Preparation of Single-Somite Explants from Zebrafish Embryos
In This Article
Summary
We present a protocol for isolating single somites from zebrafish embryos, the dynamics of which can be followed in culture for several hours by fluorescence time-lapse microscopy, thus providing a methodology to quantify tissue-scale shape changes at single-cell resolution.
Abstract
The body axis of vertebrate embryos is periodically subdivided into 3D multicellular units called somites. While genetic oscillations and molecular prepatterns determine the initial length-scale of somites, mechanical processes have been implicated in setting their final size and shape. To better understand the intrinsic material properties of somites, a method is developed to culture single-somite explant from zebrafish embryos. Single somites are isolated by first removing the skin of embryos, followed by yolk removal and sequential excision of neighboring tissues. Using transgenic embryos, the distribution of various sub-cellular structures can be observed by fluorescent time-lapse microscopy. Dynamics of explanted somites can be followed for several hours, thus providing an experimental framework for studying tissue-scale shape changes at single-cell resolution. This approach enables direct mechanical manipulation of somites, allowing for dissection of the material properties of the tissue. Finally, the technique outlined here can be readily extended for explanting other tissues such as the notochord, neural plate, and lateral plate mesoderm.
Introduction
Much of the vertebrate adult musculoskeletal system emerges from embryonic somites, which form in a periodic and rhythmic manner along the body axis of embryos1,2. Somites are three dimensional (3D) multicellular units typically consisting of an inner core of mesenchymal cells and a peripheral epithelial layer surrounded by a fibronectin-rich extracellular matrix3. The morphology of somites i.e., their size and shape, is in part determined by the segmentation clock and downstream molecular prepatterns. However, over the last decade, it has emerged that mechanical cues and forces also play a role in regulating the segmentation clock4, in addition to facilitating somite formation5,6,7, and ensuring increased precision of somite lengths following initial somite formation8.
Tissue mechanics can be studied directly in vivo with the availability of new tools9, however, to obtain a complete picture underlying the physical processes, the intrinsic material properties of tissues need to be simultaneously studied. The protocol described here provides a simple approach to prepare single somites, whose physical properties from cellular to tissue scales can be studied in isolation from the embryo. While several protocols exist for preparing explants at similar developmental stages10,11,12,13,14, to our knowledge, this is the first protocol that describes isolation of single somites. The protocol is straightforward to implement and requires only basic equipment available in most zebrafish labs working with embryos.
To aid in teasing apart the role of mechanics in morphological somite formation, a method is developed to culture single-somite explant from zebrafish embryos, which can be used to probe intrinsic material properties of somites.
Protocol
This protocol involves use of live vertebrate embryos younger than 1-day post-fertilization. All experiments were carried out using embryos derived from freely mating adults, and thus are covered under the general animal experiment license of the EPFL granted by the Service de la Consommation et des Affaires Vétérinaires of the canton of Vaud - Switzerland (authorization number VD-H23).
1. Before dissection
- Obtain embryos from a cross of heterozygous transgenic lines of interest. For explanting two to three somites, only a few embryos are typically needed. In this protocol, one embryo was used for explanting one somite.
- Prepare 1x E3 buffer from two 50x stocks: stock 1 contains 0.458 mM Na2HPO4, 0.042 mM KH2PO4, 4.084 mM NaCl, 0.128 mM KCl and stock 2 contains 0.33 mM CaCl2, 0.33 mM MgSO4. Prepare 500 mL of 1x E3 buffer by mixing 480 mL of distilled water with 10 mL each of the two 50x stocks.
- If dissecting on the same day, raise embryos in 25 mL of E3 medium in a Petri dish at 33 °C until three-somite stage. If dissecting the following morning, raise embryos in E3 medium at 28 °C until shield stage before transferring to 19 °C for overnight incubation.
- For identifying the shield stage or the different somite stages, use a standard lab stereomicroscope with an adjustable magnification range from approximately 0.67x to 4.5x.
- Assemble tools and reagents needed for dissection.
- Aliquot Leibovitz's L-15 medium in 50 mL tubes and store in fridge. We recommend using the same aliquot from the fridge for a maximum of two rounds.
- Prepare 50 mL of 2% agarose in L-15 medium by using a magnetic stirrer and keeping the temperature at 85 °C. Once dissolved, store the solution at room temperature.
- Prepare 25 mL of 2% low-melting agarose in L-15 medium by using a magnetic stirrer and keeping the temperature at 85 °C. Once dissolved, aliquot the solution in 1.5 mL tubes and store at 4 °C. This will be used for preparing the imaging chamber.
- Pour molten 2% agarose into a 20 mm Petri dish until one-third of the depth of the dish is filled and allow the agarose solution to solidify. Dissections will be performed in these Petri dishes. Prepare the Petri dishes with agarose coating well in advance and store at 4 °C.
- Assemble the following tools: Pasteur pipette for transfer of embryos, a pair of forceps for dechorionating embryos, a pair of fine forceps for removal of skin of embryos, a micro knife for making incisions in embryos, fire-polished glass pipette for transferring explants to imaging chamber.
- Sterilize the glass pipette in 100% ethanol for 15 min and rinse twice by pipetting 5 mL of L-15 medium up and down.
- Prepare an eye-lash tool by sticking an eyelash or hair from the eyebrow to a glass capillary.
- Use a fluorescent stereoscope with an adjustable magnification range from approximately 0.63x to 6.3x and standard GFP and RFP filters to sort positive transgenic embryos and transfer to a separate Petri dish with E3 medium.
2. Single-somite explant preparation
- Transfer 2 or 3 embryos to an agarose-coated dish (made in step 1.5.4) filled with 10 mL of L-15 medium. Ensure that the dish along with L-15 medium is pre-warmed at 28 °C in an incubator for 30 min before transferring embryos.
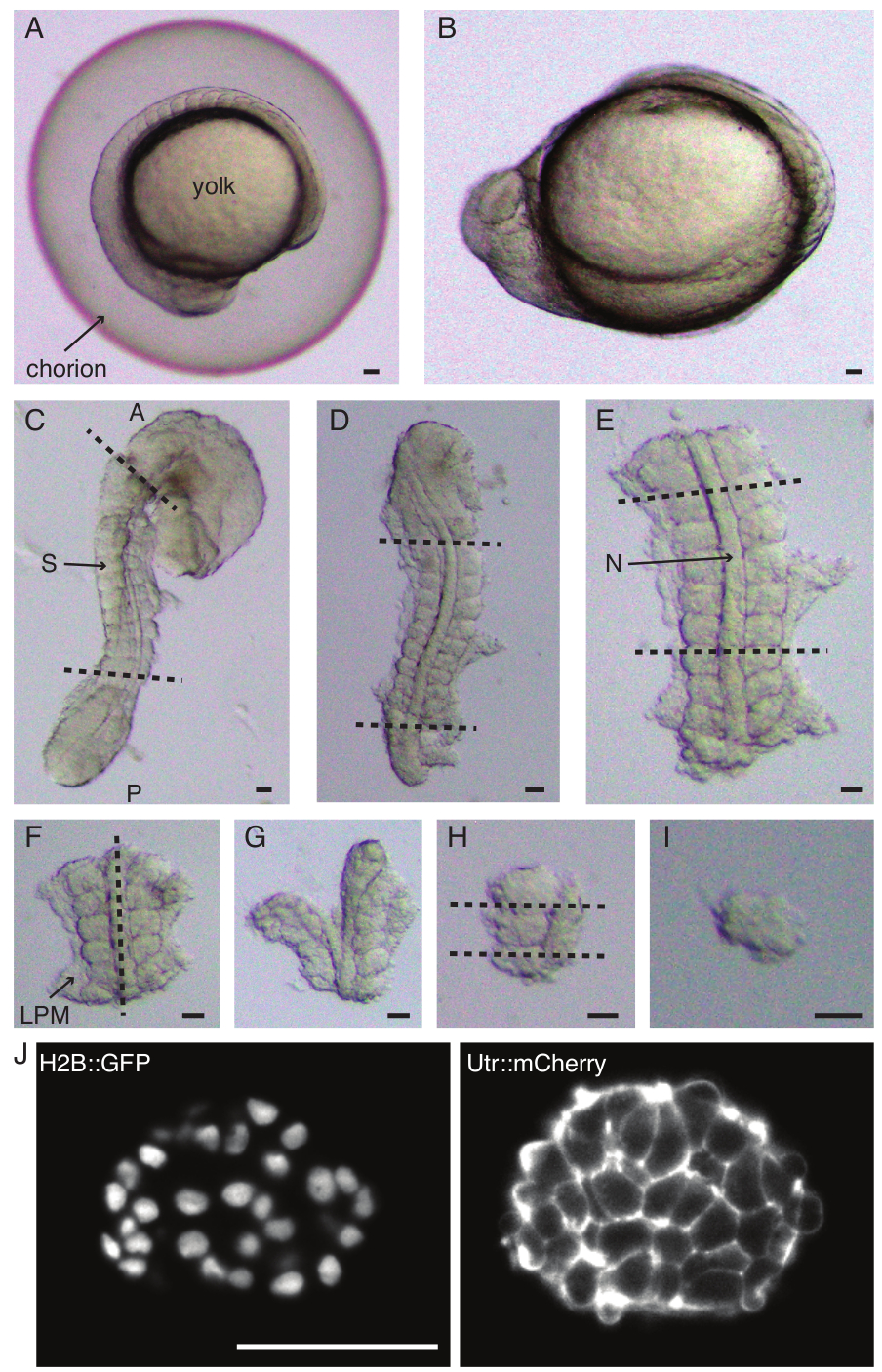
- Carefully dechorionate (Figure 1 A,B) an embryo with a pair of forceps. Hold the chorion with one of the forceps and use the second forceps to pinch and pull on the chorion close to where the first forceps is positioned.
- Repeat this step one more time to open the chorion widely after which the embryo can be gently pushed out. Ensure that the embryo is not damaged or squeezed during dechorionation.
- Orient the embryo on its lateral side and hold on to the skin in between the head and tail of the embryo with one of the fine forceps. Use the second fine forceps to pinch on the skin close to where the first forceps is still positioned.
- Move the two forceps away from each other, while holding on to the skin, thus creating a large opening. Release one of the forceps from the skin and move it close to where the second forceps is held and repeat the skin removal process until the skin is removed in somite-forming regions.
NOTE: It is critical to completely remove the skin surrounding the somites that will be explanted, else it gets difficult to do finer dissections in the later steps of the protocol. - Use a micro knife to scrape off the yolk (Figure 1C - explant with skin removed and yolk scraped off) and make a series of incisions as follows:
- Slice through the embryo with the first cut anterior to the first morphological somite in the embryo and the second cut posterior to the most recently formed somite (Figure 1D). Slice once more posterior and anterior to somites of interest (Figure 1E).
- Use the micro knife to scrape off any remaining yolk from the explanted regions of the embryo. Try and remove as much of the yolk as possible to reduce the possibility of explants getting stuck to the micro knife. If this happens, use an eye-lash tool to move the explant back to solution while keeping the micro knife immersed in the medium.
- Position the explant dorsally and make a slice along the anteroposterior axis through the notochord, close to somites on one side (Figure 1F,G).
- Take the explant without the notochord and slice through the lateral plate mesoderm (LPM) close to the somites.
NOTE: This step can be exchanged with the previous step i.e., it works equally well to slice through the LPM first followed by slicing through the notochord.
- Choose the somite to be explanted and make two slices posterior and anterior to the somite respectively (Figure 1H).
- Finally, rotate the somite by 90° using the eye-lash tool to visualize the neural plate still attached to the somite. Make a slice in between the somite and the neural plate, thus releasing a single somite (Figure 1I).
NOTE: The neural plate can also be removed immediately after step 2.6.4, i.e., after removal of the notochord and the LPM and before dissecting a somite from neighboring somites.
3. Imaging single-somite explants
NOTE: Here, the procedure for imaging single-somite explants using a single-view light-sheet microscope is described. As an alternative, the explanted single somites can also be imaged in a confocal microscope, which is more widely available or even in a widefield microscope if exclusively interested in following overall morphology of explants.
- Prepare an imaging chamber as follows:
- Fill a 1 mL syringe with silicone rubber formulation. Cut a 200 µL pipette tip about 1 cm away from the mouth and insert the syringe into the pipette tip.
- Manually wrap the membrane holder with a pre-cut fluorinated ethylene propylene (FEP) membrane strip (Figure 2A-C) and fix the imaging chamber on to the membrane holder, which fit snugly into each other (Figure 2D).
- Add silicone rubber formulation along the intersection of the chamber and the membrane and dry it overnight, which allows the membrane to stick to the chamber. Remove the membrane holder 1 h before the explant experiment.
- Heat an aliquot containing 0.5 mL of 2% low-melting agarose made with L-15 medium at 75 °C for 10 min. Add 160 µL of the heated low-melting agarose to the center of the chamber and place an imaging mold in the agarose (Figure 2E,F) and hold it upright until the agarose solidifies.
- Add another 160 µL of heated low-melting agarose to the two ends of the mold and transfer the entire unit to 4 °C for 30 min.
- Add 600 µL of L-15 medium to the chamber and remove the mold from the chamber with a forceps. Wait until the chamber with L-15 medium returns to room temperature.
- Transfer the explanted somite with a polished glass pipette by pipetting to the imaging chamber and use an eye-lash tool to position the explant in the center of the chamber.
- Align the imaging lasers as per manufacturer's instructions and carefully transfer the imaging chamber to the microscope equipped with an incubation chamber to maintain the explants at 28 °C.
- Perform a second round of laser alignment with regions of interest from the transgenic line, followed by setting up the desired z-stack, time interval and duration for time-lapse imaging. For this experiment, acquire 70 z-slices with a spacing of 2 µm and a frame interval of 2 min.
- Perform dual-colored light-sheet imaging (Figure 1J) of the explants with 561 nm (10% laser power, 100 ms exposure) and 488 nm lasers (10% laser power, 100 ms exposure). In this protocol, the signal was collected with a 25x/1.1 NA objective and through 561/25 nm and 525/50-25 bandpass filters respectively onto a sCMOS camera.
Results
Explants allow for quantification of large-scale shape changes in 3D. To illustrate this, we explanted somite four (N = 3) from early zebrafish embryos obtained from a cross between Utr::mCherry (Tg(actb2:mCherry-Hsa.UTRN); e119Tg) and H2B::GFP (Tg(h2az2a:h2az2a-GFP); kca6Tg) heterozygous lines. Utrophin is an actin-binding protein, and the Utr::mCherry line shows the distribution of filamentous actin structures. This transgene was used here as a marker for cell outlines. H2B::GFP is a fluorescently-tagged histone and consequently marks the distribution of the chromatin, providing an effective description of nuclear location and shape, as well as mitotic figures that highlight cell division.
We assessed the success of the explant protocol during imaging. We observed that in a somite damaged during dissection either the integrity of the somite tissue is compromised, with many cells dissociating and extruding from the explanted somite, and/or many cells dying, which can be noted by the presence of fragmented nuclei in the nuclear channel. Successful explants remained healthy for 4-6 h after which changes in somite integrity were observed with cells dissociating from the explant and dying.
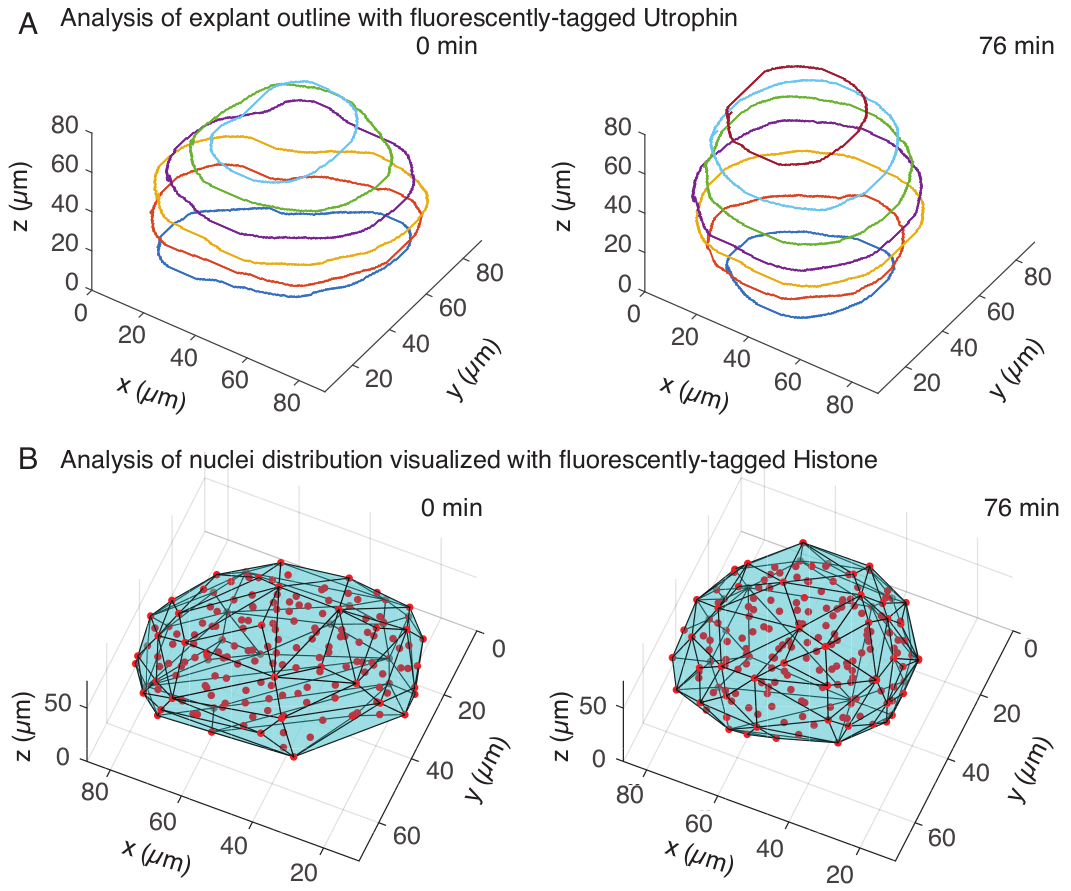
In the utrophin channel, we performed manual segmentation of explants in MATLAB (R2018b) on z-slices spaced every 10 µm. We developed a custom algorithm in MATLAB for segmentation, which is freely available for download (https://github.com/sundar07/SomSeg). In the algorithm, the file, frame and z-slice to be segmented can be set, followed by a user prompt, which allows for manual drawing of an outline around the region of interest. This was repeated for multiple z-slices and plotted, which showed rounding up of explants over time in 3D (Figure 3A). In addition, complementary information on tissue shape using the nuclear channel was also obtained. For this, we used Mastodon (version 1.0.0-beta-19, https://github.com/mastodon-sc/mastodon), a FIJI15 plugin, to obtain nuclei centroid positions through spot detection. We first converted the tif files from the microscope to xml/hdf5 format in FIJI, following which a new project was opened using the Mastodon plugin. In the plugin, we chose the spot detection option, where we defined a region of interest that covered the explant and used the difference of Gaussian detector with a diameter of 5 µm and a quality factor of 25 for detecting spots. We then transferred the nuclei centroid positions to MATLAB and used an in-built function (convhull) to obtain a convex hull (Figure 3B), which characterized the geometry of the explant. This similarly showed rounding of explants over time (Figure 3B). The nuclei data additionally allows for quantifying cell movements by tracking in 3D and a change in cell number over time. On the other hand, the utrophin channel allows for quantification of changes in cell shapes as explants become rounder. Together, these parameters are valuable for characterizing intrinsic material properties of somites, which aids in developing effective physical descriptions of tissue-scale shape changes.
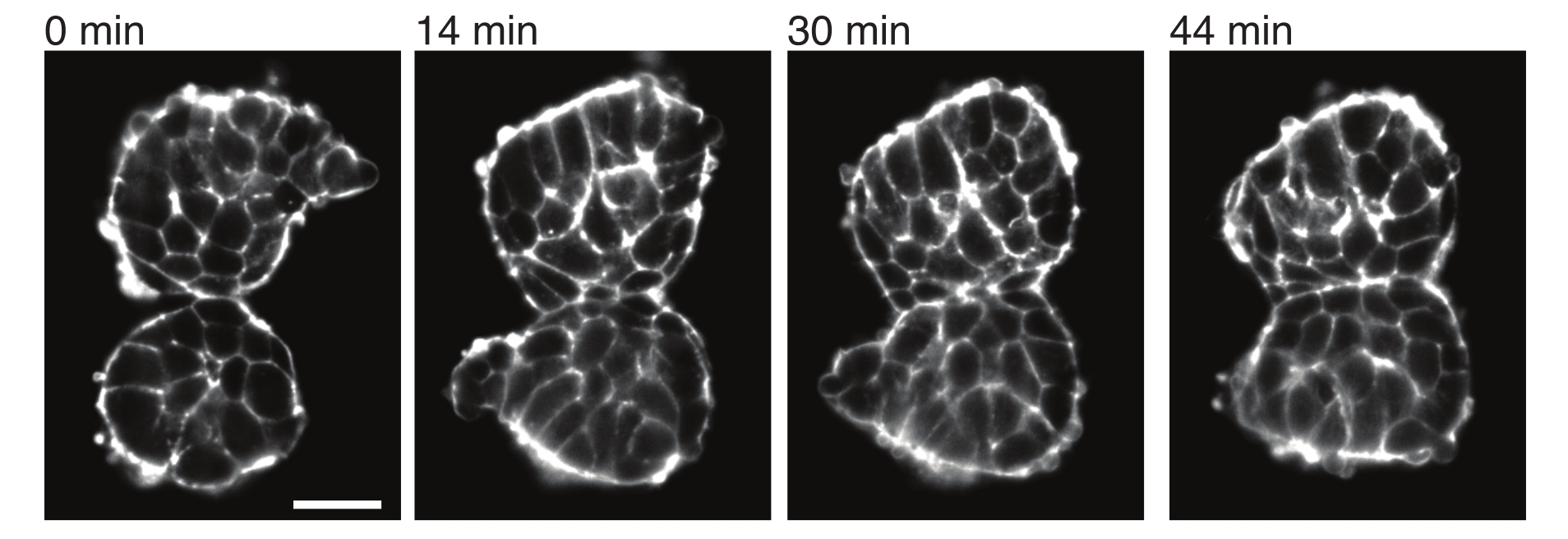
Explants also allow for characterizing contact stresses with neighboring tissues in a quantitative manner. To illustrate this, we manually isolated two somites and placed them in close proximity and observed their dynamics (N = 2). For this experiment, the orientation of the somites with respect to the in vivo body axes was not tracked. Interestingly, over time, the explanted somites adhered to each other along one surface, while the free surfaces i.e., regions away from the contact site rounded up (Figure 4). This suggests that adhesive forces overcome stresses generated by surface tension at contact sites in explants. This can be further characterized by following shape changes as described above and by quantifying contact angles between the two tissues over time in 3D. Thus, the explants provide an attractive system for quantifying competing forces in action that lead to specific tissue shapes, the implications of which can then be explored in vivo.

Figure 1: Preparation of single-somite explants. Zebrafish embryo (A) is first dechorionated (B), followed by removal of skin and yolk (C). Somite-containing region of the embryo is then selected by removing rest of the tissues (D, E). Regions around the somite of interest are then serially removed (F-H) to ultimately isolate a single somite (I), followed by time-lapse imaging using a light-sheet microscope (middle z-section of somite shown here) (J). Abbreviations: S = somite; N = notochord; LPM = lateral plate mesoderm; A = anterior; P = posterior. Dashed lines indicate cut positions. Scale bar = 50 µm in all panels. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Assembly of light-sheet imaging chamber. (A-D) A FEP membrane strip (dimensions in (B)) is wrapped around the membrane holder, fitted on to the imaging chamber and the membrane is glued to the imaging chamber. (E-F) The following day, the membrane holder is removed and an imaging mold (dimensions in (E)) is placed in the center of the imaging chamber in low-melting agarose. The entire unit is kept at 4 °C for 30 min, following which the mold is removed and the chamber with the trough is used for imaging explants. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: 3D analysis of tissue shapes. (A) Cell outlines were visualized with fluorescently-tagged utrophin (Utr::mCherry) and nuclei were visualized with fluorescently-tagged histone (H2B::GFP). The outlines of the explant were manually segmented at multiple depths using MATLAB and shown here. (B) Nuclei (red) were detected in the same explant using Mastodon, a FIJI plugin, and subjected to a convex hull (cyan), which informs on the geometry of the explant. Note rounding up of explants is evident in both analyses. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Explanted somites adhere to each other. Two somites isolated from embryos and manually placed in close proximity tend to adhere over time (N = 2). Multiple z-slices with a frame interval of 2 min were acquired using a light-sheet microscope and middle sections of somites from selected time points are shown here. Cell outlines were visualized with fluorescently-tagged Utrophin (Utr::mCherry). Scale bar = 25 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The somitogenesis field has been dominated by studies of the segmentation clock's role in setting segment lengths during embryo development. However, its equally important to consider the role of tissue mechanics in determining final somite morphologies. The protocol described here allows for explanting single somites, whose intrinsic physical properties can be studied in isolation from the embryo. However, the manual preparation limits the numbers of somites that are typically prepared to four to six per imaging session. Since the protocol involves careful fine dissections of several tissues in embryos without the use of any enzymes to facilitate tissue removal, it may take a few weeks of practice to master the dissection process. In our hands the critical steps in the protocol involve careful removal of skin and yolk from the explants, which prevents explants at different stages of the protocol from sticking to the tools used, which in turn enables easier dissections to serially remove tissues surrounding a somite.
The dissection protocol can be stopped at intermediate steps to obtain somites attached to just one of the surrounding tissues or to obtain explants of groups of somites. This provides a powerful method to serially strip down tissues and study the impact of neighboring tissues in facilitating shape changes in somites, or in the neighboring tissues. On the other hand, using this protocol, individual explanted tissues can be allowed to adhere and mechanically self-organize, as demonstrated by placing two explanted somites in close proximity.
Explants prepared by this method do not require any added ingredients in the buffer or constraints to ensure survival for periods of time up to several hours. However, one can envision culturing them in hydrogels and following their dynamics in the presence of external constraints, which could serve as a model for the contact stresses somites encounter in vivo. Furthermore, explants allow for directly probing their material properties through atomic force microscopy, pipette aspiration, or by using micro-robotic tools16. Finally, we expect that this method can be easily adapted towards culturing and studying other developmental tissues at similar stages such as the notochord, neural plate and lateral plate mesoderm.
Disclosures
The authors declare no competing interests.
Acknowledgements
We thank members of the Oates lab for comments on the protocol and the fish facility of the École polytechnique fédérale de Lausanne (EPFL). In particular, we acknowledge Laurel Ann Rohde for valuable tips on skin and yolk removal in the dissection protocol; Arianne Bercowsky Rama for building an efficient pipeline for processing light-sheet data sets through Mastodon; Jean-Yves Tinevez for building the open-source Mastodon software; Marko Popović for tips on data analysis; Chloé Jollivet, Guillaume Valentin and Florian Lang for extensive support in the fish facility; Petr Strnad and Andrea Boni for building the light-sheet microscope and for tips on light-sheet imaging. This work was supported by EPFL and S.R.N. was supported by a Long-Term Human Frontier Science Program postdoctoral fellowship (LT000078/2016).
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma | 9012-36-6 | For coating bottom of petri dishes |
| Agarose, low gelling temperature | Sigma | 39346-81-1 | For preparing Viventis imaging chamber |
| Camera | Andor | Andor Zyla 4.2 Plus | For image acquisition in the light-sheet microscope |
| Detection objective | Nikon | Nikon CFI75 Apo LWD 25x/1.1 NA | For imaging explants |
| FEP membrane strip | Lohmann Technologies UK Ltd | Dupont FEP Fluorocarbon film, 200A | For preparing Viventis imaging chamber |
| Fine forceps | Dumont | Dumont 5SF 11252-00 | For removal of skin of embryos |
| Forceps | Dumont | Dumont 55 | For dechorionating embryos |
| Leibovitz's L-15 medium | Gibco | 21083-027 | Explant culture medium |
| Light-sheet microscope | Viventis | LS1 live | For imaging explants |
| Micro knife | Fine Science Tools | 10318-14 | For making incisions in embryos |
| Silicone rubber formulation | Wacker Chemie AG | Silpuran 4200 | For preparing Viventis imaging chamber |
References
- Oates, A. C., Morelli, L. G., Ares, S. Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development. 139 (4), 625-639 (2012).
- Pourquié, O. Segmentation of the vertebrate spine: From clock to scoliosis. Cell. 145 (5), 650-663 (2011).
- Naganathan, S. R., Oates, A. C. Patterning and mechanics of somite boundaries in zebrafish embryos. Seminars in Cell & Developmental Biology. 107, 170-178 (2020).
- Hubaud, A., Regev, I., Mahadevan, L., Pourquié, O. Excitable Dynamics and Yap-Dependent Mechanical Cues Drive the Segmentation Clock. Cell. 171 (3), 668-682 (2017).
- Dias, A. S., de Almeida, I., Belmonte, J. M., Glazier, J. A., Stern, C. D. Somites without a clock. Science. 343 (6172), 791-795 (2014).
- Nelemans, B. K. A., Schmitz, M., Tahir, H., Merks, R. M., Smit, T. H. Somite Division and New Boundary Formation by Mechanical Strain. iScience. 23 (4), 100976 (2020).
- Grima, R., Schnell, S. Can tissue surface tension drive somite formation. Developmental Biology. 307 (2), 248-257 (2007).
- Naganathan, S. R., Popovic, M., Oates, A. C. Left–right symmetry of zebrafish embryos requires somite surface tension. Nature. 605, 516-521 (2022).
- Campàs, O. A toolbox to explore the mechanics of living embryonic tissues. Seminars in Cell & Developmental Biology. 55, 119-130 (2016).
- Langenberg, T., Brand, M., Cooper, M. S. Imaging brain development and organogenesis in zebrafish using immobilized embryonic explants. Developmental Dynamics: An Official Publication of The American Association of Anatomists. 228 (3), 464-474 (2003).
- Henry, C. A., Poage, C. T., McCarthy, M. B., Campos-Ortega, J., Cooper, M. S. Regionally autonomous segmentation within zebrafish presomitic mesoderm. Zebrafish. 2 (1), 7-18 (2005).
- Picker, A., Roellig, D., Pourquié, O., Oates, A. C., Brand, M. Tissue micromanipulation in zebrafish embryos. Methods in Molecular Biology. 546, 153-172 (2009).
- Manning, A. J., Kimelman, D. Tbx16 and Msgn1 are required to establish directional cell migration of zebrafish mesodermal progenitors. Developmental Biology. 406 (2), 172-185 (2015).
- Simsek, M. F., Özbudak, E. M. A 3-D Tail Explant Culture to Study Vertebrate Segmentation in Zebrafish. Journal of Visualized Experiments:JoVE. (172), e61981 (2021).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Özkale, B., et al. Modular soft robotic microdevices for dexterous biomanipulation. Lab on a Chip. 19 (5), 778-788 (2019).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ISSN 2578-2746
Copyright © 2025 MyJoVE Corporation. All rights reserved
We use cookies to enhance your experience on our website.
By continuing to use our website or clicking “Continue”, you are agreeing to accept our cookies.