Visualizing Ocular Morphogenesis by Lightsheet Microscopy Using rx3:GFP Transgenic Zebrafish
In This Article
Summary
Here, a protocol is provided for time-lapse imaging of ocular morphogenesis using a commercially available lightsheet microscope and an image processing workstation to analyze the resulting data. This protocol details the procedures for embryo anesthesia, embedding in low melting temperature agarose, suspension in the imaging chamber, setting up the imaging parameters, and finally analyzing the imaging data using image analysis software.
Abstract
Vertebrate eye development is a complex process that begins near the end of embryo gastrulation and requires the precise coordination of cell migration, proliferation, and differentiation. Time-lapse imagining offers unique insight to the behavior of cells during eye development because it allows us to visualize oculogenesis in vivo. Zebrafish are an excellent model to visualize this process due to their highly conserved vertebrate eye and their ability to develop rapidly and externally while remaining optically transparent. Time-lapse imaging studies of zebrafish eye development are greatly facilitated by use of the transgenic zebrafish line Tg(rx3:GFP). In the developing forebrain, rx3:GFP expression marks the cells of the single eye field, and GFP continues to be expressed as the eye field evaginates to form an optic vesicle, which then invaginates to form an optic cup. High resolution time lapse imaging of rx3:GFP expression, therefore, allows us to track the eye primordium through time as it develops into the retina. Lightsheet microscopy is an ideal method to image ocular morphogenesis over time due to its ability to penetrate thicker samples for fluorescent imaging, minimize photobleaching and phototoxicity, and image at a high speed. Here, a protocol is provided for time-lapse imaging of ocular morphogenesis using a commercially available lightsheet microscope and an image processing workstation to analyze the resulting data. This protocol details the procedures for embryo anesthesia, embedding in low melting temperature agarose, suspension in the imaging chamber, setting up the imaging parameters, and finally analyzing the imaging data using image analysis software. The resulting dataset can provide valuable insights into the process of ocular morphogenesis, as well as perturbations to this process as a result of genetic mutation, exposure to pharmacological agents, or other experimental manipulations.
Introduction
Embryonic development is a complex process that requires the precise coordination of many different events. The formation of the vertebrate eye begins in the developing forebrain, where a portion of the cells are specified as the eye field. These cells will evaginate toward the surface ectoderm, giving rise to two bilateral optic vesicles1,2,3,4,5,6,7,8,9,10. Contact with the surface ectoderm then induces an invagination of the optic vesicle into an optic cup. The surface ectoderm will give rise to the anterior structures of the eye, such as the lens and cornea, while the optic cup will give rise to the neural retina and retinal pigmented epithelium1,2,3,5,6,7,8,9,10,11,12. Disruptions in this process can lead to congenital defects such as microphthalmia, anophthalmia, and coloboma (MAC). At this time, there are no options to correct these defects3,5,6,7,9,10,11,12. Further studies of the mechanisms of ocular morphogenesis and the problems that can lead to MAC will provide a foundation of knowledge that will potentially lead to treatments. One powerful tool to investigate the dynamic behaviors of cells during eye development is time-lapse imagining, which allows this process to be visualized and characterized in vivo and in real time.
Zebrafish (Danio rerio) are an excellent model to visualize early ocular development using time-lapse imaging. They have a highly conserved vertebrate eye and possess the ability to develop rapidly and externally while remaining optically transparent1. Zebrafish provide a great resource for time-lapse imaging due to these characteristics that mammalian models lack. Time-lapse imaging studies of zebrafish eye development are greatly facilitated by use of the transgenic zebrafish line Tg(rx3:GFP)13. Rx3 (Retinal homeobox protein 3) is a transcription factor essential for eye development14. Rx3 is the first of the three rx genes in the zebrafish to be expressed, starting its expression mid-gastrulation, approximately 8 h post fertilization (hpf)15,16. The rx3:GFP transgene can be visualized in the developing forebrain starting at the 1 somite stage (ss), approximately 10 hpf15,17,18,19,20. In the developing forebrain, rx3:GFP expression marks the cells of the single eye field, and GFP (green fluorescent protein) continues to be expressed through the remainder of ocular morphogenesis. High resolution time lapse imaging of rx3:GFP expression, therefore, allows us to track the single eye field through time as it develops into the retina17,18,20.
Time-lapse imaging studies of zebrafish development have primarily been performed using confocal or Lightsheet microscopy. Confocal microscopy is advantageous in that it allows for the precise imaging of samples along the z-axis and reduces fluorescent background signal. However, it is limited by the amount of time it takes to acquire an image, sample position rigidity, and its propensity toward photobleaching and phototoxicity of live samples. Lightsheet microscopy is an ideal method to image ocular morphogenesis over time due to its ability to penetrate thicker samples for fluorescent imaging, increased flexibility in sample orientation, minimized photobleaching and phototoxicity, and imaging at a high speed21,22,23,24,25,26,27,28,29,30,31. The spatial resolution achievable with current light sheet microcopy systems is approximately 250-500 nanometers (nm). Although this is not significantly different from what can be obtained with confocal microscopy, the sample can be freely rotated and imaged from multiple angles, improving both imaging depth and resolution, and offering much greater flexibility for in vivo time lapse imaging experiments than the confocal platform27,32. For these reasons, Lightsheet microscopy is quickly becoming the favored method for time-lapse imaging studies of zebrafish development. This protocol describes the steps of quantifying oculogenesis through the imaging of Rx3:GFP transgenic zebrafish using a commercially available Lightsheet microscope33 and details a pipeline for image analysis using the arivis software platform.
Protocol
All experiments involving the use of zebrafish were carried out in accordance with protocols established by the University of Kentucky Institutional Animal Care and Use Committee (IACUC).
1. Sample preparation
- Set up a mating pair of Tg(Rx3:GFP) zebrafish in a gated cross tank the night before imaging is planned. The following morning pull the gate as the lights come on in the fish facility34,35.
NOTE: The mating pair of fish selected should be between 4 months to 2 years in age and are easily distinguished as male or female based on body shape and coloration35. - Check the crosses every half hour for embryos and note what time embryos are first visualized in the bottom of the cross tank. Transfer the embryos to a Petri dish and maintain the embryos at 28.5 °C for approximately 10 h to develop to the 1-2 somite stage (ss). Begin to image at the 1-2 somite stage (ss).
- To screen for the presence of somites, observe the embryos under a stereoscope at 10 hpf and count the number of somites1.
NOTE: Any embryos that are beyond the single somite stage should not be used for this experiment. - Out of the embryos that are at the single somite stage, screen for GFP expression using a fluorescence adapter in combination with a stereomicroscope to confirm the presence of the Rx3:GFP transgene. Once 3-5 GFP positive individuals have been identified, use fine forceps to dechorionate the embryos and transfer them into a small Petri dish containing E3 embryo buffer with a glass pipette35.
- Anesthetize the embryos by transferring them into 0.5 mL E3 embryo buffer (pH 7) containing 0.168 mg/mL Tricaine (MS222) in a microcentrifuge tube35,36. Spontaneous muscle movements begin as early as 17 hpf37. Ensure that embryos are anesthetized to image beyond this timepoint.
- Embed the embryos in 1% low melting temperature agarose in 0.168 mg/mL Tricaine and E3.
NOTE: This is the optimal concentration of low melting temperature agarose to allow for the embryo to be held in place but remain pliable enough to permit the growth of the embryo over the imaging time-course30,31. - Prepare a 10 mL solution of 2% low melting temperature agarose in E3 buffer. Heat it in a microwave oven to dissolve agarose using 15 s intervals, to prevent the solution from boiling over. Allow the solution to cool enough to hold without discomfort but not so much that it solidifies, and not cause harm to the embryos. Once the agarose is sufficiently cool, add 0.5 mL to the embryos in the tube of Tricaine in E3 and gently pipet the solution and embryos to mix.
- Using a 1 mm glass capillary and polytetrafluoroethylene (PTFE) plunger, pull up the embryos in the agarose solution into the capillary. Make sure to pull a total of 3-5 embryos (multiple embryos) into the capillary to increase the likelihood of having a well-positioned embryo.
NOTE: Due to the round nature of the embryos at this timepoint, it is challenging to guarantee a specific orientation in the capillary. Ideally, the body of the embryo will be positioned laterally in the capillary, allowing for the greatest ease of positioning within the microscope. - Let the agarose solidify over a period of 30-60 s at room temperature. Place the capillary in a beaker of 0.168 mg/mL Tricaine in E3 buffer until ready to image (Figure 1A).
NOTE: The excess 2% low melting temperature agarose solution can be allowed to solidify and subsequently reheated in future experiments. - Place the capillary into the sample holder (Figure 1B-F) as described in step 2.5.
2. Zeiss Lightsheet Z.1 setup
- Switch on each component of the microscope and the computer in the following order: 1) System, 2) PC, then 3) Incubation (Figure 2A).
- Place the 20x imaging objective and the 10x illumination objective into the microscope chamber. Match the objective settings in the Zen Software interface under the Maintain tab.
- Slide the chamber (Figure 2C) into the housing on the track (Figure 2B) with the tubing facing out. The tubing connects to the appropriate ports on the right as shown in Figure 2E.
- Attach the extension line to the syringe with the luer-lock mechanism (Figure 2D); fill it with Tricaine in E3 and place it in the holder attached to the right of the microscope35. Connect the extension attached to the syringe filled with Tricaine in E3 to the bottom right of the chamber with the luer-lock mechanism. Push the plunger to fill the chamber with the Tricaine/E3 buffer. Close the door to the chamber.
- Place the capillary with the sample into the capillary sample holder. The capillary sample holder comprises two rubber sleeves, a metal sample holder disc, a metal stem, and a metal cap (Figure 1B). Click the metal sheath into the center of the metal disc (Figure 1C). Place the two rubber stoppers into the sheath, with the slits facing the ends of the sheath followed by the capillary. Slide the capillary through the middle of the rubber stoppers. Fasten it in place with the metal cap once the marker is at the base of the metal sheath (Figure 1D).
- Place the capillary sample holder onto the top of the microscope, with the white marks aligning (Figure 1E,F). Close the lid.
- Click on the Locate Capillary button on the software interface (Figure 3A). Use the ErgoDrive control panel, a manual device that controls the capillary orientation (Figure 3E), to move the capillary and position it just above the objective (Figure 3B).
- Open the lid, and gently push on the plunger until the section of agarose containing the embryo is hanging below the capillary bottom and is in front of the objective (Figure 3C).
- Turn off Locate Capillary and click on the Locate Sample button (Figure 3A). This switches the view from the sample chamber's webcam to the microscope objective (Figure 3D). Use this view to adjust the position of the sample more precisely. Turn off Locate Sample (Figure 3A).
- Switch over to the Acquisition tab. Check the boxes for Z-Stack and Time Series.
- In the Acquisition Mode parameters window, choose the Dual Side Lightsheet setting and check the boxes for Online Dual Side Fusion and Pivot Scanning.
- In the Channels window, choose 488 channel, set the laser power to 1, and the exposure time to 7.5 ms.
- Next, click on the Continuous button to get a live view of the embryo. Use the ErgoDrive control panel to adjust the position of the embryo until the eye field is directly facing the camera. Continue adjusting the left and right lightsheets in the Channels parameters until the eye field is sufficiently in focus.
- Set the Z-Stack parameters using the ErgoDrive control panel to move through the Z-plane. Set the first and the last Z-Positions around 500 µm beyond the last detectable fluorescent signal. This leaves room for the eye field to remain in frame as the embryo grows throughout the time-lapse imaging session. After setting the range of the Z-Stack, click on the Optimal button to set the step size to 0.477 µm, the optimal setting.
- Set the Incubation parameters by checking the box for the Peltier Unit to keep the temperature at 28 °C. In the Time Series window, choose the frequency and time interval to acquire images. In this protocol, the parameters were set to image every 5 min, for a total of 166 intervals.
- Click on the Start Experiment button. Choose the folder to save the image set. Set the image prefix and hit Save to start the imaging.
NOTE: The microscope will now run through each image set at the interval specified. - After the time-lapse imaging session has been completed, send the stage to the load position, and remove the capillary sample holder. Take apart the capillary sample holder in the reverse order that it was put together and use the plunger to remove the sample and excess agarose from the capillary. Open the chamber door. Use the syringe to remove the chamber liquid from the chamber, disconnect and remove the chamber, then rinse with water and air dry.
3. Image analysis
- Open the arivis Vision4D software program.
- Click on File, then choose Import file. Select all .czi files from the time-lapse imaging session and open. The next window opens with the options on how to import the files; select Z-stacks as Frames to order the z-stacks in the order obtained. Once the files have been imported, the software saves as a single .sis file. To specify the location for the file to be saved, select the folder prior to clicking on Import.
- After the file is imported, arivis is now ready to render a video of the time-lapse images. Click on the 4D Viewer Cube in the bottom-left corner and the Scale Bar icon (Figure 4D-E). Click on the video icon to bring the Storyboard taskbar to the bottom of the screen (Figure 4C).
- In the Storyboard taskbar, choose Add Keyframe Sequence (Figure 4F). Specify the duration of the video in seconds, uncheck the Create Rotation box, and check the Use Time Progression to include the specific timepoints in the video. The software displays these parameters to the right of the Storyboard taskbar for any adjustments at any time. Save the Storyboard to apply the same parameters to multiple image sets (Figure 4F).
- Click on Export Movie to save a video of the time-lapse imaging (Figure 4F). Specify the movie export settings, including the file name and location, video format (.mp4), video resolution (1,080 p), framerate (60 FPS), and data resolution (1,297 x 1,297 x 784). Add timestamps here, if desired. Once these parameters are set, choose Record (Figure 4F).
- Steps 3.4 and 3.5 can be repeated with modification to Step 3.4 to create rotation videos at specific timepoints. When adding a keyframe sequence to the storyboard, check the Create Rotation box, and uncheck Use Time Progression. Then, follow the same instructions as in step 3.5.
- To render a high-resolution image at any orientation at any individual time point, select the Camera icon in the 4D Viewer to obtain a high-resolution image (Figure 4C).
- To build a pipeline for analysis, choose the Flask icon to access the Analysis panel (Figure 4B). In the Analysis panel, click on the Analysis Operations dropdown menu. As an example, the protocol demonstrates below the sequence of operations for a volume analysis pipeline. Run or undo each step of the pipeline individually to fine tune each parameter (Table 1).
- Click on the blue triangle at the top of the pipeline to allow the pipeline to run.
NOTE: This will take some time depending on the speed of the computer. Once the pipeline has been optimized, the pipeline can be exported and imported to arivis with ease and applied to multiple datasets in a batch analysis. - To access batch analysis, click on Batch Analysis under the Analysis tab.
- After the pipeline runs, a window pulls up with all of the objects found. Access this manually through the Table icon (Figure 4B). At the top of this window, click on the box labeled Feature Columns to pull up a list of features that can provide information about the object of interest. For the eye field volume analysis, these features include Surface Area, Volume (Volume, VoxelCount), Intensities #1 (Mean), Attributes (Id, Type), and Time Point (First). Click on Export to export the data to a spreadsheet.
Representative Results
The dataset displayed here was imaged using the protocol described above. A Tg(Rx3:GFP) embryo was imaged starting at the 1 somite stage (ss) through 24 hpf, a total time period of 14 h, with the images acquired at 5 min intervals. Time-lapse imaging allows for easy selection and comparison of any time-point that shows a phenotype of interest. Figure 5 demonstrates a set of high-resolution images that were rendered from the dorsal vantage point at select developmental time points. The pipeline run in arivis Vision4D builds a mask that represents the developing eye as identified by fluorescent signal. In Figure 5 and Videos 1-6, the mask can be visualized in comparison to the fluorescent rendering of the developing eye. Additionally, Table 2 displays the volume data from the developing eye at every imaging point. This dataset includes the segment name, id, volume in both µm3 and voxel count, the mean fluorescence intensity of the object, the time point the object was identified, and the object's surface area in µm2. It is important to note that when the eye field separates into two optic vesicles (starting at Timepoint 64), there remains a third region that is Rx3:GFP positive in the forebrain, which will contribute to the hypothalamus38,39,40 (Figure 5D-M). This shows up in the volume data represented in Table 2 (highlighted in yellow starting at Timepoint 71) and can easily be separated out from the optic vesicles, since it is much smaller in volume than either optic vesicle.

Figure 1: Sample preparation. (A) Positioning of embryos in a glass capillary. The arrow points to an embryo in the capillary. (B) Glass capillary and capillary holder parts. (C) Partially assembled capillary holder. (D) Fully assembled capillary holder. (E) Lightsheet mounting chamber. The arrow indicated the white line used to orient the capillary holder. (F) Capillary holder properly mounted in the Lightsheet. The arrow shows the matching white lines, indicating proper orientation of the capillary holder. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Lightsheet imaging set-up. (A) Switchboard to turn on the Lightsheet, computer, and incubation unit. The numbers indicate the order of operations. (B) Lightsheet objective chamber. (C) Imaging chamber. (D) Syringe and tubing that will be connected to the imaging chamber. (E) The imaging chamber properly positioned within the objective chamber with all of the tubes connected to the appropriate ports to the right. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Sample positioning. (A) Locate Capillary and Locate Sample buttons in Zen Software as indicated by the arrows. (B) The positioning on the glass capillary. The arrow indicates the edge of the glass capillary positioned just above the lens of the objective. (C) The embryo suspension beyond the glass capillary. The arrow indicated the embryo suspended in agarose beneath the glass capillary in front of the objective's lens. (D) View of the embryo through the objective. (E) The ErgoDrive control panel. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Important icons for navigating arivis Vison4D. Each panel has the icon function identified from left to right. (A) Open, Save, Close. (B) Analysis Panel, Show Objects Table, Open Track Editor. (C) Copy current viewer content as an image into clipboard, Toggle Bookmarks, Create a high-resolution image for the current view, Toggle Storyboard. (D) Show Measure Box, Show Orientation Cross, Show Legend, Show Scale Bar. (E) Show as 2D Viewer, Show as Gallery Viewer, Show as 4D Viewer, Show as Info Viewer, Show as Projection Viewer. F) Refresh all Keyframes, Add Keyframe, Add Keyframe sequence, Insert Keyframe, Remove all Keyframes, Export Movie, Load Storyboard, Save Storyboard, Adjust the target time of the entire movie, First Keyframe, Play, Pause, Stop, Last Keyframe. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: High-resolution images and eye field masks. (A-M) A set of high-resolution images that were rendered from the dorsal vantage points; (A'-M') the eye field masks for each corresponding timepoint. Each image set is notated by the time it was acquired from the start of imaging and the corresponding developmental stage in either somite stage (ss) or hours post fertilization (hpf). Please click here to view a larger version of this figure.
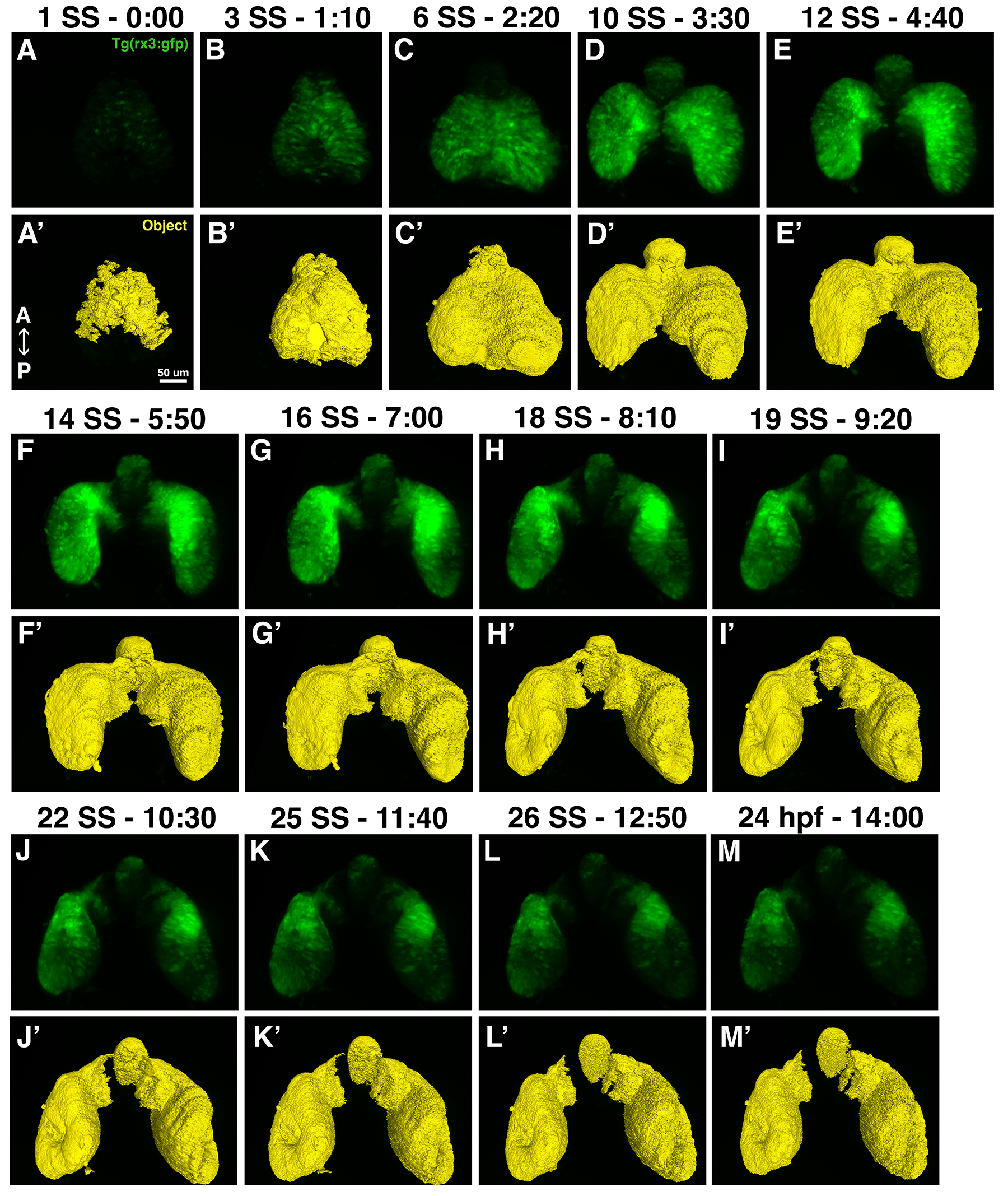{kind=link}
Video 1: Time-lapse video of Tg(rx3:GFP) zebrafish embryo from 1 ss-24 hpf. Please right click here to download the video (save link as).
Video 2: Time-lapse video of Tg(rx3:GFP) zebrafish embryo from 1 ss-24 hpf with the eyefield as identified by the arivis Vision4D pipeline. Please right click here to download the video (save link as).
Video 3: 360° rotation of a Tg(rx3:GFP) zebrafish embryo at 1 ss. Please right click here to download the video (save link as).
Video 4: 360° rotation of a Tg(rx3:GFP) zebrafish embryo eye field mask at 1 ss. Please right click here to download the video (save link as).
Video 5: 360° rotation of a Tg(rx3:GFP) zebrafish embryo at 24 hpf. Please right click here to download the video (save link as).
Video 6: 360° rotation of a Tg(rx3:GFP) zebrafish embryo eye field mask at 24 hpf. Please right click here to download the video (save link as).
Table 1: arivis Vision4D pipeline for volume analysis of the developing eye field. Please click here to download this Table.
Table 2: Volume and surface area of the developing eye field acquired by arivis Vision4D. The rows pertaining to the presumptive hypothalamus are highlighted in yellow to distinguish them from the optic vesicles. Please click here to download this Table.
Discussion
In this protocol, the Lightsheet microscope was used to perform time-lapse imaging of eye development and the resulting data were analyzed. The resulting dataset can provide valuable insights into the process of ocular morphogenesis, as well as perturbations to this process as a result of genetic mutation, exposure to pharmacological agents, or other experimental parameters. Here the protocol demonstrated how this dataset can be obtained and provided an example of how to analyze the volume of the eye field through early development. This data was found to be reproducible and consistent (less than 10% variation in volume) across biological replicates, bearing in mind that slight differences in embryo staging prior to the start of the run can lead to some variation in final volume measurements.
Care should be taken in the initial positioning of the embryo in the capillary and in positioning the embedded embryo in front of the objective. Orientation plays an important role in preventing the embryo from growing and moving out of the view of the objective. The embryos have a round shape at 10 hpf, which makes it challenging to guarantee a specific orientation in the capillary. Ideally, the body of the embryo will be positioned laterally in the capillary. Loading multiple embryos in the capillary will increase the likelihood of having a well-positioned embryo.
In this procedure, the embryo is embedded in agarose in order to suspend it in front of the imaging and illumination objectives. Choosing the correct concentration of the low melting temperature agarose is critical. Too high of a concentration will constrict the embryo and prevent it from properly developing; too low of a concentration will result in the agarose falling apart and not holding the embryo. The concentration optimal for this protocol is a final concentration of 1% low melting temperature agarose30,31.
Another element that should be taken into consideration is the level of saturation. As the eyefield grows and differentiates, the strength of the Rx3:GFP signal intensifies. Therefore, when setting the initial imaging parameters, the exposure and laser power should be reduced to undersaturate the image. This will prevent the image from becoming oversaturated as the Rx3:GFP gets brighter over time. Modifications can be made to correct for undersaturation in image processing but oversaturation cannot be corrected after the images have been acquired.
There are a few additional modifications that can be made to this protocol that may be advantageous to some projects that are not described in this paper. For example, it is possible to set up Multiview imaging in the image acquisition set up. This parameter would allow multiple embryos at different positions along the y-axis to be sequentially imaged at each time interval. While adding complexity to the data set, it would increase the rate of data collection. Additionally, in terms of image processing, it is possible to quantify the eye field by other parameters. Here, we described how to quantify the data in terms of the eye field volume. Alternatively, a pipeline could be made to quantify and track individual cells or determine the rate of optic vesicle evagination.
As previously mentioned, both confocal and Lightsheet microscopy have been used to perform time-lapse imaging studies of zebrafish. Lightsheet was specifically chosen for this project due to its superior ability to image through a thick (>1 mm) sample, because it is equipped with an incubation unit to maintain an ideal temperature environment for the zebrafish embryo, and because its ability to image at a faster rate than confocal microscopy allows for image acquisition at the numerous time intervals required for this protocol no accompanying damage or photobleaching of the embryo21,22,23,24,25,26,27,28,29,30,31. It is also important to note that the Lightsheet microscope is equipped to image the signal from multiple fluorophores. The Lightsheet microscope used in this study has solid state laser excitation lines at 405, 445, 488, 515, 561, and 638 nm, which could be useful for imaging transgenic embryos expressing more than one fluorescent reporter transgene.
While this protocol details instructions for image acquisition analysis specifically using the Lightsheet Z.1 Dual Illumination Microscope System and arivis Vision4D analysis software, there are other commercially available Lightsheet microscopes made by Leica, Olympus, and Luxendo, as well as image analysis software by Imaris, that could be used to achieve similar results. The selection of equipment and software for this protocol was determined by the availability at our institution.
In summary, it is anticipated this protocol will provide a solid starting point for conducting time-lapse imaging using Lightsheet microscopy, and for image quantification of early eye development in zebrafish.
Acknowledgements
Research reported in this publication was supported by the Office of The Director of the National Institutes of Health (NIH) under Award Number S10OD020067 and by NIH award R01EY021769 (to A.C.M.). We are grateful for the assistance of Doug Harrison and Jim Begley in the Arts & Sciences Imaging Center at the University of Kentucky, and to Lucas Vieira Francisco and Evelyn M. Turnbaugh for expert zebrafish care.
Materials
| Name | Company | Catalog Number | Comments |
| 60mL Syringe Luer-Lok Tip | BD | 309653 | |
| Agarose, Low Melting Temperature | Promega | V2111 | |
| arivis Vision4D Software | arivis | https://www.arivis.com/en/imaging-science/arivis-vision4d | |
| Dumoxel N3C Forceps | Dumont | 0203-N3C-PO | |
| E3 fish buffer (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) | |||
| Glass Capillary – Size 2 (~1mm) | Zeiss | Black/701904 | |
| Heidelberger Extension Line 100cm | B. Braun | 4097262 | |
| Light & Filter Set – Royal Blue | Night Sea | SFA-LFS-RB | |
| Petri Dish (100 x 15 mm) | VWR | 25384-302 | |
| Stereomicroscope Fluorescence Adapter | NightSea | ||
| Teflon Tipped Plunger – Size 2 | Zeiss | #701997 | |
| Tg(rx3:GFP) zebrafish | This line was established by Rembold et al.13 | ||
| Tricaine (MS222) | Sigma | A-5040 | |
| Z.1 Lightsheet | Zeiss | ||
| Zen Software | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
References
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253-310 (1995).
- Li, Z., Joseph, N. M., Easter, S. S. The morphogenesis of the zebrafish eye, including a fate map of the optic vesicle. Developmental Dynamics. 218 (1), 175-188 (2000).
- Chow, R. L., Lang, R. A. Early eye development in vertebrates. Annual Review of Cell and Developmental Biology. 17, 255-296 (2001).
- Wilson, S. W., Houart, C. Early steps in the development of the forebrain. Developmental Cell. 6 (2), 167-181 (2004).
- Picker, A., et al. Dynamic coupling of pattern formation and morphogenesis in the developing vertebrate retina. PLoS Biology. 7 (10), 1000214 (2009).
- Fuhrmann, S. Eye morphogenesis and patterning of the optic vesicle. Current Topics in Developmental Biology. 93, 61-84 (2010).
- Kwan, K. M., et al. Bin A complex choreography of cell movements shapes the vertebrate eye. Development. 139 (2), 359-372 (2012).
- Heermann, S., Schütz, L., Lemke, S., Krieglstein, K., Wittbrodt, J. Eye morphogenesis driven by epithelial flow into the optic cup facilitated by modulation of bone morphogenetic protein. eLife. 4, 05216 (2015).
- Eckert, P., Knickmeyer, M. D., Schütz, L., Wittbrodt, J., Heermann, S. Morphogenesis and axis specification occur in parallel during optic cup and optic fissure formation, differentially modulated by BMP and Wnt. Open Biology. 9 (2), (2019).
- Yoon, K. H., Fox, S. C., Dicipulo, R., Lehmann, O. J., Waskiewicz, A. J. Ocular coloboma: Genetic variants reveal a dynamic model of eye development. American Journal of Medical Genetics Part C: Seminars in Medical Genetics. 184 (3), 590-610 (2020).
- Sidhaye, J., Norden, C. Concerted action of neuroepithelial basal shrinkage and active epithelial migration ensures efficient optic cup morphogenesis. eLife. 6, 22689 (2017).
- Pillai-Kastoori, L., Wen, W., Morris, A. C. Keeping an eye on SOXC proteins. Developmental Dynamics. 244, 367-376 (2015).
- Rembold, M., et al. Individual cell migration serves as the driving force for optic vesicle evagination. Science. 313, 1130-1134 (2006).
- Loosli, F., et al. Loss of eyes in zebrafish caused by mutation of chokh/rx 3. EMBO Reports. 4 (9), 894-899 (2003).
- Chuang, J. C., Mathers, P. H., Raymond, P. A. Expression of three Rx homeobox genes in embryonic and adult zebrafish. Mechanisms of Development. 84 (1-2), 195-198 (1999).
- Cavodeassi, F., et al. Early stages of zebrafish eye formation require the coordinated activity of Wnt11, Fz5, and the Wnt/β-catenin pathway. Neuron. 47 (1), 43-56 (2005).
- Ivanovitch, K., Cavodeassi, F., Wilson, S. W. Precocious acquisition of neuroepithelial character in the eye field underlies the onset of eye morphogenesis. Developmental Cell. 27 (3), 293-305 (2013).
- Ebert, A. M., Childs, S. J., Hehr, C. L., Cechmanek, P. B., McFarlane, S. Sema6a and Plxna2 mediate spatially regulated repulsion within the developing eye to promote eye vesicle cohesion. Development (Cambridge). 141 (12), 2473-2482 (2014).
- Emerson, S. E., et al. Identification of target genes downstream of semaphorin6A/plexinA2 signaling in zebrafish. Developmental Dynamics. 246 (7), 539-549 (2017).
- Hehr, C. L., Halabi, R., McFarlane, S. Polarity and morphogenesis of the eye epithelium requires the adhesion junction associated adaptor protein Traf4. Cell Adhesion and Migration. 12 (5), 489-502 (2018).
- Jemielita, M., Taormina, M. J., Delaurier, A., Kimmel, C. B., Parthasarathy, R. Comparing phototoxicity during the development of a zebrafish craniofacial bone using confocal and light sheet fluorescence microscopy techniques. Journal of Biophotonics. 6 (11-12), 920-928 (2013).
- Huisken, J., Swoger, J., Del Bene, F., Wittbrodt, J., Stelzer, E. H. K. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science. 305 (5686), 1007-1009 (2004).
- Reynaud, E. G., Kržič, U., Greger, K., Stelzer, E. H. K. Light sheet-based fluorescence microscopy: More dimensions, more photons, and less photodamage. HFSP Journal. 2 (5), 266-275 (2008).
- Icha, J., et al. Using light sheet fluorescence microscopy to image zebrafish eye development. Journal of Visualized Experiments:JoVE. (110), e53966 (2016).
- Keller, P. J., Dodt, H. U. Light sheet microscopy of living or cleared specimens. Current Opinion in Neurobiology. 22 (1), 138-143 (2012).
- Pampaloni, F., Chang, B. J., Stelzer, E. H. K. Light sheet-based fluorescence microscopy (LSFM) for the quantitative imaging of cells and tissues. Cell and Tissue Research. 360 (1), 129-141 (2015).
- Pantazis, P., Supatto, W. Advances in whole-embryo imaging: A quantitative transition is underway. Nature Reviews Molecular Cell Biology. 15 (5), 327-339 (2014).
- Park, O. K., et al. 3D light-sheet fluorescence microscopy of cranial neurons and vasculature during zebrafish embryogenesis. Molecules and Cells. 38 (11), 975-981 (2015).
- Royer, L. A., et al. Adaptive light-sheet microscopy for long-term, high-resolution imaging in living organisms. Nature Biotechnology. 34 (12), 1267-1278 (2016).
- Keller, P. J., Schmidt, A. D., Wittbrodt, J., Stelzer, E. H. K. Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy. Science. 322 (5904), 1065-1069 (2008).
- Keller, P. J., et al. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nature Methods. 7 (8), 637-642 (2010).
- Santi, P. A. Light sheet fluorescence microscopy: A review. Journal of Histochemistry and Cytochemistry. 59 (2), 129-138 (2011).
- Reynaud, E. G., Peychl, J., Huisken, J., Tomancak, P. Guide to light-sheet microscopy for adventurous biologists. Nature Methods. 12 (1), 30-34 (2014).
- Avdesh, A., et al. Regular care and maintenance of a Zebrafish (Danio rerio) laboratory: An introduction. Journal of Visualized Experiments: JoVE. (69), e4196 (2012).
- Westerfield, M. . The Zebrafish Book. A Guide for the laboratory use of zebrafish (Danio rerio)., 5th Edition. , (2007).
- Hirsinger, E., Steventon, B. A versatile mounting method for long term imaging of zebrafish development. Journal of Visualized Experiments: JoVE. (1199), e55210 (2017).
- Saint-Amant, L., Drapeau, P. Time course of the development of motor behaviors in the zebrafish embryo. Journal of Neurobiology. 37 (4), 622-632 (1998).
- Cavodeassi, F., Ivanovitch, K., Wilson, S. W. Eph/Ephrin signalling maintains eye field segregation from adjacent neural plate territories during forebrain morphogenesis. Development (Cambridge). 140 (20), 4193-4202 (2013).
- Muthu, V., Eachus, H., Ellis, P., Brown, S., Placzek, M. Rx3 and Shh direct anisotropic growth and specification in the zebrafish tuberal/anterior hypothalamus. Development (Cambridge). 143 (14), 2651-2663 (2016).
- Rojas-Muñoz, A., Dahm, R., Nüsslein-Volhard, C. chokh/rx3 specifies the retinal pigment epithelium fate independently of eye morphogenesis. Developmental Biology. 288 (2), 348-362 (2005).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved