is a validated crystallization system of the target protein reproducibly delivering crystals with diffraction properties to considerably high resolution, typically better than 2 Å.
An exclusive benefit of CFS compared to all other fragment screening methodologies is the provision of detailed 3D information about the binding mode of the identified fragments. This structural information is absolutely crucial for the rational optimization of the fragment hits to higher-affinity binders. Established elaboration strategies are growing, merging, and linking fragment hits5. Thereby relatively high ligand efficiency is provided from the start, and the introduction of unnecessary or spatially not suitable groups can be avoided, thus reducing chemical synthesis costs. All in all, CFS has unrivaled advantages as a starting strategy for drug design.
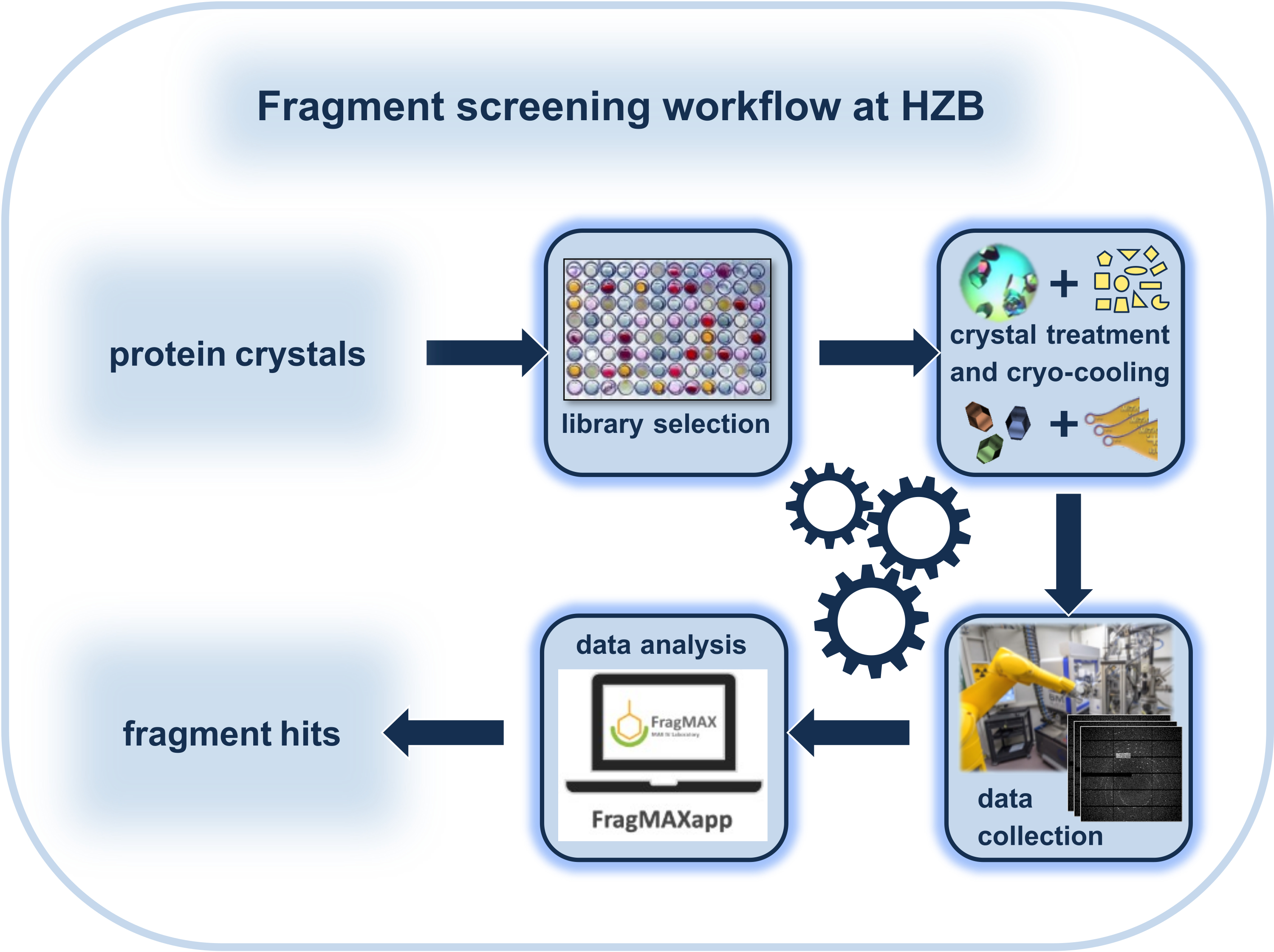
Given that a particular biological target meets the high requirements of CFS regarding crystal quality, there are some main factors that maximize the chances for a successful outcome of such screening campaigns. It depends on the quality of the fragment library used, on an efficient workflow to carry out the experiments before the diffraction experiment, on synchrotron beamlines with sufficient automation and data collection speed, as well as on ways and means for largely automated data processing and analysis. Here, the complete workflow from the crystal soaking experiments to the hit identification is presented, in the way it is successfully established at the macromolecular crystallography beamlines at BESSY II (Figure 1). The facility is open to academic and industrial users for collaboration. Additionally, academic users of EU-countries outside Germany can straightforwardly apply for funding via the iNEXT Discovery project.
There are indispensable prerequisites to be able to start a CFS campaign and conduct the protocol outlined in this work: well-diffracting crystals of the target protein are available that can be reproducibly grown in large numbers, that are stable at ambient temperature, and that were grown using a crystallization cocktail without highly volatile ingredients. Another prerequisite is the suitability of the crystal lattice for the experiment. In an appropriate lattice, the interesting sites of the target protein must be exposed towards the solvent channels and thus accessible. Another preceding step that is optional but nevertheless highly recommended to ensure success in the workflow of the CFS campaign is the optimization of the soaking condition for the experiment. Vital benchmark statistics here are the diffraction power of the crystal and the relevant data quality indicators, which are determined during the data scaling procedure. Typical factors to optimize are DMSO-tolerance, buffer concentration and cryo-protectant. Although not a strict prerequisite as further detailed below, DMSO as a co-solvent can help to increase fragment solubilization. Typical tests should include soaking of 0, 3, 6, or 10% (v/v) DMSO overnight. An increase of the buffer concentration to 200 or 300 mM helps to prevent loss in diffraction quality due to occasional pH-shifting effects arising from the high fragment concentrations to be used. Finally, it is decisive to find out whether and which additional cryoprotectant is required and if it can be already included in the soaking condition. In many cases, however, an additional cryo-protectant is not needed, because DMSO itself can act as a cryo-protectant. If so, this will save one handling step in the final experiment. Most crystals need less cryo-protectant if flash-cooled on appropriately sized loops, minimizing or avoiding surrounding mother liquor as much as possible. However, in rare cases, a layer of the mother liquor is indeed necessary to prevent damage to the crystal upon flash cooling.
The number of hits obtained in a CFS campaign is not only dependent on the druggability of the target protein and the suitability of the crystal lattice (see above), but it is also dependent on the quality of the library. Library quality comprises two aspects: the selection of the compounds for the library and the confectioning of the compounds, (i.e., in which physical form they are presented for the experiment). For compound selection different strategies can be employed. Most library designs include the maximization of the chemical diversity of the fragments. A strategic focus could be to include the chemical tractability of the fragments for follow-up design, which has been applied for instance in the Diamond-SGC-iNEXT poised library10. Yet another strategic focus for library design could be to maximize the representation of commercially available chemical space of fragments by shape- and pharmacophore-based clustering, as has been exemplified by the F2X libraries developed at HZB11. More specifically, the 1103-membered F2X-Universal Library and representative 96-compound subset for initial CFS campaigns, which is called F2X-Entry Screen, have been developed and the F2X-Entry Screen has been validated successfully11. The F2X-Entry Screen is the primary choice for CFS campaigns at HZB. Subsequently, larger campaigns can then be carried out using the F2X-Universal Library or the 1056-membered EU-OPENSCREEN fragment library12 that is also being offered at HZB. At present, these libraries are available for users of the macromolecular crystallography beamlines of the BESSY II synchrotron in Berlin free-of-charge on the basis of a collaboration contract. That also applies to users via iNEXT Discovery proposals. Moreover, the F2X-Entry Screen is available to all interested scientists on the basis of a material transfer agreement.
With respect to the physical presentation of a library, two approaches are commonly adopted: the fragments are either used as DMSO stock solutions or the fragments are dried and immobilized on ready-to-use plates. At HZB, both the F2X-Entry Screen and the non-volatile compounds of the F2X-Universal Library are presented as dried-on compounds in a 3-lens 96-well MRC low profile crystallization plate. The presentation of the fragments immobilized in crystallization plates has two vital advantages: Firstly, it allows transport of the screening plates to the user's home lab. Therefore, the soaking and crystal handling steps of the workflow presented here (steps 1-3) can be carried out anywhere. Secondly, DMSO-free solution can be employed. DMSO-sensitive targets can thus be screened easily, largely retaining expected hit rates11. However, DMSO does increase fragment solubility, hence it is worthwhile to check the DMSO tolerance of a crystal system of choice beforehand as outlined above.
The protocol outlined below will describe a typical experiment with a 96-compound screen such as the F2X-Entry Screen. For that, approximately 250 crystals need to be prepared in time to be used freshly. It is highly advisable to prepare the soaks for all 96 compounds in duplicate. It is recommended, but optional, to prepare additional mock-soaks that will later help with data analysis using the pan-data density analysis (PanDDA) approach for hit identification13. Mock-soaks are defined as soaking experiments on protein crystals using the same soaking solution as the fragment soaks for the same incubation time, but no fragments are present. If the soaking solution is equal to the crystallization condition, the crystals may be directly harvested from the crystallization plate.
Dependent on the capabilities of the robotic sample changer, different puck formats may have to be used. At the moment, samples for the HZB-operated beamline BL14.1 need to be prepared in Unipuck format, samples for the HZB-operated beamline BL14.2 need to be prepared in SPINE puck format. In this protocol, preparation in Unipuck format is assumed.
Protocol
1.Soaking crystals
- Take the screening plate (here, an F2X-Entry Screen plate, Figure 2) from the -20 °C freezer and place it onto the bench/table for about 30 min to pre-warm it to room temperature, thus avoiding condensation moisture.
- Arrange the working place with two closely arranged microscopes and all tools needed (Figure 3A). The materials are listed in the Table of Materials.
- Choose 3-4 loops of the appropriate size for transfer of the crystals to be soaked and place them close to the microscopes.
- Fill the glass spot plate cavities with de-ionized or distilled water.
- Prepare 5 mL of soaking solution.
- Cut open the bag of the screening plate pre-warmed to room temperature.
- Remove the lid and the foil from the screening plate, while keeping the plate placed on the bench/table.
- Decant the 5 mL of soaking solution in the reagent reservoir.
- Fill each of the 96 reservoirs with 40 µL of soaking solution using the 12-channel pipette.
- Place the EasyAccess Frame on top of the screening plate and secure it with the included clamps by sliding them onto the left and right side of the device.
NOTE: The EasyAccess Frame is a special device for handling multiple crystals, which was developed at the HZB14. It enables easy access to each well by shifting the movable tiles while protecting the other wells from evaporation. - Place the screening plate (incl. the EasyAccess Frame) under the first microscope and the crystallization plate including the crystals to be soaked under the second microscope.
- Slide open well A1 of the screening plate by moving the respective acrylic glass tile of the EasyAccess Frame either with a finger or the supplied pen tool.
- Add 0.4 µL of soaking solution from the reservoir to the fragment containing well (upper left lens) using a fresh pipette tip. Check through the microscope that the drop covers the dried-on fragment, so it can dissolve.
NOTE: Alternatively, this step can be carried out using a pipetting robot before the assembly of the EasyAccess Frame. This way the soaking drops of all wells could be placed in one automatic procedure. However, the authors recommend adding the soaking solution directly before the soaking step as described to ensure that the fragment solubilizes slowly and in the presence of the crystal. This avoids that the crystal experiences a sudden shock upon transfer to a drop with a high fragment concentration. - Under the second microscope, cut open the sealing foil of the crystallization plate at one of the wells that contains the target crystals.
- Transfer two crystals using an appropriately sized loop mounted on the crystal wand to the well A1 of the screening plate under the first microscope.
- Wash the loop in the prepared glass spot plate and dry it by gently touching the tissue. Do this after every transfer to avoid cross contamination with fragment containing soaking solutions.
- Use the microscope to check that the crystals have been properly placed.
- Move on to the next well (e.g., B1).
- Repeat steps 1.13-1.18 with all 96 wells of the screening plate until each soaking drop contains two crystals.
- Remove the screening plate (incl. the EasyAccess Frame) from under the microscope and place it onto the bench/table.
- Remove the EasyAccess Frame from the screening plate.
- Seal the screening plate with sealing foil and place it in the crystallization incubator or cupboard, respectively, where the crystals were grown.
- Incubate for the optimized soaking time. Overnight is usually convenient.
- (optional) Preparation of approximately 40 apo crystals (i.e., mock soaking)
- Take an MRC 3-lens 96-well low-profile crystallization plate and fill two columns with 40 µL of soaking solution per well using the 12-channel pipette.
- Place the EasyAccess Frame on top of the crystallization plate and secure it with the included clamps by sliding them onto the left and right side of the device.
- Slide open the acrylic glass tile of well A1.
- Place 0.4 µL of soaking solution in each of the two left lenses of the well.
- Transfer 2-3 crystals to each drop. After each transfer, wash the loop in the prepared glass spot plate and dry by gently touching the tissue.
- Move to the next well (e.g., B1).
- Repeat steps 1.24.4-1.24.6 until about 40 crystals are ready for incubation.
- Remove the crystallization plate (incl. EasyAccess Frame) from under the microscope onto the bench/table and remove the EasyAccess Frame.
- Seal the crystallization plate with sealing foil and place it in the aforementioned crystallization incubator or cupboard.
- Incubate for the same time as the screening plate.
2.Harvesting crystals
- Take out the incubating plate(s) from the incubator or cupboard, respectively.
- Arrange the working place with one microscope and all tools needed (Figure 3B). The materials are listed in the Table of Materials.
- Prepare a Unipuck foam dewar with 3 Unipuck lids (i.e., sample enclosures) and fill it with liquid nitrogen (LN2).
NOTE: Observe the appropriate safety precautions for working with LN2 (i.e., wear safety goggles and use suitable protective equipment). It is best to get fresh LN2 several times during the session in order to avoid water condensation in the LN2 storage can. Through the entire following procedure, make sure the LN2 level in the foam dewar is always reaching the upper edge of the dewar. Also ensure that the LN2 is ice-free; frequently replace the LN2 (e.g., once every 45 min), or latest if ice starts to accumulate. Then, fill the second foam dewar and transfer Unipucks to it. Empty the icy foam dewar and remove residual ice and moisture with the blow dryer. - Remove the foil from the screening plate and place the EasyAccess Frame on top.
- Slide open well A1.
- Harvest two crystals from the drop and flash-cool them in LN2 (one by one) by plunging with a fast vertical movement into the LN2 and then inserting the sample in the proper puck position. Take relevant notes on the sample tracking sheet.
- Cryoprotection step (if necessary for the target crystals). In such case, perform this step instead of 2.6.
- Place 0.4 µL of soaking solution including cryo-protectant on the lower left lens of the well.
- Pull the loop with a crystal mounted from the drop in the upper left lens slowly through the solution in the lower left lens while keeping the crystal in the loop, and then flash-cool in LN2. Harvest two crystals in this way.
NOTE: In steps 2.6 and 2.7, make sure that the time the crystal is in the loop and exposed to air is kept very short. The plunging (i.e., the vertical drop of the sample in the LN2-filled dewar) should be performed as fast as possible. This ensures high sample quality and prevention of ice rings in the data. Track the samples (i.e., note if crystals have damages, etc.) to prioritize either duplicate for the following X-ray measurements, use the template for that. Even if crystals have cracks, “hairs” or other defects due to the soaking, they can still be used and should always be harvested. In case crystals broke into several pieces, two of the biggest/best looking pieces should be harvested. Figure 5 shows some examples of how such crystals can look like. All the shown crystals gave still useful datasets in the respective campaign11, underlining that it is worth to harvest crystals after soaking treatment, even if substantial morphologic changes occurred.
- Go to the next well and repeat steps 2.5 - 2.6./2.7 until all three pucks are filled.
- Add the Unipuck bases on top of the lids after pre-cooling them in LN2.
- Store the Unipucks in storage racks in a transport dewar or storage dewar.
- Repeat the preceding steps until all the wells of the screening plate have been processed.
- (optional) If mock-soaked crystals were prepared, harvest them in a similar fashion as described beforehand.
NOTE: If two crystals for each of the 96 conditions of the screening plate could be flash-cooled, there will be space for 32 mock-soaked apo crystals, to fill up the 14 Unipucks. - Store the Unipucks in LN2 until the measurement.
3.Data collection
- Transfer the Unipucks to beamline BL14.1. If SPINE pucks have been used in step 2, transfer them to beamline BL14.2.
- Carry out standard measurements on the beamline using the specific recommendations given below. Details about the facility and the experiment control program MXCuBE2 have been presented previously15,16. Figure 4 shows the interior of the experimental hutches of beamlines BL14.1 and BL14.2 as well as an example screenshot of the MXCuBE2 control software at beamline BL14.1.
- To maximize time efficiency and throughput, skip the collection of test images. The sample-to-detector distance will be fixed to a value that is suitable for the upper resolution limit of the crystal system determined in earlier experiments. If the data collection strategy was not optimized beforehand, collect 1800 images of 0.2 degrees each with an exposure time of 0.1 s per image.
- Ideally, test the data collection strategy in prior experiments using mock-soaked apo crystals. For higher symmetry space groups, 1200 images or even 900 images (i.e., 240° or 180°, respectively) will already give complete datasets with good statistics, independent of the starting angle of data collection.
NOTE: Higher redundancy and finer slicing can yield superior data quality17. However, using this “enough but not more” strategy proposed here is an excellent trade-off between quality, data collection time, as well as computational requirements for analysis later on. In the described way, 200 data collections in 24 hours are well possible at beamlines BL14.1 and BL14.2. Nevertheless, samples should be prioritized. - First collect diffraction datasets for one sample per fragment condition, based on the prioritization in step 2.6./2.7 (i.e., collect the data for the higher prioritized duplicate).
- For those experiments in 3.2.3 where data collection failed, diffraction was lost or severe ice rings occurred, collect data for the second duplicate sample of the respective fragment condition.
- Collect diffraction datasets of apo crystals (if prepared according to steps 1.24 and 2.12).
- Collect diffraction datasets of the remaining duplicates of each fragment condition.
- In the MXCuBE2 program, match the dataset identifiers of a CFS campaign to the following pattern: <protein>-<library>-[ABCDEFGH][01][0123456789][ab] (e.g., MyProtein-F2XEntry-B05a, where “B05” stands for the well (i.e., the fragment condition in the screen) and the following “a” for the first duplicate.)
4.Data treatment
- For data analysis of the CFS campaign, use FragMAXapp, a web-based solution to control a multiplex analysis for processing auto-refinement and PanDDA hit evaluation of CFS data18 (Lima et al. FragMAXapp, unpublished data). In the FragMAXapp version deployed at HZB the following programs/pipelines are available: XDSAPP19, Xia2-DIALS and Xia2-XDS20, fspipeline7, DIMPLE21, Phenix LigFit22, PanDDA13,23. Use a well refined input model of the target protein as input for automatic refinement; otherwise perform meticulous refinement of one high resolution mock-soaked crystal that was collected during the campaign.
NOTE: A key element for hit identification is PanDDA. Details are explained in the respective publications13,23. In brief, PanDDA automatically calculates electron density maps of a set of data sets in a CFS campaign. These are then assumed as non-binding fragment conditions and averaged to generate the so-called ground state model. The ground state model is then used to derive local discrepancies between each electron density map and the ground state map, using voxel-associated Z-scores. Then, for areas of high Z-scores a so called PanDDA-map is created by fine-tuned subtraction of ground state density from the respective map. This largely enhances the visibility of fragment binding events. - To maximize the outcome of PanDDA, use a two-step approach. Firstly, performing a PanDDA run (pandda.analyse) with standard settings. Even if mock-soaked crystals have been collected, their identity will not be included as a parameter (which is possible nonetheless) in order to enable an unbiased generation of the ground state model by PanDDA from all available data. Afterwards, the output data is evaluated by the user via a so-called PanDDA inspection in Coot24. Here, hits with relatively high confidence should be noted, concluding the first step.
- Secondly, re-run the pandda.analyse step excluding the preliminary hits (determined in the first step) from the ground state model via the --exclude_from_characterisation="<list-of-bound-dataset-ids>" command line option. Further details are described on the PanDDA help pages (https://pandda.bitbucket.io/). This way, datasets that are clear hits and thus would obscure the ground state model if included are disregarded. This leads to an improved ground state model and thus to improved results overall. Finally, a thorough PanDDA inspection is performed to complete the hit identification.
NOTE: FragMAXapp includes also an output option to save the modeled bound states or prepare data for PDB submission, for further detail see FragMAX webpages (https://fragmax.github.io/).
Representative Results
As part of the previously reported validation campaigns of the F2X-Entry Screen11, three campaigns were conducted at the BioMAX beamline at MAX IV and one campaign was conducted at beamline BL14.1 at HZB. In the latter campaign, a particular set of F2X-Entry Screen conditions using a soaking condition that did not contain DMSO was screened against the protein-protein complex of yeast Aar2 and the RNaseH-like domain of yeast Prp8 (AR). The selected set of conditions comprises the hits that were found in an earlier campaign of the F2X-Entry Screen against AR in a soaking condition containing DMSO11, (i.e., in the campaign performed at HZB those hits were re-screened in the absence of DMSO). Figure 7 shows an overview of the hits obtained after analyzing the data with the FragMAXapp combination of XDSAPP for processing, fspipeline for auto-refinement and subsequent hit finding using PanDDA.

Figure 1: Schematic representation of the workflow of a crystallographic fragment-screening (CFS) experiment with a focus on the special environment at the Helmholtz-Zentrum Berlin. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Formulation and packaging of the F2X-Entry Screen. The 96-compound screen is available on a 3-lens 96-well MRC low-profile plate, sealed with foil and vacuum-packed. The 96 compounds of the screen are dried from DMSO solutions in two of the three lenses of each well. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Photography of the CFS workbench in the HZB preparation lab. Assemblies of necessary tools for A) soaking and for B) crystal harvesting are displayed. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Data collection end stations and control software. A) Photograph of the experimental hutch of HZB-MX beamlines BL14.1 (left) and BL14.2 (right)15. B) Screenshot of the MXCuBE2 experiment control interface16 used at BL14.1 for diffraction data collection. At BL14.2 a very similar interface is used. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Photographic snapshots of some crystalline samples in cryogenic environment before data collection. This illustrates the variability of morphologies of the crystals after performing the fragment soaking and crystal harvesting. The photographs were taken on the BioMAX beamline (MAX IV synchrotron, Lund, Sweden) for AR samples collected there as part of the F2X-Entry Screen validation11. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Screenshot of the FragMaxApp18 installed at the HZB for convenient data analysis. More details in Lima et al., FragMAXapp, unpublished data. Please click here to view a larger version of this figure.
{kind=link}

Figure 7: Overview of the results of the CFS campaign F2X-Entry vs. AR (without DMSO). The AR protein complex is shown in cartoon view, with Aar2 colored in gray and the RNaseH-like domain of Prp8 colored in blue. The fragment hits of the campaign are colored in element colors (C - yellow, O - red, N - blue, S - orange, Cl - light cyan). Please click here to view a larger version of this figure.
{kind=link}
Sample Tracking Sheet Template. Please click here to download this file.
Discussion
For a successful CFS campaign, it is vital to adhere to the described prerequisites (see Introduction). A reliable crystallization system is needed for the reproducible growth of many well-diffracting crystals, and a well-refined structure is needed as the input apo model for automated refinement. It is also important to check that the target site on the protein (active site, or interface area) is accessible for fragments in the crystal lattice. It is crucial to optimize the soaking conditions beforehand to ensure that the soaking does not significantly deteriorate the crystal quality. Neglecting these aspects will very likely lead to a suboptimal experiment, which will be of limited use and will, in the worst case, require a repetition of the entire experiment.
The protocol described above outlines the procedures that are followed during a standard CFS campaign. If all prerequisites are met, at least 90% of all soaked crystals should display diffraction to high resolution in a diffraction experiment. If this is not the case, the soaking times may be shortened to a few hours or even minutes. Due to the good solubility of most of the fragments, this should suffice to obtain decent occupancy values. Also, a typical CFS campaign will result in a hit rate of roughly 10% or above. For the F2X-Entry Screen validation campaigns11 and ongoing user campaigns with the same library even higher hit rates have been observed (20% and above, data not shown).
A general caveat of crystallographic fragment screening is the presence of crystallographic contact sites. These could either occlude a priori known active sites (to be checked before the screening, see above), or these contact sites also often provide pockets and hot spots where fragments can bind. Such fragment hits will be artifacts of the crystallization lattice and will likely not bind to the protein in solution. These events tend to occur more often in soaking experiments than in co-crystallization experiments (probably due to the higher fragment concentrations employed in soaking experiments). However, according to previous experience, they generally constitute only a minor portion of the hits obtained. For example, in the F2X-Entry Screen validation campaign using endothiapepsin (EP) and the spliceosomal protein-protein complex of Prp8RNaseH and Aar2 (AR), most of the hits occurred in promising sites11. For EP, 27 out of the 37 observed binding events were located in the active site (i.e., the peptide cleft of this protease). The 10 remote binding events comprise two solvent exposed binding events and eight crystal contact binding events (corresponding to five unique hits). Excluding those crystal contact hits would still reflect an overall rate of 24% unique hits for the EP campaign. It is also important to notice that binding events remote of a known active site (except crystal contact binders) could also potentially be interesting (e.g., revealing new hot spots or allosteric sites of the protein). For the AR campaign (in the same publication), of the 23 observed binding events, seven were located at crystal contacts, one was located at the direct interface of the two proteins, seven were located at known protein-protein interactions sites with other binding partners of the larger biological context (hence different assembly stages of the spliceosome), eight binding events revealed two hot spots on AR of yet unknown function and one being at a solvent exposed surface of Prp8RnaseH. Therefore, excluding the events at crystal contacts and the Prp8RnaseH singleton, the number of potentially useful binding events is 15 (corresponding to 14 unique hits) thus a hit rate of 15.6%. These hits can be starting points for design of protein-protein interaction modulators or for tool compounds aimed to explore the two discovered Aar2 hot spots. Taken together, also in line with conducted user campaigns, often only a minor portion of hits in crystallographic fragment screening must be disregarded as artifacts. However, this will also be largely target dependent.
If the hit rate is significantly lower, this may indicate one of the following problems related to the target protein. For instance, in a CFS campaign against a viral cysteine protease a hit rate of only 3% was observed (data not shown). It turned out that the protein used was likely chemically modified in its active site. In such a case, a different protein preparation may solve the problem. If crystals are very DMSO intolerant, the F2X-Entry Screen may also be used without DMSO, although the results may differ to some degree. Most of the hits obtained in the presence of DMSO will also show up in its absence. There will also be some hits that cannot be observed in the absence of DMSO, even though they can be observed in its presence. And finally, there will be some that only show up in the absence of DMSO.
The most severe difficulty occurs if the protein undergoes an induced-fit motion upon substance binding. Most likely, the crystal lattice will not tolerate the protein motion and the crystals will disintegrate. In such a case, the only choice is to resort to co-crystallization of the protein and the fragments. This may, however, lead to new crystal forms. Therefore, much of the automation of the entire process will not work efficiently anymore. Luckily, in most CFS campaigns conducted at the HZB so far, this kind of problem has not been encountered. It may be, that the weak binding of a fragment, does not provide enough energy to induce a protein motion, in particular if the crystallized conformation is stabilized by crystal packing forces.
Another serious limitation of the method which the authors have encountered so far is when the crystallization cocktail (and thus the soaking solution) contains volatile compounds. Then it becomes close to impossible to perform all the crystal handling in a meaningful way.
Different proteins may contain druggable sites to a greater or lesser extent. For example, protein-protein interactions are usually mediated by extended flat surfaces that are more difficult to target. The fragment binding hit rate will therefore likely depend on the structure of the protein’s molecular surface. In an extreme case, a protein might not contain any suitable surface hot spots that serve as target sites for fragment binding. Thus, despite a meticulously performed experiment, no fragment hits will result from the screening. However, the authors have so far not encountered such a situation.
In principle, using the protocol outlined above, the crystal soaking and harvesting part of a CFS campaign can be performed in any laboratory that is equipped for crystal handling. This distinguishes the methodology at HZB from other CFS facilities and can be an advantage in some cases. For example, if the crystals cannot be easily re-produced at another site or if the travelling of the experimenters is limited (e.g., in a world-wide pandemic situation), users at HZB are therefore provided with the entire equipment (pucks, tools, EasyAccess Frame, sample holders, etc.) as a portable set.
However, the requirements for large numbers of sample holders and cryogenic storage capacities are still more conveniently met at dedicated CFS facilities. Moreover, the need for collection of many diffraction data sets strongly advocates for localizing these facilities close to beamlines which are geared towards a high sample throughput. Examples for this are the beamlines I04-1 at the Diamond Light Source and the associated XChem facility in UK8,25, the MASSIF beamlines at the ESRF in France26 or the FragMAX facility at the BioMAX beamline at MAX IV in Sweden18.
In the future, it could be envisioned to design CFS experiments without the need for crystal handling altogether. First advances in this direction have been reported. For instance, by acoustic liquid transfer allowing the mixing of both the crystal-containing solutions and the fragment solutions directly on mesh-type sample holders27. Another approach was used for XFEL-based ligand-screening. In a proof-of-principle experiment, a crystal slurry was prepared in batch, and soaking and diffraction data collection were performed on a silicon fixed target chip28. However, these approaches are still under development and far from being applicable to a wide range of protein targets or feasible for CFS facilities as a routine.
With the protocol in this work detailed instructions to successfully perform CFS campaigns straight-forwardly at HZB (and elsewhere) have been outlined and general guidance and useful hands-on tips in preparing and conducting such experiments with higher chances for success have been given. Ultimately, better odds and success rates in CFS screening largely contribute to efficiently providing starting points for downstream development of tool compounds or drug candidates.
Acknowledgements
We thank the numerous user groups that have performed CFS campaigns at the HZB. Their feedback led to the incremental improvement of our workflow. We want to thank the drug design group at the University of Marburg and the FragMAX group at MAX IV, as the close collaborations were the foundation for several developmental leaps for improved CFS. We are thankful for the support by the German Federal Ministry of Education and Science (BMBF), via the projects Frag2Xtal and Frag4Lead (numbers 05K13M1 and 05K16M1). We are additionally grateful for support via iNEXT-Discovery, project number 871037, funded by the Horizon 2020 program of the European Commission.
Materials
| Name | Company | Catalog Number | Comments |
| 1 µL pipet | Eppendorf | EP3123000012 | |
| 12 channel pipet, 100 µL | Eppendorf | EP4861000791 | |
| Blow dryer | TH-Geyer | 9.106 788 | |
| Crystal containing crystallization plates | Contains crystals to be soaked | ||
| Crystallization incubator | Providing constant temperature for crystallization experiment, at HZB: 20°C | ||
| Dual Thickness MicroLoops (LD) of different aperture sizes | MiTeGen | various, e.g. M5-L18SP-75LD | 250 loops in the appropiate size needed for the protocol, can be provided by HZB |
| EasyAccess Frame | HZB | The EasyAccess Frame is a special device for handling multiple crystals, which was developed at the HZB (Barthel et al., 2021). | |
| F2X-Entry Screen plate | HZB | Developed F2X-Entry Screen (Wollenhaupt et al., 2020) | |
| Glas spot plate | VWR | MARI1406506 | |
| Liquid nitrogen | At least a filled up 5 L can | ||
| Liquid nitrogen storage can | n.a. | n.a. | |
| Magentic crystal wand | MiTeGen | M-R-1013198 | |
| Microscopes | Leica | n.a. | |
| MRC 3-lens 96-well low profile crystallization plate | SwissCI | 3W96TLP-UVP | For mock-soaked crystals (optional) |
| Reagent reservoir | Carl Roth | EKT6.1 | 25 ml volume |
| Sample tracking template | https://www.jove.com/files/ftp_upload/62208/TemplateCFSHZBSampleTracking. xlsx | ||
| Scalpel | B. Braun | BA825SU | |
| Sealing foil for microtiter plates | GreinerBioOne | 676070 | |
| Shelved puck shipping canes (for Unipucks) | MiTeGen | M-CP-111-065 | 2 canes made of aluminum; can be provided by the HZB |
| Soaking solution | At least 5 ml are needed | ||
| Soaking solution including cryo-protectant, 150µL | Only needed if soaking solution is not cryo-protectant already | ||
| Tissues | Roth (Kimberly Clark Professional) | AA64.1 | |
| Transport dewar (Whartington dry shipper) | MiTeGen | TW-CX100 | 2 Travel dewars for storage of the 2 unipuck canes, alternatively a storage dewar of type VHC35 or similar could be used. |
| Unipuck foam dewars with lid | MiTeGen | M-CP-111-022 | two foam dewars especially suited for unipuck handling described in the protocol if SPINE pucks are used, different foam dewars might have to be applied. |
| Unipuck starter set | MiTeGen | M-CP-UPSK001 | Can be provided by the HZB |
| Unipucks | MiTeGen | M-CP-111-021 | 14 unipucks; can be provided by the HZB |
References
- Erlanson, D. A., Fesik, S. W., Hubbard, R. E., Jahnke, W., Jhoti, H. Twenty years on: the impact of fragments on drug discovery. Nature Reviews Drug Discovery. 15 (9), 605-619 (2016).
- Hall, R. J., Mortenson, P. N., Murray, C. W. Efficient exploration of chemical space by fragment-based screening. Progress in Biophysics and Molecular Biology. 116 (2-3), 82-91 (2014).
- Erlanson, D. A. Introduction to fragment-based drug discovery. Topics in Current Chemistry. 317, 1-32 (2012).
- Scott, D. E., Coyne, A. G., Hudson, S. A., Abell, C. Fragment-based approaches in drug discovery and chemical biology. Biochemistry. 51 (25), 4990-5003 (2012).
- Lamoree, B., Hubbard, R. E. Current perspectives in fragment-based lead discovery (FBLD). Essays in Biochemistry. 61 (5), 453-464 (2017).
- Schiebel, J., et al. Six Biophysical Screening Methods Miss a Large Proportion of Crystallographically Discovered Fragment Hits: A Case Study. ACS Chemical Biology. 11 (6), 1693-1701 (2016).
- Schiebel, J., et al. High-Throughput Crystallography: Reliable and Efficient Identification of Fragment Hits. Structure. 24 (8), 1398-1409 (2016).
- Krojer, T., et al. The XChemExplorer graphical workflow tool for routine or large-scale protein-ligand structure determination. Acta Crystallographica Section D: Structural Biology. 73 (3), 267-278 (2017).
- Radeva, N., et al. Active Site Mapping of an Aspartic Protease by Multiple Fragment Crystal Structures: Versatile Warheads to Address a Catalytic Dyad. Journal of Medicinal Chemistry. 59 (21), 9743-9759 (2016).
- Cox, O. B. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chemical Science. 7 (3), 2322-2330 (2016).
- Wollenhaupt, J., et al. F2X-Universal and F2X-Entry: Structurally Diverse Compound Libraries for Crystallographic Fragment Screening. Structure. 28 (6), 694-706 (2020).
- . EU OPENSCREEN fragment library Available from: https://www.eu-openscreen.eu/services/compound-collection/fragment-library.html (2020)
- Pearce, N. M., et al. A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nature Communications. 8, 15123 (2017).
- Barthel, T., Huschmann, F. U., Wallacher, D., Klebe, G., Weiss, M. S., Wollenhaupt, J. Facilitated crystal handling using a simple device for evaporation reduction in microtiter plates. Journal of Applied Crystallography. 54, (2021).
- Mueller, U., et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. The European Physical Journal Plus. 130 (7), 141 (2015).
- Oscarsson, M., et al. MXCuBE2: The dawn of MXCuBE collaboration. Journal of Synchrotron Radiation. 26 (2), 393-405 (2019).
- Mueller, M., Wang, M., Schulze-Briese, C. Optimal fine φ-slicing for single-photon-counting pixel detectors. Acta Crystallographica Section D: Biological Crystallography. 68 (1), 42-56 (2012).
- Lima, G. M. A., et al. FragMAX: The fragment-screening platform at the MAX IV Laboratory. Acta Crystallographica Section D Structural Biology. 76 (8), 771-777 (2020).
- Sparta, K. M., Krug, M., Heinemann, U., Mueller, U., Weiss, M. S. XDSAPP2.0. Journal of Applied Crystallography. 49 (3), 1085-1092 (2016).
- Winter, G. xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography. 43 (1), 186-190 (2010).
- Winn, M. D., et al. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D: Biological Crystallography. 67 (4), 235-242 (2011).
- Terwilliger, T. C., Klei, H., Adams, P. D., Moriarty, N. W., Cohn, J. D. Automated ligand fitting by core-fragment fitting and extension into density. Acta Crystallographica Section D: Biological Crystallography. 62 (8), 915-922 (2006).
- Pearce, N. M., Krojer, T., Von Delft, F. Proper modelling of ligand binding requires an ensemble of bound and unbound states. Acta Crystallographica Section D Structural Biology. 73 (3), 256-266 (2017).
- Emsley, P., Lohkamp, B., Scott, W. G., Cowtan, K. Features and development of Coot. Acta Crystallographica Section D: Biological Crystallography. 66 (4), 486-501 (2010).
- Collins, P. M., et al. Chapter Eleven - Achieving a Good Crystal System for Crystallographic X-Ray Fragment Screening. Methods in Enzymology. 610, 251-264 (2018).
- Bowler, M. W., et al. MASSIF-1: a beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. Journal of Synchrotron Radiation. 22 (6), 1540-1547 (2015).
- Cuttitta, C. M., et al. Acoustic transfer of protein crystals from agarose pedestals to micromeshes for high-throughput screening. Acta Crystallographica Section D Biological Crystallography. 71 (1), 94-103 (2015).
- Moreno-Chicano, T., et al. High-throughput structures of protein-ligand complexes at room temperature using serial femtosecond crystallography. IUCrJ. 6 (6), 1074-1085 (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved