A subscription to JoVE is required to view this content. Sign in or start your free trial.
Production of Adeno-Associated Virus Vectors in Cell Stacks for Preclinical Studies in Large Animal Models
In This Article
Summary
Here we provide a detailed procedure for large-scale production of research-grade AAV vectors using adherent HEK 293 cells grown in cell stacks and affinity chromatography purification. This protocol consistently yields >1 x 1013 vector genomes/mL, providing vector quantities appropriate for large animal studies.
Abstract
Adeno-associated virus (AAV) vectors are among the most clinically advanced gene therapy vectors, with three AAV gene therapies approved for humans. Clinical advancement of novel applications for AAV involves transitioning from small animal models, such as mice, to larger animal models, including dogs, sheep, and nonhuman primates. One of the limitations of administering AAV to larger animals is the requirement for large quantities of high-titer virus. While suspension cell culture is a scalable method for AAV vector production, few research labs have the equipment (e.g., bioreactors) or know how to produce AAV in this manner. Moreover, AAV titers are often significantly lower when produced in suspension HEK 293 cells as compared to adherent HEK293 cells. Described here is a method for producing large quantities of high-titer AAV using cell stacks. A detailed protocol for titering AAV as well as methods for validating vector purity are also described. Finally, representative results of AAV-mediated transgene expression in a sheep model are presented. This optimized protocol for large-scale production of AAV vectors in adherent cells will enable molecular biology laboratories to advance the testing of their novel AAV therapies in larger animal models.
Introduction
Gene therapy utilizing adeno-associated virus (AAV) vectors has made huge strides over the past three decades1,2. Demonstrated improvements in a diverse range of genetic diseases, including congenital blindness, hemophilia, and diseases of the musculoskeletal and central nervous system, have brought AAV gene therapy to the forefront of clinical research3,4. In 2012, the European Medicines Agency (EMA) approved Glybera, an AAV1 vector expressing lipoprotein lipase (LPL) for the treatment of LPL deficiency, making it the first marketing authorization for a gene therapy treatment in either Europe or the United States5. Since then, two additional AAV gene therapies, Luxturna6 and Zolgensma7, have received FDA approval, and the market is expected to expand quickly over the next 5 years with as many as 10-20 gene therapies expected by 20258. Available clinical data indicate that AAV gene therapy is a safe, well-tolerated, and efficacious modality making it one of the most promising viral vectors, with above 244 clinical trials involving AAV registered with ClinicalTrials.gov. The increasing interest in clinical applications involving AAV vectors requires robust and scalable production methods to facilitate the evaluation of AAV therapies in large animal models, as this is a critical step in the translational pipeline9.
For AAV vector production, the two main requirements are the AAV genome and the capsid. The genome of wild-type (wt)-AAV is single-stranded DNA that is approximately 4.7 kb in length10. The wt-AAV genome comprises inverted terminal repeats (ITRs) found at both ends of the genome, which are important for packaging, and the rep and cap genes11. The rep and cap genes, necessary for genome replication, assembly of the viral capsid, and encapsulation of the genome into the viral capsid, are removed from the viral genome and provided in trans for AAV vector production12. The removal of these genes from the viral genome provides room for therapeutic transgenes and all the necessary regulatory elements, including the promoter and polyA signal. The ITRs remain in the vector genome to ensure proper genome replication and viral encapsulation13,14. To improve the kinetics of transgene expression, AAV vector genomes can be engineered to be self-complementary, which mitigates the need for conversion from single-stranded to double-stranded DNA conversion during AAV genome replication, but reduces the coding capacity to ~2.4 kb15.
Beyond AAV genome design, capsid serotype selection determines the tissue and cell tropism of the AAV vector in vivo2. In addition to tissue tropism, different AAV serotypes have been shown to display different gene expression kinetics16. For example, Zincarelli et al.17 classified different AAV serotypes into low expression serotypes (AAV2, 3, 4, 5), moderate expression serotypes (AAV1, 6, 8), and high expression serotypes (AAV7 and 9). They also categorized AAV serotypes into slow-onset expression (AAV2, 3, 4, 5) or rapid-onset expression (AAV1, 6, 7, 8, and 9). These divergent tropisms and gene expression kinetics are due to amino acid variations in the capsid proteins, capsid protein formations, and interactions with host cell receptors/co-receptors18. Some AAV capsids have additional beneficial characteristics such as the ability to cross the blood-brain barrier following intravascular administration (AAV9) or reside in long-living muscle cells for durable transgene expression (AAV6, 6.2FF, 8, and 9)19,20.
This paper aims to detail a cost-effective method for producing high-purity, high-titer, research-grade AAV vectors for use in preclinical large animal models. Production of AAV using this protocol is achieved using dual-plasmid transfection into adherent human embryonic kidney (HEK)293 cells grown in cell stacks. Furthermore, the study describes a protocol for heparin sulfate affinity chromatography purification, which can be used for AAV serotypes that contain heparin-binding domains, including AAV2, 3, 6, 6.2FF, 13, and DJ21,22.
A number of packaging systems are available for the production of AAV vectors. Among these, the use of a two-plasmid co-transfection systems, in which the Rep and Cap genes and Ad helper genes (E1A, E1B55K, E2A, E4orf6, and VA RNA) are contained within one plasmid (pHelper), has some practical advantages over the common three-plasmid (triple) transfection method, including reduced cost for plasmid production23,24. The AAV genome plasmid containing the transgene expression cassette (pTransgene), must be flanked by ITRs, and must not exceed ~4.7 kb in length. Vector titer and purity can be affected by the transgene due to potential cytotoxic effects during transfection. Assessment of vector purity is described herein. Vectors produced using this method, which yield a 1 x 1013 vg/mL for each, were evaluated in mice, hamsters, and ovine animal models.
Table 1: Composition of required solutions. Necessary information, including percentages and volumes, of components needed for various solutions throughout the protocol. Please click here to download this Table.
Protocol
1. Double plasmid transfection of HEK293 cells in cell stacks
- Thaw a cryo-vial of HEK293 cells in a bead bath set at 37 °C.
NOTE: Pre-warm complete DMEM to 37 °C while cells are thawing to ensure the cold temperature does not shock cells when plating. Ensure the cells have a low passage number, ideally less than 20, to ensure optimal growth and transfection efficiency. Ensure that the cells are certified to be mycoplasma-free. - Transfer the contents of the cryo-vial dropwise into a 15 mL conical tube containing 10 mL of pre-warmed complete DMEM and centrifuge the cells at 500 x g for 5 min.
- Aspirate the media, and then resuspend the HEK293 cells in 20 mL of pre-warmed complete DMEM. Seed the cells in a 15 cm plate and incubate at 37 °C, with 5% CO2.
- Split the cells from one 15 cm plate into three for seeding in the cell culture chamber.
- Once cells are 80% confluent, aspirate the media and gently wash the plate with 3 mL of PBS to not disrupt the monolayer. Then, aspirate PBS and add 3 mL of trypsin.
- Incubate for 2 min at 37 °C until the cells lift from the plate, and then neutralize trypsin by adding 7 mL of complete DMEM to the plate.
- Collect all the media and cells into a 15 mL tube and pellet the cells by centrifuging at 500 x g for 5 min.
- Aspirate the supernatant from the 15 mL tube and resuspend the cell pellet in 3 mL of complete DMEM. Add 1 mL to each 15 cm plate containing 20 mL of complete DMEM; gently rock the plates to distribute the cells evenly, and incubate at 37 °C, with 5% CO2.
- Once the cells are 80% confluent, repeat steps 1.4.1 and 1.4.2. Collect the supernatant in 50 mL conical tubes, and gently invert the tube to ensure the cells are homogeneous.
- Determine the cell density by mixing 10 µL of the samples of cells with 10 µL of trypan blue and adding the mixture to a cell counting slide for analysis in the cell counter.
- Mix 1 L of pre-warmed complete DMEM with the needed cell suspension to seed the cell culture chamber (surface area of 6360 cm2) with 1 x 104 cells/cm2. Pour the cell mixture into the cell culture chamber and gently rotate to evenly distribute the cells across each monolayer (Figure 1) and incubate at 37 °C, with 5% CO2.
- In addition to the cell culture chamber, plate a 15 cm plate with 1 x 104 cells/cm2 as a reference for confluency.
- Following ~65-h incubation, check the reference plate for confluency-ideally ~80%-90% confluent.
NOTE: Pre-warm complete DMEM for adding to the cell culture chamber at 37 °C.

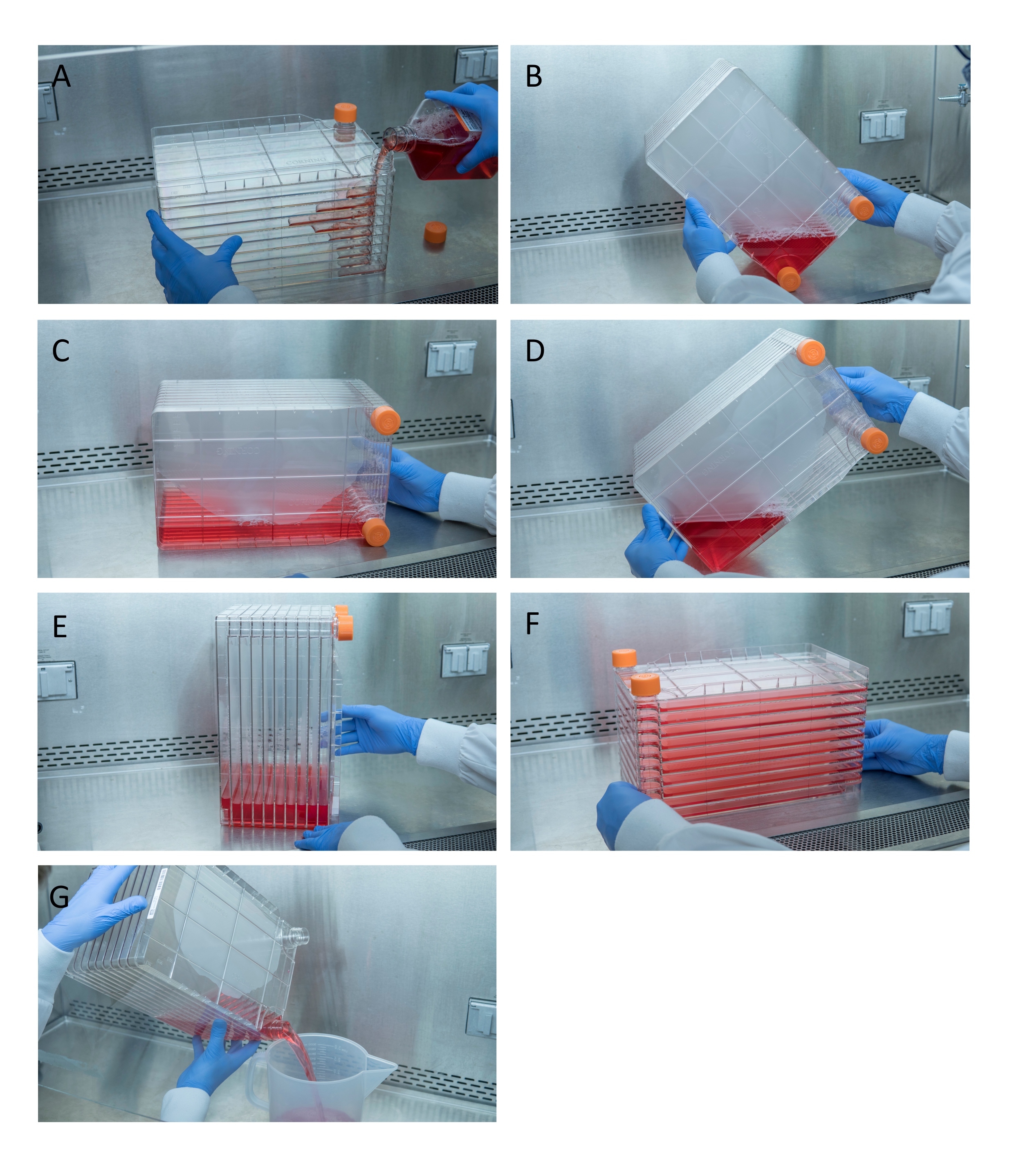
Figure 1: Maneuvering of cell stack for cell seeding and transfection. For seeding cell stack, start by removing one of the vent caps and pouring in 1 L of pre-warmed complete DMEM with needed quantity of HEK293 cells (A). Evenly distribute cells and media by tightening both vent caps and bring all media to the corner of the cell stack with one of the vent caps and place it in that corner (B), place the cell stack on its side (C), and then turn the cell stack 90° (D) so that the vent ports are up (E). Gently lower the cell stack to its normal horizontal position and ensure all the chambers of the cell stack are completely covered in media (F). When transfecting, unscrew both vent caps and slowly pour out old media into a waste sterile waste container for even flow not to disturb the monolayer of the cells (G). Please click here to view a larger version of this figure.
{kind=link}
- Prepare polyethyleneimine (PEI)/DNA mixture at a concentration ratio of 3:1 (w/w).
- Prepare the DNA mixture in a 50 mL conical tube by adding 475 µg of pTrangene and 1425 µg of pHelper to 40 mL of reduced-serum medium to create a 3:1 ratio of pHelper:pTrangene.
NOTE: The PEI/DNA mixture calculator can be found using Table 2. - Add 5.7 mL of PEI (1 g/L) to the reduced-serum medium and the DNA mixture dropwise. Then, vortex briefly and incubate for 10 min at room temperature.
NOTE: As PEI/DNA incubates at room temperature, it will become slightly cloudy.
- Prepare the DNA mixture in a 50 mL conical tube by adding 475 µg of pTrangene and 1425 µg of pHelper to 40 mL of reduced-serum medium to create a 3:1 ratio of pHelper:pTrangene.
- After 8 min of PEI/DNA incubation, remove the media from the cell culture chamber.
NOTE: Ensure to loosen both orange caps to maintain a smooth flow of media to avoid dislodging of cells. - Add PEI/DNA to 1 L of pre-warmed complete DMEM, and slowly pour the mixture into the cell culture chamber port. Distribute the liquid evenly to all the rows (Figure 1) and incubate for 72 h at 37 °C, with 5% CO2.
2 Harvesting AAV and chemical lysis of the transfected HEK293 cells
- Shake the cell culture chamber vigorously to dislodge the cells until the media appears cloudy from dislodged cells and pour into four 500 mL centrifuge tubes.
- Centrifuge the tubes at 18,000 x g for 30 min at 4 °C to pellet the cells. Pour the clarified supernatant into 1 L polyethylene terephthalate copolyester (PETG) bottle.
NOTE: If one does not have access to a high-speed centrifuge, centrifuge at 12,000 x g for 40 min. The pelleted cells may not be solid at this speed and will slide as pouring out supernatant. - Resuspend the cell pellets in 500 mL centrifuge tubes with 50 mL of lysis buffer and incubate for 60 min at 37 °C.
- Centrifuge the tubes at 18,000 x g for 30 min, and then transfer the supernatant into the same 1 L PETG bottle. Discard the pelleted cell debris.
NOTE: Purify the clarified supernatant immediately and store at 4 °C for up to 72 h. For longer-term storage, store at -80 °C. Do not store at -20 °C.
3 AAV Vector purification using heparin affinity chromatography
- Remove the crude lysate from -80 °C and leave at 4 °C overnight to thaw. Once thawed, use a 0.22 µM filter to filter the crude lysate.
- To passivate the centrifugal concentrator, add 4 mL of filter pre-treatment buffer to a centrifugal concentrator for each heparin sepharose column being used. Passivate the centrifugal concentrator at room temperature for 2-8 h. Set up passivation immediately before the purification steps.
- Set up the tubing and the pump (Figure 2).
- Place the tubing in a peristaltic pump and run 20 mL of 1 M NaOH. Next, run 50 mL of molecular grade water, and then run 50 mL of basal DMEM.
- Attach 5 mL heparin sepharose column to tubing and run 25 mL of basal DMEM to remove the preservative.
- Run 0.2 µM of the filtered crude lysate through the column at a flow rate of 1-2 drops/s.

Figure 2: Set up for peristaltic pump for AAV purification. Run the tubing from the crude lysate, through the peristaltic pump, and into the heparin matrix column. Please click here to view a larger version of this figure.
{kind=link}
NOTE: Ensure not to introduce bubbles or allow the column to run dry, as this will compromise the column and prevent the elution of AAV. Discard the column if it runs dry and use a new column for the remainder of crude lysate.
- Load all of the crude lysate onto the heparin column and use the following solutions to wash the column.
- Wash using 50 mL of 1x Hank's Balanced Salt Solutions (HBSS) without Mg2+ and Ca2+.
- Wash using 15 mL of 0.5 % N-Lauroylsarcosine in HBSS without Mg2+ and Ca2+.
- Wash using 50 mL of HBSS without Mg2+ and Ca2+.
- Wash using 50 mL of HBSS with Mg2+ and Ca2+
- Wash using 50 mL of 200 mM NaCl/HBSS with Mg2+ and Ca2+.
- Elute 5 x 5 mL (25 mL total) with 300 mM of NaCl/HBSS with Mg2+ and Ca2+ and label the elutions as E1-E5 (each elution is of 5 mL).
- Concentrating the virus using a centrifugal concentrator
- Spin the centrifugal concentrator containing the pre-treatment buffer at 900 x g for 2 min. Discard the flow-through.
- Wash the centrifugal concentrator filter with 4 mL of HBSS with Mg2+ and Ca2+ and centrifuge at 1000 x g for 2 min; discard the flow-through.
- Add the elution E2 to the centrifugal concentrator. Spin at 1000 x g for 5 min and discard the flow-through.
- Finish adding E2 and then add E3 to the centrifugal concentrator and spin at 1000 x g for 5 min until the concentrated virus is approximately 1 mL.
NOTE: Avoid centrifuging the vector such that the volume is below the level of the filter. Do not concentrate E1, E4, or E5 in the centrifugal concentrator, as they contain very little to no vector and contain contaminants. - Remove the concentrated virus from the centrifugal concentrator using a p200 filtered tip and place it into a sterile 1.5 mL centrifuge tube.
- Rinse the centrifugal concentrator with 200 µL of HBSS with Mg2+ and Ca2+ to dislodge any remaining AAV from the filter. Pipette up and down vigorously multiple times (for ~30 s) to dislodge any virus adhered to the membrane and place in the 1.5 mL centrifuge tube with the remainder of the virus. Mix the tube well.
- Aliquot 5 µL for DNA extractions and store the purified vector at -80 °C.
- Wash the column using 25 mL of 2 M NaCl. Further, use 25 mL of 0.1% Triton X-100, pre-heated to 37 °C to wash the column. Next, wash the column using 50 mL of sterile dH2O, and then wash using 25 mL of 20% ethanol.
- Ensure the column membrane is fully saturated in 20% ethanol, as this is the storage solution. Seal the column with plugs provided and store at 4 °C.
- Store the tubing in 1 M NaOH.
NOTE: If cleaned properly, heparin sepharose columns can be re-used up to five times.
4 AAV genomic DNA extraction
- Prepare the reaction mix mentioned in Table 3 in a PCR tube for DNase treatment.
| Component | Volume |
| Purified AAV vector | 5 μL |
| 10x DNase Buffer | 2 μL |
| DNase | 1 μL |
| ddH2O | 12 μL |
| Final Volume | 20 μL |
Table 3: DNase treatment master mix formula. Recommended components and volumes required for DNase treatment of AAV viral vectors during DNA extraction.
- Vortex the PCR tube to mix and pulse the PCR tube to spin down the contents.
- Using a thermocycler, incubate at 37 °C for 20 min followed by 75 °C for 15 min to heat inactivate the DNase.
- Add 5 µL of Proteinase K.
- Using a thermocycler, incubate at 50 °C for 60 min and then at 95 °C for 30 min to heat inactivate Proteinase K.
- Use a DNA clean-up kit to remove potential contaminants.
NOTE: This step was performed using a commercially available blood and tissue clean up kit (Table of Materials).- Add 200 µL of AL buffer (Blood and tissue clean up kit, Table of Materials) to the PCR tube containing the DNase/Proteinase K treated vector.
- Vortex the PCR tube and incubate at 56 °C for 10 min in a thermocycler.
- Pipette the liquid from the PCR tube into a sterile spin column sitting in a collection tube.
- Add 200 µL of 100% ethanol to the column and mix thoroughly by vortexing.
- Centrifuge at 6,000 x g for 1 min and discard the flow through.
- Add 500 µL of buffer AW1 (Blood and tissue clean up kit, Table of Materials) to the spin column.
- Centrifuge at 6,000 x g for 1 min and discard the flow through.
- Add 500 µL of buffer AW2 (Blood and tissue clean up kit, Table of Materials) to the spin column.
- Centrifuge at 15,000 x g for 3 min and discard the flow through.
- Place the spin column into a sterile 1.5 mL centrifuge tube and add 200 µL of buffer AE (Blood and tissue clean up kit, Table of Materials) directly to the spin column membrane.
- Incubate at room temperature for 1 min.
- Centrifuge at 6,000 x gfor 1 min to elute the DNA.
- Store the DNA at -20 °C.
5 Titration of AAV vector genomes using quantitative polymerase chain reaction and a Simian Virus 40 (SV40) probe
NOTE: Perform all qPCR work in a PCR hood using filtered pipette tips to avoid external DNA contamination. If the AAV genome does not encode an SV40 polyA sequence, use a probe against the ITR described elsewhere25. Ensure the plasmid DNA selected as standard contains SV40 polyA sequence.
- Stock standard preparation
- Dilute the stock plasmid DNA standard (pTransgene plasmid containing SV40 polyA sequence) to a final concentration of 10 µg/µL and store at -20 °C in 6 µL aliquots.
- Determine the copy number present in plasmid DNA standard using the following online calculator26.
NOTE: Use a plasmid DNA used for the standard produced by a commercial vendor to ensure quality and correct concentration. Prepare a large quantity of standard (e.g., 10 mL) to conduct bridging studies when transitioning to a newly prepared standard.
- Prepare the following reagent mix mentioned in Table 4 for both the samples and the standard in a 1.5 mL centrifuge tube.
NOTE: Prepare sufficient overage of master mix. See Table 5 for primer/probe sequences.
| Component | Volume |
| Universal qPCR master mix (2X) | 10 μL |
| Molecular grade water | 4.5 μL |
| 40x SV40 polyA primer/probe | 0.5 μL |
| Final Volume | 15 μL |
Table 4: qPCR master mix for AAV titration. Recommended components and volumes required for qPCR of DNA extracted from AAV viral vectors.
| Component | Sequence |
| Forward primer | 5’-AGCAATAGCATCACAAATTTCACAA-3’ |
| Reverse primer | 5’-CCAGACATGATAAGATACATTGATGAGTT-3’ |
| Probe | /56-FAM/AGCATTTTT/Zen/TTCACTGCATTCTAGTTGTGGTTTGTC/3IABkFQ |
Table 5: Primer sequences against the SV40 polyA DNA sequence. Sequences of the primers and probe used for qPCR titration, which bind to specific areas of AAV viral vectors that contain the SV40 polyA sequence.
- Pipette the master mix up and down to mix.
- Set up the dilution plate.
- Use a clear 96-well plate to prepare standard and sample dilutions, add 45 µL of molecular grade water to each well in every other column starting with column 1 (columns 1, 3, 5, 7, 9, and 11).
- Add 5 µL of the standard to well A1 and pipette to mix.
- Use a new filtered pipette tip to create a 1/10 dilution from well A1 to B1.
- Continue a series of 10-fold dilutions down the column until reaching G1.
- Do not add to H1, as this will act as a negative control.
- Apply the first sample (S1) by adding it to well A3, forming a 1/10 dilution. Pipette this mixture and transfer 5 µLto well B3. Discard the pipette tip after this transfer.
- With a new pipette tip, mix the solution in well B3 and form a 1/100 dilution. Transfer 5 µLof this mixture to well C3 and discard the tip after the transfer.
- With a new pipette tip, pipette up and down the solution in well C3 to make a 1/1000 dilution. Discard the tip.
- Continue diluting samples without adding any samples to columns 2, 4, 6, 8, 10, or 12.
- Once all samples are diluted, mix the contents in wells of column 1, and then transfer 20 µL to column 2.
- Repeat this for columns 3, 5, 7, 9, and 11 to create replicates of each standard and sample dilution. Refer to Figure 3 for plate layout.
NOTE: When following plate set-up in Figure 3, samples diluted in rows G and H will only have 1/10 and 1/100 dilutions.

Figure 3: Plate layout for qPCR AAV titration. Blue indicates the placement of the serial dilution of the standard; green indicates the placement of the negative control; purple indicates the placement of the dilution of the samples. Each standard, negative, or sample is added in replicate. An example for the concentration of the standard has been added to show the dilution series of the standard, and placement of sample dilutions have been added to their respective wells. Please click here to view a larger version of this figure.
{kind=link}
- Titration by SV40 polyA detection-based qPCR
- Add 15 µL of qPCR master mix to each well of a white-semi skirted 96-well qPCR plate.
- Transfer 5 µL of each sample from the clear 96-well plate to the white-semi skirted 96 well qPCR plate.
- Use a multichannel pipette to ensure adequate mixing of the qPCR master mix and sample.
- Seal the plate with a sealing film and centrifuge the qPCR plate at 1500 x g for 30 s.
- Run the qPCR reaction on plate-based real-time PCR amplification and detection instrument, using the suggested conditions in Table 6.
| Section | Cycles | Time | Temperature | Description |
| Pre-incubation | 1x | 5 min | 95 °C | DNA denaturation. |
| Amplification | 38x | 15 s | 95 °C | Amplification of DNA. Settings can be modified if using alternative primers with different annealing temperatures. |
| 60 s | 60 °C | |||
| Cooling | 1x | 60 s | 40 °C | Plate Cooling. End of run. |
Table 6: Thermocycler protocol for hydrolysis probe-based qPCR titration. Recommended thermocycler protocol for use of probe-based qPCR titration of DNA extracted purified AAV vectors.
NOTE: For qPCR AAV titration worksheet see Table 7.
- Data analysis to determine AAV genome copy numbers.
- Fill in the spreadsheet data cells (Table 7A) with the concentration values obtained from the qPCR run for both standard and sample dilutions.
- Use concentration values from Table 7A to produce a standard curve (Table 7B).
NOTE: The standard curve will be shown as a natural logarithm (y = a ln(x) + b) along with R2 efficiency. A standard curve must have an efficiency close to 100 % and R2 close to 1.0 (≥0.99). - Fill in the slope efficiency by filling in this online calculator27.
NOTE: An efficiency between 90%-110% is acceptable. If the efficiency of qPCR is outside of this range, re-run the qPCR. - Use concentration values from Table 7A to average the dilutions of each sample and determine the standard deviation of each sample (Table 7C).
NOTE: Exclude dilutions from samples that are more than one standard deviation away from the average of the sample dilutions. - Using the mean concentration of each dilution, multiply by the dilution factor, and then divide by five to get the vector genomes (vg)/µL of each sample (Table 7C).
- Calculate the vg/mL of each sample by multiplying the mean of each sample's concentrations by 80,000 (Table 7C).
- Average the vg/mL of each dilution to produce the final vg/mL of each sample (Table 7C).
NOTE: The user must divide the mean concentration of each dilution by a factor five to account for the 5 µL loaded into each well for the qPCR run to produce the concentration in vg/µL. The factor of 80,000 accounts for the transition from each samples' mean concentration value to vg/mL. First, the mean of each sample's concentration value must be multiplied by 2 to account for the single-stranded genomes, as the primer-probe set only quantifies positive-sense, single-stranded DNA (ssDNA), and the AAV genome exists in an approximate 1:1 ratio between positive and negative-sense ssDNA25,28. The mean of each sample's concentration value must be multiplied x40 to account for the sample dilution from 5 µL of purified vector (section 4.1) to 200 µL of extracted DNA (section 4.6.12). Lastly, the mean of each sample's concentration value must be multiplied x1000 to convert from vg/µL to vg/mL.
6 Assessment of vector quality and purity
- Quality Control - Western Blot
- Prepare a 12% SDS PAGE gel.
- Perform polyacrylamide gel electrophoresis.
NOTE: Load 6 x 1010 vg of samples per well. - Transfer the proteins to polyvinylidene difluoride (PVDF) membrane.
- Blocking PVDF membrane
- Remove the membrane from the transfer apparatus and rinse in 0.1% PBST to remove loose acrylamide.
- Place the membrane in blocking solution for at least 1 h at room temperature or overnight at 4 °C.
NOTE: Blocking buffer can be further supplemented with 2% goat serum.
- Incubation with primary antibody
- Decant the blocking buffer and add the primary antibody, an anti-AAV mouse monoclonal antibody at a 1:200 dilution.
- Incubate overnight at 4 °C.
- Decant the primary antibody and wash five times with 0.1% PBST for 5 min at room temperature with agitation.
- Incubation with secondary antibody
- Decant the wash solution and add HRP conjugated secondary antibody, diluted at a 1:7500 in blocking buffer, and incubate for 1 h at room temperature with agitation.
- Decant the secondary antibody and wash five times with 0.1% PBST for 5 min at room temperature with agitation.
- Perform a final wash with PBS at room temperature with agitation.
- Detect the proteins using enhanced chemiluminescent (ECL) substrate.
- Image the gel to visualize the viral proteins (VP1, VP2, and VP3 subunits) (Figure 4).

Figure 4: Western blot showing AAV capsid proteins. Lane A; MW ladder, Lane B; AAV6.2FF-hIgG01, Lane C; AAV6.2FF-hIgG02, Lane D; AAV6.2FF-hIgG03, and Lane E; AAV6.2FF-hIgG04. 6 x 1010 vg of each AAV6.2FF-hIgG was loaded into their respective lanes. Please click here to view a larger version of this figure.
{kind=link}
- Purity Control - SDS PAGE and Coomassie Stain
- Prepare SDS-PAGE gel and samples as described from step 6.1.1 and 6.1.2.
- Fix the gel in fixing solution for 1 h or overnight with gentle agitation. Change the fixing solution once during the first hour.
- Stain the gel in staining solution for 2-4 h with gentle agitation.
- Destain the gel with a destaining solution. Replenish the destaining solution several times until the background of the gel is fully destained (4-24 h).
- Store the destained gel in a storage solution.
- Image the gel to visualize all proteins stained by Coomassie staining solution.

Figure 5: Coomassie-stained gel. Lane A; MW ladder, Lane B; AAV6.2FF-hIgG01, Lane C; AAV6.2FF-hIgG02, Lane D; AAV6.2FF-hIgG03, Lane E; AAV6.2FF-hIgG04, Lane F; AAV6.2FF-hIgG05, and Lane G; AAV6.2FF-hIgG06. 6 x 1010 vg of each AAV6.2FF-hIgG was loaded into their respective lanes. Please click here to view a larger version of this figure.
{kind=link}
- Alternative purity control assay - HEK293 host cell protein detection ELISA
- Perform the HEK293 host cell protein detection via ELISA as per the manufacturer's instructions.
NOTE: Use dilutions of 5 x 10-2 and 1 x 10-3 for purified rAAV samples. Once TMB is added to the well, incubate away from light. Linear regression cannot be used to analyze the results. - Perform a point-to-point analysis, cubic spline, or four-parameter logistic fit method to interpolate concentrations of unknowns and multiply by the dilution factor to determine original sample concentration.
- Perform the HEK293 host cell protein detection via ELISA as per the manufacturer's instructions.
Results
Translation from small rodent models to larger animal models and eventual clinical application presents a significant challenge due to the large amount of AAV required to transduce larger animals and achieve therapeutic effects. To compare transduction efficiency of the rationally designed AAV6.2FF capsid, previously demonstrated a 101-fold increase in transduction efficiency in murine muscle cells compared to AAV63, mice, hamsters, and lambs were all administered AAV6.2FF expressing a human monoc...
Discussion
The production of recombinant AAV (rAAV) vectors described in this paper uses common materials, reagents, and equipment found in majority of the molecular biology research labs and facilities. This paper allows for high-quality in vitro and in vivo grade rAAV to be produced by the reader. Above all, this protocol for rAAV production, compared to more tedious protocols involving cesium chloride purification, is efficient and avoids the use of ultracentrifugation. Once HEK293 cells have been transfected, ...
Disclosures
Sarah K. Wootton is an inventor on a US patent US10806802B2 for the AAV6.2FF capsid.
Acknowledgements
Amira D. Rghei, Brenna A. Y. Stevens, Sylvia P. Thomas, and Jacob G. E. Yates were recipients of Ontario Veterinary College Student Stipends as well as Ontario Graduate Scholarships. Amira D. Rghei was the recipient of a Mitacs Accelerate Studentship. This work was funded by the Canadian Institutes for Health Research (CIHR) Project Grant (#66009) and a Collaborative Health Research Projects (NSERC partnered) grant (#433339) to SKW.
Materials
| Name | Company | Catalog Number | Comments |
| 0.22 μm filter | Millipore Sigma | S2GPU05RE | |
| 0.25% Trypsin | Fisher Scientific | SM2001C | |
| 1-Butanol | Thermo Fisher Scientific | A399-4 | CAUTION. Use under a laminar flow hood. Wear gloves |
| 10 chamber cellstack | Corning | 3271 | |
| 1L PETG bottle | Thermo Fisher Scientific | 2019-1000 | |
| 30% Acrylamide/Bis Solution | Bio-Rad | 1610158 | |
| 96-well skirted plate | FroggaBio | FS-96 | |
| Adhesive plate seals | Thermo Fisher Scientific | 08-408-240 | |
| Ammonium persulfate (APS) | Bio-Rad | 161-0700 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Blood and Tissue Clean up Kit | Qiagen | 69506 | Use for DNA clean up in section 4.6 of protocol |
| Bromophenol blue | Fisher Scientific | B392-5 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Cell Culture Dishes | Greiner bio-one | 7000232 | 15 cm plates |
| Culture Conical Tube | Thermo Fisher Scientific | 339650 | 15 mL conical tube |
| Culture Conical Tube | Fisher Scientific | 14955240 | 50 mL conical tube |
| Dulbecco's Modified Eagle Medium (DMEM) with 1000 mg/L D-glucose, L-glutamine | Cytiva Life Sciences | SH30022.01 | |
| ECL Western Blotting Substrate | Thermo Fisher Scientific | 32209 | |
| Ethanol | Greenfield | P016EA95 | Dilute ethyl alcohol(95% vol) to 20% for section 3.7.4 and 70% for section 6.1.1.1 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | SH30396.03 | |
| Glacial acetic acid | Fisher Scientific | A38-500 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Glycerol | Fisher Scientific | BP229-1 | |
| Glycine | Fisher Scientific | BP381-500 | |
| HBSS with Mg2+ and Ca2+ | Thermo Fisher Scientific | SH302268.02 | |
| HBSS without Mg2+ and Ca2+ | Thermo Fisher Scientific | SH30588.02 | |
| HEK293 cells | American Tissue Culture Collection | CRL-1573 | Upon receipt, thaw the cells and culture as described in manufacturer’s protocol. Once cells have been minimally passaged and are growing well, freeze a subfraction for future in aliquots and store in liquid nitrogen. Always use cells below passage number 30. Once cultured cells have been passaged more than 30 times, it is recommended to restart a culture from the stored aliquots |
| HEK293 host cell protein ELISA kit | Cygnus Technologies | F650S | Follow manufacturer’s instructions |
| Heparin sulfate column | Cytiva Life Sciences | 17040703 | |
| Kimwipe | Thermo Fisher Scientific | KC34120 | |
| L-glutamine (200 mM) | Thermo Fisher Scientific | SH30034.02 | |
| Large Volume Centrifuge Tube Support Cushion | Corning | CLS431124 | Support cushion must be used with large volume centrifuge tubes uless the centrifuge rotor has the approriate V-bottom cushions |
| Large Volume Centrifuge Tubes | Corning | CLS431123-6EA | 500 mL centrifuge tubes |
| MgCl2 | Thermo Fisher Scientific | 7791-18-6 | |
| Microcentrifuge tube | Fisher Scientific | 05-408-129 | 1.5 mL microcentrifuge tube, sterilize prior to use |
| Molecular Grade Water | Cytiva Life Sciences | SH30538.03 | |
| N-Lauroylsarcosine sodium salt | Sigma Aldrich | L5125 | CAUTION. Wear gloves |
| NaCl | Thermo Fisher Scientific | BP35810 | |
| Optimem, reduced serum medium | Thermo Fisher Scientific | 31985070 | |
| Pasteur pipets | Fisher Scientific | 13-678-20D | Sterilize prior to use |
| PBS (10x) | Thermo Fisher Scientific | 70011044 | Dilute to 1x for use on cells |
| Penicillin-Streptomycin Solution | Cytiva Life Sciences | SV30010 | |
| pHelper plasmid | De novo design or obtained from plasmid repository | NA | |
| Pipet basin | Thermo Fisher Scientific | 13-681-502 | Purchase sterile pipet basins |
| Polyethylene glycol tert-octylphenyl ether (Triton X-100) | Thermo Fisher Scientific | 9002-93-1 | CAUTION. Wear gloves |
| Polyethylenimine (PEI) | Polyscience | 24765-1 | Follow manufacturer’s instructions to produce a 1L solution. 0.22μm filter and store at 4°C |
| Polypropylene semi-skirted PCR Plate | FroggaBio | WS-96 | |
| Polysorbate 20 (Tween 20) | Thermo Fisher Scientific | BP337-100 | CAUTION. Wear gloves |
| polyvinylidene difluoride (PVDF) membrane | Cytiva Life Sciences | 10600023 | Use forceps to manipulate. Wear gloves. |
| Primary antibody | Progen | 65158 | |
| Protein Ladder | FroggaBio | PM008-0500 | |
| Proteinase K | Thermo Fisher Scientific | AM2546 | |
| pTrangene plasmid | De novo design or obtained from plasmid repository | NA | Must contain SV40 polyA in genome to be compatible with AAV titration in section 5.0 |
| Pump tubing | Cole-Parmer | RK-96440-14 | Optimize length of tubing and containment of virus in fractions E1-E5 |
| RQ1 Dnase 10 Reaction Buffer | Promega | M6101 | Use at 10x concentration in protocol from section 4.0 |
| RQ1 Rnase-free Dnase | Promega | M6101 | |
| Sample dilutent | Cygnus Technologies | I700 | Must be purchased separately for use with HEK293 host cell protein ELISA kit |
| Secondary antibody, HRP | Thermo Fisher Scientific | G-21040 | |
| Skim milk powder | Oxoid | LP0033B | |
| Sodium dodecyl sulfate (SDS) | Thermo Fisher Scientific | 28312 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Sodium hydroxide (NaOH) | Thermo Fisher Scientific | SS266-4 | |
| SV40 polyA primer probe | IDT | Use sequence in Table X for quote from IDT for synthesis | |
| Tetramethylethylenediamine (TEMED) | Thermo Fisher Scientific | 15524010 | CAUTION. Use under a laminar flow hood. Wear gloves |
| Tris Base | Fisher Scientific | BP152-5 | |
| Trypan blue | Bio-Rad | 1450021 | |
| Ultra-Filter | Millipore Sigma | UFC810024 | Ultra-4 Centrifugal 10K device must be used, as it has a 10000 molecular weight cutoff |
| Universal Nuclease for cell lysis | Thermo Fisher Scientific | 88702 | |
| Universal qPCR master mix | NEB | M3003L | |
| Whatman Paper | Millipore Sigma | WHA1001325 | |
| β-mercaptoethanol | Fisher Scientific | 21985023 | CAUTION. Use under a laminar flow hood. Wear gloves |
| CAUTION: Refer to the Materials Table for guidelines on the use of dangerous chemicals. |
References
- Hastie, E., Samulski, R. J. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success-a personal perspective. Human Gene Therapy. 26 (5), 257-265 (2015).
- Wang, D., Tai, P. W. L., Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature Reviews Drug Discovery. 18 (5), 358-378 (2019).
- Nathwani, A. C., et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. The New England Journal of Medicine. 371 (21), 1994-2004 (2014).
- Kuzmin, D. A., et al. The clinical landscape for AAV gene therapies. Nature Reviews Drug Discovery. 20 (3), 173-174 (2021).
- Ylä-Herttuala, S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (10), 1831-1832 (2012).
- FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss (2020)
- FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. FDA News Release. FDA Available from: https://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease (2020)
- Statement from FDA Commissioner Scott Gottlieb, MD and Peter Marks, MD Ph.D., Director of the Center for Biologics Evaluation and Research on new policies to advance development of safe and effective cell and gene therapies. FDA Available from: https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-and-peter-marks-md-phd-director-center-biologics (2020)
- Asokan, A., Schaffer, D. V., Samulski, R. J. The AAV vector toolkit: poised at the clinical crossroads. Molecular Therapy: The Journal of the American Society of Gene Therapy. 20 (4), 699-708 (2012).
- Rose, J. A., Hoggan, M. D., Shatkin, A. J. Nucleic acid from an adeno-associated virus: chemical and physical studies. Proceedings of the National Academy of Sciences of the United States of America. 56 (1), 86-92 (1966).
- Lusby, E., Fife, K. H., Berns, K. I. Nucleotide sequence of the inverted terminal repetition in adeno-associated virus DNA. Journal of Virology. 34 (2), 402-409 (1980).
- Masat, E., Pavani, G., Mingozzi, F. Humoral immunity to AAV vectors in gene therapy: challenges and potential solutions. Discovery Medicine. 15 (85), 379-389 (2013).
- Ling, C. Enhanced Transgene Expression from Recombinant Single-Stranded D-Sequence-Substituted Adeno-Associated Virus Vectors in Human Cell Lines In Vitro and in Murine Hepatocytes In Vivo. Journal of Virology. 89 (2), 952-961 (2014).
- Cathomen, T., Stracker, T. H., Gilbert, L. B., Weitzman, M. D. A genetic screen identifies a cellular regulator of adeno-associated virus. Proceedings of the National Academy of Sciences of the United States of America. 98 (26), 14991-14996 (2001).
- McCarty, D. M. Self-complementary AAV vectors; advances and applications. Molecular Therapy. 16 (10), 1648-1656 (2008).
- Aschauer, D. F., Kreuz, S., Rumpel, S. Analysis of transduction efficiency, tropism and axonal transport of aav serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLOS One. 8 (9), 76310 (2013).
- Zincarelli, C., Soltys, S., Rengo, G., Rabinowitz, J. E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Molecular Therapy. 16 (6), 1073-1080 (2008).
- Pillay, S., et al. Adeno-associated virus (AAV) serotypes have distinctive interactions with domains of the cellular AAV receptor. Journal of Virology. 91 (18), (2017).
- Merkel, S. F. Trafficking of adeno-associated virus vectors across a model of the blood-brain barrier; a comparative study of transcytosis and transduction using primary human brain endothelial cells. Journal of Neurochemistry. 140 (2), 216-230 (2017).
- van Lieshout, L. P., et al. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Molecular Therapy. Methods & Clinical Development. 9, 323-329 (2018).
- Wu, Z., Asokan, A., Grieger, J. C., Govindasamy, L., Agbandje-McKenna, M., Samulski, R. J. single amino acid changes can influence titer, heparin binding, and tissue tropism in different adeno-associated virus serotypes. Journal of Virology. 80 (22), 11393-11397 (2006).
- Liu, J., Moon, Y. -. A. Simple purification of adeno-associated virus-DJ for liver-specific gene expression. Yonsei Medical Journal. 57 (3), 790-794 (2016).
- Grimm, D., Kern, A., Rittner, K., Kleinschmidt, J. A. Novel tools for production and purification of recombinant adeno-associated virus vectors. Human Gene Therapy. 9 (18), 2745-2760 (1998).
- Kimura, T., et al. Production of adeno-associated virus vectors for in vitro and in vivo applications. Scientific Reports. 9 (1), 13601 (2019).
- Aurnhammer, C., et al. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Human Gene Therapy Methods. 23 (1), 18-28 (2012).
- . Paramyxoviridae: The viruses and their replication. Fields Virology Available from: https://www.scholars.northwestern.edu/en/publications/paramyxoviridae-the-viruses-and-their-replication (1996)
- Boussif, O., et al. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proceedings of the National Academy of Sciences of the United States of America. 92 (16), 7297-7301 (1995).
- Kaludov, N., Brown, K. E., Walters, R. W., Zabner, J., Chiorini, J. A. Adeno-associated virus serotype 4 (AAV4) and AAV5 Both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of Virology. 75 (15), 6884-6893 (2001).
- Burnham, B., et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Human Gene Therapy Methods. 26 (6), 228-242 (2015).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Backovic, A., et al. Capsid protein expression and adeno-associated virus like particles assembly in Saccharomyces cerevisiae. Microbial Cell Factories. 11, 124 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved