Ortho- and Ectopic Zebrafish Xeno-Engraftment of Ocular Melanoma to Recapitulate Primary Tumor and Experimental Metastasis Development
In This Article
Summary
Here, we present a protocol to establish versatile orthotopic and ectopic zebrafish xenograft models for ocular melanoma to assess the growth kinetics of the primary tumor, dissemination, extravasation and distant, peri-vascular metastasis formation and the effect of chemical inhibition thereon.
Abstract
There are currently no animal models for metastatic ocular melanoma. The lack of metastatic disease models has greatly hampered the research and development of novel strategies for the treatment of metastatic ocular melanoma. In this protocol we delineate a quick and efficient way to generate embryonic zebrafish models for both the primary and disseminated stage of ocular melanoma, using retro-orbital orthotopic and intravascular ectopic cell engraftment, respectively. Combining these two different engraftment strategies we can recapitulate the etiology of cancer in its totality, progressing from primary, localized tumor growth under the eye to a peri-vascular metastasis formation in the tail. These models allow us to quickly and easily modify the cancer cells prior to implantation with specific labeling, genetic or chemical interference; and to treat the engrafted hosts with (small molecular) inhibitors to attenuate tumor development.
Here, we describe the generation and quantification of both orthotopic and ectopic engraftment of ocular melanomas (conjunctival and uveal melanoma) using fluorescently labelled stable cell lines. This protocol is also applicable for engraftment of primary cells derived from patient biopsy and patient/PDX derived material (manuscript in preparation). Within hours post engraftment cell migration and proliferation can be visualized and quantified. Both tumor foci are readily available for imaging with both epifluorescence microscopy and confocal microscopy. Using these models, we can confirm or refute the activity of either chemical or genetic inhibition strategies within as little as 8 days after the onset of the experiment, allowing not only highly efficient screening on stable cell lines, but also enables patient directed screening for precision medicine approaches.
Introduction
Metastatic dissemination is considered the main cause of death of ocular melanoma; currently there is no viable treatment regime for disseminated ocular melanoma1,2. Furthermore, there are no animal models available for ocular melanoma that reflects the metastatic disease. To bridge this gap, we generated two distinct zebrafish models that recapitulate either primary tumor formation or the early stages of metastatic dissemination, thus readily allowing the study of these normally difficult to study processes 3. The micro-metastasis models allow the analysis of the last phases of metastatic spread, including homing, colonization and extravasation. Genetic or chemical interventions at this stage and beyond could potentially provide a powerful handhold in the treatment of metastatic ocular melanoma.
The use of the zebrafish larvae as a recipient of xeno- and allografts is supported by the intrinsic strengths of this species, such as its optical transparency at the early stages of development (or its entire life-cycle for casper mutants4), high fecundity and ex utero fertilization5. High transcriptional homology in vertebrates ensures the retention of core signaling mechanisms between the zebrafish and humans and therefore high potential translatability of results 6, although genetic approaches are sometimes marred or complicated due to the teleost genome duplication 7. Recent developments have underscored the importance of zebrafish xenograft models as pre-clinical "avatars" of human disease8, effectively yielding a multitude of personalized cancer therapy models for the pre-clinical evaluation of treatment strategies from a single zebrafish experiment 9.
Considering the lack of animal models and the concordant lack of treatment options for metastatic ocular melanoma, our models provide a quick and easy translational platform to screen both genetic alterations (cancer cell intrinsic) or develop chemical intervention strategies in a pre-clinical setting. Within the same model we can visualize and measure cancer cell growth kinetics, engraftment rate/metastatic potential, and cell homing on a whole animal level using low level magnification in a stereo fluorescent microscope, and make similar measurements using medium or high magnification confocal microscopic analysis to dissect different steps of ocular melanoma progression at subcellular resolution 10.
Here, we describe comprehensive and detailed protocols for: the generation of fluorescently labeled cancer cells using highly optimized lentiviral transduction11; subsequent intravenous and retro-orbital (RO) engraftments of these cells into 2 days post fertilization (dpf) zebrafish larvae to generate ectopic and orthotopic models respectively; followed by data acquisition and analysis. These methods although comprehensive for the applications described herein can be modified to engraft cells in the hind brain cavity, liver and perivitellin space when required (solely by changing the injection site, or time of injection)12,13.
As a proof-of-concept we elaborated upon the findings of Pontes et al. 2018, where we showed a dose and cell intrinsic mutation specific response of conjunctival melanoma cell lines in the zebrafish model 14. We elaborated upon these findings by showing the efficacy of BRAF V600E mutation-specific inhibitor vemurafenib in both metastatic and primary conjunctival melanoma models.
Protocol
All animal experiments were approved by Animal Experiments Committee (Dier Experimenten Commissie, D.E.C.) under license AVD1060020172410. All animal were maintained in accordance with local guidelines using standard protocols (www.ZFIN.org).
1. Preparation
- Reagents
- Prepare egg water: 0.6 mg/L final concentration sea salt.
- Prepare 5 mg/mL Tricaine 25x stock: Mix 5 g of Tricaine (ethyl 3-aminobenzoate methanesulfonate or MS-222) powder, 900 mL of demineralized water, and 21 mL of 1 M Tris (pH 9). Adjust to pH 7 and fill up to 1 L. Tricaine can be stored at 4 °C for short term (up to six months) or can be stored at room temperature for a month at room temperature when protected from sunlight.
- Prepare 1.5% (w/v) agarose in egg water: 1.5 g in 100 mL of DPBS. Microwave to dissolve.
- Prepare 1% (w/v) low-melting agarose in egg water: 1.5 g in 100 mL of DPBS. Microwave to dissolve.
- Prepare 2% (w/v) PVP40 stock in DPBS: 1 g of PVP40 in 50 mL of DPBS. Vortex and incubate at 37 °C to facilitate dissolving. Store at room temperature.
- Use DMSO. It is often used as the solvent in drug treatments and should be stored at 2-8 °C the dark.
- Use TrypLE, a synthetic trypsin replacement that is less damaging to the cells and allows for the gentle dispersion of strongly adherent cells.
- Prepare Dulbecco's phosphate buffered saline (DPBS) without Mg2+ and Ca2+ for washing the cells. The lack of Ca2+ impairs cell-cell adhesion through cadherins.
- Prepare lentiviral plasmids: psPAX2 (plasmid #12260) and pMD2.G (plasmid #12259) gifted by Didier Trono and either a GFP (Plasmid #106172) or tdTomato (Plasmid #106173) encoding transfer plasmid (Addgene).
- Use LipodD293: Highly efficient HEK293T optimized transfection reagent.
- Agarose dish
NOTE: When using dishes that have been stored for a long time make sure to add a small volume of egg water to the dishes before starting injection (this will prevent the fish from drying out too fast).- Prepare 1.5% (w/v) agarose coated dishes (agarose dissolved in egg water).
- Use immediately, or store at 4 °C in inverted position.
2. Needles
NOTE: Make sure that the capillaries have been calibrated on the filament used. When switching either the filament or the capillary, determine the ramp value of the capillaries on the filament used (see needle puller manual).
- One glass capillary will yield two micro injection needles. Before making needles, check the structural integrity of the filament (2.5mm box filament) of the needle puller.
- Make sure that both filament and capillary are calibrated to get the corresponding ramp value. When the filaments structural integrity is compromised (i.e., uneven, holes, molten etc.), change the filament.
- Use the following program (Needle #99, Heat=ramp+15, pull=95, velocity=60, time=90). Store the needles in a designated Petri dish (containing either clay or tape to stick the needles to)
3. Generation of lentiviral particles
NOTE: To prevent a waste of time and resources a quick tumorigenicity check can be performed prior to lentiviral transduction. This is done to ensure that the cell line to be used is sufficiently tumorigenic in the zebrafish model, to this end the cells can be stained with a CMdiI (or analogous tracer) as described in Liverani et al. 2017 15.
- Plate HEK 293t cells one day prior transfection to achieve a confluency of approximately 70% (routinely done by splitting a full flask to the same volume culture flask at a dilution 1:3 one day prior).
- At the day of transfection, co-transfect the required packaging plasmids psPAX2 and pMD2.G viral envelope expressing plasmid along with either a GFP (Plasmid #106172) or tdTomato (Plasmid #106173) encoding the transfer plasmid. The exact amount of plasmid used is specified in Table 1.
NOTE: Both psPAX2 and pMD2.G were gifted by Didier Trono (Addgene plasmid #12260 and #12259, respectively).

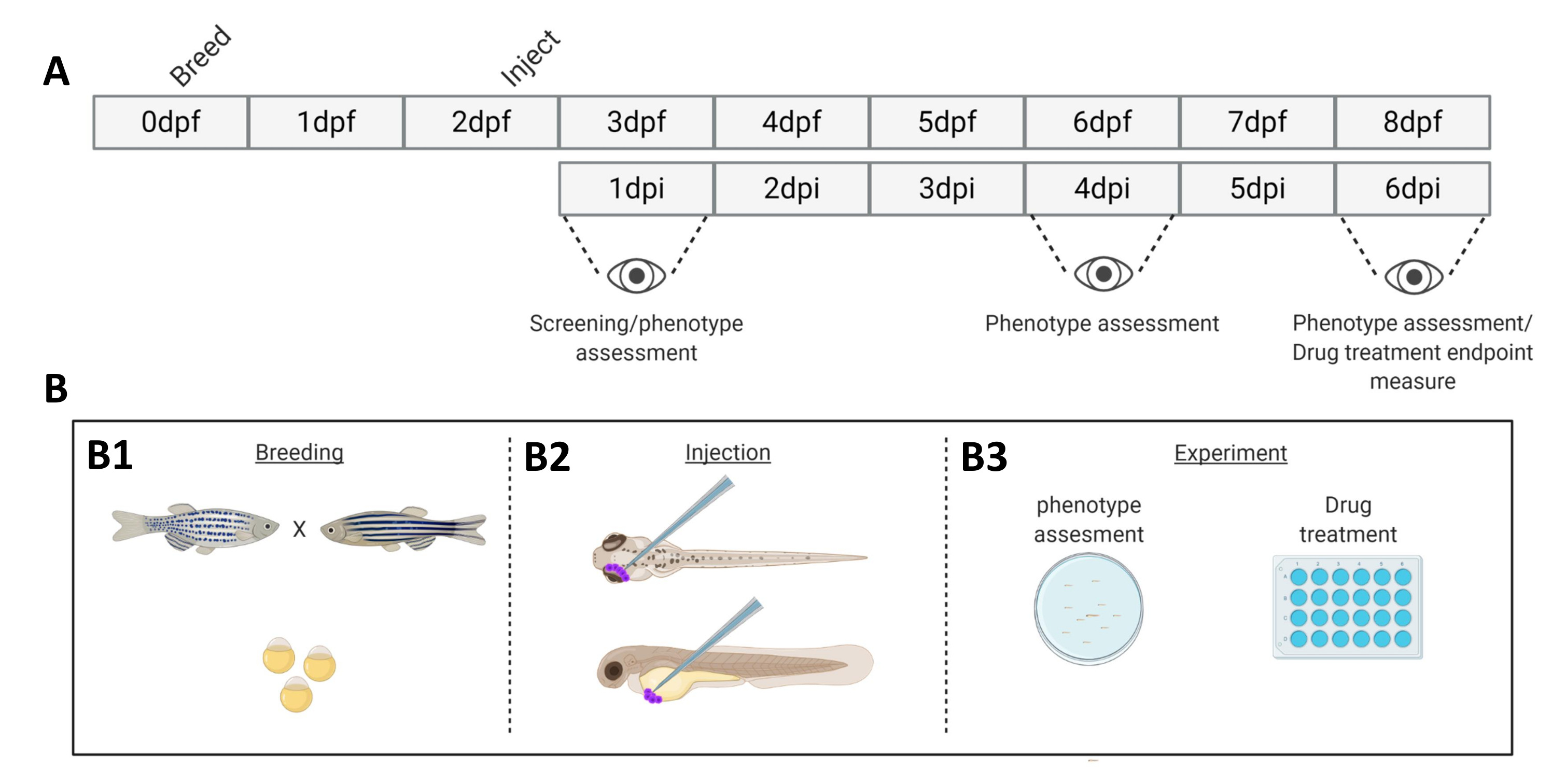
Figure 1. Schematic representation of the described zebrafish engraftment system. A) The timeline of the approach, with breeding the zebrafish at day 0 (B1). The fish are harvested in the morning after crossing the fish (day 1). After 48-54 hours the fish have largely hatched (shedding their chorion) and the fish are injected (retro-orbitally or systemically, B2) after cleaning the water of the chorion debris (day 2). The larvae are subsequently screened using a stereo fluorescent microscope and all larvae displaying unwanted phenotypes are discarded (day 3). Depending on the goal of the experiment either the larvae are imaged over time (B3, engraftment kinetics, imaged at 1-, 4- and 6-days post injection (dpi)) or the fish are randomized and entered into experimental groups, treated with drugs and compared to vehicle control (drug screening, imaged at 6 dpi). Please click here to view a larger version of this figure.
{kind=link}
- Mix all plasmids together in 500 µL of serum free medium, to allow complete mixing of all plasmids. Add 32 µL of LipoD293 reagent to 500 µL of serum-free DMEM, and vortex to mix completely. Mix both volumes together thoroughly. Allow the plasmids and the lipoD293 to complex for 20 minutes.
- Add dropwise to a 75 cm2 cell culture flask containing 70% confluent HEK293T cells containing 9 mL of complete culture medium. Add the transfection mixture directly to the cell layer using a serological pipette (flask in horizontal orientation).
- Replace medium with 20 mL of fresh complete DMEM 16 hours post-transfection. Harvest supernatant after 72 hours post transfection. Aliquot viral supernatant in 1 mL aliquots and store at -80 °C. The lentiviral supernatant is stable at -80 °C for at least 1 year.
4. Lentiviral transduction
- Before lentiviral transduction, establish a kill curve when using a selectable lentiviral construct.
- For the kill curve, plate the cell line to be transduced in a 12 well plate (confluence approximately 10-20%). Add a dose curve of the selectant (approximate concentrations for kill curves: puromycine 0.5-10 µg/mL, blasticidin 1-20 µg/mL, geneticin (G418) 100-2000 µg/mL, hygromycin 100-2000 µg/mL).
- Change the medium every three days to assure a stable concentration of the chosen selectant.
- Add 1 mL of lentiviral supernatant to 9 mL of culture medium, containing a final concentration of 8 µg/mL polybrene on 20-40% confluent cells. Volumes can be scaled down, while maintaining this ratio of supernatant/medium.
- 16-24 hours post transduction, exchange the medium. When required, repeat the former step to enhance phenotype penetrance (check fluorescence to decide if another transduction is required).
- 48 hours post transduction, select the cells using the antibiotic corresponding to the resistance marker incorporated into the lentiviral cassette. The concentration to use for the selection of the transduced cell population should kill the wild-type population within 7 days after application of the selectant (i.e., allowing the transduced cells to outgrow the wildtype population).
- Apply viral supernatant in different multiplicities of infection (MOI's) to ensure that the transduction and the genetic lesions incurred by the cellular genome does not negatively affect cell viability or tumorigenicity.
5. Breeding zebrafish
- On day 0, 2 days prior to engraftment of cancer cells, mate adult zebrafish in "family cross" fashion at room temperature (Figure 1).
- Remove the tank of zebrafish from the housing system (maintained at 28.5 °C).
- Separate the fish into small breeding clusters at a 1:1 ratio male: female, with 10 fish per cluster. Place the fish in small breeding tanks, in water drawn from the housing system, above a slanted grate (slanted, to mimic the shallows wherein zebrafish would naturally spawn).
NOTE: Induced by the decline in temperature from 28.5°C to room temperature (25°C) and the entrance into the next light phase of the dark/light cycle the fish will spawn. - Subsequently, remove the adults and transfer into their housing tank.
- Collect the eggs and wash with egg water using a strainer. Divide the eggs to approximately 75-100 per dish and maintain at 28.5°C.
- Approximately 6 hours post collection, clean the dishes of dead or malformed embryos.
- The next morning, exchange the egg water and again clean the dishes of dead embryos.
6. Harvesting cells
NOTE: Proper cell preparation is key to the implantation procedure, using a superfluous amount of cells allows for easier downstream processing. The third centrifugation step is critical, as this will leave you with only the cell pellet, the remaining PBS stuck on the sides of the centrifuge tube greatly exceeds the final resuspension volume.
- Prewarm all media and solutions used in cell culture in a 37 °C water bath before use.
- Add 2 mL of TryplE per 75 cm2 culture flask or 1 mL per 25 cm2 flask and incubate until all cells are rounded. For most cell lines 2-5 minutes should be sufficient. For highly epithelial cells or fibroblastic cells 5-10 minutes should allow for proper detachment (insufficient trypsinization will hinder downstream processes, and facilitates cell aggregation during implantation).
- Gently tap the side of the flask to dislodge remaining cells.
- Add up to the original culture volume of complete medium. Pipette up and down gently but thoroughly with a serological pipette to shear cell clumps into single cell suspension. Do not generate foam during this process as foam is indicative of mechanical shearing of the cells.
- Transfer into a sterile 15 mL tube and centrifuge for 5 minutes at 200 x g at room temperature. Aspirate supernatant and add 1 mL of sterile PBS. Carefully and thoroughly resuspend the cells using a sterile 1000 µL pipette.
- Remove 20 µL cell suspension for counting and transfer the remaining cell suspension to the centrifuge. Centrifuge for 4 minutes at 200 x g at room temperature.
- CRITICAL STEP: Remove all PBS, centrifuge for 30 s at 200 x g at room temperature, and remove the remaining PBS.
- Dilute the cells to 250 cells/nL in 2% polyvinylpyrrolidon 40 (PVP40, 2% (w/v) in DPBS) as follows:

(for example,
- Thoroughly resuspend the cells, while preventing the formation of air bubbles (cells can be kept for at least 2 hours in 2% PVP40 without loss of tumorigenic potential).
7. Xenograft modeling
All experiments should be performed in compliance with local animal welfare regulations.
Depending on the application two main variations in experimental design are classified as a phenotype assessment (7.1 the pre-screening stage) and secondly 7.2 a screen where either the cells have been modified prior to engraftment or 7.3 where the embryos are treated with a chemical inhibitor.
- Pre-screening and determination of tumorigenic potential
- Engraft zebrafish larvae of interest (WT, transgenic or reporter line) at 2 dpf with a varying number of fluorescent cells (i.e., 200, 400, 600 ±100).
- Screen larvae 16-24 hours after injection to remove outliers (extremely high or low cell numbers in circulation for the ectopic model, or cells inside the head for the orthotopic model) and remove wrongly engrafted fish. Indicate nr of larvae per experimental group for group analysis vs kinetic analysis of the same larvae.
- Monitor the zebrafish larvae at regular intervals (1,2,4,6 days post injection (dpi)) and image 20 individuals (as described in steps 9 and 10), out of a pool of ±50 larvae.
- Monitor general phenotype and disease progressions and subsequently quantify with ImageJ (measuring integrated density of the fluorophore signal in the cancer cells).
- Plot the data to visualize the cancer cell growth kinetics within the zebrafish (Figure 3).
- Modify cells a priori (knock down or knock out of a gene of interest) and engraft into zebrafish.
- Engraft fish and remove all unwanted phenotypes (per condition).
- Image the individuals at 1 dpi (20 larvae per group). Individuals can be imaged at set intervals (1,2,4 and 6 dpi).
- At 6dpi after imaging, euthanize the fish by overdosing with tricaine (10-fold over dosing at 0.4 mg/mL) and discard on absorbent paper lining a funnel.
- Treat fish with drugs after engraftment.
- Prior to drug application on engrafted zebrafish, determine the maximum tolerated dose (MTD) on zebrafish (titrate down from 10 µM- 0.150 nM, using the highest volume of solvent as a negative control) we have set the MTD as the concentration where >80% of individuals survive the entire treatment.
- One day post injection, remove the unwanted phenotypes.
- Randomly divide the fish into groups (36-48 individuals/ condition) and maintain in a 24 wells plate with 6 larvae per well in 1 mL of egg water.
- Apply drugs 24 hours after engraftment. As a control use the same amount of solvent (DMSO, EtOH etc.) at the highest volume applied for an experimental group.
- Start drug treatment at the maximum tolerated dose. Change the egg water containing drug every other day. Remove egg water and dead larvae as completely as possible during every change.
8. Injection
NOTE: Use a pneumatic pulse controller coupled to a compressed air line, supplying pressure in surplus of 100 psi. This allows for enough pressure to both inject (≈20 psi) and to eject possible cell aggregates (≈100 psi). The starting pressure and time should be approximately 200 ms at 20 psi. If either has to be decreased more than 50% at the start of the injection either the cell suspension is too fluid (cell or PVP40 concentration too low) or needle opening is too large.
- Carefully remove a capillary needle from its container. Break the needle to form an opening of ø20 µm, using a fine watchmakers' forceps.
- Carefully and thoroughly resuspend the cells using a 20 µL pipette tip. Pipette cell suspension into the open glass capillary needle using a long (microloader) tip. Load the needle into the micro manipulator.
- Place ~20-40 larvae anesthetized in 0.04 mg/mL tricaine on an agarose dish using a transfer pipette. Remove excess moisture to immobilize the larvae using a transfer pipette. The larvae will mostly be oriented in a lateral fashion due to the presence of a still relatively large yolk sac.
- Inject the larvae with approximately 200, 400 and 600 cells via the Duct of Cuvier (doC) for ectopic model.
- Similarly, inject larvae retro-orbitally (RO). To yield the orthotopic model (injecting 100 ±50 cells), modify the pneumatic pulse length on the picopump (start at ~20 psi, 200 ms and adjust accordingly). During injection ensure that the larvae do not dry out. Make sure that all (or most) larvae are injected.
- Flush off injected larvae with fresh egg water and transfer to a labelled clean Petri dish (pooling up to 150 individuals per dish). Repeat this process until sufficient larvae are injected.
- After engraftment, maintain the fish at 34 °C in a humidified incubator, where 34 °C is the highest temperature readily tolerated by zebrafish and allows for efficient engraftment of mammalian cancer cells.
NOTE: In general, with injection of single cell lines in both doC and RO we have observed an approximate death due to mechanical damage of <5% (mechanical damage kills the larvae between 1-16 hours post injection).
9. Screening
- Using a stereo-fluorescence microscope, screen the fish for the appropriate phenotype 1 hour post implantation when comparing cells modified a priori (or 1 day post implantation, when screening drugs, before the random assignment into treatment groups).
- Larvae implanted through the doC should have cells in the tail between 1 hour and 16 hours post implantation. Remove all other fish, including fish that display abnormality, from the injected pool.
NOTE: Larvae implanted retro-orbitally should have cells only in the interstitium behind the eye, larvae that have cells spread throughout the head or body are removed from the pool. - Clean positively screened larvae and randomly assign to experimental groups.
- After engraftment, maintain fish at 34 °C in a humidified incubator and monitor daily. Hematogenous dissemination of cells implanted through the doC is almost instantaneous, whereas metastatic spread of cells implanted in the RO cavity will spread after 2-4 days.
10. Epifluorescent imaging of zebrafish larvae
- Anesthetize zebrafish larvae with 0.2 mg/mL tricaine, either by adding tricaine to the water of the fish or by moving a sub-population of fish from the maintenance dish to a dish containing 0.2 mg/mL tricaine.
- Keep zebrafish in a dish with tricaine until they remain stationary, until stimulation of the lateral line does not induce flight behavior.
- Transfer fish to an agarose covered Petri dish, approximately 10 per dish. Remove the majority of the water though gently raising one end of the dish (allowing the water to gently pool in the lower end of the Petri dish). If done carefully all fish will align, tails facing downwards.
- Image all fish from the top of the dish to the bottom. Then wash the fish off with egg water into a dish without tricaine.
- Repeat until enough individuals are imaged.
- Then transfer the larvae either back to the 34 °C or cull (at 6 dpi) through overdosing with tricaine (i.e., 0.5 mg/mL, incubating for 10 min, prior to discarding on absorbent paper lining a funnel).
11. Confocal imaging of (engrafted) zebrafish larvae
- Anesthetize zebrafish with 0.2 mg/mL tricaine as described previously.
- Place a glass bottom confocal dish under a stereo microscope and focus on the bottom of the dish. Transfer 5-10 larvae to a glass bottom confocal dish. Remove as much water as possible.
- Cover the larvae with 42 °C, 1% low melting agarose dissolved in egg water. Make sure that the agarose has cooled down to at least 42 °C before use; higher temperatures might harm or kill the larvae.
- Using the stereomicroscope, quickly but gently orient the larvae pushing it down, using a trimmed down micro loader tip. If a ventral orientation is required, hold the larvae in place with the tongs of a watchmaker's forceps (without touching the embryo).
- While the agarose sets make fine adjustments to the orientation of the larvae. Allow the larvae to set completely before transferring to the confocal microscope.
12. Setting the confocal microscope
- Switch on the green (488 nm) and red (564 nm) excitation laser lines. Place the confocal dish in the holder of the confocal microscope. Using the epifluorescence, move the light bundle to coalesce with the first fish (setting x and y). Through the ocular set the focus to coincide with the center of the larvae (setting z).
- Set 700 gain on both fluorescent channels, 1-5% laser power. Increase laser power and decrease offset to approximate full dynamic range. Do not over saturate the signal, but enhance the signal to merely show a few saturated pixels.
- When capturing a stitch, set the start and end of the larvae along one axis (either x or y), if set along one axis a whole embryo can be imaged in 1 x 4 segments and can be post processed into one image using ImageJ.
- After imaging, remove the larvae from the agarose by gently tearing it around the embedded larvae using watchmaker's forceps. Otherwise, euthanize the larvae by overdosing with undiluted tricaine, covering the agarose with a layer of tricaine and incubating 10 minutes.
13. Data analysis
- Open the individual data sets in ImageJ/Fiji (i.e., control, drug A, drug B, drug A+B) separately, starting with vehicle control.
- Open the analysis macro (annotated script available) (http://doi.org/10.5281/zenodo.4290225).
- In brief the macro analysis does the following: concatenates all open images (one condition); splits the images into the separate channels comprising the image; closes all accessory channels (leaving the cancer cell channel); runs a thresholding algorithm, on the entire concatenated sequence; measures integrated density of each individual image; and saves the measures as an excel sheet in the root folder.
- Run the macro analysis on all conditions.
- Combine measurements (in general at least n=2*20) and remove outliers (Q-test in Graph pad Prism v8).
- Normalize measurements either to solvent control or to day 1 (dependent on the type of experiment, the former for a drug inhibition experiment and the latter for a growth kinetics experiment). Express measurements as normalized cancer cell burden (y axis) over time or condition (x axis) as shown in Figure 3 and Figure 4, respectively.
Results
We have provided step by step instructions for a fast and easy approach to progress from a novel cell line to its analysis. We start with the over expression of a fluorescent tracer using a lentiviral overexpression cassette (steps 3 and 4). This is followed by cell preparation to ensure the least possible dead volume while injecting, allowing to inject high cell numbers into both doC and retro-orbital space (steps 6 and 7). Subsequently, we perform semi-high throughput data acquisition using stereo-fluorescent microscopy and higher magnification confocal microscopy for qualitative analysis of whole-body cancer cell dissemination (Figure 2 and steps 10, 11 and 12). Care has to be taken when acquiring data, as to ensure the reproducibility for both stereo and confocal microscopic imaging, the generic settings and standardization are delineated (steps 11 and 12). Data analysis is discussed (using imageJ/Fiji) 16, along with standardization using imageJ macros (step 13).
In step 3 we mentioned the transient labelling of (cancer) cells to perform a quick pre-screening to assess the tumorigenic potential of a new cancer cell line. One important caveat is that although easy to use and long living, the transient stain described herein has the possibility to form artefacts (i.e., care has to be taken to ensure that cell fragments can be distinguished from whole cells as was performed extensively by Fior and colleagues 9). In our experience the formation of these artefacts is directly linked to the extreme stability of the stain and the brightness (even after cell death), where cell fragments are dispersed and taken up by immune cells, which could subsequently be falsely concluded to derive from active metastasis.
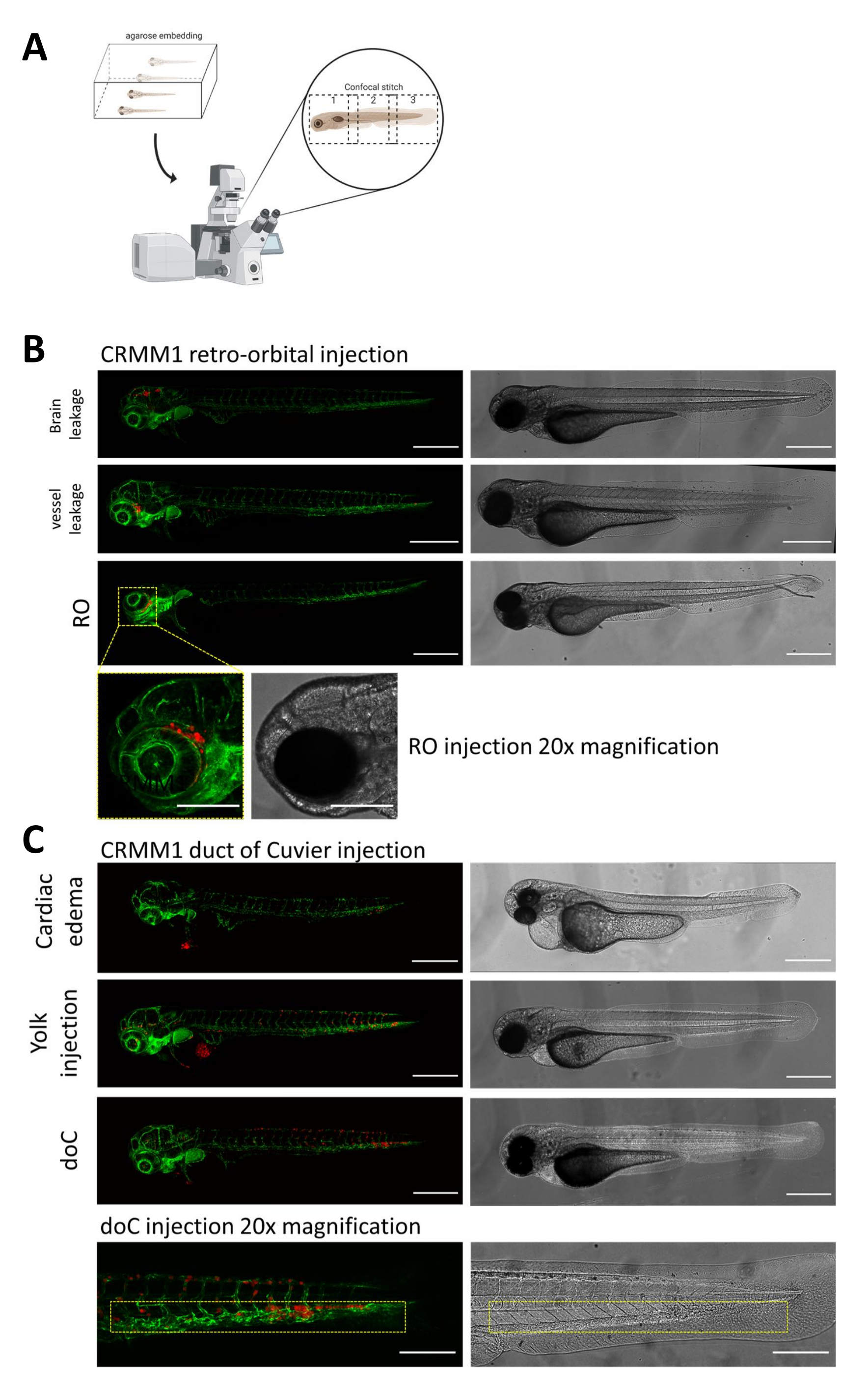
In both described models, the systemic engraftment through the doC and the localized engraftment in the retro-orbital space, thorough screening of the larvae one day after injection is of paramount importance. As shown in Figure 2B all larvae that display mechanical displacement of the engrafted cells into the head area (beyond the retro-orbital site) in the retro-orbital model and cells in the yolk sac, or displaying an edema in the doC injected pool should be removed. All negatively selected phenotypes are displayed as high-resolution confocal stitches in Figure 2, but can be readily seen and removed through stereo microscopical observation.
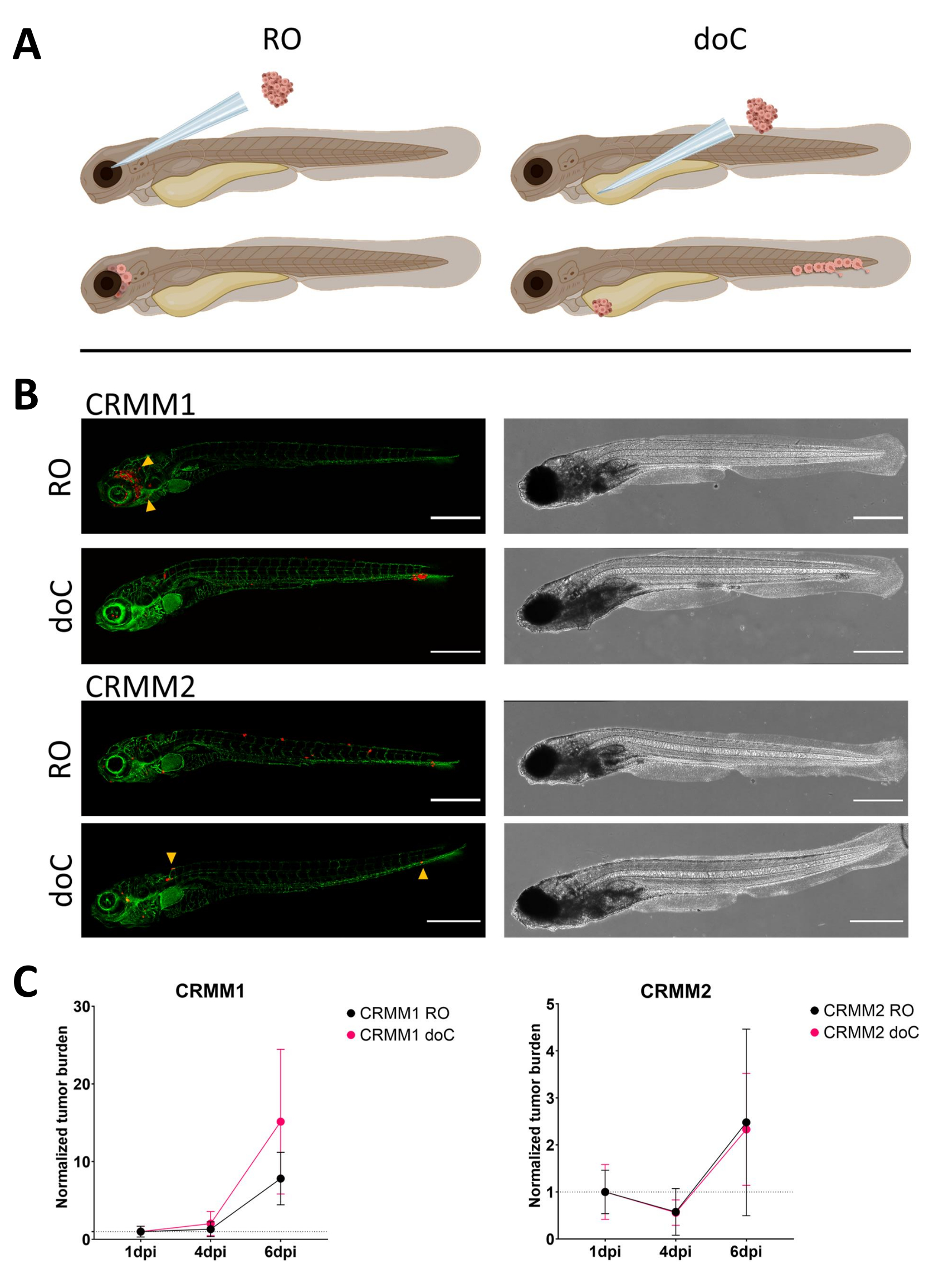
Over time cells will both migrate and proliferate. For the retro-orbital model, we observed infiltration into neighboring tissues for CRMM1, but we observed less proliferation for CRMM2. We strikingly did observe distant metastasis arising between 2-4 dpi in some individuals (20%), where we measured a significant difference at 6 dpi, as shown in Figure 4. For both cell lines, we tested the proliferative potential when injected in both sites. For CRMM1 there was a significant (p<0.0001) increase in cancer cell number for or at the injection sites, when displayed as normalized tumor cell burden, normalizing to day one for each model (7.8-fold increase, ±3.2 for the RO model and an increase of 15-fold ±8,8 for the doC model). CRMM2 did not display significant growth when normalized to day one for each individual model (2.4-fold increase, ±1.9- and 2.3-fold increase, ±1.14 for the RO and doC). CRMM1 was found to readily proliferate in both retro-orbital tissue and the caudal hematopoietic tissue after engraftment. Cell line CRMM2 was less proliferative in both models, but interestingly was found to be capable of distant metastasis when injected in the retro-orbital space as shown in Figure3B,C.
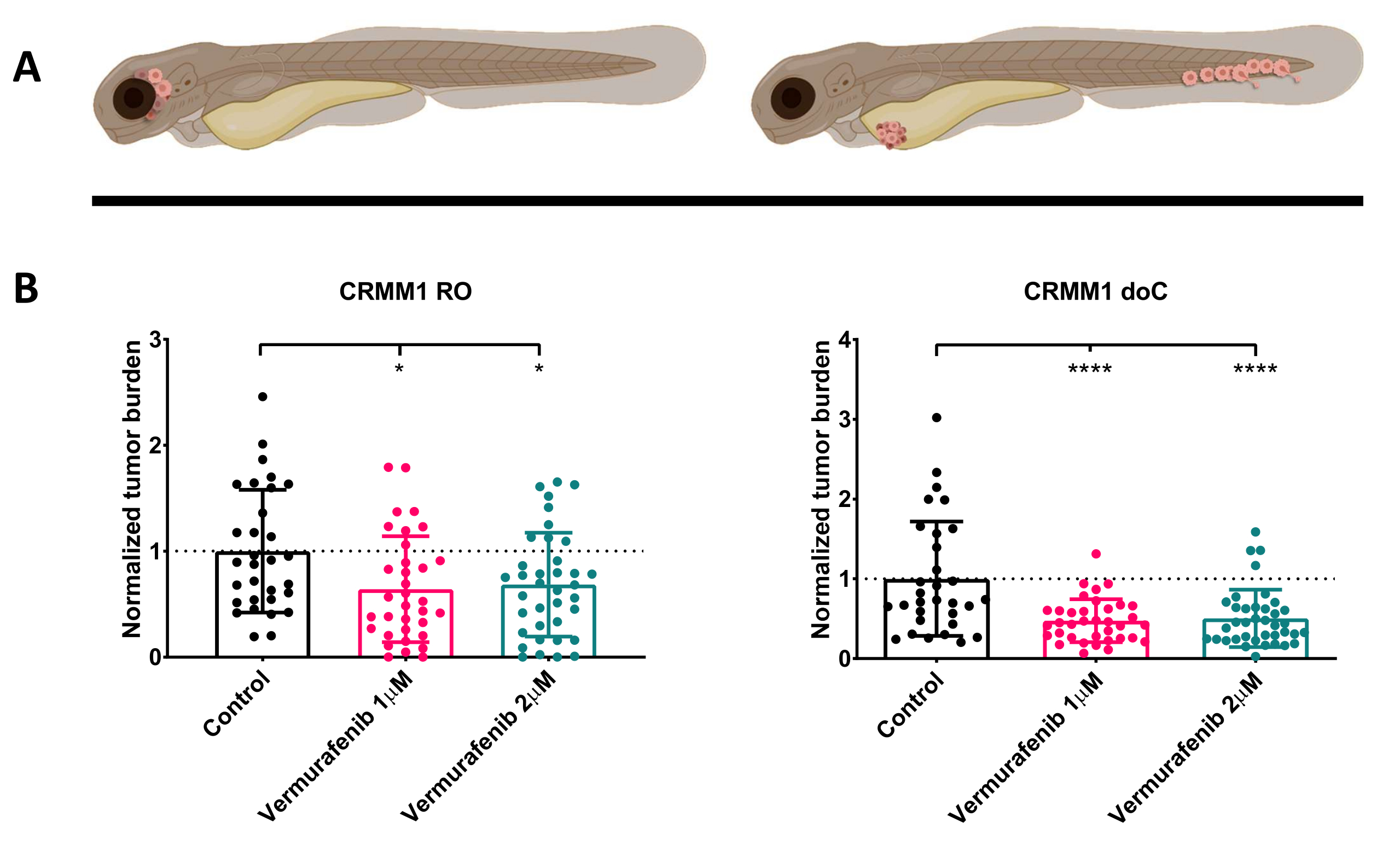
After screening the injected larvae at 1 dpi and randomly assigning the individuals to either treatment or control groups, the fish were treated for 6 days, changing the water containing Vemurafenib (this inhibitor can readily be interchanged for any other titrated antitumor compound). We chose to elaborate upon the previously published hematogenous conjunctival melanoma dissemination model engrafting CRMM114, by testing Vemurafenib's efficacy on orthotopically engrafted CRMM1. CRMM1 showed a strong significant reduction of the Vemurafenib treated ectopically engrafted group (P<0.0001) and a stunted yet significant response for the orthotopically engrafted model (p<0.05) as shown in Figure 4.

Figure 2. Phenotypic assessment and screening after injection. A) Schematic depiction of zebrafish xenograft confocal stitch generation, yielding seamless, high resolution images after integration of subsequent confocal projection. Here zebrafish xenografts are embedded in 1% low melting agarose and mounted on a glass bottom confocal dish (as described in step 11.3). B) All possible outcomes of retro-orbital and duct of Cuvier engraftment are displayed injected in green fluorescent blood vessel reporter zebrafish (TG:fli:GFP), with cells stained through lentiviral over expression of tdTomato). We denote the correct engraftment at 1 dpi (RO panel) and the unwanted phenotypes (both brain leakage and blood vessel leakage). The latter two populations must be removed to ensure they do not confound downstream experimental findings. C) The unwanted phenotypes for the hematogenous engraftment through the duct of Cuvier (doC) are outlines where cardiac edematous larvae (Cardiac edema) and larvae with cells leaking into the yolk sac (Yolk injection) must be removed to prevent interference with downstream measurements. The correctly injected larvae are entered into experimental groups as described in step 7.1. (All images acquired at 1 dpi, using a confocal microscope, scale bars 200 µm. Yellow boxes indicate metastatic sites for both RO and doC engraftments, head region and caudal hematopoietic tissue, respectively). Please click here to view a larger version of this figure.
{kind=link}

Figure 3. Comparative analysis of conjunctival melanoma cell lines CRMM1 and CRMM2 show differential metastatic and growth capacity. A) Schematic representation of injection models, retro-orbital model (RO) and hematogenous engraftment model (doC) the fish used are TG(fli:GFP) green blood vessel reporters, with cells over expressing tdTomato shown in red. B) Representative phenotypes of fish engrafted with CRMM1 and CRMM2, CRMM1 displays efficient engraftment (both RO and doC) and small scale invasion into the tissue surrounding the RO engraftment site (RO, yellow arrowheads). CRMM2 exhibits a remarkably lower engraftment efficiency for both engraftment models, but shows distant metastasis when injected retro-orbitally (as shown in RO, denoted by the arrowheads). (All images acquired at 6 dpi, a confocal microscope, scale bars 200 µm. Yellow arrowheads indicate metastatic sites for both RO and doC engraftments, head region and caudal hematopoietic tissue respectively). C) Kinetic engraftment plots for both CRMM1 and CRMM2, comparing both engraftment models to day 1 (normalizing to day 1), there is a significant (p<0.0001) increase in normalized tumor burden for cell line CRMM1(between 1 dpi and 6 dpi) where there is a (non-significant) upward trend for CRMM2. CRMM1 reveals a significant difference between RO and doC growth, where the doC model shows a higher tumor expansion rate (approximately 2-fold higher for the doC engrafted larvae). Graphs display the mean and standard error of the mean (SEM). All groups were normalized to 1 dpi for each individual condition. Please click here to view a larger version of this figure.
{kind=link}

Figure 4. BRAF V600E inhibitor Vemurafenib significantly inhibits both RO and doC conjunctival melanoma engrafted zebrafish larvae. A) Schematic representation of zebrafish phenotypes, RO and doC models. B) Both RO and doC engrafted larvae, injected with conjunctival melanoma cell line CRMM1 display a significant reduction of normalized tumor burden (p<0.05 and P<0.001 respectively). The doC engrafted zebrafish models indicate an enhanced drug response and a dose independent relationship to drug inhibition, indicating a possible saturation of inhibition). Graphs show the mean and standard error of the mean (SEM), All groups were normalized to control for each individual cell line. Please click here to view a larger version of this figure.
{kind=link}
| Reagent | Volume |
| psPAX2 | 1.71 pmol (12.14µg) |
| pMD2.G | 0.94 pmol (3.66µg) |
| Transfer Plasmid* | 1.64 pmol (calculate exact volume) |
Table 1.
Discussion
Here, we have defined a meticulous approach to model primary and metastatic ocular melanoma in zebrafish xenografts. By combining both a localized, orthotopic injection and a systemic, ectopic injection models we have recapitulated the etiology of carcinogenesis for a cancer where no animal models were previously available. The inherent transparency of the early zebrafish larva allows the tracking of fluorescently labelled cancer cells on a whole animal level, ensuring the easy visualization of potential metastatic sites17. Moreover high magnification confocal microscopical analysis allows us to track cells at a subcellular resolution10.
We have provided step by step instructions for a fast and easy approach to progress from a novel cell line to establishment of the xenograft and its analysis. We start with the overexpression of a fluorescent tracer using a lentiviral overexpression cassette (step 3 and 4) followed by cell preparation to ensure the least possible dead volume while injecting. This enables the injection of high cell numbers into both doC and retro-orbital space (step 7 and 8). Then we perform semi-high throughput data acquisition using stereo-fluorescent microscopy and higher magnification confocal microscopy for qualitative analysis of whole-body cancer cell dissemination (Figure 2 and step 9 and 10). Care has to be taken when acquiring data, as to ensure the reproducibility for both stereo and confocal microscopic imaging, the generic settings and standardization are delineated (steps 11 and 12). Data analysis is discussed (using imageJ/Fiji) 16, along with standardization using ImageJ macros (step 13).
In step 3 we mention the transient labelling of (cancer) cells to perform a quick pre-screen to assess the tumorigenic potential of a new cancer cell line. One important caveat is that although easy to use and long living, the transient stain described herein has the possibility to form artefacts (e.g., care has to be taken to ensure that cell fragments can be distinguished from whole cells as was performed extensively by Fior and colleagues 9). In our experience the formation of these artefacts is directly linked to the extreme stability of the stain and the brightness (even after cell death), where cell fragments are dispersed and taken up by immune cells, which could subsequently be falsely concluded to derive from active metastasis.
Using these models, we simulated primary tumor development by physically confining the engrafted cells within the retro-orbital interstice. Subsequent thorough screening at 1 day post engraftment ensures that cells found at distant site later in the experiment have actively metastasized (intravasated and disseminated, ultimately to extravasate at the metastatic niche). Engraftment through the doC, the embryonic common cardinal vein, allows for easy and highly reproducible implantation of large quantities for cells (at a surplus of 600 cells when properly concentrated), effectively circumventing the primary stages of the metastatic cascade (intravasation) and allowing us to focus on the later stages of the metastatic cascade (adhesion, extravasation and outgrowth). Although powerful tools when used properly, both models should be monitored extensively during the first day post engraftment to ensure that no false positive conclusions are drawn during the later stages of the experiment.
In line with previous publications we have shown that conjunctival melanoma lines readily form metastatic colonies after dissemination throughout the zebrafish blood circulation system14. Here we report the expanding of the engraftment repertoire with the retro-orbital injection as an orthotopic model, and the subsequent active metastasis to the caudal hematopoietic tissue of the cell line CRMM2. Subsequently we report the efficacy of BRAF V600E specific inhibitor Vemurafenib also on the primary form of conjunctival melanoma when modelled in zebrafish larvae.
Using the aforementioned methods, a skilled researcher is capable of generating in excess of hundreds of engrafted larvae per day (approximately 200 per hour) of either model proposed. In a timescale of two weeks a drug can be both titrated for maximum tolerated dose, and screened on established xenograft model. From start to finish, using a non-transduced cell line, to having a drug sensitivity profile in the zebrafish model can be achieved within a month (given that the injected cell line is tumorigenic within the zebrafish model). In our hands as little as 20 larvae per experiments and two biological repeats have reproducibly yielded robust drug inhibition, when two individual experiments conflict (or do not yield statistically significant growth inhibition) a third biological repeat can be conducted.
Through minor adjustments, these models have allowed us to quickly adapt these implantation strategies for glioblastoma (hind brain cavity injection), breast cancer (doC injection) and osteo sarcoma (doC) among others 18,19,20,21. These models can subsequently be utilized for both basic research and pre-clinical screening of both single drugs and combinatorial drug strategies. Recently, we described different administration regimes of drugs and their photo activation using these models 13.
Disclosures
None.
Acknowledgements
This work was supported by funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 667787 (UM Cure 2020 project, www.umcure2020.org). The Chinese Scholarship Council is kindly acknowledged for a PhD grants to J.Y.
Materials
| Name | Company | Catalog Number | Comments |
| 2.5mm box filament | Science products | FB255B | for pulling micro injection needles using a Sutter P97 or P1000 |
| 3mL transfer pipettes | Merck | Z350796 | for transfer and selection of zebrafish embryos |
| Agarose | Milipore | 2120 | 1.5% (w/v) in eggwater, 1.5 g in 100 mL DPBS, microwave to dissolve, for injecting and stereofluorescence imaging of zebrafish larvae |
| Capillaries: borosilicate glass outer | World precision instruments | BF100-78-10 | Borosilicate glass capillaries used for needle preparation |
| DMSO | Sigma | D8418 | Often used as solvent in drug treatments, should be stored at 2-8°C the dark. |
| DPBS | Thermo Fischer Scientific | 14190144 | Dulbecco’s phosphate buffered saline, without Mg2+ and Ca2+ for washing the cells, lack of Ca2+ impairs cell-cell adhesion through cadherins and prevents cell aggregation during injection |
| Egg water | Instant ocean | SS15-10 | 0.6 mg/L final concentration sea salt in demineralized water |
| GFP encoding lentiviral transfer plasmid | Addgene | Plasmid #106172 | Generated in Snaar lab, available at Addgene |
| Hek293T | ATCC | CRL-3216 | Stable cell line for generating lentiviral particles, contains SV40-T antigen required for the generation of lentiviral particles |
| Leica sp8 confocal | Leica | Leica TCS SP8 | automated stage confocal microscope with 405/488/514/635nm lasers |
| LipodD293 | Signagen | SL100668 | Highly efficient HEK293t optimized transfection reagent |
| Low-melting agarose | Milipore | 2070 | 1% (w/v) in eggwater 1.5 g in 100 mL DPBS, microwave to dissolve, for embedding zebrafish larvae for confocal imaging |
| Micro loader tips | Fischer scientific | 10289651 | flexible microloader tips |
| Micro manipulator | World precision instruments | M3301R | x/y/z manual micro manipulator for microinjection |
| Needle puller: P-97 or P-1000 | Sutter | P-97 | needle puller used for generating standardized micro engraftment needles |
| Nr.5 watchmakers forceps | VWR | HAMMHSC818-11 | fine watchmakers forceps used for breaking back needles |
| Picopump | World precision instruments | SYS-PV820 | pulse controller supplying pressure for microinjection |
| pMD2.G | Addgene | plasmid #12259 | Gifted by Didier Trono, 2nd generation lentiviral virulence plasmid |
| psPAX2 | Addgene | plasmid #12260 | Gifted by Didier Trono, 2nd generation lentiviral packaging plasmid |
| PVP40 | Sigma-Aldrich | PVP40 | Polyvinylpyrrolidone average mol wt 40,000) PVP40 2% (w/v) in DPBS, 1 g PVP40 in 50 mL DPBS. Vortex and incubate at 37°C to facilitate dissolving. Store at room temperature. |
| tdTomato encoding lentiviral transfer plasmid | Addgene | Plasmid #106173 | Generated in Snaar lab, available at Addgene |
| transmitted light stereo microscope | Leica | leica M50 with (MDG33 base) | leica transmitted light microscope with mirror adjustable illumination. |
| Tricaine | Sigma-Aldrich | E10521 | Ethyl 3-aminobenzoate methanesulfonate or MS-222 |
| TryplE | Thermo Fischer Scientific | 12604-01 | Synthetic trypsine replacement, less damaging to the cells and allows for the gentle dispersion of strongly adherent cells. (Thermo- |
| willco dish | WillCo wells | GWST-5040 | 50mm glass bottom dishes, allow for the embedding of up to 20 zebrafish larvae, enabling the imaging of multiple conditions in one dish due to its large optical glass surfac |
References
- Yang, J., Manson, D. K., Marr, B. P., Carvajal, R. D. Treatment of uveal melanoma: where are we now. Therapeutic Advances in Medical Oncology. 10, (2018).
- Wong, J. R., Nanji, A. A., Galor, A., Karp, C. L. Management of conjunctival malignant melanoma: A review and update. Expert Review of Ophthalmology. 9, 185-204 (2014).
- Nguyen, D. X., Bos, P. D., Massagué, J. Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer. 9, 274-284 (2009).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Zon, L. I., Peterson, R. T. In vivo drug discovery in the zebrafish. Nature Reviews Drug Discovery. 4, 35-44 (2005).
- Howe, K., et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 496, 498-503 (2013).
- Palmblad, M., et al. Parallel deep transcriptome and proteome analysis of zebrafish larvae. BMC Research Notes. 6, 428 (2013).
- Yan, C., et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell. 177, 1903-1914 (2019).
- Fior, R., et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proceedings of the National Academy of Sciences of the United States of America. 114, 8234-8243 (2017).
- Campbell, P. D., Chao, J. A., Singer, R. H., Marlow, F. L. Dynamic visualization of transcription and RNA subcellular localization in zebrafish. Development. 142, 1368-1374 (2015).
- Campeau, E., et al. A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells. PLoS One. 4, 6529 (2009).
- vander Helm, D., et al. Mesenchymal stromal cells prevent progression of liver fibrosis in a novel zebrafish embryo model. Scientific Reports. 8, 16005 (2018).
- Chen, Q., et al. TLD1433 photosensitizer inhibits conjunctival melanoma cells in zebrafish ectopic and orthotopic tumour models. Cancers. 12, (2020).
- Pontes, K. C. d. e. S., et al. Evaluation of ( fli:GFP ) Casper Zebrafish Embryos as a Model for Human Conjunctival Melanoma. Investigative Opthalmology & Visual Science. 58, 6065 (2017).
- Liverani, C., et al. Innovative approaches to establish and characterize primary cultures: an ex vivo 3D system and the zebrafish model. Biology Open. 6, 133-140 (2017).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9, 676-682 (2012).
- White, R. M., et al. Transparent Adult Zebrafish as a Tool for In Vivo Transplantation Analysis. Cell Stem Cell. 2, 183-189 (2008).
- Mercatali, L., et al. Development of a patient-derived xenograft (PDX) of breast cancer bone metastasis in a Zebrafish model. International Journal of Molecular Sciences. 17, (2016).
- Tulotta, C., et al. Imaging cancer angiogenesis and metastasis in a zebrafish embryo model. Advances in Experimental Medicine and Biology. 916, 239-263 (2016).
- Paauwe, M., et al. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clinical Cancer Research. 24, 6331-6344 (2018).
- Cao, J., et al. Overexpression of EZH2 in conjunctival melanoma offers a new therapeutic target. Journal of Pathology. 245, (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved