Hybrid De Novo Genome Assembly for the Generation of Complete Genomes of Urinary Bacteria using Short- and Long-read Sequencing Technologies
In This Article
Summary
This protocol details a comprehensive approach for the culturing, sequencing, and de novo hybrid genome assembly of urinary bacteria. It provides a reproducible procedure for the generation of complete, circular genome sequences useful in studying both chromosomal and extrachromosomal genetic elements contributing to urinary colonization, pathogenesis, and antimicrobial resistance dissemination.
Abstract
Complete genome sequences provide valuable data for the understanding of genetic diversity and unique colonization factors of urinary microbes. These data may include mobile genetic elements, such as plasmids and extrachromosomal phage, that contribute to the dissemination of antimicrobial resistance and further complicate treatment of urinary tract infection (UTI). In addition to providing fine resolution of genome structure, complete, closed genomes allow for the detailed comparative genomics and evolutionary analyses. The generation of complete genomes de novo has long been a challenging task due to limitations of available sequencing technology. Paired-end Next Generation Sequencing (NGS) produces high quality short reads often resulting in accurate but fragmented genome assemblies. On the contrary, Nanopore sequencing provides long reads of lower quality normally leading to error-prone complete assemblies. Such errors may hamper genome-wide association studies or provide misleading variant analysis results. Therefore, hybrid approaches combining both short and long reads have emerged as reliable methods to achieve highly accurate closed bacterial genomes. Reported herein is a comprehensive method for the culture of diverse urinary bacteria, species identification by 16S rRNA gene sequencing, extraction of genomic DNA (gDNA), and generation of short and long reads by NGS and Nanopore platforms, respectively. Additionally, this method describes a bioinformatic pipeline of quality control, assembly, and gene prediction algorithms for the generation of annotated complete genome sequences. Combination of bioinformatic tools enables the selection of high quality read data for hybrid genome assembly and downstream analysis. The streamlined approach for the hybrid de novo genome assembly described in this protocol may be adapted for the use in any culturable bacteria.
Introduction
The urinary microbiome is an emerging area of research that has shattered a decades long misconception that the urinary tract is sterile in healthy individuals. Members of the urinary microbiota may serve to balance the urinary environment and prevent urinary tract infection (UTI)1,2. Uropathogenic bacteria invade the urinary tract and employ diverse virulence mechanisms to displace the resident microbiota, colonize the urothelium, evade immune responses and counteract environmental pressures3,4. Urine is a relatively nutrient-limited medium characterized by high osmolarity, limited nitrogen and carbohydrate availability, low oxygenation, and low pH5,6,7. Urine is also considered to be antimicrobial, composed of high concentrations of inhibitory urea and antimicrobial peptides such as the human cathelicidin LL-378. Investigating mechanisms employed by both resident bacteria and uropathogens to colonize the urinary tract is critical to further understanding urinary tract health and developing new strategies for UTI treatment. Furthermore, as the failure of front-line antimicrobial therapies becomes more common, it is increasingly important to monitor the dissemination of mobile genetic elements carrying antimicrobial resistance determinants within populations of urinary bacteria9,10.
To investigate genotypes and phenotypes of urinary bacteria, their successful culture and subsequent whole genome sequencing (WGS) is imperative. Culture-dependent methods are necessary to detect and identify viable microbes in urine samples11. Standard clinical urine culture involves plating urine onto 5% sheep blood agar (BAP) and MacConkey agar and incubating aerobically at 35 °C for 24 h12. However, with a detection threshold of ≥105 CFU/mL13, many members of the urinary microbiota are not reported by this method. Improved culturing techniques such as Enhanced Quantitative Urine Culture (EQUC)11 employ various combinations of different urine volumes, incubation times, culture media, and atmospheric conditions to identify microbes commonly missed by standard urine culture. Described in this protocol is a modified version of EQUC, termed here Modified Enhanced Urine Culture protocol, that enables culturing of diverse urinary bacteria and uropathogens using selective media and optimal atmospheric conditions but is not inherently quantitative. The successful isolation of urinary bacteria enables the extraction of genomic DNA (gDNA) for downstream WGS and genome assembly.
Genome assemblies, complete assemblies in particular, enable the discovery of genetic factors that may contribute to colonization, niche maintenance, and virulence among both resident microbiota and uropathogenic bacteria. Draft genome assemblies contain a diverse number of contiguous sequences (contigs) that may contain sequencing errors and lack orientation information. In a complete genome assembly, both the orientation and the accuracy of every base pair have been verified14. Furthermore, obtaining complete genome sequences provides insight into genome structure, genetic diversity, and mobile genetic elements15. Short reads alone may identify the presence or absence of important genes but may not pinpoint their genomic context16. With enabling long-read sequencing technologies such as Oxford Nanopore and PacBio, generating closed de novo assemblies of bacterial genomes no longer requires strenuous methods such as manual closing of de novo assemblies by multiplex PCR17,18. The combination of Next Generation short-read sequencing and Nanopore long-read sequencing technologies allows the facile generation of accurate, complete, and closed bacterial genome assemblies at relatively low costs19. Short-read sequencing produces accurate yet fragmented genome assemblies generally consisting of an average of 40-100 contigs, while Nanopore sequencing generates long reads of about 5-100 kb in length that are less accurate but can serve as scaffolds to join contigs and resolve genomic synteny. Hybrid approaches utilizing both short-read and long-read technologies can produce accurate and complete bacterial genomes19.
Described here is a comprehensive protocol for the isolation and identification of bacteria from human urine, genomic DNA extraction, sequencing, and complete genome assembly using a hybrid assembly approach. This protocol provides a special emphasis on the steps necessary to properly modify reads generated by short-read and long-read sequencing for the accurate assembly of a closed bacterial chromosome and extrachromosomal elements such as plasmids.
Protocol
Bacteria were cultured from urine collected from consenting women as part of institutional review board-approved studies 19MR0011 (UTD) and STU 032016-006 (UTSW).
1. Modified enhanced urine culture
NOTE: All culture steps must be carried out under sterile conditions. Sterilize all instruments, solutions, and media. Clean the work area with 70% ethanol, then set up a Bunsen burner and work carefully close to the flame to reduce the chances of contamination. Alternately, a class II biosafety cabinet may be used to maintain a sterile environment. Wear appropriate personal protective equipment (PPE) to avoid exposure to potentially pathogenic microbes.
- Plating glycerol-stocked urine and colony isolation
- Thaw glycerol-stocked urine at room temperature (RT). Once thawed, vortex the sample for 5 s to mix. In sterile microcentrifuge tubes, prepare 1:3 and 1:30 dilutions of the urine in sterile 1x Phosphate-Buffered Saline (PBS) to a final volume of 100 µL.
NOTE: Glycerol-stocked urine is prepared by mixing 500 µL of undiluted urine and 500 µL of 50% sterile glycerol in cryovials and storing at -80 °C. - Pre-warm agar plates at 37 °C for 15 min before use. Please see Figure 1 for media types and culture conditions suitable to common urinary bacterial genera. Mix the diluted urine well by pipetting before plating, plate 100 µL of the diluted urine on the desired agar plate and spread the sample using sterile glass beads. Plate 100 µL of the 1x PBS diluent on a separate plate as a no growth control.
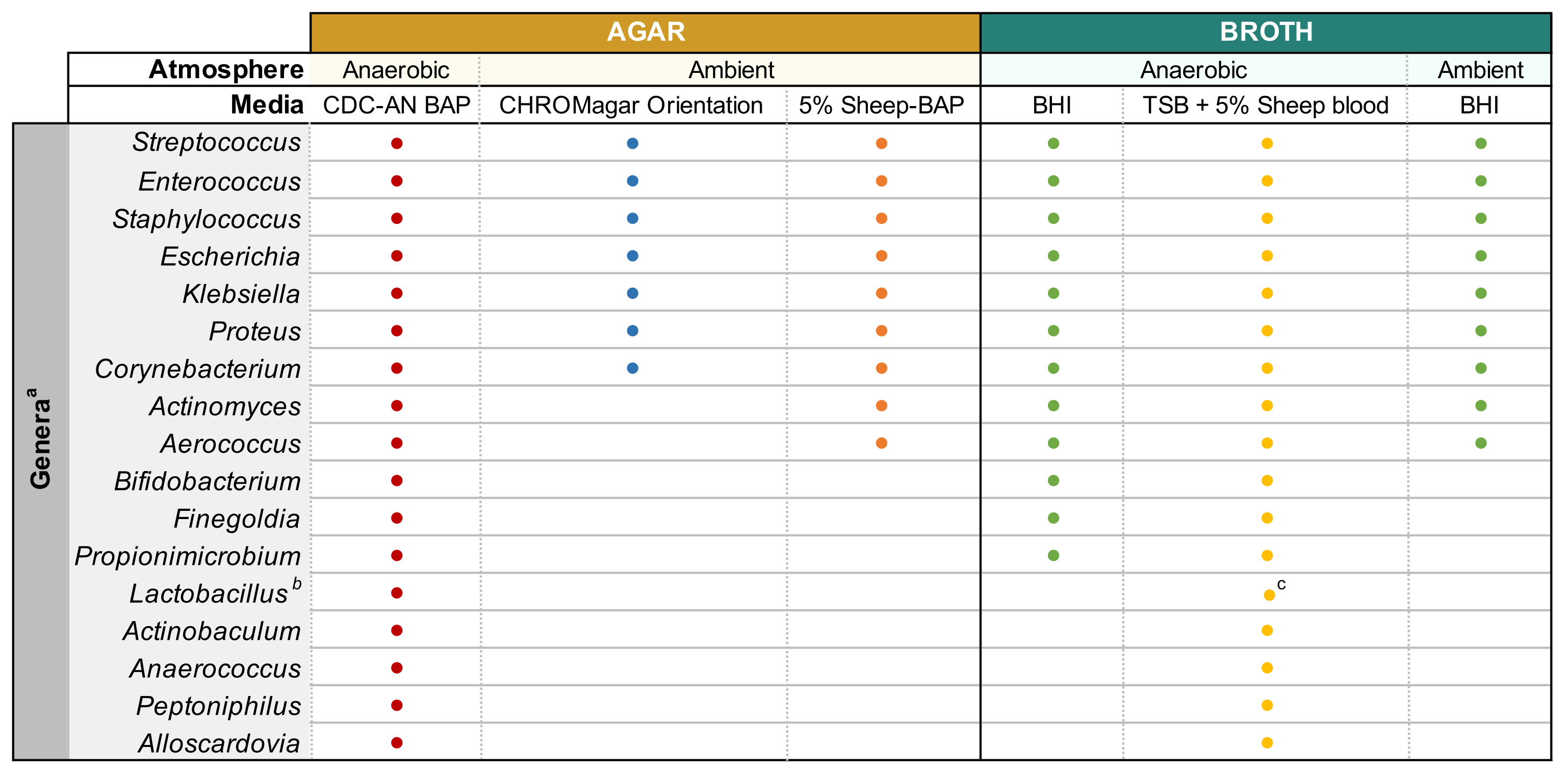
NOTE: If attempting to culture common uropathogenic species (e.g., Escherichia coli, Klebsiella spp., Enterococcus faecalis, etc.), it is recommended to use chromogenic agar (Table of Materials) as it allows easy identification of uropathogenic bacterial species (Figure 1). Colistin Nalidixic Acid (CNA) or MRS agar are useful for isolating fastidious Gram-positive species (e.g., Lactobacillus spp.) from urine known to contain Gram-negative uropathogens, which may outcompete the fastidious species in non-selective agars. - Incubate the plate inverted in the desired atmospheric condition at 35 °C for a period of 24 h for uropathogens and 3-5 days for fastidious bacteria (Figure 1).
- After the incubation period, remove the plates from the incubator. From each plate, pick the colonies that exhibit a unique color, morphology, or hemolytic patterns.
- Re-streak the bacterial colony using a sterile loop onto the corresponding agar and incubate the plate inverted for 2-5 days in the desired atmosphere to obtain well-isolated colonies.
NOTE: If utilizing BAP for primary culture, patching colonies on chromogenic agar may provide useful information about the heterogeneity of the bacterial population in the sample.
- Thaw glycerol-stocked urine at room temperature (RT). Once thawed, vortex the sample for 5 s to mix. In sterile microcentrifuge tubes, prepare 1:3 and 1:30 dilutions of the urine in sterile 1x Phosphate-Buffered Saline (PBS) to a final volume of 100 µL.
- Culturing in liquid broth and glycerol-stocking bacterial isolates
- Once the isolated colonies that match the morphology of the parent colony are obtained, pick a single colony and inoculate into 3 mL of liquid broth using a sterile inoculation loop. Refer to Figure 1 for broth capable of supporting the growth of common urinary microbiota genera. Seal the agar plates with parafilm and store them at 4 °C for 2-4 days. Incubate liquid cultures in the desired atmospheric conditions for 1-5 days until the culture is visibly turbid.
- After growth is observed, vortex the culture, and then add 1 mL of the overnight culture to 500 µL of sterile 50% glycerol in a 2 mL cryovial; seal and gently mix by inversion. Prepare two glycerol stocks for each colony (one serves as a backup) and store at -80 °C.
2. Identification of bacterial species by 16S rRNA gene Sanger sequencing
NOTE: Microbial identity can be alternatively confirmed using Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF)20.
- Colony-Polymerase Chain Reaction (PCR)
- Prepare a 25 µL of the PCR reaction in PCR tubes by adding 12.5 µL of 2x Taq Polymerase Master Mix, 0.5 µL of 10 µM 8F primer, 0.5 µL of 10 µM 1492R primer (Table of Materials), and 11.5 µL of nuclease-free water21.
NOTE: If performing PCR for multiple samples, make a reaction master mix of Taq Polymerase mix, primers, and sterile nuclease-free water. Then aliquot 25 µL into each PCR tube. - To perform colony-PCR, swipe a well-isolated colony from the re-streak using a sterile toothpick or pipette tip. Resuspend the colony in the PCR reaction mix prepared in step 2.1.1. Gently mix. Collect the liquid at the bottom of the tube by a quick spin at 2000 x g.
NOTE: Ensure the sample is free of air bubbles. Include a no-template control (NTC) sample containing the PCR reaction mix alone. - Place the sample tubes in the thermocycler and run the following program: 95 °C for 3 min; 40 cycles of: 95 °C for 30 s, 51 °C for 30 s, and 72 °C for 1 min 30 s; 72 °C for 10 min; hold at 10 °C.
- Prepare a 25 µL of the PCR reaction in PCR tubes by adding 12.5 µL of 2x Taq Polymerase Master Mix, 0.5 µL of 10 µM 8F primer, 0.5 µL of 10 µM 1492R primer (Table of Materials), and 11.5 µL of nuclease-free water21.
- Gel extraction and species identification
- Upon completion of the PCR run, check the PCR product on a 1% agarose gel prepared in 0.5x Tris-Borate-EDTA (TBE) buffer. Prior to casting the gel, add ethidium bromide (EtBr). Then, cast the gel using combs for wells that hold at least 20 µL sample volume.
CAUTION: EtBr is an intercalating agent suspected to be carcinogenic. Always wear gloves and PPE when handling it and dispose of materials containing EtBr according to the institution's guidelines. - When the gel is set, place the gel in the electrophoresis tank filled with 0.5x TBE buffer and remove the comb. Load the 1 kb ladder in the first well and 10-20 µL of the PCR reaction into subsequent wells. Run at 100-140 V until resolved. Visualize the gel under UV light and confirm the presence of a clearly defined band at ~1.5 kb that is absent in the NTC well.
CAUTION: UV rays are harmful to skin and eyes, use an appropriate guard when visualizing the gel and wear appropriate PPE.
NOTE: Colony PCR may be unsuccessful for some bacteria; proceeding with PCR from isolated gDNA is an alternate option22. - Excise the ~1.5 kb bands using a razor and transfer the gel cuttings into clean microcentrifuge tubes. Proceed with gel extraction protocol as per the manufacturer's instructions (Table of Materials). Measure the concentration of the purified DNA by microvolume spectrophotometer.
NOTE: A concentration >10 ng/µL is desirable, and A260/280 between 1.7-2.0 is acceptable. - Prepare two Sanger sequencing reactions for each sample, one using the 8F and the other using the 1492R primer in nuclease-free water according to the guidelines of any chosen Sanger sequencing service.
- Once the sequencing data is received, upload the DNA sequences to the NCBI Basic Local Alignment Search Tool (BLAST) website (blast.ncbi.nlm.nih.gov/Blast.cgi), choose Nucleotide BLAST (blastn), select the rRNA/ITS database 16S ribosomal RNA sequences (Bacteria and Archaea), and run the Megablast program. The isolate may be identified by the highest quality hit to a reference from the database.
NOTE: Some bacterial species exhibit high identity in their 16S rRNA sequences and may be indistinguishable by this method alone. Speciation will require DNA homology and biochemical analyses to confidently distinguish members of the same genus23.
- Upon completion of the PCR run, check the PCR product on a 1% agarose gel prepared in 0.5x Tris-Borate-EDTA (TBE) buffer. Prior to casting the gel, add ethidium bromide (EtBr). Then, cast the gel using combs for wells that hold at least 20 µL sample volume.
3. Extraction of genomic DNA (gDNA)
NOTE: This section utilizes reagents and spin-columns provided in the gDNA extraction kit referenced in the Table of Materials for the high yield extraction of quality genomic DNA from diverse bacterial species. Provided below are recommended modifications and instructions.
- Prepare kit reagents per manufacturer's instructions.
- Prepare 3-10 mL cultures in appropriate sterile broth (Figure 1) by inoculating bacteria from well-isolated colonies into the media and incubating at the temperature and atmospheric pressure noted in Figure 1 until sufficient growth is observed.
- After incubation, measure the optical density at 600 nm (OD600) of the culture using a spectrophotometer24.
- Prepare the sample for quantification by diluting overnight cultures in 1:10 ratio. Include a blank of the sterile culture media for measurement as well. Calculate the optical density by subtracting the blank reading from the sample reading and multiplying by the dilution factor of ten.
- Using the OD600 measurement and a pre-established OD600 to CFU/mL ratio for the species, calculate how many milliliters of culture are necessary to obtain 2 x 109 cells.
- Centrifuge the required culture volume for 5 min at 5000 x g to pellet. Aspirate the supernatant and resuspend the pellet in 200 µL cold TE buffer (pre-chill on ice at the beginning of the procedure).
- Centrifuge the sample for 2 min at 5000 x g. Remove the supernatant, and then resuspend the pellet in 180 µL of Enzymatic Lysis Buffer (ELB) and add 20 µL of pre-boiled RNase A (10 mg/mL). For efficient lysis of Gram-positive bacteria, add 18 µL of mutanolysin (25 kU/mL). Vortex well, and then incubate the samples at 37 °C on rotator for 2 h.
NOTE: It is recommended to utilize the ELB described in the manufacturer's protocol for both Gram-positive and Gram-negative bacteria. - Proceed according to the manufacturer's instructions.
NOTE: Repeat the elution steps for one or two more times to obtain additional gDNA yield, if desired. - Assess the quality of extracted gDNA as instructed in section 4 and store gDNA at 4 °C if it will be used within 1 week. Alternatively, keep gDNA at -20 °C for long-term storage.
4. Assessing the quality of extracted gDNA
- To assess the quality by gel electrophoresis, prepare 1% agarose gel as described in subsection 2.2. Prepare the sample in a clean tube: mix 1-2 µL of extracted gDNA and 3 µL of 2x loading dye on parafilm. Run the gel once loaded, and then visualize it under UV light.
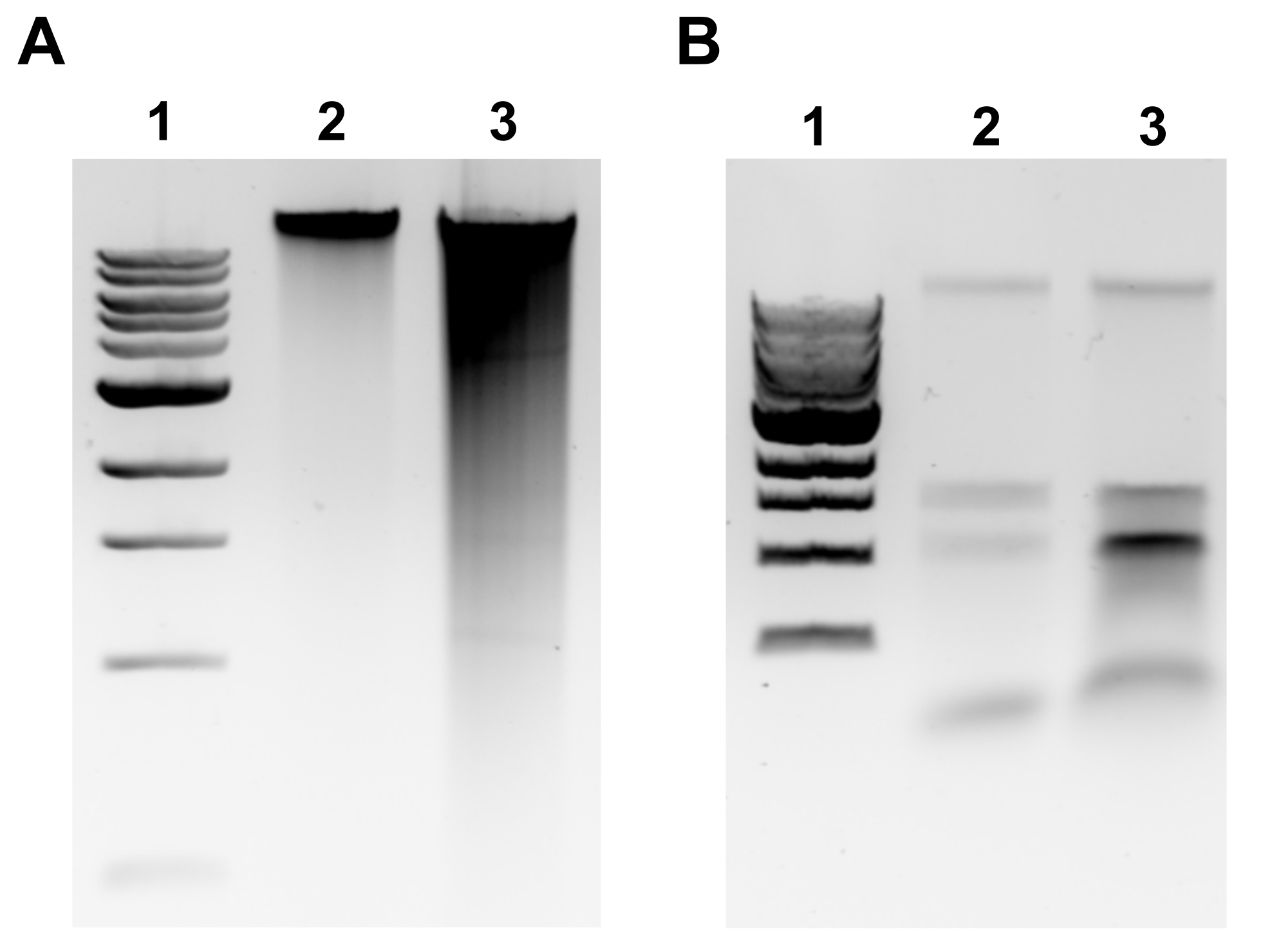
NOTE: Successful gDNA extraction will be evident by a discrete band at the top of the gel and minimal smearing (Figure 2A). Smearing is indicative of shearing. If no gDNA band is evident and/or smearing is substantial, repeat gDNA extraction. Consider reducing incubation times in RNase A and Proteinase K. If two bands around 1.5-3 kb are observed, this suggests RNA contamination (Figure 2B). Prepare fresh RNase A and repeat extraction. - To assess the quality by microvolume spectrophotometer, measure gDNA concentration and absorbance ratio A260/280 by microvolume spectrophotometer. Concentrations >50 ng/µL and A260/280 between 1.7-2.0 are acceptable.
NOTE: Low gDNA yield may be due to low input, high input, contamination of nucleases, insufficient lysis. Absorbance ratios above the range indicate RNA contamination. Repeat extraction if the gDNA quality is poor. - To assess the quality by fluorometer, follow the manufacturer's instructions to quantify gDNA concentration using High-Sensitivity assay kit and fluorometer instrument (Table of Materials). Concentration >50 ng/µL is desirable.
5. Paired-end next generation short-read sequencing and library preparation
NOTE: Short-read sequencing may be performed on various instruments at distinct read lengths and orientations. 150 bp (300 cycle) paired-end sequencing is recommended for bacterial WGS. Both library preparation and sequencing may be outsourced to core facilities or commercial laboratories.
- Prepare sequencing library according to the manufacturer's instructions (Table of Materials). Follow the manufacturer's recommended final loading library concentration; however, a recommended modification is to load the pooled library at 1.8 pM for optimal read generation on NextSeq instruments.
- Although optional, utilize a Bioanalyzer (Table of Materials) to assess the pooled library fragment distribution and ensure that the fragment size is 600 bp on average.
6. Nanopore MinION sequencing library preparation
- Prepare the sequencing library according to the manufacturer's protocol (Table of Materials). Using two barcode expansion kits allows for multiplexing of up to 24 samples on a single flow cell. It is recommended to perform library preparation in two parts, 12 samples at a time when multiplexing 24 samples. All 24 samples may be pooled as described below.
NOTE: Samples may be stored at 4 °C overnight upon finishing Native Barcode Ligation - this provides a stopping point in the protocol, if necessary. At the end of the Native barcode ligation section of the library preparation protocol, it is recommended to pool equimolar amounts of each sample up to the maximum DNA mass (ng) possible.- To do so, quantify all the samples following barcode ligation using a fluorometer (Table of Materials) per manufacturer's instructions. Estimate the volume of the sample with the lowest dsDNA concentration, and then calculate the total dsDNA found in this sample. Use this number to determine the equimolar amounts of all the other samples that will be pooled together.
NOTE: Because the equimolar calculation will maximize the amount of pooled dsDNA and thus yield a high-volume pool (>65 µL), cleanup is necessary to concentrate the pool.
- To do so, quantify all the samples following barcode ligation using a fluorometer (Table of Materials) per manufacturer's instructions. Estimate the volume of the sample with the lowest dsDNA concentration, and then calculate the total dsDNA found in this sample. Use this number to determine the equimolar amounts of all the other samples that will be pooled together.
- dsDNA pool cleanup and concentration
- Add 2.5x volume of paramagnetic beads (Table of Materials) to the DNA pool, and then gently flick the tube to mix the contents. Place the tube in the rotator for 5 min at RT. Spin down the sample at 2000 x g and pellet on a magnet.
- Add 250 µL freshly prepared 70% ethanol (in nuclease free water), taking care not to disturb the pellet. Aspirate the ethanol and repeat the ethanol wash once.
- After the second aspiration, spin down the sample at 2000 x g and place it back on the magnet. Pipette off any residual ethanol and allow the sample to dry for approximately 30 s.
- Remove the tube from the magnet and resuspend the pellet in 60-70 µL of nuclease free water. Incubate at RT for 2 min. Pellet the sample on the magnet until the elute is clear, and then remove the elute and transfer into a clean 1.5 mL microcentrifuge tube.
- Quantify the concentrated pool using a fluorometer, and then prepare an aliquot to proceed to the adapter ligation step: prepare 700 ng of the sample in 65 µL final volume. Retain the remainder of the pool at 4 °C for a second run to be completed once the first run is finished.
- Proceed with adapter ligation as instructed by the manufacturer and load the sample on the flow cell. Start the sequencing run.
NOTE: Aspirate air and ~200 µL of storage buffer from the flow cell priming port prior to the sample loading. This is critical for the successful flow cell priming and sample loading. Use a p1000 pipette and tips when drawing and depositing solutions through the priming port of the flow cell.
- Sequence the library according to the manufacturer's instructions.
- Open the operating software for sequencing and click on Start. Input a name for the experiment, a recommended nomenclature includes the run date and user's name. Click on Continue to Kit Selection, select the appropriate library prep kit and barcode expansion pack(s) used, and then click on Continue to Run Options.
- Adjust the run length to 48 h if planning to prepare sufficient library for a second run (otherwise leave at default 72 h). Click on Continue to Basecalling.
- Check the basecalling option Config: Fast Basecalling and make sure that Barcoding is set to Enabled so that output FASTQ files will be trimmed of the barcode sequences and demultiplexed into separate directories based on barcode. Click on Continue to Output.
- Choose where to save output sequencing data. Expect approximately 30-50 Gb of data if only saving FASTQ output and >500 Gb of data if also saving FAST5 output. Uncheck the Filtering option Qscore: 7 | Readlength: Unfiltered if planning to proceed with filtering described in section 7.2, otherwise leave checked and adjust Readlength to 200.
- Click on Continue to Run Setup and review all the settings. If the settings are correct, click on Start, otherwise click on Back and make any necessary adjustments.
- If desired, the flow cell may be washed per the manufacturer's instructions and reloaded with the remaining pool. Repeat the steps in 6.2 for the remaining pool once the first run is complete and the flow cell has been washed.
NOTE: When setting up the second run, adjust the Bias voltage to -250 mV per the manufacturer's recommendations for flow cells previously used in runs over 48 h.
7. Assessing and preparing reads
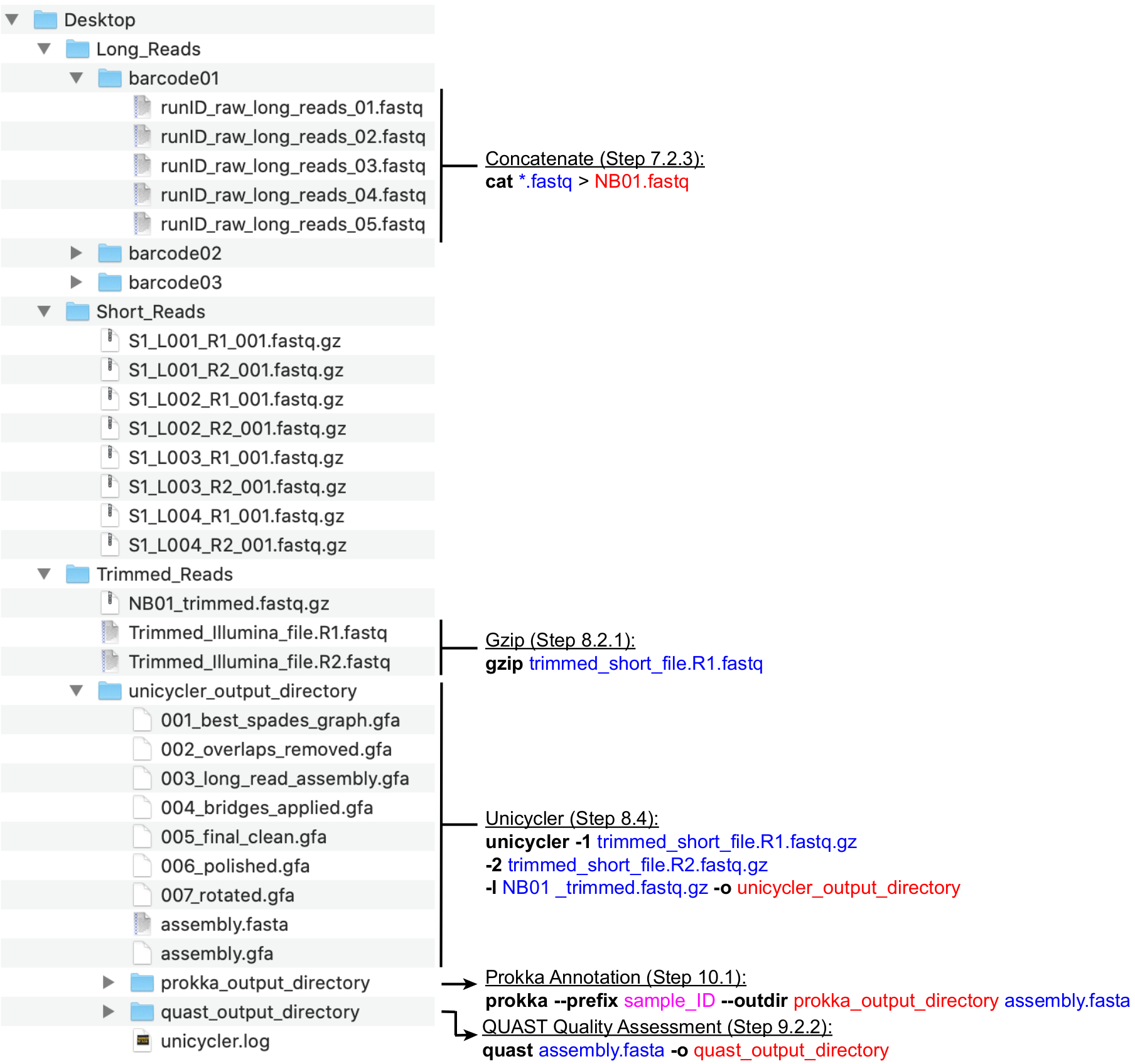
NOTE: A recommended directory structure is depicted in Figure 4. Create the directories found in the Desktop, namely, Long_Reads, Short_Reads and Trimmed_Reads, prior to proceeding with the computation steps below.
- Short reads (Figure 3)
NOTE: Short reads are generated in the FASTQ format. The files contain 4000 maximum reads per FASTQ. These are often zipped (.gz archive) and organized into multiple files. Depending on the platform, barcodes are typically trimmed. Some programs accept files in the zipped format, others may require their extraction prior to importing. The reads must pass quality control (QC) steps to ensure data accuracy during genome assembly. If CLC Genomics Workbench is not available, alternate programs may be used to trim and QC short reads such as Trimmomatic25 or Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) for trimming and FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) for evaluating read quality. Average short read coverage, estimated by multiplying number of reads by average read length and dividing by the genome size, is recommended to be >100x.- Open Genomics Workbench software (Table of Materials) and import all paired-end short-read FASTQ files. Paired files will be generated automatically.
- Create a new folder under CLC_Data by clicking on the New at the top toolbar and selecting Folder... to store the files. Name the folder as desired, a recommended convention is using the sample ID. Save all the output from the following steps to this folder.
- At the top toolbar, click on the Import button and select Illumina... Navigate to and select all short-read files that correspond to the sample. Make sure that the paired reads option is selected and uncheck the Remove failed reads option. Click on Next, select Save, and click on Next again. Choose to save the imported files in the new folder created in the previous step and click on Finish.
- Create a sequence list of all paired files for the isolate; this will concatenate read data into a single file for simplicity of analysis.
- At the top toolbar, click on the New button and select Sequence List... On the directory list on the left, select the files to be concatenated and use the arrows to move those into the selected files list on the right. Click on Next, select Save, and click on Next again. Choose to save the sequence list and click on Finish.
- Once the sequence list is generated, immediately rename it with the sample ID.
- Run the QC for Sequencing Reads tool on the sequence list: This procedure will assess the overall quality parameters of the reads generated by short-read NGS.
- Search for the QC for Sequencing Reads tool in the toolbox menu (bottom-left window). Double-click on the tool, and then choose the sequence list to be analyzed and click on Next.
- Ensure all the output options are checked and choose Save under Results Handling. Click on Next and specify to save the output files, and then click on Finish.
- Run the Trim Reads tool on the sequence list: Trimming will be done based on quality, length, and ambiguity. This process assumes the barcodes used in sequencing have been trimmed prior to this step.
- Search for the Trim Reads tool in the toolbox (bottom-left window). Double click on Trim Reads, and then choose the sequence list to be analyzed and click on Next.
- Quality trimming: set the quality score limit to 0.01 and leave ambiguous nucleotides at 2. Click on Next.
NOTE: Parameters may be adjusted at user discretion; these are the recommended settings. - Uncheck Automatic Read-Through Adapter Trimming (only do this if adapters have been trimmed from the reads prior to import into CLC). Click on Next and check Discard Reads Below Length, use default 15.
- Click on Next, check Create Report, and then choose Save. Click on Next and specify where to save the output files. Click on Finish.
- Export the trimmed sequence list: subsequent hybrid assembly and analysis will be completed outside of CLC and requires trimmed short-read files to be exported.
- From the directory navigation on the top left, choose the trimmed file generated in step 7.1.4, and then click on Export at the top toolbar. Select Fastq for the export file type and click on Next. Check Export Paired Sequence List to Two Files. Then, click on Next and choose the Trimmed_Reads directory to export the files to. Click on Finish. Ensure that the trimmed short-read files were exported successfully as two files (R1 and R2) with the extension .fastq.
NOTE: The trimmed sequence list must be exported into two files, typically designated by CLC as R1 and R2. This is critical as downstream hybrid assembly requires short-read data input to be set up as such. - Rename the exported files, please refrain from the use of spaces and special characters in file names. For simplicity a recommended format is trimmed_short_file.R1.fastq.
- From the directory navigation on the top left, choose the trimmed file generated in step 7.1.4, and then click on Export at the top toolbar. Select Fastq for the export file type and click on Next. Check Export Paired Sequence List to Two Files. Then, click on Next and choose the Trimmed_Reads directory to export the files to. Click on Finish. Ensure that the trimmed short-read files were exported successfully as two files (R1 and R2) with the extension .fastq.
- Open Genomics Workbench software (Table of Materials) and import all paired-end short-read FASTQ files. Paired files will be generated automatically.
- Long (MinION) reads (Figure 3)
NOTE: The following pipeline for the preparation of Long (MinION) sequencing reads for hybrid assembly utilizes NanoFilt and Nanostat programs26 executed by the command-line. Install the tools prior to proceeding and be familiar with the basics of UNIX in order to execute these commands. Default terminals and Bash Shell are recommended. A lesson guide for common terminal commands and usage is found at Software Carpentry27. The instructions below assume that the files generated will be named with the barcode nomenclature (NB01, NB02, etc.) and are saved in the Long_Reads directory. Alternatively, read filtering can be accomplished using MinKNOW when setting up the sequencing run. Average long read coverage is recommended to be >100x. Recommended average read length is >2000 bp; therefore, the number of long reads needed is lower than the number of short reads.- Create new directories for each barcode used in the run (barcode01, barcode02, etc.) within the Long_Reads directory (Figure 4). Copy all of the .fastq files that correspond to each barcode into the appropriate folder. Combine all .fastq files for each barcode from every run.
- Open Terminal and navigate to the barcode directories within the Long_Reads directory using the cd command: cd Desktop/Long_Reads/barcode01
- Concatenate all .fastq files per barcode into a single .fastq file by executing the following command: cat *.fastq > NB01.fastq
NOTE: This command combines all of the reads from each of the FASTQ files into one large, single FASTQ named NB01.fastq. - Use NanoStat to assess read quality of the sample by executing the following command: NanoStat --fastq NB01.fastq
- Record the results by copying the output into a text or Word file for future reference.
- Use NanoFilt to filter MinION reads discarding reads with Q < 7 and length < 200 by executing the command: NanoFilt -q 7 -l 200 bp NB01.fastq | gzip > NB01 _trimmed.fastq.gz
- Run NanoStat on the trimmed file generated in step 7.2.6 by executing the command: NanoStat --fastq NB01 _trimmed.fastq.gz
- Record the results by copying the output into a text or Word file and compare to the results from step 7.2.4 to ensure that the filtering was successful (Table 1).
- Repeat steps 7.2.2 to 7.2.8 for each barcode used in the sequencing run.
NOTE: The NB01_trimmed.fastq.gz file generated in step 7.2.6 will be used for hybrid assembly.
8. Generating hybrid genome assembly
NOTE: The following assembly pipeline utilizes Unicycler19,28,29,30 to combine short and long reads prepared in sections 7.1 and 7.2 (Figure 3). Install Unicycler and its dependencies and execute the commands below. Short-read files exported in step 7.1.5 are assumed to be named trimmed_short_file.R1.fastq and trimmed_short_file.R2.fastq for simplicity.
- Organize the short-read files and long-read files into a single directory named Trimmed_Reads. The directory must contain the following:
- A .fastq.gz file for trimmed long reads (generated in step 7.2.6).
- Two .fastq files (R1 and R2) for trimmed short reads (generated in step 7.1.5).
- Navigate to the directory Trimmed_Reads that stores the read files using the cd command in Terminal: cd Desktop/Trimmed_Reads
- Once in the correct directory, zip the two short read files so they are also in the .fastq.gz format by executing the following command: gzip trimmed_short_file.R1.fastq
- Repeat step 8.2 for both R1 and R2. Check that all the read files are now in the .fastq.gz format and verify that all the files match the same isolate.
- Begin the hybrid assembly using Unicycler by running the following command:
unicycler -1 trimmed_short_file.R1.fastq.gz -2 trimmed_short_file.R2.fastq.gz -l NB01 _trimmed.fastq.gz -o unicycler_output_directory
NOTE: -o specifies the directory in which the Unicycler output will be saved, Unicycler will create this directory once the command is executed; do not generate the directory beforehand. Run time varies by computational power of the computer used as well as genome size and the number of reads. This may take anywhere from 4 h to 1 or 2 days. This protocol was performed on a CentOS Linux 7 machine with 250 Gb RAM, Intel Xeon (R) CPU with 2.5 GHz 12 practical cores and 48 virtual cores. Alternatively, personal computers with 16 Gb RAM and 2.6 GHz 6-core processors can compute these assemblies at a longer processing time. - When the run is complete, review the unicycler.log file to ensure no errors - record the number, size, and status (complete, incomplete) of the contigs generated.
- If incomplete contigs are identified (denoted as incomplete in the Unicycler log), re-run Unicycler in bold mode by adding the following flag to the command in step 8.4: --mode bold.
NOTE: Bold mode will lower the quality threshold accepted for long read bridges during assembly; this may yield a complete assembly, but the assembly quality may be diminished. It is recommended to utilize bold mode only when necessary and as preliminary evidence for contig joining to be later confirmed by PCR.
- If incomplete contigs are identified (denoted as incomplete in the Unicycler log), re-run Unicycler in bold mode by adding the following flag to the command in step 8.4: --mode bold.
9. Assessing assembly quality
NOTE: The following protocol utilizes Bandage31 and QUAST32, two programs that must be set up prior to use (Figure 2 and Figure 4). Bandage does not require installation once downloaded and QUAST requires familiarity with basic command-line usage. It is also recommended to assess genome completeness using Benchmarking Universal Single-Copy Orthologs (BUSCO)33.
- Bandage: Click on File. Then, choose Load Graph and select the assembly.gfa file that was saved to unicycler_output_directory generated by Unicycler in step 8.4. Once loaded, click on the Draw Graph button on the left-hand toolbar and look at how the contigs (called nodes) are connected and organized to evaluate if the assembly is complete (Figure 5).
NOTE: Complete assemblies are represented by single circular contigs linked at both ends (Figure 5A,B). Incomplete assemblies have multiple contigs linked together or are linear (Figure 5C). Small linear contigs may not be incomplete as they may indicate linear extrachromosomal elements. Coverage, also called depth, will be noted in bandage and represents the relative abundance of the contigs to the chromosome, normalized in Unicycler to 1x. - QUAST
- Within the Terminal, navigate to the folder that stores the Unicycler output using the cd command: cd Desktop/Trimmed_Reads/unicycler_output_directory
NOTE: Spaces are not permitted in the path to where the assembly is located, i.e., no directories leading to the Unicycler output can have spaces in their name. Alternatively, copy the assembly.fasta file to the Desktop for easy access. - Run QUAST by executing the following command: quast assembly.fasta -o quast_output_directory
- Review the reports generated by QUAST in the output directory quast_output_directory.
- Within the Terminal, navigate to the folder that stores the Unicycler output using the cd command: cd Desktop/Trimmed_Reads/unicycler_output_directory
10. Genome annotation
NOTE: The below annotation pipeline utilizes Prokka34, a command-line tool that must be installed prior to usage. Alternatively, use Prokka through the automated GUI K-Base (Table of Materials) or annotate genomes via the web server RAST35. If depositing genomes into NCBI, they will be automatically annotated using the Prokaryotic Genome Annotation Pipeline (PGAP)36.
- Navigate within the Terminal to the folder that stores the Unicycler output using the cd command (see step 9.2.1). Then, run Prokka by executing the following command: prokka --prefix sample_ID --outdir prokka_output_directory assembly.fasta
NOTE: --prefix will name all output files based on the specified sample_ID. --outdir will create an output directory with the specified name where all Prokka output files will be saved; do not create an output directory for Prokka beforehand. - Review the annotations by opening the .tsv table and/or by uploading the .gff file generated into a sequence analysis software to visualize and analyze the annotations (Figure 6).
- Specific types of annotations can be generated depending on genetic factors of interest. It is recommended to start with the user-friendly tools on the Center for Genomic Epidemiology (www.genomicepidemiology.org/) web server for preliminary analysis37,38,39,40,41. Additional tools for detection of CRISPR-cas systems and prophage are available (Figure 3)42,43.
11. Suggested practices for data democratization
- When possible, deposit all the raw read data as well as assembled genomes in a public repository such as NCBI Sequence Read Archive (SRA) and Genbank. Genomes are automatically annotated via the PGAP pipeline during the NCBI deposition process.
Representative Results
This protocol has been optimized for the culture and sequencing of urinary bacteria belonging to the genera listed in Figure 1. Not all urinary bacteria are culturable by this method. Culture media and conditions are specified by the genus in Figure 1. Exemplary gel electrophoresis assessments of gDNA integrity are depicted in Figure 2. An overview of the bioinformatics pipeline for sequencing read processing, genome assembly, and annotation is described in Figure 3. A guide for computational directory structure is provided in Figure 4 to both simplify protocol understanding and provide framework for successful organization. Furthermore, included are representative complete genomes of two Klebsiella spp., K. pneumoniae and K. oxytoca, that were generated by this protocol. A representation of these assemblies is provided in Figure 5 and also includes an additional incomplete example K. pneumoniae genome. A detailed overview of each fully annotated complete genome is shown in Figure 6. Finally, a summary of sequencing read statistics is provided in Table 1 to offer a broad understanding of raw and trimmed data sufficient for the generation of high-quality closed genome assemblies. Additionally, key parameters of the two representative complete Klebsiella spp. genomes are listed. Genomes and raw data were deposited in Genbank under the BioProject PRJNA683049.

Figure 1: Modified enhanced urine culture of diverse urinary genera. Chart for the agar and liquid broth that may be used to culture diverse urinary genera. All culturing is suggested to be performed at 35 °C as described in subsection 1.1. Circles represent media appropriate for culturing a particular genus, colors were arbitrarily selected to distinguish one media type from another. CDC-AN BAP (red), CDC Anaerobe Sheep Blood Agar; 5% Sheep-BAP (orange), Sheep Blood Agar; BHI (green), Brain Heart Infusion; TSB (yellow), Tryptic Soy Broth; CHROMagar Orientation (blue). aGardnerella vaginalis should be cultured on HBT Bilayer G. vaginalis Selective agar in microaerophilic atmosphere and under special broth culture requirements44. bLactobacillus iners should be cultured on 5% Rabbit-BAP plates and NYCIII broth in microaerophilic atmosphere. cLactobacillus spp. may be cultured on MRS in microaerophilic conditions. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Genomic DNA extraction agarose gel images. Representative gel images depicting gDNA extraction outcomes. (A) Lane 1: 1 kb ladder, Lane 2: intact gDNA representing successful extraction, Lane 3: smearing indicating fragmented gDNA. (B) Lane 1: 1 kb ladder, Lanes 2 & 3: rRNA contamination denoted by two bands between 1.5 kb and 3 kb. Please click here to view a larger version of this figure.
{kind=link}

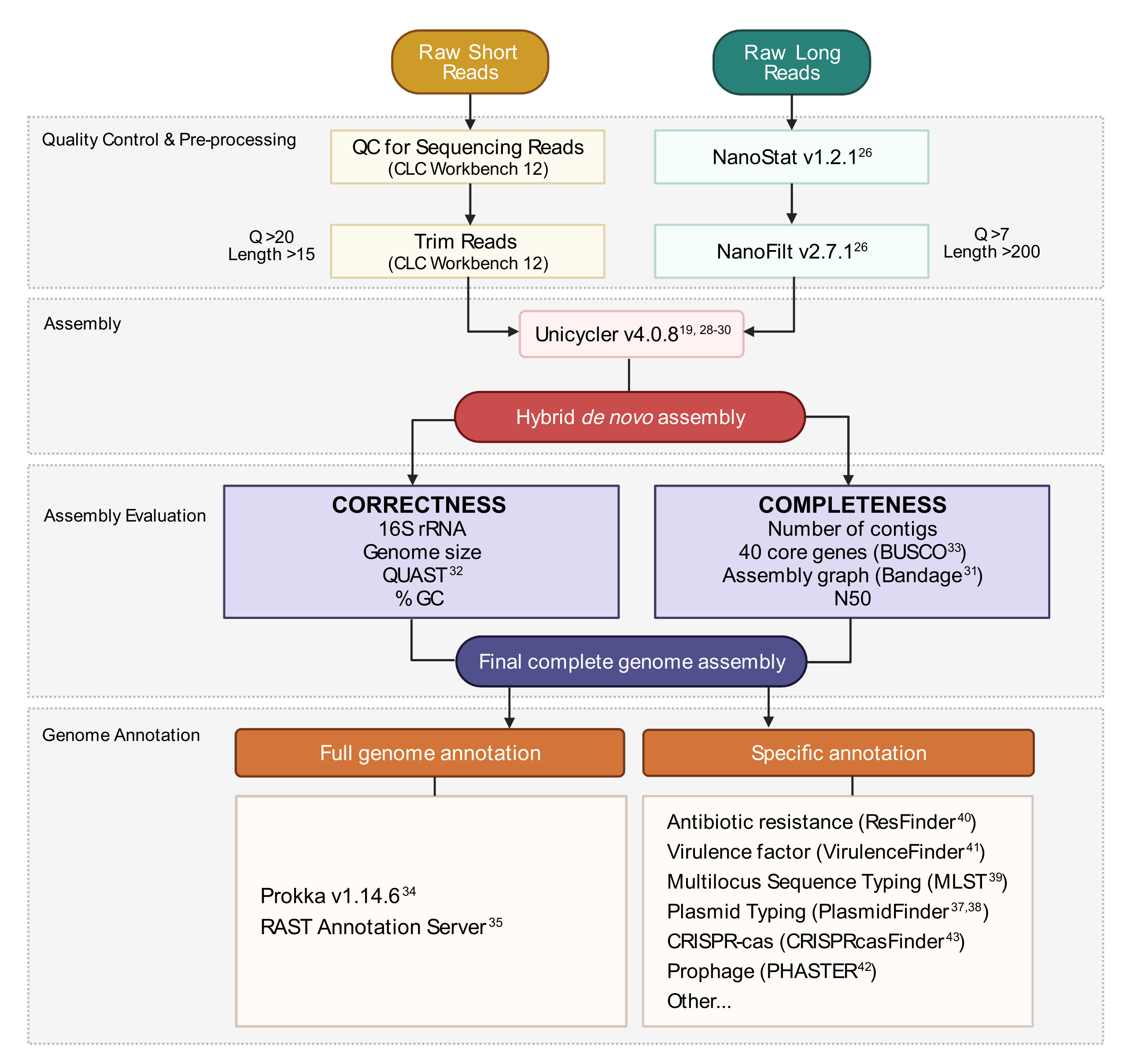
Figure 3: Hybrid genome assembly workflow. Schematic of steps from read quality control and pre-processing to assembly annotation. Read trimming removes ambiguous and low-quality reads. Q-score and length parameters are indicated and represent the reads that are retained. Assembly utilizes both short and long reads to generate a hybrid de novo genome assembly. Assembly quality is evaluated based on completeness and correctness using specified tools and parameters. The final genome assembly is annotated for all genes and specific loci of interest. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Bioinformatics directory structure guide. A schematic of recommended directory and file organization for the processing of short and long reads, hybrid assembly, and genome annotation and QC. Key command-line data processing steps are highlighted next to corresponding files and directories. Eliciting commands and flags (bold), input files (blue), output files or directories (red), user input such as file naming convention (magenta). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Genome assembly graphs by Bandage. Representative complete genome assembly graphs of (A) Klebsiella oxytoca KoPF10 and (B) Klebsiella pneumoniae KpPF25 and incomplete genome assembly of (C) Klebsiella pneumoniae KpPF46. The complete genome of KoPF10 demonstrates a single closed chromosome and the complete genome of KpPF25 consists of a closed chromosome and five closed plasmids. The incomplete chromosome of KpPF46 consists of two interconnected contigs. Unicycler hybrid de novo assembly generates an assembly graph that is visualized by Bandage. The assembly graph provides a simplistic schematic of the genome, indicating closed chromosome or plasmids by a linker connecting two ends of a single contig. The presence of more than one interconnected contig indicates incomplete assembly. Contig size and depth can be noted in Bandage as well. Please click here to view a larger version of this figure.
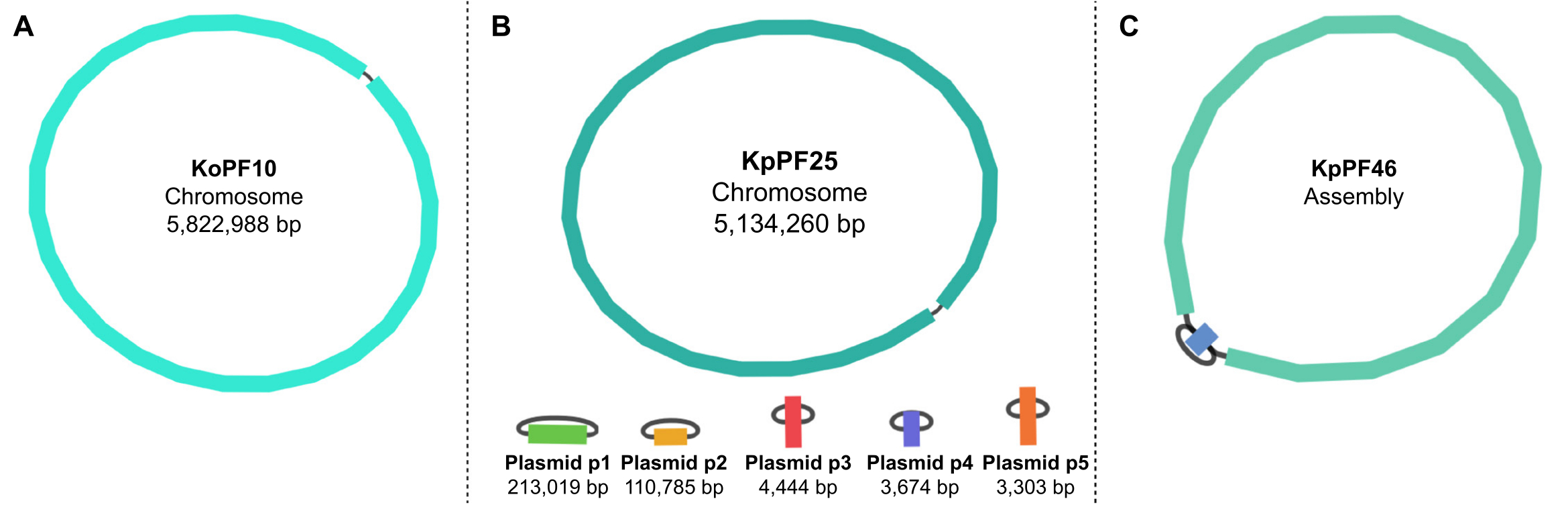{kind=link}

Figure 6: Complete genome maps of annotated hybrid assemblies. Assembly maps generated by Geneious Prime for the complete genome of (A) K. oxytoca KoPF10 and (B) K. pneumoniae KpPF25 showing annotated genes denoted by colored arrows along plasmid backbones. Chromosomes only show rRNA and tRNA genes for simplicity. Genome annotations were performed using Prokka as indicated in section 10 of this protocol. Please click here to view a larger version of this figure.
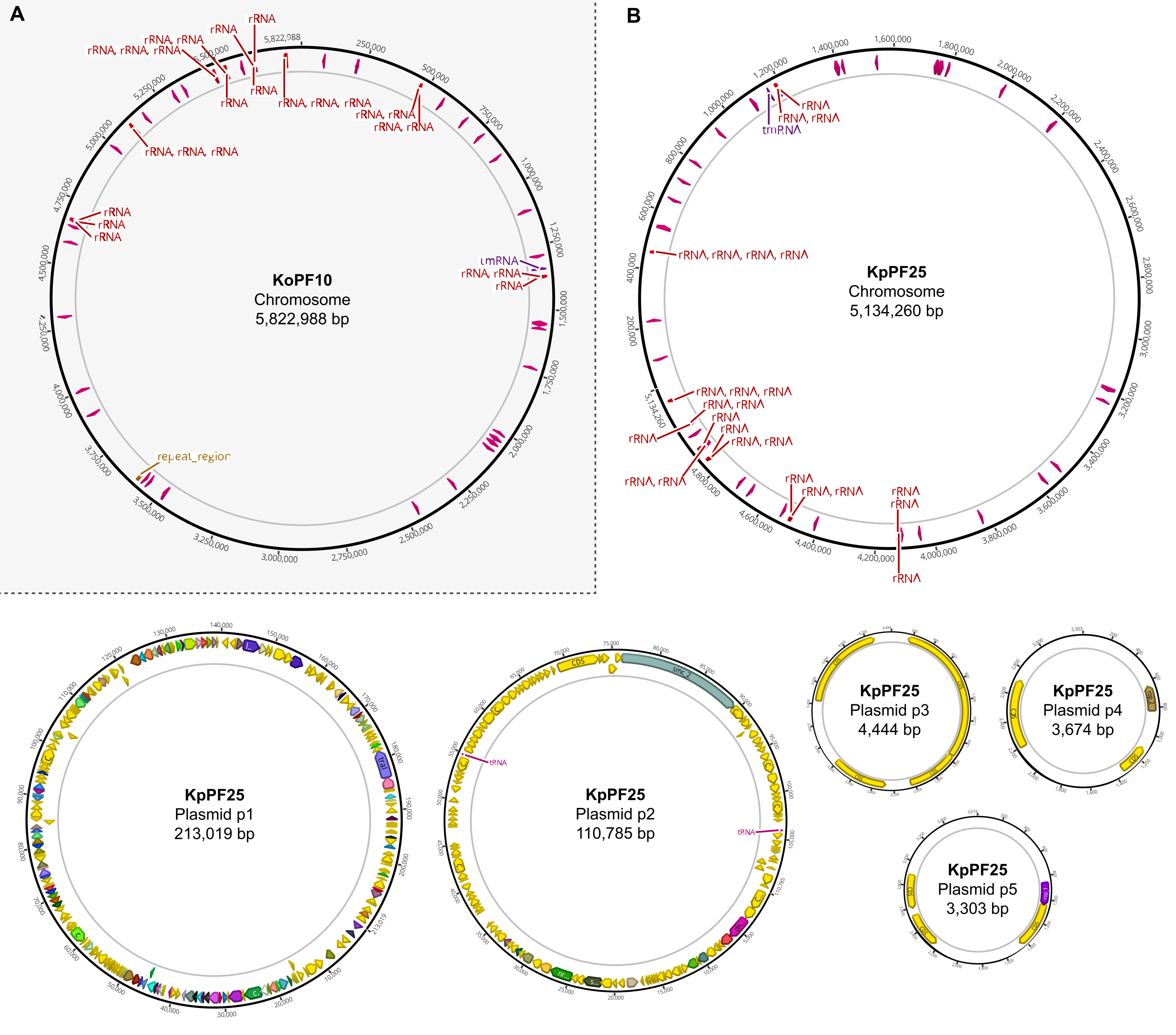{kind=link}

Table 1: Representative Klebsiella spp. complete assembly characteristics. Assembly parameters of K. oxytoca strain KoPF10 and K. pneumoniae strain KpPF25. Accession numbers for the deposited data on NCBI are provided. Number of reads both prior to and after trimming are specified for both sequencing technologies. N50 is provided for long reads only since short reads are of a controlled length. Plasmid replicon predicted using PlasmidFinder v2.1 Enteroebacteriaceae database with parameters set to 80% identity and 60% length. a MLST, Multilocus Sequence Type. b CDS, Coding Sequences. c Plasmid replicon predicted using PlasmidFinder v2.1 Enterobacteriaceae database with parameters set to 80% identity and 60% length. d Oxford Nanopore Technologies (ONT) deposited read data. e Illumina deposited read data. Please click here to download this Table.
Discussion
The comprehensive hybrid genome assembly protocol described here offers a streamlined approach for the successful culturing of diverse urinary microbiota and uropathogens, and the complete assembly of their genomes. Successful WGS of bacterial genomes begins with the isolation of diverse and sometimes fastidious microbes in order to extract their genomic DNA. To date, existing urine culture protocols either lack the necessary sensitivity to detect many urinary species or involve lengthy and extensive approaches that require extended time and resources11. The Modified Enhanced Urine Culture approach described offers a simplified yet comprehensive protocol for the successful isolation of bacteria belonging to 17 common urinary genera, including potentially pathogenic or beneficial commensal species, and both facultative and obligate aerobic or anaerobic bacteria. This in turn provides the necessary starting material for accurate sequencing and assembly of bacterial genomes and for critical phenotypic experiments, which contribute to the understanding of urinary health and disease. Furthermore, this modified culture approach provides for a more defined clinical diagnosis of viable microorganisms found in urine specimens and allows for their biobanking for future genomic studies. However, this protocol is not without limitations. It may require long incubation times depending on the organism as well as use of resources such as a hypoxia chamber or controlled incubators that may not be readily available. The use of anaerobic GasPaks offers an alternative solution but these are costly and do not always produce a sustained and controlled environment. Finally, culture bias as well as sample diversity may allow for particular organisms and uropathogens to outcompete fastidious bacteria. Despite these limitations, a culture of diverse urinary bacteria is made possible by this approach.
Genomic sequencing has gained popularity with the advancement of Next Generation Sequencing technologies which tremendously increased both the yield and accuracy of sequencing data14,15. Coupled with the development of algorithms for data processing and de novo assembly, complete genome sequences are at the fingertips of novice and expert scientists alike15,45. Knowledge of overall genome organization provided by complete genomes offers important evolutionary and biological insights, including gene duplication, gene loss, and horizontal gene transfer14. Additionally, genes important to antimicrobial resistance and virulence are often localized on mobile elements, which are typically not resolved in draft genome assemblies15,16.
The protocol herein follows a hybrid approach for the combination of sequencing data from short-read and long-read platforms to generate complete genome assemblies. While focused on urinary bacterial genomes, this procedure may be adapted to diverse bacteria from various isolation sources. Critical steps in this approach include following adequate sterile technique and utilizing appropriate media and culture conditions for the isolation of pure urinary bacteria. Furthermore, the extraction of intact, high-yield gDNA is essential for generating sequencing data free of contaminating reads that may hamper assembly success. Subsequent library preparation protocols are critical for the generation of quality reads of sufficient length and depth. Therefore, it is of utter importance to handle gDNA with care during library preparation for long-read sequencing in particular, as this technology's greatest benefit is the generation of long reads with no theoretical upper length limit. Also outlined are sections for the appropriate quality control (QC) of sequencing reads that eliminates noisy data and improves assembly outcome.
Despite successful DNA isolation, library preparation, and sequencing, the nature of genomic architecture of some species may still provide an obstacle for the generation of a closed genome assembly45,46. Repetitive sequences often complicate assembly computation and despite long read data, these regions may be resolved with low confidence, or not at all. Long reads thus have to be on average longer than the largest repeat region in the genome or coverage must be high (>100x)19. Some genomes may remain incomplete and require manual approaches for completion. Nevertheless, hybrid assembled incomplete genomes are typically composed of fewer contigs than short-read draft genomes. Adjusting default parameters of the assembly algorithm or following more stringent cutoffs for read QC may help. Alternatively, one suggested approach is to map long reads to the incomplete regions in search of evidence for the most likely assembly path, and then confirm the path utilizing PCR and Sanger sequencing of the amplified region. Mapping reads using Minimap2 is suggested and Bandage offers a useful tool for the visualization of mapped reads along assembled contigs providing evidence for contig linkage47.
An additional challenge to generating complete genomes lies in familiarity and comfort with command-line tools. Many bioinformatic tools are developed to offer computational opportunities to any user; however, their utilization relies on an understanding with the basics of UNIX and programming. This protocol aims to provide sufficiently detailed instructions to enable individuals without prior command-line experience to generate closed genome assemblies and annotate them.
Acknowledgements
We thank Dr. Moutusee Jubaida Islam and Dr. Luke Joyce for their contributions to this protocol. We would also like to recognize the University of Texas at Dallas Genome Center for their feedback and support. This work was funded by the Welch Foundation, award number AT-2030-20200401 to N.J.D., by the National Institutes of Health, award number R01AI116610 to K.P., and by the Felecia and John Cain Chair in Women's Health, held by P.E.Z.
Materials
| Name | Company | Catalog Number | Comments |
| Equipment: | |||
| Bioanalyzer 2100 | Agilent | G29398A | Optional but recommended |
| Centrifuge | Eppendorf | -- | Any centrifuge for spinning conicals and microcentrifuge tubes (e.g. Models 5810R/5424R) |
| Electrophoresis | BioRad Laboratories | 1645070 | |
| Gel Imaging System | BioRad Laboratories | ChemiDoc models | |
| Incubator | ThermoFisher Scientific | -- | Any CO2 Incubator (e.g. Thermo Forma model 3110) |
| Magnetic Rack | New England BioLabs | S15095 | 12-tube rack |
| MinION | Oxford Nanopore Technologies | -- | |
| Nanodrop | ThermoFisher Scientific | ND-ONE-W | |
| NextSeq 500 | Illumina | SY-415-1002 | Other Illumina models are acceptable |
| Plate Reader | BioTek | -- | Synergy H1 |
| Qubit fluorometer | ThermoFisher Scientific | Q33238 | |
| Rotator | Benchmark Scientific | H2024 | |
| Thermocycler | ThermoFisher Scientific | -- | Any thermocycler for PCR reactions (e.g. ProFlex PCR system) |
| Materials: | |||
| 10X Phosphate Buffered Saline (PBS) | Fisher Scientific | BP3991 | |
| 10X TBE buffer | -- | -- | 1M Tris,1M Boric Acid,0.2M EDTA (pH 8.0) |
| 1429R primer | Sigma Aldrich (Custom oligos) | -- | GGTTACCTTGTTACGACTT |
| 1kb Ladder | VWR | 101228-494 | |
| 1M Tris-Cl (pH 7.5) | ThermoFisher Scientific | 15567027 | |
| 6x Loading dye | Fisher Scientific | NC0783588 | |
| 8F primer | Sigma Aldrich (Custom oligos) | -- | AGAGTTTGATCCTGGCTCAG |
| Agar | Fisher Scientific | BP1423-2 | |
| Agarose | BioRad Laboratories | 63001 | |
| AMPure XP Beads | Beckman Coulter | A63880 | |
| Anaerobe Pouch System - GasPak EZ | BD Diagnostic Systems | B260683 | |
| Boric Acid | Fisher Scientific | A73-500 | |
| Brain Heart Infusion Broth | BD Diagnostic Systems | 212304 | |
| CDC Anaerobe 5% Sheep Blood Agar | BD Diagnostic Systems | L007357 | |
| CHROMagar Orientation | BD Diagnostic Systems | PA-257481.04 | |
| DNeasy Blood & Tissue | QIAGEN | 69504 | |
| DreamTaq Master Mix | ThermoFisher Scientific | K1081 | |
| Dry Anaerobic Indicator Strips | BD Diagnostic Systems | 271051 | |
| EDTA | Fisher Scientific | S311-500 | |
| Ethanol 200 Proof | Sigma Aldrich | E7023 | For molecular biology |
| Ethidium Bromide | ThermoFisher Scientific | BP130210 | |
| Flow cell priming kit | Oxford Nanopore Technologies | EXP-FLP002 | |
| Flow cell wash kit | Oxford Nanopore Technologies | EXP-WSH003 | |
| Gel Extraction Miniprep Kit | BioBasic | BS654 | |
| Ligation sequencing kit | Oxford Nanopore Technologies | SQK-LSK109 | |
| Lysozyme | Research Products International Corp | L381005.05 | |
| Mutanolysin | Sigma Aldrich | M9901-5KU | |
| Native barcoding expansion 1-12 | Oxford Nanopore Technologies | EXP-NBD104 | |
| NEB Blunt/TA Ligase Master Mix | New England BioLabs | M0367L | |
| NEBNext FFPE DNA Repair Mix | New England BioLabs | M6630L | |
| NEBNext quick ligation buffer | New England BioLabs | B6058S | |
| NEBNext Ultra II End repair / dA-tailing module | New England BioLabs | E7546L | |
| Nextera DNA CD Indexes | Illumina | 20018708 | |
| Nextera DNA Flex Library Prep - (M) Tagmentation | Illumina | 20018705 | |
| Nuclease-free water | Sigma Aldrich | W4502 | |
| Qubit 1X dsDNA HS Assay Kit | ThermoFisher Scientific | Q33230 | |
| Qubit Assay Tubes | ThermoFisher Scientific | Q32856 | |
| Quick T4 DNA Ligase | New England BioLabs | E6056L | |
| R9 Flow cell | Oxford Nanopore Technologies | FLO-MIN106D | |
| RNase A | ThermoFisher Scientific | EN0531 | |
| Sheep Blood | Hemostat Laboratories | DS13250 | |
| TE buffer | -- | -- | 10mM Tris, 1mM EDTA (pH 8.0) |
| Triton X-100 | Sigma Aldrich | T8787 | |
| Tryptic Soy Broth | BD Diagnostic Systems | 211825 | |
| Software & Bioinformatic Tools: | |||
| Bandage | -- | -- | https://rrwick.github.io/Bandage/ |
| Center for Genomic Epidemiology | -- | -- | http://www.genomicepidemiology.org/ |
| CLC Genomics Workbench 12 | QIAGEN | -- | |
| CRISPRcasFinder | -- | -- | https://crisprcas.i2bc.paris-saclay.fr/ |
| FastQC | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Geneious Prime | Geneious | -- | |
| gVolante (BUSCO) | -- | -- | https://gvolante.riken.jp/ |
| Kbase Prokka Wrapper | -- | -- | https://kbase.us/applist/apps/ProkkaAnnotation/annotate_contigs/release |
| Minimap2 | -- | -- | https://github.com/lh3/minimap2 |
| MinKNOW | Oxford Nanopore Technologies | -- | |
| NanoFilt | -- | -- | https://github.com/wdecoster/nanofilt |
| NanoStat | -- | -- | https://github.com/wdecoster/nanostat |
| PHASTER | -- | -- | https://phaster.ca/ |
| Prokka | -- | -- | https://github.com/tseemann/prokka |
| QUAST | -- | -- | http://quast.sourceforge.net/quast |
| Trim Galore | -- | -- | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Trimmomatic | -- | -- | http://www.usadellab.org/cms/?page=trimmomatic |
| Unicycler | -- | -- | https://github.com/rrwick/Unicycler#necessary-read-length |
References
- Brubaker, L., Wolfe, A. The urinary microbiota: a paradigm shift for bladder disorders. Current Opinion in Obstetrics & Gynecology. 28 (5), 407-412 (2016).
- Neugent, M. L., Hulyalkar, N. V., Nguyen, V. H., Zimmern, P. E., De Nisco, N. J. Advances in understanding the human urinary microbiome and its potential role in urinary tract infection. mBio. 11 (2), (2020).
- Klein, R. D., Hultgren, S. J. Urinary tract infections: microbial pathogenesis, host-pathogen interactions and new treatment strategies. Nature Reviews. Microbiology. 18 (4), 211-226 (2020).
- Horsley, H., et al. Enterococcus faecalis subverts and invades the host urothelium in patients with chronic urinary tract infection. PLoS One. 8 (12), 83637 (2013).
- Reitzer, L., Zimmern, P. Rapid growth and metabolism of uropathogenic Escherichia coli in relation to urine composition. Clinical Microbiology Reviews. 33 (1), 00101-00119 (2019).
- Snyder, J. A., et al. Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infection and Immunity. 72 (11), 6373-6381 (2004).
- Ipe, D. S., Horton, E., Ulett, G. C. The basics of bacteriuria: Strategies of microbes for persistence in urine. Frontiers in Cellular and Infection Microbiology. 6, 14 (2016).
- Babikir, I. H., et al. The impact of cathelicidin, the human antimicrobial peptide LL-37 in urinary tract infections. BMC Infectious Diseases. 18 (1), 17 (2018).
- Jancel, T., Dudas, V. Management of uncomplicated urinary tract infections. The Western Journal of Medicine. 176 (1), 51-55 (2002).
- Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P & T. 40 (4), 277-283 (2015).
- Price, T. K., et al. The clinical urine culture: Enhanced techniques improve detection of clinically relevant microorganisms. Journal of Clinical Microbiology. 54 (5), 1216-1222 (2016).
- Kass, E. H. Asymptomatic infections of the urinary tract. Transactions of the Association of American Physicians. 69, 56-64 (1956).
- Garcia, L. S. . Clinical microbiology procedures handbook. 3rd edn. , (2010).
- Fraser, C. M., Eisen, J. A., Nelson, K. E., Paulsen, I. T., Salzberg, S. L. The value of complete microbial genome sequencing (you get what you pay for). Journal of Bacteriology. 184 (23), 6403-6405 (2002).
- Chen, Z., Erickson, D. L., Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genomics. 21 (1), 631 (2020).
- Greig, D. R., Dallman, T. J., Hopkins, K. L., Jenkins, C. MinION nanopore sequencing identifies the position and structure of bacterial antibiotic resistance determinants in a multidrug-resistant strain of enteroaggregative Escherichia coli. Microbial Genomics. 4 (10), 000213 (2018).
- Carraro, D. M., et al. PCR-assisted contig extension: stepwise strategy for bacterial genome closure. Biotechniques. 34 (3), 626-628 (2003).
- Tettelin, H., Radune, D., Kasif, S., Khouri, H., Salzberg, S. L. Optimized multiplex PCR: efficiently closing a whole-genome shotgun sequencing project. Genomics. 62 (3), 500-507 (1999).
- Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Computational Biology. 13 (6), 1005595 (2017).
- Singhal, N., Kumar, M., Kanaujia, P. K., Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Frontiers in Microbiology. 6, 791 (2015).
- Turner, S., Pryer, K. M., Miao, V. P., Palmer, J. D. Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. The Journal of Eukaryotic Microbiology. 46 (4), 327-338 (1999).
- Weisburg, W. G., Barns, S. M., Pelletier, D. A., Lane, D. J. 16S ribosomal DNA amplification for phylogenetic study. Journal of Bacteriology. 173 (2), 697-703 (1991).
- Janda, J. M., Abbott, S. L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. Journal of Clinical Microbiology. 45 (9), 2761-2764 (2007).
- Stevenson, K., McVey, A. F., Clark, I. B. N., Swain, P. S., Pilizota, T. General calibration of microbial growth in microplate readers. Science Reports. 6, 38828 (2016).
- Bolger, A. M., Lohse, M., Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30 (15), 2114-2120 (2014).
- De Coster, W., D'Hert, S., Schultz, D. T., Cruts, M., Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 34 (15), 2666-2669 (2018).
- Wilson, G., et al. The UNIX Shell. Zenodo. , (2019).
- Bankevich, A., et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 19 (5), 455-477 (2012).
- Vaser, R., Sovic, I., Nagarajan, N., Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Research. 27 (5), 737-746 (2017).
- Walker, B. J., et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 9 (11), 112963 (2014).
- Wick, R. R., Schultz, M. B., Zobel, J., Holt, K. E. Bandage: interactive visualization of de novo genome assemblies. Bioinformatics. 31 (20), 3350-3352 (2015).
- Gurevich, A., Saveliev, V., Vyahhi, N., Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 29 (8), 1072-1075 (2013).
- Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31 (19), 3210-3212 (2015).
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30 (14), 2068-2069 (2014).
- Aziz, R. K., et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 9, 75 (2008).
- Tatusova, T., et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Research. 44 (14), 6614-6624 (2016).
- Carattoli, A., Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods in Molecular Biology. 2075, 285-294 (2020).
- Carattoli, A., et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrobial Agents and Chemotherapy. 58 (7), 3895-3903 (2014).
- Larsen, M. V., et al. Multilocus sequence typing of total-genome-sequenced bacteria. Journal of Clinical Microbiology. 50 (4), 1355-1361 (2012).
- Bortolaia, V., et al. ResFinder 4.0 for predictions of phenotypes from genotypes. The Journal of Antimicrobial Chemotherapy. 75 (12), 3491-3500 (2020).
- Joensen, K. G., et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. Journal of Clinical Microbiology. 52 (5), 1501-1510 (2014).
- Arndt, D., et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Research. 44 (1), 16-21 (2016).
- Couvin, D., et al. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Research. 46 (1), 246-251 (2018).
- Totten, P. A., Amsel, R., Hale, J., Piot, P., Holmes, K. K. Selective differential human blood bilayer media for isolation of Gardnerella (Haemophilus) vaginalis. Journal of Clinical Microbiology. 15 (1), 141-147 (1982).
- Nagarajan, N., Pop, M. Sequence assembly demystified. Nat Reviews. Genetics. 14 (3), 157-167 (2013).
- Phillippy, A. M., Schatz, M. C., Pop, M. Genome assembly forensics: finding the elusive mis-assembly. Genome Biology. 9 (3), 55 (2008).
- . Unicycler Wiki Available from: https://github.com/rrwick/Unicycler/wiki (2017)
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved