Chemical Cartography Approaches to Study Trypanosomatid Infection
* These authors contributed equally
In This Article
Summary
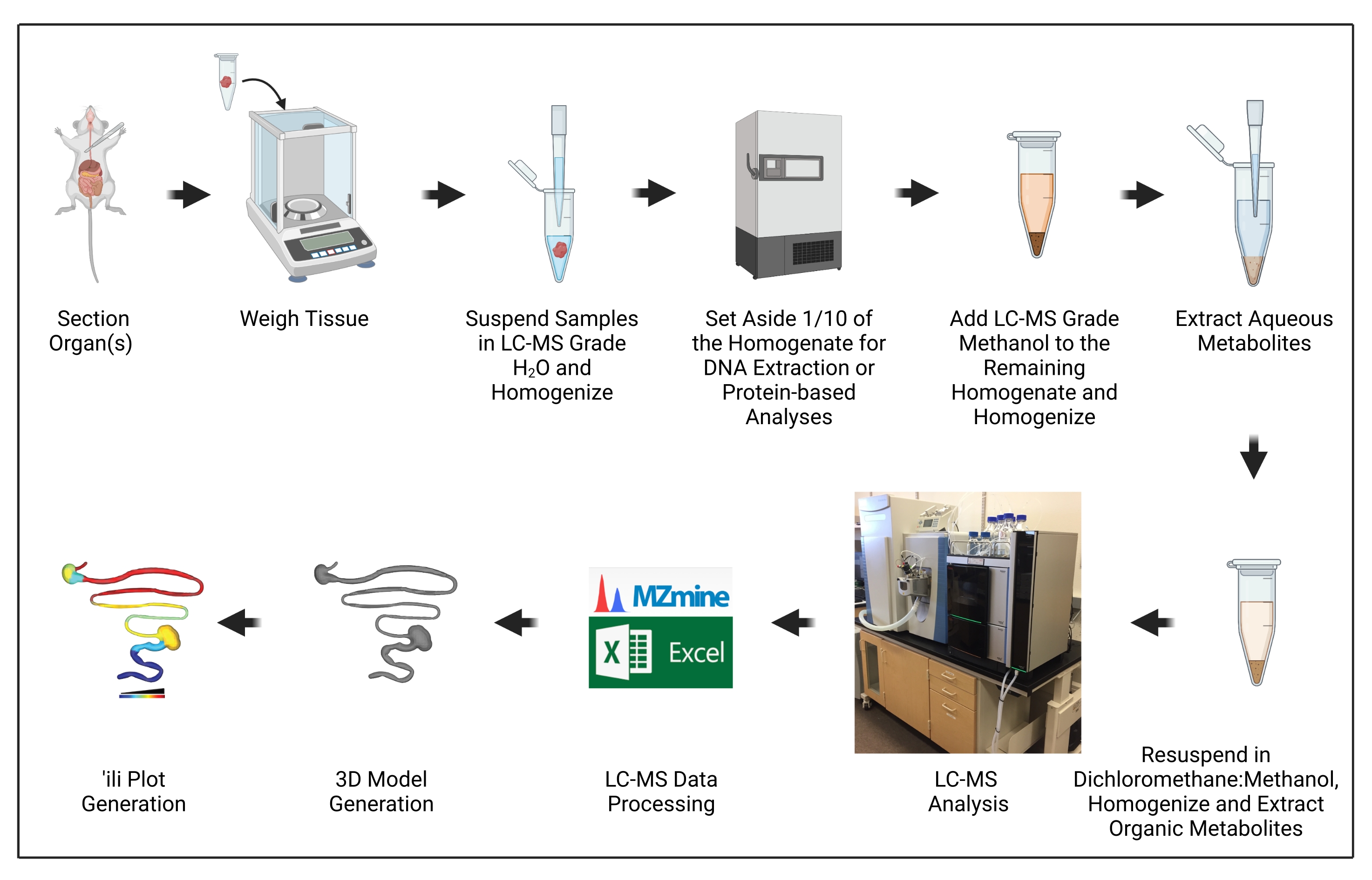
This protocol describes the steps to generate a 3D model of metabolite distribution during trypanosomatid infection, including sample collection, metabolite extraction, an overview of liquid chromatography-tandem mass spectrometry data acquisition, 3D model generation, and finally, data visualization.
Abstract
Pathogen tropism and disease tropism refer to the tissue locations selectively colonized or damaged by pathogens, leading to localized disease symptoms. Human-infective trypanosomatid parasites include Trypanosoma cruzi, the causative agent of Chagas disease; Trypanosoma brucei, the causative agent of sleeping sickness; and Leishmania species, causative agents of leishmaniasis. Jointly, they affect 20 million people across the globe. These parasites show specific tropism: heart, esophagus, colon for T. cruzi, adipose tissue, pancreas, skin, circulatory system and central nervous system for T. brucei, skin for dermotropic Leishmania strains, and liver, spleen, and bone marrow for viscerotropic Leishmania strains. A spatial perspective is therefore essential to understand trypanosomatid disease pathogenesis. Chemical cartography generates 3D visualizations of small molecule abundance generated via liquid chromatography-mass spectrometry, in comparison to microbiological and immunological parameters. This protocol demonstrates how chemical cartography can be applied to study pathogenic processes during trypanosomatid infection, beginning from systematic tissue sampling and metabolite extraction, followed by liquid chromatography-tandem mass spectrometry data acquisition, and concluding with the generation of 3D maps of metabolite distribution. This method can be used for multiple research questions, such as nutrient requirements for tissue colonization by T. cruzi, T. brucei, or Leishmania, immunometabolism at sites of infection, and the relationship between local tissue metabolic perturbation and clinical disease symptoms, leading to comprehensive insight into trypanosomatid disease pathogenesis.
Introduction
Trypanosomatid parasites consist of Leishmania species, African trypanosomes (Trypanosoma brucei), and American trypanosomes (Trypanosoma cruzi). Leishmania protozoa cause leishmaniasis, which includes self-healing and self-limited localized cutaneous leishmaniasis, mucocutaneous leishmaniasis in which the mucosal tissues of the mouth, nose, and throat become damaged, and visceral leishmaniasis with parasite tropism to the visceral organs causing fever and hepatosplenomegaly1,2. T. brucei causes Human African trypanosomiasis (HAT), also known as sleeping sickness, mainly reported in African countries3. The clinical signs and symptoms include hepatosplenomegaly, fever, headache, musculoskeletal pains, lymphadenopathies, and anemia in the hemo-lymphatic stage when parasites localize to the bloodstream and lymphatics. This is followed by the meningo-encephalitic stage, where parasites localize to the central nervous system and cause sleep disturbance, behavioral alteration, and eventually fatal comas4. T. cruzi causes Chagas disease, endemic in the Americas. Infected individuals experience an initial acute stage, usually asymptomatic, with broad parasite tropism. About 10%-30% of infected individuals experience chronic stage symptoms after decades of infection, characterized by megaoesophagus, megacolon, and cardiovascular complications5,6.
Metabolomics studies small molecular species (50-1,500 Da), including biological compounds from primary or secondary metabolism and externally-derived compounds such as drugs or food-derived molecules. In the context of host-pathogen interactions, metabolomics can explore the impact of infection on host metabolite environments, crucial in accessing the effect of the pathogen on the host. It can also assess pathogen adaptations to the host nutritional and immunological environment7,8,9. Mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy are common metabolomics tools used to identify, quantify, and characterize metabolites. This "omics" approach can also be applied to biomarker discovery and drug development10,11.
Given the specific tissue tropism of trypanosomatid parasites, spatial metabolomics analyses can enable significant insight into the pathogenesis of the diseases they cause. Mapping the spatial distribution of metabolites revealed metabolites locally affected by chronic Trypanosoma cruzi infection in mouse heart tissue and acute and long-term Trypanosoma cruzi infection in the mouse gastrointestinal tract6,12,13. Specifically, 3D chemical cartography demonstrated a disconnect between parasite persistence and metabolic alterations in the heart tissue of chronically Trypanosoma cruzi-infected mice. Metabolism was most perturbed in lower and apical segments of the heart, matching with sites of Chagas disease symptoms (cardiac apical aneurysms). Metabolite families perturbed by infection at specific cardiac sites and correlated to disease severity include acylcarnitines and glycerophosphocholines12,13,14. In the gastrointestinal tract, persistent metabolic alterations concurred with sites of Chagas disease symptoms: esophagus and colon. In contrast, metabolism is re-normalized at sites not associated with Chagas disease symptoms, such as the small intestine. Metabolites locally perturbed by infection in the gastrointestinal tract include acylcarnitines, glycerophosphocholines, kynurenine, tryptophan, and cholic acid. In addition, these analyses enabled the identification of a new metabolic mechanism of tolerance to Chagas disease6. Applying these methods to the study of cutaneous leishmaniasis revealed significant metabolic perturbations at the site of the lesion, but also specific metabolic changes in lesion-adjacent, macroscopically healthy tissue. For example, glutamine was depleted at the lesion site, whereas glycerophosphocholines in the m/z (mass to charge ratio) 200-299, 400-499, 500-599, and 600-699 were significantly increased at the lesion site. PC (O-34:1) was only increased at lesion-adjacent sites15.
The goal of this manuscript is to demonstrate the steps necessary to generate 3D models of metabolite distribution ("chemical cartography") as applied to trypanosomatid parasite infection models (Figure 1). This approach builds on several critical advances in the context of metabolomics and metabolomics data processing, particularly the development of 'ili software to plot metabolomics data onto 3D models easily16.
Protocol
All animal experiments described were approved by the University of Oklahoma or the University of California San Diego Institutional Animal Care and Use Committee. All steps handling infectious material were performed inside a biosafety cabinet (class II, type A2) and according to local regulations.
1. Tissue collection
- Infect appropriate trypanosomatid infection animal models, generally mice or hamsters.
NOTE: There is a considerable variety of mouse models for trypanosomatid infection, depending on the desired symptoms to be produced, speed of disease progression, disease severity, etc. The users can choose at their convenience.- For cutaneous leishmaniasis infection models, infect subcutaneously in the footpad or intradermally in the ear15.
- For visceral leishmaniasis infection models, infect intravenously1,17.
- For Chagas disease and sleeping sickness infection models, infect intraperitoneally6,12,18,19. Determine the parasite dose to use based on the parasite strain and planned time points.
- Plan sectioning positions.
- Plan to generate sections with a minimum of 10 mg of tissue per section. It is best to use 30-50 mg.
- For Chagas disease infection models, plan to collect cardiac and gastrointestinal segments systematically.
- For cutaneous leishmaniasis infection models, collect lesional tissue and lesion-adjacent samples.
- For visceral leishmaniasis infection models, plan to collect spleen and multiple liver lobes. Additional tissue sites such as adipose tissue may also be of interest to collect.
CAUTION: Samples from infected animals must be handled under the appropriate, institutionally-approved biosafety protocol. This will generally involve personal protective equipment (PPE) requirements and only opening tubes and collecting samples inside a biosafety cabinet (class II, type A2).
- Label and weigh tubes for homogenization.
- Use the tube type appropriate for the available homogenization system. For a TissueLyser, use 2 mL microcentrifuge tubes.
- Euthanize mice at the desired infection timepoints using isoflurane overdose as approved by institutional IACUC or according to IACUC-approved protocol.
- Section tissue systematically as planned, with one section per tube.
- Remember to wash sample collection equipment between samples with extraction solvent (50% methanol in this protocol).
- Keeping tube lid open, immediately snap freeze samples in liquid nitrogen.
CAUTION: Do not close tubes until any liquid nitrogen entered the tube has wholly evaporated to prevent tubes from exploding as nitrogen expands. Take adequate steps to ensure that the skin does not come in contact with liquid nitrogen (use cryo gloves and forceps to hold the tubes). Wear a safety face shield. - Store the tubes on dry ice until all the desired samples have been collected.
- Pause point: store samples at -80 °C until ready to do extractions.
- Weigh tubes to determine tissue sample weight. Record in a spreadsheet.
- Keep the samples frozen during the weighing process: keep tubes on dry ice, rapidly weigh, put back on dry ice right away. Do not allow the samples to thaw while weighing.
2. Metabolite extraction
NOTE: Only LC-MS grade liquids and reagents must be used throughout. This method was adapted from Reference20.
- Prepare all extraction solvents (LC-MS grade H2O, LC-MS grade methanol, spiked with 4 µM of sulfachlorpyridazine, LC-MS grade dichloromethane: methanol spiked with 2 µM of sulfachlorpyridazine) in 1 L glass bottles, using dedicated glassware.
NOTE: Volume to be prepared should be calculated based on sample weights, considering 500 µL of water per 50 mg of sample, 500 µL of methanol spiked with 4 µM of sulfachlorpyridazine per 50 mg of sample and 1,000 µL of prechilled dichloromethane: methanol spiked with 2 µM of sulfachlorpyridazine per 50 mg of sample, increasing the calculated volume by 10% to allow for pipetting inaccuracy. Store extraction solvent at 4 °C at least overnight to pre-chill. - Perform water-based homogenization of the tissue samples as per the steps mentioned below.
- Add one 5 mm stainless steel bead (see Table of Materials) to each of the 2 mL microcentrifuge tubes containing tissue samples, using a bead dispenser. Keep the tubes on ice.
- Make one blank tube containing LC-MS grade H2O that will go through all the steps and serve as an extraction blank. Use the average H2O volume from sample extractions. Add chilled LC-MS grade H2O to the frozen tissue samples.
- Normalize water volume to tissue weight by adding 500 µL of water / 50 mg of sample, using the sample weights calculated at step 1.7.
CAUTION: If handling biohazardous samples, continue to follow the appropriate, institutionally-approved biosafety protocol. This will generally involve requirements for personal protective equipment (PPE) and opening tubes only inside the biosafety cabinet (class II, type A2).
- Normalize water volume to tissue weight by adding 500 µL of water / 50 mg of sample, using the sample weights calculated at step 1.7.
- Homogenize samples at 25 Hz speed for 3 min using a tissue homogenizer (see Table of Materials).
- Close tubes tightly to avoid spilling the reagents during processing.
- Collect about 1/10th of the homogenization volume for DNA extraction, qPCR, protein-based analyses, or other analyses (if desired). Store in a microcentrifuge tube or 96-well plate (depending on the volume collected) for up to 6 months at -80 °C, if DNA extraction experiments are to be performed on a different day. Longer storage durations may be possible but have not been tested.
- Perform DNA extractions using any standard commercial kit for mammalian DNA extraction (see Table of Materials) from tissues as described in Reference13. Quantify DNA yield and store the extracted DNA at -20 °C. Acceptable DNA quantity and quality for qPCR has been observed even up to 3 years later.
NOTE: This step can be performed on frozen homogenate on a subsequent day from metabolite extraction. - Perform qPCR as described in Reference13, using 180 ng of extracted DNA.
NOTE: This can be performed on DNA collected on a previous day and frozen at -20 °C.- In the case of studies on T. cruzi infection, use the following primers: ASTCGGCTGATCGTTTTCGA and AATTCCTCCAAGCAGCGGATA to quantify parasite levels21 and the following primers to normalize to host DNA levels: TCCCTCTCATCAGTTCTATGGCCCA and CAGCAAGCATCTATGCACTTAGACCCC22 (see Table of Materials).
NOTE: The recommended qPCR cycles are as follows: denature at 95 °C for 10 min; perform 40 cycles at 95 °C for 30 s, and then 58 °C for 60 s, and finally 72 °C for 60 s. Perform melting curve analysis as appropriate for the available thermocycler. Process data using the ΔΔCt method23 to obtain relative parasite load between sampling sites. Absolute quantification can be obtained by comparing sample-derived ΔΔCt values to a standard curve generated from known amounts of parasites, spiked into uninfected tissue samples, and extracted as in steps 2.2 to 2.2.4.1.
- In the case of studies on T. cruzi infection, use the following primers: ASTCGGCTGATCGTTTTCGA and AATTCCTCCAAGCAGCGGATA to quantify parasite levels21 and the following primers to normalize to host DNA levels: TCCCTCTCATCAGTTCTATGGCCCA and CAGCAAGCATCTATGCACTTAGACCCC22 (see Table of Materials).
- Perform protein-based characterization of immune responses using multiplexed cytokine kits or standard commercial ELISA kits (see Table of Materials) as described in Reference13 on the stored homogenate.
- Perform DNA extractions using any standard commercial kit for mammalian DNA extraction (see Table of Materials) from tissues as described in Reference13. Quantify DNA yield and store the extracted DNA at -20 °C. Acceptable DNA quantity and quality for qPCR has been observed even up to 3 years later.
- Save at least half of the 500 µL of the homogenization volume for metabolite extraction.
- Perform aqueous metabolite extraction.
NOTE: Solvent selection can be adapted based on the chemical properties of metabolites of interest.- Add ice-cold LC-MS grade methanol spiked with 4 µM of sulfachlorpyridazine to the homogenate to achieve a final concentration of 50% methanol with 2 µM of sulfachlorpyridazine in water.
CAUTION: Methanol is flammable and hazardous. Use appropriate safety procedures, including handling inside a fume hood or biosafety cabinet. - Homogenize samples in a tissue homogenizer at 25 Hz speed for 3 min. Centrifuge samples at 16,000 x g at 4 °C for 10 min.
- Collect an equal volume of supernatant into a 96-well-plate. Select the volume to match the smallest volume of methanol + water combined across all samples. This is the aqueous fraction.
- Set aside any remaining aqueous homogenate supernatant at -80 °C as a backup.
- Keep the solid residue on ice while collecting the supernatants.
- Dry aqueous extraction supernatant until dry (~3 h or overnight). Use maximum speed and no heating.
- Freeze the dried 96-well-plate at -80 °C.
- Add ice-cold LC-MS grade methanol spiked with 4 µM of sulfachlorpyridazine to the homogenate to achieve a final concentration of 50% methanol with 2 µM of sulfachlorpyridazine in water.
- Perform organic metabolite extraction.
NOTE: Solvent selection can be adapted based on the chemical properties of metabolites of interest.- Add 1,000 µL per 50 mg of the sample of prechilled dichloromethane: methanol spiked with 2 µM of sulfachlorpyridazine to the solid residue from step 2.3.4.
CAUTION: Use appropriate safety procedures when handling solvents, including handling inside a fume hood with a good flow rate. - Homogenize samples in a tissue homogenizer at 25 Hz speed for 5 min. Centrifuge the samples at 16,000 x g at 4 °C for 10 min.
- Collect an equal volume of supernatant into a 96-well-plate. Select the volume to match the smallest volume of dichloromethane: methanol, across all samples. This is the organic fraction.
- Store the pellet at -80 °C as a backup. Store the remaining organic extract at -80 °C as a backup. Air-dry the organic extract in a fume hood overnight.
- Freeze the dried 96-well-plate at -80 °C.
- Add 1,000 µL per 50 mg of the sample of prechilled dichloromethane: methanol spiked with 2 µM of sulfachlorpyridazine to the solid residue from step 2.3.4.
3. LC-MS data acquisition
- Resuspend aqueous and organic extracts into 60 µL each of 50% methanol + 2 µM of sulfadimethoxine, and combine. Sonicate for 10 min; then, centrifuge for 10 min and transfer the supernatant to a clean 96-wellplate. Seal with zone-free plate seal and place the plate in an LC autosampler.
CAUTION: Use appropriate safety procedures when handling solvents, including handling inside a fume hood with a good flow rate. - Connect appropriate mobile phases to the LC system (see Table of Materials).
NOTE: For positive mode reversed-phase LC, authors recommend LC-MS-grade H2O + 0.1% formic acid as mobile phase A and LC-MS-grade acetonitrile + 0.1% formic acid as mobile phase B with a flow rate of 0.5 mL/min and a 7.5 min LC gradient. Recommended gradient steps are as published in Reference6: 0-1 min, 2% B; 1-2.5 min, linear increase to 98% B; 2.5-4.5 min, hold at 98% B; 4.5-5.5 min, linear decrease to 2% B; 5.5-7.5 min, hold at 2% B. - Ensure that the instrument is clean. Calibrate MS in both positive and negative mode.
- Perform MS performance evaluation as appropriate for the instrument.
- Create MS run sequence.
- Start with 2 blanks, 2 standards (6-mixes), and 5 pooled quality controls (QC) in a dilution series, beginning at 2 µL of injection volume and increasing stepwise to 30 µL injection volume.
- Randomize the sample order.
- After every 12 samples, run a blank, and then a pooled QC.
- Connect C8 LC column (1.7 µm particle size, 100 Å pore size, 50 x 2.1 mm length x internal diameter) and monitor for leaks and excessive backpressure. Fix issues as per instrument standard operating procedure.
- Start MS run sequence and collect data-dependent LC-MS/MS data.
NOTE: For a Q-Exactive Plus MS instrument, use Heated Electrospray Ionization and data-dependent MS2 acquisition (top 5) in positive mode, a resolution of 70,000 for MS1 and 17,500 for MS2, AGC Target of 1E6 for MS1 and 2E5 for MS2, maximum IT of 100 ms for both MS1 and MS2, scan range to 100-1500 m/z for MS1, and MS2 isolation window of 1 m/z. Set sheath gas to 35, aux gas to 10 and sweep gas to 0, spray voltage to 3.8 kV, capillary temperature to 320 °C, S-lens RF level to 50, and aux gas temperature to 350 °C.- Verify the data quality: check initial blanks (confirm lack of major peaks), standards (confirm the presence of expected peaks and symmetric peak shape), and QCs (confirm the presence of expected peaks, peak shape, and expected peak intensity).
- Periodically monitor MS run during the run sequence.
- Once the run is finished, check the data for any missed injections or other errors.
- Store the LC column as recommended by the manufacturer. Remove and store samples at -80 °C.
- Upload raw data to the data repository.
NOTE: MassIVE (massive.ucsd.edu) is recommended to enable the downstream link to molecular networking for metabolite annotations24,25.
4. LC-MS data processing
- Convert raw files to open format (.mzXML or .mzML) using MSConvert26.
- Upload raw data and mzXML or mzML data to the data repository.
- Generate feature table. There are multiple tools to do so (MZmine, MS-DIAL, openMS, XCMS, etc.27,28,29,30).
NOTE: MZmine is recommended because it is free, open-source, and can directly import mzXML files after MSconvert, has graphical user interface options for processing data and monitoring the impact of parameter selection, and can directly export to GNPS for molecular networking24.
NOTE: Follow tool documentation and use parameters appropriate for the available instrument. Additional details can also be found in Reference31.- Export feature table in .csv format.
5. 3D model generation
- Hand-draw 3D model de novo to scale as per the steps mentioned below.
- Take a picture of the organ of interest.
- Perform the following in SketchUp software (see Table of Materials) as mentioned below.
- Delete the default picture of a man that appears when the software opens.
- Click on File > Import to import a picture of the organs of interest.
- Click on the Lines tool and select the Freehand option. Use the pencil tool to trace and draw the outlines of the organs of interest. Ensure to close the line by drawing all the way back to the starting point. Once the line has been successfully closed, the drawn area will automatically appear shaded.
- Select the Push/Pull tool and pull up on the shaded area to convert the drawing from 2D to 3D.
- Delete the organ picture: select the Eraser button, and then right-click on the picture and select Erase.
- Export the file in .dae format: File > Export > 3D Model.
- Improve the realism of the model as per the steps mentioned below.
- Import the model into MeshLab software (see Table of Materials): open MeshLab and select File > Import Mesh. Select the .dae model generated at the previous step. A Pre-Open Options menu will pop up. Select OK.
- Select Wireframe on the top menu. Then, select: Filters > Remeshing, Simplification and Reconstruction > Subdivision surfaces: Midpoint. Leave all values as default and select Apply twice. Visually inspect the wireframe view of the model to ensure it is finely gridded. Close the pop-up menu.
- Export the model in .stl format: File > Export Mesh As, and then select STL File Format (*.stl) in the Files of Type dropdown menu. Click on Save. Select OK in the next pop-up menu.
- Open the Meshmixer software (see Table of Materials). Click on the Import (+) button. Select the .stl file generated at the previous step. Use the Sculpt > Brushes > Drag and Sculpt > Brushes > Inflate tools to pull out the model's surfaces that need to be rounded out.
- Once the model has the desired appearance, save it in .stl format: File > Export. Name the file as desired and select STL Binary Format (*.stl) in the Save as type dropdown menu. Click on Save. If a pop-up menu appears, click on Continue.
6. 'ili plot generation
- Obtain coordinates for the positions in the 3D model that correspond to the sampling sites.
- Open the 3D model from step 5.1.3.5 in the MeshLab software: File > Import Mesh. Select the model generated in step 5.1.3.5. Click on OK in the Post-Open Processing pop-up window.
- To obtain x, y, and z coordinates for each sampling spot: select the PickPoints tool, and then right-click at regularly spaced intervals across the 3D model surface. Once all the desired coordinates have been selected, click on the top-most Save button in the Form pop-up window.
NOTE: This will export the coordinates in .pp file format. This file can be opened in spreadsheet software. - In spreadsheet software: Open the .pp file generated in step 6.1.2. Adjust data display using Data > Text to Columns > Delimited. Click on Next, and then select Space, and click on Finish.
- Reformat so that only numerical values remain in the spreadsheet cells by selecting Home > Find & Select > Replace. In the Find what box, enter: y=". Leave the Replace with box empty. Click on Replace All, and then on OK. Repeat for x=" and for z=" and for " />. Values are now ready for step 6.2.
- Make the 'ili feature table. This method was adapted from Reference16.
- In a spreadsheet software, build the feature table. Rows correspond to each position and columns to data.
NOTE: The first columns must be the position name (sample name), followed by x, y, and z coordinates of the sampling spots obtained at step 6.1.2 (with column headers x, y, z). The fifth column must be titled "radius". - Paste the appropriate metadata and metabolite feature abundance in the subsequent spreadsheet columns.
- In column "radius", enter the desired size of the sampling spots to be visualized on the model. Determine the values for radius empirically: enter 1 as default, and then assess whether radius or coordinates need to be adjusted in step 6.3. Save the file in .csv format (File > Save As), and then select CSV (Comma delimited) (*.csv) in the dropdown menu. Name the file as desired. Click on Save.
- In a spreadsheet software, build the feature table. Rows correspond to each position and columns to data.
- Open the data in 'ili (software developed by16).
- Open the 'ili website (ili.embl.de). Select Surface. Drag and drop the created 3D model into the browser window. Drag and drop the created feature table into the same browser window.
- Use the legend at the bottom-right corner to project the desired data column on the 3D model. Ensure that the spots and radii selected in step 6.2.1 match the sampling sites. If necessary, adjust values in the feature table, or choose additional coordinates in the MeshLab (see step 6.1.2).
NOTE: Visualization can also be improved by selecting the Spots > Border Opacity and setting the slider to its maximal value. - Consecutively select each data column to assess the distribution of this metabolite feature on the 3D model.
NOTE: This approach can also be used to visualize only specific metabolite features of interest, for example, those with p < 0.05 or a certain fold change between infected and uninfected samples, as determined in external statistical analysis tools. Features to visualize may also be selected outside of 'ili through machine learning approaches such as a random forest.
- Perform linear/log data visualization as per the steps mentioned below.
- Using the Mapping tab at the top right of the page, select Linear or Logarithmic in the Scale drop-down menu.
- Set the same scale for all plots if visualizing multiple features.
NOTE: The website automatically chooses the minimum and maximum for each data column to display. - To set the scale manually, deselect the Auto Min/Max option and enter the desired scaling. All data will now be displayed within the same scale.
- Change the color scale of the data (grayscale, blue-white-red, etc.).
- Change the color scale to the desired color scheme in the Mapping menu using the Color Map drop-down menu. Ensure that the selected color scheme is color-blind-friendly.
- To change the color of the 3D model or the background color, use Color and Background options under the 3D menu.
- Hide 3D axes by unselecting the Show the Origin box under the 3D menu.
- Save the image by screenshotting, using the snipping tool, or the shortcut key Crtl + S for Windows or Linux and
 + S on OS X.
+ S on OS X.
Representative Results
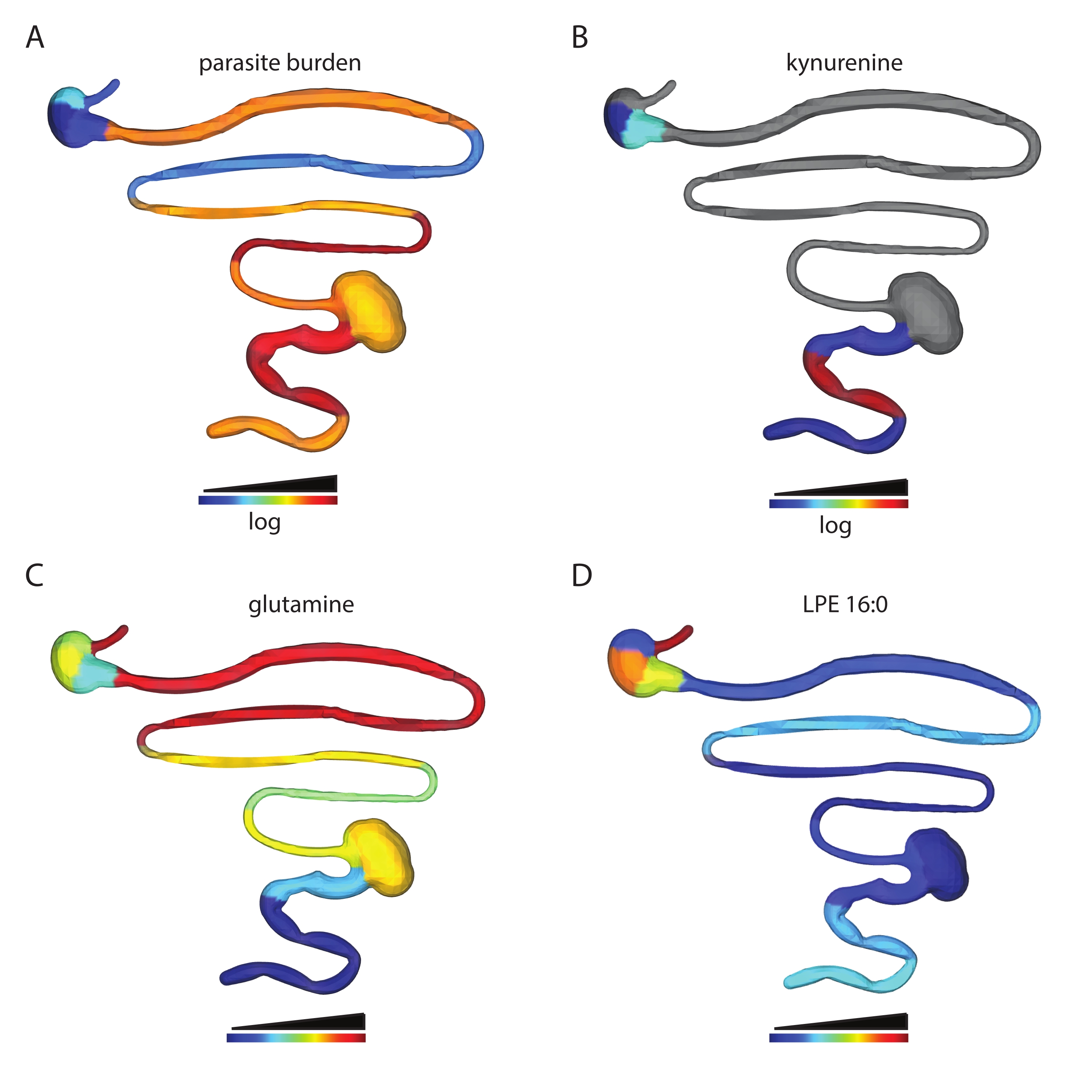
The number of metabolite features obtained depends on the tissue type analyzed and data processing parameters. For example, this protocol has been used to analyze the spatial impact of T. cruzi infection on the gastrointestinal tract metabolome in a mouse model of T. cruzi infection. In prior work, male C3H/HeJ were injected intraperitoneally with 1,000 CL + luc T. cruzi parasites32,6. Animals were euthanized 12 or 89 days post-infection, and a chemical cartography analysis of 13 contiguous segments of the gastrointestinal tract was performed as described in this protocol. This analysis led to a feature table of 5,502 features, which were then visualized into 3D using the steps described in this protocol. This approach enables the visualization of metabolite features in individual animals that are high at the site of high parasite load (kynurenine, Figure 2B vs. parasite load, Figure 2A), of metabolites with differential distribution across tissue regions (glutamine, Figure 2C) and metabolite features that are found at comparable levels across small and large intestines (LPE 16:0 Figure 2D). Kynurenine was selected for visualization because of its known relationship to inflammation and prior publications on the ability of kynurenine-derived metabolites to regulate T. cruzi load33. Random forest-based machine learning models had previously revealed an association between kynurenine levels and infection status6. Glutamine was selected for a visualization based on previous publications demonstrating a relationship between in vitro glutamine availability and T. cruzi drug sensitivity34. Differential distribution was confirmed using logistic regression, p < 0.05. LPE 16:0 was selected after visual inspection of the data to discover metabolite features found at comparable levels across tissue sites.

Figure 1: Protocol overview. The illustration was created with BioRender.com. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Chemical cartography analysis. Male C3H/HeJ mice were injected intraperitoneally with 1,000 CL+luc T. cruzi parasites32. Animals were euthanized 12 or 89 days post-infection, and the gastrointestinal tract was collected and sectioned systematically (step 1)6. Metabolites were extracted as in step 2 and analyzed by LC-MS/MS. 3D model generation was performed using the SketchUp software (step 5), and data were plotted in 3D as in step 6. (A) Parasite distribution in a specific mouse, 12 days post-infection. (B) Kynurenine metabolite distribution in the same mouse, 12 days post-infection. (C) Mean glutamine distribution across infected mice, 89 days post-infection. (D) Comparable levels of m/z 454.292 retention time 2.929 min, annotated as 2-hexadecanoyl-sn-glycero-3-phosphoethanolamine (LPE 16:0), in the same mouse as in A and B in the small intestine and colon. Samples and data were generated in6. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Understanding trypanosomatid infections is essential to guide novel drug development and treatment approaches. This chemical cartography method is uniquely poised to provide actionable insights into the relationship between metabolism and trypanosomatid disease pathogenesis, thus addressing this translational need.
Only LC-MS grade solvents are recommended during metabolite extraction and MS analyses, to lessen background contamination. Polymeric contamination35, commonly derived from paraffin film and/or other plastics36,37,38, must be avoided where possible. Parafilm, in particular, must never be used. These aspects are crucial since LC-MS data quality depends on the materials used during sample preparation and metabolite extraction. Data quality should be ensured before generating 'ili plots. In addition, generating these comprehensive spatial metabolomics maps requires the collection of all adjacent tissue samples and metabolite extraction from all collected samples to avoid gaps in these maps. Collection procedures, logistics of metabolite extraction and LC-MS analysis, and costs should thus be considered and planned accordingly.
This protocol can be modified to meet user needs in multiple ways. For example, the polarity and solubility of solvents used during metabolite extraction will influence what metabolites are detected39. To maximize the diversity of detected metabolite features for untargeted chemical cartography analyses, combining multiple extraction steps and solvents is recommended. For example, this method utilizes dichloromethane, methanol, and water as extraction solvents because they enable accurate detection of nonpolar and polar molecules20,40. However, these solvents are not universally suitable for every MS experiment, and researchers should select extraction solvents based on the goals of their project. Likewise, different LC-MS/MS conditions can be used, such as replacing reversed-phase chromatography with normal phase chromatography. Alternative columns can also be used for reversed-phase data collection instead of C8 chromatography, though empirically, C8 chromatography is more robust to tissue lipids and has a lower clogging frequency. Conceptually, these protocols can also be applied to other mass spectrometry methods such as gas chromatography-mass spectrometry, etc.
An alternative approach is mass spectrometry imaging. Indeed, unlike mass spectrometry imaging approaches, liquid chromatography-mass spectrometry does not inherently preserve spatial information10. Chemical cartography approaches bridge this gap by including sampling location at the time of project conceptualization, in the sample metadata, and at data processing steps. A strength of this chemical cartography approach, unlike mass spectrometry imaging, is the ability to provide confident annotations (Metabolomics Standards Initiative level 1 or level 2 annotation confidence41), unlike mass spectrometry imaging where the bulk of applications rely on accurate mass only for annotation. Mass spectrometry imaging will enable fine-grained spatial mapping, sometimes down to the single-cell level, e.g.,42,43. In contrast, chemical cartography approaches enable large-scale cross-organ mapping of metabolite distribution without requiring highly specialized whole-animal cryosectioning skills. Chemical cartography provides complementary evidence to the many spatial transcriptomic approaches being developed, e.g.,44, with the advantage of focusing on the 'omics layer closest to the phenotype'45. Alternative methods for parasite load quantification include measuring bioluminescence at the time of sample collection6. Fine segments could also be collected to enable confocal or electron microscopy to assess localized parasite burden and tissue damage. The water homogenate, which is used for cytokine quantification in this protocol and prior publications13, could also be used to quantify protein-based markers of tissue damage.
There are also multiple ways to obtain 3D models suitable to plot the resulting LC-MS data. In addition to the method suggested here, models can be purchased pre-made from various online vendors. Ensure that the terms of use match with the intended usage, especially concerning publication. Models for large organs can be generated de novo using 3D scanners according to scanner instructions. Alternatives such as MATLAB exist for generating and visualizing 3D models for chemical cartography46, but they were primarily implemented before the development of 'ili16. MATLAB is a data analysis and programming tool suite offering a wide variety of applications across many fields. However, MATLAB is neither free nor open-source, and it requires familiarity with MATLAB interfaces, especially considering MATLAB was not developed for processing mass spectrometry data. This proposed method's alternatives, namely, SketchUp, Meshlab, and 'ili, are freely accessible, user-friendly, and offer similar functions as MATLAB for chemical cartography purposes.
This method is robust concerning sample preparation and metabolite extraction. Troubleshooting is most often necessary at the LC-MS data acquisition step. This is beyond the scope of this article. Readers are directed to excellent publications on LC-MS data acquisition troubleshooting, including20,47. Likewise, the complexities of metabolite annotation are beyond the scope of this method's focus on 3D model generation. Useful references on this topic include24,25,48,49.
While this method effectively explores disease pathogenesis, there are limitations to this approach, some of which are common across any metabolomics experiment. One such limitation is the low annotation rate of LC-MS features50, which is contingent upon reference spectral libraries' availability and quality. A further limitation is that this protocol does not preserve mRNA due to the incompatibility of RNA preservation reagents such as RNAlater with LC-MS/MS analysis. However, the protein quality is adequate for downstream analyses and thus can replace mRNA-based analyses.
A chemical cartography approach to infection pathogenesis directly reflects how bacterial, viral, or parasitic infections develop in organ systems and cause localized disease. Analyzing these regional subsamples and generating 3D models ultimately conveys how metabolites function across three-dimensional space, shedding light on these previously unrecognized spatial dimensions of molecular biology. Using this protocol, for example, metabolite localization was compared to Trypanosoma cruzi parasite load. Results clarified the relationship between the pathogen and host tissue and also demonstrated the metabolic dynamics of Chagas disease symptom progression6. Chemical cartography methods have also been applied to various topics, such as human-built environment interaction51,52,53, the chemical makeup of organ systems like human skin46 and lungs54, and plant metabolism and environment interactions55. Future applications can involve assessing localized disease tolerance and resilience, or the relationship between local metabolite levels, pathogen tropism, and disease tropism in models beyond trypanosomatid infection. This approach should also have broad applicability to expand current pharmacokinetics protocols, to assess the relationship between local tissue drug levels and drug metabolism vs. overall metabolic context, tissue damage, and pathogen clearance. Overall, chemical cartography allows unique explorations of metabolite distributions in various sample types, with applications including disease pathogenesis, human health, human-environment interactions, and microbial dynamics.
Acknowledgements
Laura-Isobel McCall, PhD, holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. The authors further wish to acknowledge support from NIH award number R21AI148886, a pilot grant from the Oklahoma Center for Respiratory and Infectious Diseases (OCRID) under NIH award number P20GM103648, and start-up funds from the University of Oklahoma (to LIM). The content is solely the authors' responsibility and does not necessarily represent the official views of the funders. The authors also wish to thank the developers of the tools used in this protocol. All relevant publications have been cited, where applicable.
Materials
| Name | Company | Catalog Number | Comments |
| 1.5 mL Eppendorf tubes | NA | NA | any brand, as available |
| 1 L bottle, pyrex | NA | NA | any brand, as available |
| 1 L graduated cylinder, pyrex | NA | NA | any brand, as available |
| 2 mL SafeLock Eppendorf tubes | VWR | 20901-540 | use the appropriate tube model for the available tissue homogenizer |
| 3D model (de-novo generated according to protocol steps, or purchased) | NA | NA | as appropriate for system under investigation |
| 5 mm stainless steel bead | Qiagen | 69989 | |
| 96 well plate | Fisher | 3252449 | |
| AATTCCTCCAAGCAGCGGATA primer | NA | NA | any brand, as available; published in Piron, M. et al. Development of a real-time PCR assay for Trypanosoma cruzi detection in blood samples. Acta tropica. 103 (3), 195–200 (2007). |
| analytical balance | NA | NA | any brand, as available |
| ASTCGGCTGATCGTTTTCGA primer | NA | NA | any brand, as available; published in Piron, M. et al. Development of a real-time PCR assay for Trypanosoma cruzi detection in blood samples. Acta tropica. 103 (3), 195–200 (2007). |
| benchtop centrifuge with microcentrifuge, falcon tube and 96-well-plate capacity | NA | NA | any brand, as available |
| biosafety cabinet | NA | NA | class II, type A2; any brand, as available |
| CAGCAAGCATCTATGCACTTAG ACCCC primer | NA | NA | any brand, as available; published in Cummings, K.L., Tarleton, R.L. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Molecular and biochemical parasitology. 129 (1), 53–59 (2003). |
| camera | NA | NA | any brand, as available. A cellphone camera is adequate for this protocol |
| chemiluminescent-capable imaging system | NA | NA | any system, as available |
| cotton balls | NA | NA | any brand, as available |
| cryogloves | VWR | 97008-208 | replace with any brand, as available |
| dissection scissors | NA | NA | any brand, as available |
| dry ice | NA | NA | any brand, as available |
| extra-length forceps | NA | NA | any brand, as available |
| flammable-grade refrigerator | NA | NA | any brand, as available |
| freezer storage boxes for microcentrifuge tubes | NA | NA | any brand, as available |
| fume hood | NA | NA | any brand, as available |
| high-resolution mass spectrometer | NA | NA | any brand, as available, such as ThermoFisher Q-Exactive Plus (catalog number 0726030) |
| ice bucket | NA | NA | any brand, as available |
| ili software | ili.embl.de | NA | |
| isoflurane | Covetrus | 29405 | |
| large tupperware | NA | NA | any brand, as available; large enough to comfortably contain mouse, cotton ball |
| LC-MS grade acetonitrile | Fisher Optima | A955-4 | |
| LC-MS grade dicholoromethane | Fisher Optima | D151-4 | |
| LC-MS grade formic acid | Fisher Optima | A11750 | |
| LC-MS grade methanol | Fisher Optima | A456-4 | |
| LC-MS grade water | Fisher Optima | W64 | |
| liquid chromatography column | Phenomenex | 00B-4499-AN | may be changed to other brands and models as appropriate for the metabolites of interest |
| liquid chromatography column guard cartridge | Phenomenex | AJ0-8784 | may be changed to other brands and models as appropriate for the metabolites of interest |
| liquid chromatography column guard cartridge holder | Phenomenex | AJ0-9000 | may be changed to other brands and models as appropriate for the metabolites of interest |
| liquid nitrogen | NA | NA | any brand, as available |
| luciferin | Goldbio | LUCK-1G | |
| MeshLab software | https://www.meshlab.net/ | NA | |
| Meshmixer software | https://www.meshmixer.com/ | NA | |
| MS calibrant | NA | NA | appropriate one for available instrument |
| MS data processing software | NA | NA | multiple options available; authors recommend MZmine |
| MSConvert software | http://proteowizard.sourceforge.net/ | NA | |
| Nanodrop | ThermoFisher | ND-ONE-W | other nanodrop models are also suitable |
| p1000 pipet tips | NA | NA | use the appropriate brand to fit available pipettors |
| p1000 pipettor | NA | NA | any brand, as available |
| p20 pipette tips | NA | NA | use the appropriate brand to fit available pipettors |
| p20 pipettor | NA | NA | any brand, as available |
| p200 pipette tips | NA | NA | use the appropriate brand to fit available pipettors |
| p200 pipettor | NA | NA | any brand, as available |
| personal protective equipment (gloves, lab coat, safety glasses/goggles; faceshield) | NA | NA | any brand, as available |
| Q-Plex Mouse Cytokine - Screen (16-Plex) | Quansys biosciences | 110949MS | can replace with other protein-based cytokine assays such as other commercial cytokine ELISA kits |
| Quick-DNA Miniprep Plus Kit (200 preps) | Zymo | D4069 | replace with any brand of mammalian DNA extraction kit, as available |
| real-time thermocycler | NA | NA | any brand, as available |
| salt shaker or tea infuser | NA | NA | any brand; to contain isoflurane-soaked cotton ball and prevent contact with mouse skin |
| SketchUp software | https://www.sketchup.com/ | NA | |
| specimen forceps | NA | NA | any brand, as available |
| speedvac with microcentrifuge tube and 96-well-plate capacity | NA | NA | any brand, as available |
| spreadsheet software | https://www.microsoft.com/en-us/microsoft-365/excel | NA | can replace with other spreadsheet management software, as applicable |
| sulfachloropyridazine | Sigma | S9882-100G | |
| sulfadimethoxine | Sigma | S7007-10G | |
| Sybr green qPCR reaction mix | Fisher | A25780 | can replace with other Sybr green qPCR reaction mixes, as desired |
| TCCCTCTCATCAGTTCTAT GGCCCA primer | NA | NA | any brand, as available; published in Cummings, K.L., Tarleton, R.L. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Molecular and biochemical parasitology. 129 (1), 53–59 (2003). |
| tissue homogenizer | NA | NA | any brand, as available; for example, Qiagen TissueLyser II, catalog number 85300, with TissueLyser Adapter Set (2 x 24), catalog number 69982 |
| tissue samples | NA | NA | from appropriate infection model |
| TissueLyser single-bead dispenser | Qiagen | 69965 | |
| UHPLC | NA | NA | any brand, as available, such as ThermoFisher Vanquish (catalog number IQLAAAGABHFAPUMBHV) |
| ultra-low temperature freezer (-80) | NA | NA | any brand, as available |
| ultrasonic bath | NA | NA | any system, as available |
| wet ice | NA | NA | any brand, as available |
| zone-free sealing film | VWR | 490007-390 |
References
- McCall, L. -. I., et al. Adaptation of Leishmania donovani to and visceral environments: in vivo selection and proteomic analysis. Journal of Proteome Research. 14 (2), 1033-1059 (2015).
- McCall, L. -. I., Zhang, W. -. W., Matlashewski, G. Determinants for the development of visceral leishmaniasis disease. PLoS Pathogens. 9 (1), 1003053 (2013).
- de Castro Neto, A. L., da Silveira, J. F., Mortara, R. A. Comparative analysis of virulence mechanisms of trypanosomatids pathogenic to humans. Frontiers in Cellular and Infection Microbiology. 11, 669079 (2021).
- Franco, J. R., Simarro, P. P., Diarra, A., Jannin, J. G. Epidemiology of human African trypanosomiasis. Clinical Epidemiology. 6, 257-275 (2014).
- Ottilie, S., et al. Rapid chagas disease drug target discovery using directed evolution in drug-sensitive yeast. ACS Chemical Biology. 12 (2), 422-434 (2017).
- Hossain, E., et al. Mapping of host-parasite-microbiome interactions reveals metabolic determinants of tropism and tolerance in Chagas disease. Science Advances. 6 (30), (2020).
- Parab, A. R., McCall, L. -. I. Tryp-ing up metabolism: Role of metabolic adaptations in kinetoplastid disease pathogenesis. Infection and Immunity. 89 (4), (2021).
- Lewis, C. M., McCall, L. -. I., Sharp, R. R., Spicer, P. G. Ethical priority of the most actionable system of biomolecules: the metabolome. American Journal of Physical Anthropology. 171 (2), 177-181 (2020).
- Liu, Z., Ulrich vonBargen, R., McCall, L. I. Central role of metabolism in Trypanosoma cruzi tropism and Chagas disease pathogenesis. Current Opinion in Microbiology. 63, 204-209 (2021).
- Newsom, S. N., McCall, L. -. I. Metabolomics: Eavesdropping on silent conversations between hosts and their unwelcome guests. PLoS Pathogens. 14 (4), 1006926 (2018).
- Wishart, D. S., et al. HMDB: the Human Metabolome Database. Nucleic Acids Research. 35, 521-526 (2007).
- Dean, D. A., et al. Spatial metabolomics identifies localized chemical changes in heart tissue during chronic cardiac Chagas disease. PLoS Neglected Tropical Diseases. 15 (10), 0009819 (2021).
- McCall, L. -. I., et al. Mass spectrometry-based chemical cartography of a cardiac parasitic infection. Analytical Chemistry. 89 (19), 10414-10421 (2017).
- Hoffman, K., et al. Alterations to the cardiac metabolome induced by chronic infection relate to the degree of cardiac pathology. ACS Infectious Diseases. 7 (6), 1638-1649 (2021).
- Parab, A. R., et al. Dysregulation of glycerophosphocholines in the cutaneous lesion caused by Leishmania major in experimental murine models. Pathogens. 10 (5), 593 (2021).
- Protsyuk, I., et al. 3D molecular cartography using LC-MS facilitated by Optimus and 'ili software. Nature Protocols. 13 (1), 134-154 (2018).
- McCall, L. -. I., et al. Targeting ergosterol biosynthesis in Leishmania donovani: Essentiality of Sterol 14alpha-demethylase. PLOS Neglected Tropical Diseases. 9 (3), 0003588 (2015).
- Tear, W. F., et al. Selectivity and physicochemical optimization of repurposed Pyrazolo[1,5-]pyridazines for the treatment of human African trypanosomiasis. Journal of Medicinal Chemistry. 63 (2), 756-783 (2020).
- Klug, D. M., et al. Hit-to-lead optimization of benzoxazepinoindazoles as human African trypanosomiasis therapeutics. Journal of Medicinal Chemistry. 63 (5), 2527-2546 (2020).
- Want, E. J., et al. Global metabolic profiling of animal and human tissues via UPLC-MS. Nature Protocols. 8 (1), 17-32 (2013).
- Piron, M., et al. Development of a real-time PCR assay for Trypanosoma cruzi detection in blood samples. Acta Tropica. 103 (3), 195-200 (2007).
- Cummings, K. L., Tarleton, R. L. Rapid quantitation of Trypanosoma cruzi in host tissue by real-time PCR. Molecular and Biochemical Parasitology. 129 (1), 53-59 (2003).
- Livak, K. J., Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 25 (4), 402-408 (2001).
- Nothias, L. -. F., et al. Feature-based molecular networking in the GNPS analysis environment. Nature Methods. 17 (9), 905-908 (2020).
- Wang, M., et al. Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nature Biotechnology. 34 (8), 828-837 (2016).
- Chambers, M. C., et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 30 (10), 918-920 (2012).
- Tsugawa, H., et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nature Methods. 12 (6), 523-526 (2015).
- Sturm, M., et al. OpenMS - an open-source software framework for mass spectrometry. BMC Bioinformatics. 9, 163 (2008).
- Pluskal, T., Castillo, S., Villar-Briones, A., Oresic, M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 11, 395 (2010).
- Smith, C. A., Want, E. J., O'Maille, G., Abagyan, R., Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Analytical Chemistry. 78 (3), 779-787 (2006).
- . FBMN with MZmine Available from: https://ccms-ucsd.github.io/GNPSDocumentation/featurebasedmolecularnetworking-with-mzmine2/ (2021)
- Lewis, M. D., et al. Bioluminescence imaging of chronic Trypanosoma cruzi infections reveals tissue-specific parasite dynamics and heart disease in the absence of locally persistent infection. Cellular Microbiology. 16 (9), 1285-1300 (2014).
- Knubel, C. P., et al. 3-Hydroxy kynurenine treatment controls T. cruzi replication and the inflammatory pathology preventing the clinical symptoms of chronic Chagas disease. PloS One. 6 (10), 26550 (2011).
- Dumoulin, P. C., Vollrath, J., Tomko, S. S., Wang, J. X., Burleigh, B. Glutamine metabolism modulates azole susceptibility in amastigotes. eLife. 9, (2020).
- Keller, B. O., Sui, J., Young, A. B., Whittal, R. M. Interferences and contaminants encountered in modern mass spectrometry. Analytica Chimica Acta. 627 (1), 71-81 (2008).
- Weaver, R., Riley, R. J. Identification and reduction of ion suppression effects on pharmacokinetic parameters by polyethylene glycol 400. Rapid Communications in Mass Spectrometry: RCM. 20 (17), 2559-2564 (2006).
- Larger, P. J., Breda, M., Fraier, D., Hughes, H., James, C. A. Ion-suppression effects in liquid chromatography-tandem mass spectrometry due to a formulation agent, a case study in drug discovery bioanalysis. Journal of Pharmaceutical and Biomedical Analysis. 39 (1-2), 206-216 (2005).
- Furey, A., Moriarty, M., Bane, V., Kinsella, B., Lehane, M. Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta. 115, 104-122 (2013).
- Lu, W., et al. Metabolite measurement: Pitfalls to avoid and practices to follow. Annual Review of Biochemistry. 86, 277-304 (2017).
- Masson, P., Alves, A. C., Ebbels, T. M. D., Nicholson, J. K., Want, E. J. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Analytical Chemistry. 82 (18), 7779-7786 (2010).
- Sumner, L. W., et al. Proposed minimum reporting standards for chemical analysis. Metabolomics. 3 (3), 211-221 (2007).
- Rappez, L., et al. SpaceM reveals metabolic states of single cells. Nature Methods. 18 (7), 799-805 (2021).
- Yang, B., Tsui, T., Caprioli, R. M., Norris, J. L. Sample preparation and analysis of single cells using high performance MALDI FTICR mass spectrometry. Methods in Molecular Biology. 2064, 125-134 (2020).
- Longo, S. K., Guo, M. G., Ji, A. L., Khavari, P. A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nature Reviews. Genetics. 22 (10), 627-644 (2021).
- Patti, G. J., Yanes, O., Siuzdak, G. Innovation: Metabolomics: the apogee of the omics trilogy. Nature Reviews. Molecular Cell Biology. 13 (4), 263-269 (2012).
- Bouslimani, A., et al. Molecular cartography of the human skin surface in 3D. Proceedings of the National Academy of Sciences of the United States of America. 112 (17), 2120-2129 (2015).
- Dudzik, D., Barbas-Bernardos, C., García, A., Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. A review. Journal of Pharmaceutical and Biomedical Analysis. 147, 149-173 (2018).
- Alseekh, S., et al. Mass spectrometry-based metabolomics: a guide for annotation, quantification and best reporting practices. Nature Methods. 18 (7), 747-756 (2021).
- Chaleckis, R., Meister, I., Zhang, P., Wheelock, C. E. Challenges, progress and promises of metabolite annotation for LC-MS-based metabolomics. Current Opinion in Biotechnology. 55, 44-50 (2019).
- Viant, M. R., Kurland, I. J., Jones, M. R., Dunn, W. B. How close are we to complete annotation of metabolomes. Current Opinion in Chemical Biology. 36, 64-69 (2017).
- Kapono, C. A., et al. Creating a 3D microbial and chemical snapshot of a human habitat. Scientific Reports. 8 (1), 3669 (2018).
- Petras, D., et al. Mass spectrometry-based visualization of molecules associated with human habitats. Analytical Chemistry. 88 (22), 10775-10784 (2016).
- McCall, L. -. I., et al. Analysis of university workplace building surfaces reveals usage-specific chemical signatures. Building and Environment. 162, 106289 (2019).
- Garg, N., et al. Three-dimensional microbiome and metabolome cartography of a diseased human lung. Cell Host & Microbe. 22 (5), 705-716 (2017).
- Floros, D. J., et al. Mass spectrometry based molecular 3D-cartography of plant metabolites. Frontiers in Plant Science. 8, 429 (2017).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved