Visualizing Cytoskeleton-Dependent Trafficking of Lipid-Containing Organelles in Drosophila Embryos
In This Article
Summary
In the early Drosophila embryo, many organelles are motile. In principle, they can be imaged live via specific fluorescent probes, but the eggshell prevents direct application to the embryo. This protocol describes how to introduce such probes via microinjection, and then analyze bulk organelle motion via particle image velocimetry.
Abstract
Early Drosophila embryos are large cells containing a vast array of conventional and embryo-specific organelles. During the first three hours of embryogenesis, these organelles undergo dramatic movements powered by actin-based cytoplasmic streaming and motor-driven trafficking along microtubules. The development of a multitude of small, organelle-specific fluorescent probes (FPs) makes it possible to visualize a wide range of different lipid-containing structures in any genotype, allowing live imaging without requiring a genetically encoded fluorophore. This protocol shows how to inject vital dyes and molecular probes into Drosophila embryos to monitor the trafficking of specific organelles by live imaging. This approach is demonstrated by labeling lipid droplets (LDs) and following their bulk movement by particle image velocimetry (PIV). This protocol provides a strategy amenable to the study of other organelles, including lysosomes, mitochondria, yolk vesicles, and the ER, and for tracking the motion of individual LDs along microtubules. Using commercially available dyes brings the benefits of separation into the violet/blue and far-red regions of the spectrum. By multiplex co-labeling of organelles and/or cytoskeletal elements via microinjection, all the genetic resources in Drosophila are available for trafficking studies without the need to introduce fluorescently tagged proteins. Unlike genetically encoded fluorophores, which have low quantum yields and bleach easily, many of the available dyes allow for rapid and simultaneous capture of several channels with high photon yields.
Introduction
Vital dyes and molecular probes are powerful tools to image specific cellular structures and organelles live. In the Drosophila embryo, many different organelles display cytoskeleton-driven localization1,2,3,4, but the application of these small molecules is challenging because the eggshell is impermeable to many of them. This protocol describes a method to use fluorescent probes (FPs) in live embryos via microinjection in order to detect large-scale trafficking of organelles. The procedure covers preparation of the injection solution, egg collection and preparation of embryos, microinjection, imaging, and image analysis.
Dramatic spatial rearrangements of organelles are common in many animal oocytes, eggs, and embryos, in part because of the large size of these cells. In the Drosophila embryo, for example, lipid droplets (LDs) and yolk vesicles move toward the embryo center just before cellularization5. This motion depends on microtubules and leaves a ~40 µm region all around the periphery of the embryo depleted of the two organelles. During earlier cleavage stages, many organelles are transported by cytoplasmic flows that are driven by actin-myosin-based contractions at the embryo surface6. Although the embryos of many species exhibit similar rearrangements, the Drosophila embryo is particularly suited for following these processes by imaging because it develops externally at standard laboratory 'room temperature', is relatively transparent, small enough to fit on most microscope setups, and can be manipulated using powerful genetic tools.
For some organelles, fluorescently tagged proteins are available that specifically label these structures. For example, LSD-2 (also known as dPLIN2) is a protein that in embryos specifically targets LDs7. Fly lines are available that carry either inducible transgenes encoding a fusion between Green Fluorescent Protein (GFP) and LSD-28 or a gene trap in which yellow fluorescent protein (YFP) is inserted into the coding region of the endogenous LSD-2 gene9,10. However, this approach has limitations, including that these fusion proteins have low quantum yields and tend to bleach easily. In addition, labeling multiple different structures simultaneously can be challenging: for many organelles, only one type of fluorescent tag (often GFP or mCherry) is currently available, so imaging two organelles at the same time may require new transgenes or insertions; also, even if compatible tags are available, introducing them into a single strain can require time-consuming crosses. It also makes using the many powerful genetic resources less convenient, e.g., if two organelle markers, a Gal4 driver, and an inducible RNAi construct all have to be present in the same mother.
In principle, these limitations can be overcome with the use of FPs, including vital dyes (e.g., LysoTracker to mark lysosomes), molecular probes (e.g., SiR-tubulin to label microtubules), and fluorescently labeled biological molecules (e.g., C12 BODIPY to probe fatty acid metabolism11). From use in cultured cells, they are typically well-validated as powerful tools for probing cellular biology. FPs are versatile, have superior photo properties, and are compatible with fluorescent proteins. Multiple dyes can be mixed and applied simultaneously, often with the benefits of separation into the violet/blue and far-red areas of the spectrum and small Stokes shifts, preventing channel bleed through. Small Stokes shifts allow for simultaneous capture of multiple imaging channels, enabling the tracking of several organelles at once. Finally, they can equally be applied to labeling organelles in the embryos of other Drosophila species or even other insects where no fluorescently tagged proteins may be available.
However, most of these FPs cannot traverse the elaborate eggshell of the Drosophila embryo. It consists of five layers: three outer chorionic layers (chorion) that prevent mechanical damage plus a waxy layer surrounding the vitelline membrane that creates a chemical barrier12. For simplicity, the combination of the waxy layer and the vitelline membrane will be referred to as the "vitelline membrane" below. To bypass the eggshell, this protocol adapts an established embryo microinjection approach to introduce FPs into the Drosophila embryo. The protocol describes how to monitor the cytoplasmic flow of LDs in cleavage stage embryos. It includes the preparation of injection needles and egg collection cages, the process of egg collection, and mechanical removal of the chorion. It goes over how to microinject and image the embryos and how to analyze the bulk flow of LDs using particle image velocimetry (PIV, adapted from6). It provides advice on troubleshooting to ensure embryo survival and create the best system for imaging. Also discussed is how the protocol can be modified to simultaneously image LDs and microtubules or to apply it to the study of other organelles, including lysosomes, mitochondria, yolk vesicles, and the endoplasmic reticulum (ER).
Protocol
1. Prepare necessary materials
NOTE: These preparations are best done days or weeks ahead of time.
- Prepare injection needles.
NOTE: Needles can be stored indefinitely in a covered container. Needles must be fine enough to deliver ~700 fL while being strong enough to pierce the vitelline membrane.- Place a capillary into the capillary holder on the needle puller. Secure the capillary with the wingnuts. Align the center of the capillary with the heating element so that two symmetric needles are generated.
- Choose appropriate settings on the needle puller according to the manufacturer's instructions.
- Perform quality control using a dissecting scope.
NOTE: Needle tips should be as fine as possible. Cracked, jagged, or large-bore needle tips are discarded. - Prepare batches of 10 needles (5 capillaries worth) at a time.
- Place a deformable putty, which has been rolled up into a cylinder, at the center of the bottom of a container with a lid. Carefully press the needles into the putty such that it holds them horizontally with the needle tips safely suspended in the air to prevent damage. Cover with the lid, and store until use.
- Prepare egg collection plates. Store at 4 °C.
NOTE: Egg collection plates are small agar plates supplemented with fruit juice to encourage egg-laying. How to prepare them is described in many protocols, including13. - Prepare heptane glue.
NOTE: This glue is extracted from double-sided tape and is used to securely attach the embryos to a coverslip. It stops the embryo from floating out of focus during injection and imaging. The glue can be prepared days or weeks ahead of time.- Ball up double-sided tape to a size bigger than a golf ball and place it at the bottom of a ~50 mL glass container with a tightly sealing lid. Pack tape tightly so enough adhesive will be present.
- In a fume hood, fill the glass container with heptane to cover all the tape. Keep the container tightly closed when not in use.
CAUTION: Heptane is flammable as a liquid and vapor. It can cause eye, skin, and respiratory tract irritation. Breathing vapors may cause drowsiness, dizziness, and lung damage. Keep heptane away from sources of ignition and store it in a well-ventilated area meant for flammables and away from incompatible substances. - Let the heptane glue sit overnight or longer. For best results, place the container on a shaker or another agitator overnight to help with dissolving the adhesive.
NOTE: The tape itself will remain, but the adhesive will be dissolved in the heptane. In step 5.1.2, the glue is applied to a coverslip; heptane evaporates, leaving the glue behind. - Test the heptane glue preparation by placing a drop of it on a glass slide and making sure a sticky residue remains once the heptane has evaporated. If adhesive residue is not visible afterward, add more tape to the glass container and repeat the previous step.
NOTE: Once prepared, the glue can be used for months or even years.
- Prepare a 1 mg/mL (3.8 mM) BODIPY493/503 stock solution by diluting commercially obtained preparation in pure anhydrous DMSO. Keep it covered to protect it from light and water. Store indefinitely at -20 °C or short term at 4 °C.
2. Prepare egg collection cages
NOTE: Do this at least a day (preferably 2 or 3 days) before the planned injection(s).
- Prepare yeast paste.
NOTE: Yeast paste supplements protein and other vital nutrients not provided in the apple juice plates. It promotes egg production and egg-laying.- Add 2-10 g of dry baker's yeast, and then 1 mL of tap water to a small beaker. Mix using a spatula.
- Keep adding tap water in 1 mL increments until the desired, tooth-paste-like consistency is reached.
- Cover the mixture and store it at 4 °C.
- Set up fly cages for egg collection.
NOTE: Male and female flies of the desired genotype are required: 10-50 females less than 2 weeks old and a similar number of males. A number of different options for fly cages are available, including homemade options14,13. It is important that the size of the apple juice plates is matched to the size of the fly cages.- Place a small smear of yeast paste on an apple juice plate. Keep it covered and let it come to room temperature because flies will not lay eggs on the plate if it is too cold.
- Transfer flies to an embryo collection cage, seal with the yeasted apple juice plate, and secure the plate to the embryo collection cage.
- Allow newly transferred flies to acclimate to the cage for 1-2 days, replacing the plate with a fresh one daily. If all the yeast has been eaten by the next day, increase the amount of yeast paste for future collections.
NOTE: This is an important step to increase egg yield as well-fed females lay more eggs.
3. On the day of injection, prepare the injection solution and load it into the needle
- Use the stock solution of BODIPY 493/503 (3.8 mM in DMSO) as the injection solution.
- Load a single needle with 1 µL of the injection solution using needle loading tips.
NOTE: Strive to get the liquid all the way to the tip without trapping air bubbles. Be ready to quickly replace the loaded needle in case of accidental breakage.- Attach a loading tip to a micropipette and draw 1 µL of the injection solution into the tip. Using gloves, hold the needle in one hand with its tip pointed away to minimize the risk of breakage.
- Carefully insert the loading tip into the needle and push it close to the needle tip. Dispense the liquid near the needle tip. After removing the pipette, hold the needle vertically with its tip down until the liquid flows to the tip.
- Store the loaded needles in a separate container with putty as above (see step 1.1.5).
NOTE: Needles should be prepared in advance (up to several hours) of injection but should not be used after more than a day. - Keep needles out of ambient light to prevent bleaching of the dyes. Cover the storage container with aluminum foil without damaging the needle tips. Make sure all light is obscured.
4. Collecting embryos for injection
NOTE: The timing of collection depends on what stage of embryos the injection needs to be performed at. With the timing scheme below, the embryos at the time of preparation for injection will be 0-90 min old, which corresponds to cleavage stages15.
- On the day of injection, prepare apple juice plates with yeast, as in step 2.2.1. If N rounds of injections are planned, prepare N + 2 plates. Keep these plates at room temperature.
NOTE: In addition to the N plates to collect embryos for injection, one plate is needed as a pre-collection plate and another one to feed the flies in the cage once collections are completed. - Replace the plate on the cage with a fresh yeasted plate (pre-collection plate) and leave it on the cage for 1-2 h.
- Replace the plate on the cage with a fresh plate (collection plate). Discard the pre-collection plate, as it will contain embryos older than desired. Then, allow the flies to lay eggs for 1.5 h.
NOTE: As well-fed flies typically lay their eggs shortly after the eggs have been fertilized, a 1.5 h collection time assures that at the end of the collection, most of the embryos on the plate are 0-90 min old, i.e., are in cleavage stages. Female flies that have not been fed fresh yeast since the previous day will tend to retain fertilized eggs for some indeterminate amount of time before laying. Hence, the pre-collection plate may have embryos that were fertilized before the start of collection and thus are older than 60 min, sometimes much older. - Replace the plate on the cage with a fresh yeasted plate. Cover the collection plate so that stray flies do not lay eggs on them.
5. Prepare embryos for microinjection
- Assemble the materials needed.
- Attach a piece of double-sided tape to a glass slide. Avoid touching the tape with fingers as that reduces its stickiness.
NOTE: This slide will be used for removing the chorion and not for imaging. - Using a transfer pipette or a micropipette (p200 or p1000), place a small drop (200 µL or less) of heptane glue roughly in the center of a rectangular coverslip (60 x 25 mm). Allow the heptane to evaporate (this takes less than a min).
NOTE: This coverslip will be used to mount the embryos for injection and imaging. The dimensions are chosen to fit into an adjustable metal holder on the confocal microscope. - Assemble a desiccation chamber, a sealed chamber (e.g., a Tupperware sandwich box) containing desiccation beads. Only use desiccation beads that have not hydrated.
NOTE: To ensure the embryos take up the injection solution, the embryo must be slightly desiccated to reduce internal pressure. This is done by placing the coverslip with the dechorionated, air-exposed embryos into the desiccation chamber.
- Attach a piece of double-sided tape to a glass slide. Avoid touching the tape with fingers as that reduces its stickiness.
- Mechanically remove the chorion.
- Cover the appropriately aged embryo plate with a thin layer of Halocarbon Oil 27 to turn the eggshell transparent.
NOTE: Embryos become translucent within tens of seconds15. If this step does not occur efficiently, the apple juice plates may be too wet and need to be dried off before use. - View the plate under a dissection scope with transillumination (i.e., the light going through the plate into the eyepieces) to confirm the stage of the embryos.
- Select embryos in cleavage stages.
NOTE: Cleavage stage embryos are entirely opaque; the subsequent blastoderm stages can be recognized by a band of transparent cytoplasm all around the opaque center15. A good introduction on how to recognize various embryonic stages is available on the website (given in reference16) maintained by the Society of Developmental Biology. - Using fine tweezers, grab an embryo of the desired stage by its dorsal appendages and transfer it onto the prepared glass slide covered with a piece of double-sided tape. Place the embryo on the tape. Minimize the transfer of oil.
- As gently as possible, roll the embryo across the surface of the tape by gently nudging the embryo with the side of the tip of the tweezers. Do not poke the embryo directly with the sharp tweezer tips.
NOTE: If the force applied is too low, the embryo will not roll. If it is too high, it will burst. Finding the appropriate intermediate force requires experience and, thus, this step should be practiced beforehand. - Continue rolling the embryo until the chorion transiently adheres to the tape and cracks.
- Once the chorion cracks, keep rolling to separate the embryo (still inside the vitelline membrane) from the chorion as the chorion remains stuck to the tape.
- Confirm that the chorion is completely removed by observing the loss of the dorsal appendages from the embryo.
- Roll the embryo back onto the chorion, which is less adhesive than the tape. Gently rub the embryo with the tweezers until it attaches to the tweezers for transfer.
- Transfer the embryo to the coverslip with the heptane glue and bring it in contact with the glue, which typically detaches it from the tweezers.
- Adjust the orientation of the embryo on the coverslip.
- Embed the lateral surface of the embryo into the glue.
NOTE: The surface of the embryo embedded into the glue is the one that will be imaged. Embedding the lateral surface is the simplest, but this can be adjusted depending on personal preference or the biological question to be answered. - Orient the long axis of the embryo perpendicular to the long axis of the coverslip.
- If the embryo does not lay in the preferred orientation, clean the tweezers to remove any oil that would detach the embryo from the glue. Then, gently attempt to roll the embryo into position.
- Embed the lateral surface of the embryo into the glue.
- Prepare the desired number of embryos for injection in the manner described above.
NOTE: As time passes after removal of the chorion, the ambient air will begin to desiccate the embryos and thus the effect of step 5.3 will be uneven for the embryos on the coverslip, which were dechorionated at different times. Typically, 1-3 embryos are the most manageable.
- Cover the appropriately aged embryo plate with a thin layer of Halocarbon Oil 27 to turn the eggshell transparent.
- Desiccate embryos.
- Place the coverslip with embryos into the prepared desiccation chamber and seal it.
- Allow the embryos to desiccate for 5-12 min.
NOTE: The timing of this step is dependent on the ambient temperature and humidity and thus needs to be determined empirically. - Remove the coverslip from the chamber and place a drop of Halocarbon Oil 700 onto it, fully covering the embryos, to prevent further desiccation.
- Inspect the embryos on a dissecting scope to judge proper desiccation.
- If the embryos are just slightly shriveled, but not deflated, proceed to microinjection (step 6).
- If the embryos are not sufficiently or overly desiccated, return to step 5.2 and prepare a new batch of embryos as the Halocarbon Oil 700 cannot be removed. If the embryos were overly desiccated, shorten desiccation time for the next batch of embryos by 3 min; if they were under-desiccated, increase desiccation time by 3 min.
6. Microinject embryos
NOTE: Ensure the microinjection setup includes an inverted microscope, a micromanipulator to hold and position the injection needle, and a commercial microinjector to deliver controlled volumes.
- Power on the microinjector and input the preferred settings.
NOTE: For this protocol, it is recommended to use linear motion output and the fast setting, but many others will work. The operator should determine personal preference. - Load the needle into the micromanipulator. To prevent the needle from being damaged while loading the coverslip containing the embryos, move it out of the way into a safe position.
- Place the coverslip onto the stage, with embryos on top, toward the needle. Then, carefully move the needle back into position for injection, with its tip still well above the stage.
- Put the 4x objective into the light path. Using the focus knob on the microscope, bring the embryos into focus.
NOTE: This will be the focal plane used for injection and will be adjusted only minimally going forward. - Using the stage controls, move the embryos horizontally into the center of the field of view.
- Pan away from the embryos, keeping them at the edge of the field of view, in preparation for lowering the needle. Stay in the same focal plane to ensure the needle is lowered to the correct position.
- Lower the needle tip into the correct focal plane and ensure that it is visible in the field of view together with the embryos.
- Using the micromanipulator controls and looking from above (not yet through the eyepieces), slowly drop the needle tip into the oil, aiming for the area where the objective points to. As the oil is quite viscous, go slowly to avoid damaging the needle.
- Once the needle is visible through the eyepieces, continue using the micromanipulator controls until the needle tip is in focus and at the center of the field of view.
- By moving the stage, bring the embryos back to the center of the field of view. Use the stage control, micromanipulator controls, and focus knob to fine-tune the position of the embryo and needle tip relative to each other while both are in focus. Aim to perform injections as close to the coverslip as possible.
- Perform needle quality control.
- To ensure that the needle is functional, dispense some of the injection solution into the Halocarbon Oil 700 surrounding the embryo, visible as a bubble in the oil.
- If nothing is flowing out of the needle, gradually increase pressure at the injector. If this does not work, get a new needle.
- Inject the embryo.
NOTE: Acceptable injection volumes range from ~0.06-1 pL. The lower limit is set by what can be visualized entering the embryo. The upper limit is set by the trauma the injection exerts on the embryo.- Inject along the lateral edges of the embryo as this is the least invasive.
- Using the stage controls, move the embryo toward the needle tip until the latter gently punctures the embryo and enters it.
- At the same time, start the flow of injection solution and watch for the appearance of a transient clear spot at the site of the needle tip, which indicates a successful transfer of liquid into the embryo.
- Monitor the embryo. If the embryo resists the needle and ruptures (cytoplasm squirts out) upon entry, return to step 5 and increase the desiccation time. If the embryo is flat against the coverslip and appears floppy during the injection, return to step 5, and shorten the desiccation time.
- If no dye entered the embryo, confirm that dye can still freely flow from the needle as in step 6.7. If the dye does not flow, replace the needle. If the dye does flow, inject a new embryo, and begin releasing the dye just before the needle punctures the embryo.
NOTE: Repeated injections of the same embryo are not recommended as it ruptures the previous wound/injection site.
- Repeat until all the embryos are injected.
7. Image embryos
NOTE: This protocol utilizes a laser scanning confocal microscope.
- Place coverslip into the metal holder on the confocal microscope so that the embryos are imaged directly from below through the coverslip; above the coverslip, there is only oil and no other barrier.
- As a quality control step, image first using the epifluorescence function on the confocal microscope, with a 40x objective. Make sure the fluorophore is visible in the embryo before proceeding.
- If the desired plane of imaging is different from the site of injection, allow enough time for the dye to diffuse to the target area.
NOTE: For BOPIPY 493/503, this time typically ranges from 30-60 min. As diffusion time depends greatly on the dye, other dyes may require optimizing the site of injection and waiting time. - Use different objectives for imaging at different scales. Use a 40x objective to image all of the embryo and a 63x objective to image subsections for smaller scales.
- For live imaging during the syncytial stages before cellularization, use the following conditions to start with: image size of 512 x 512 pixels, line average of 3, frame rate of 0.1 frames per second (i.e., 1 frame acquired every 10 s). Adjust conditions according to the capabilities of the microscope employed.
NOTE: These conditions allow acquiring ~500+ images from BOPIPY 493/503 and capturing most of the motion.
8. Analyze LD flow by first using FIJI to prepare a time series of images, and then python for PIV analysis
- Optimize image acquisition.
- Perform injections of the desired dye at the appropriate stage. Repeat this step until day-to-day variation is negligible.
- Optimize image acquisition settings including magnification, zoom, resolution, and frame rate.
NOTE: There is a tradeoff between high-quality images (resolution + line averaging) and acquisition speed (frame rate).
- Prepare data in FIJI.
- Acquire a high-quality time series to be analyzed.
- Open one frame of the time series to generate a mask with FIJI. Duplicate the image. Convert the Type of the image to an 8 bit.
- Define the boundary of the embryo using the Polygon or Freehand Selection Tool. Use Clear Outside to set pixel values outside the selection to 0. Clear and then Invert within the selection to set the value of the pixels within the selection to the maximum value (255).
- Unselect, then use the Histogram command to ensure that only pixel values of 0 and 255 are present.
- Save this new image mask for the specific time series.
- Open the time series of interest. Choose which ten frames best capture the time of interest, for example, nuclear cycle 9 at the onset of the cortical contraction. Duplicate the ten frames of interest, forming a substack.
- Use the Stack to Images function to generate 10 images to analyze with PIV.
- Save the individual files in a manner that preserves their order (SeriesX_1, SeriesX_2, SeriesX_3 ...).
- Use the prepared mask and individual frames for PIV analysis, employing the provided sample python script (Supplemental data) or a custom script generated by the user.
NOTE: The provided script is based on the python application of OpenPIV17. It outputs distance in pixels which can be converted using the pixel width and frame rate to generate speeds or velocities. - Generate replicates by completing the previous steps for additional embryos and compiling the output values.
Representative Results
Following injection, the dye will be localized only at the site where the needle tip was inserted. The dye will then diffuse away from the injection site depending on its diffusive characteristics. Figure 1 shows injection of BODIPY 493/503, soon after injection (panel A) and 24 min later (panel B). After 24 min, the dye has made it to roughly the midpoint of the embryo's long axis.
Analyzing organelle motility can be achieved through dye injections and time-lapse imaging. In Figure 2, an embryo was co-injected with BODIPY 493/503 (Figure 2A) and LysoTracker Red (Figure 2B) and imaged using laser excitation at 488 and 596 nm, respectively. This embryo was then time-lapse imaged (one frame every 30 s for 30 min, 5 min analyzed). The time series was then run through PIV analysis, the output of which is shown via streamlines in Figure 2A,B. Note that the streamlines do not represent the trajectory of individual particles, but the cytoplasmic flow is inferred from analyzing all the particles in that region of the cytoplasm. Through labeling of two independent cellular structures (LDs and acidic organelles), the PIV analysis finds similar flows, with both labels converging on the central region of the embryo where the cytoplasm is flowing into the embryo's interior6.
Currently available FPs allow the labeling of many other organelles and cellular structures. Figure 3 shows the labeling of the ER via ER tracker Green. ER tracker provides a nice resolution of the nuclear envelope, allowing visualization of major cell cycle stages. ER tracker Green is imaged using 488 nm excitation.
Labeling of the mitochondria is tricky, as most dyes tested seem to be trapped in the first mitochondrion they enter. On the other hand, no sign of dye toxicity was detected, making it possible to follow labeled mitochondria through cellularization and the ectodermal nuclear cycle 15. Figure 4 shows an ectodermal cell several hours post-injection with Mitoview 633 (excitation wavelength 633 nm).
BODIPY, Lysotracker, and LipidSpot are robust and can be used to acquire ~500+ images at 512 x 512, line average 3, frame rates from 1/s to 0.1/s. ER tracker Green, SiR-tubulin, and the mitochondrial dyes mentioned are less robust and yield ~50-200 images under the same conditions.
Starting at blastoderm stages, LDs move bidirectionally along microtubules, powered by the opposite-polarity motors kinesin-1 and cytoplasmic dynein5. This motion can be visualized by co-labeling LDs and microtubules via injecting both BODIPY 493/503 and SiR-Tubulin (Figure 5). As LDs frequently reverse their direction of movement (as they switch between kinesin-1 and cytoplasmic dynein), higher frame rates during acquisition better capture critical details of LD motility.
Live imaging of autofluorescent yolk vesicles is possible without any form of dye injection (Figure 6). However, the autofluorescence is dim, and the excitation laser is phototoxic. Thus, live imaging of autofluorescent yolk has a poor signal-to-noise ratio relative to dye injection.

Figure 1: Diffusion of BODIPY 493/503 through the embryo. The dye was injected along the lateral edge toward the anterior end (top right) and diffuses from this injection site into the embryo, labeling LDs. (A) The dye has diffused through portions of the embryo adjacent to the injection site. (B) Roughly 24 min/2 nuclear cycles later, the dye has diffused past the midpoint of the embryo. Scale bar: 100 µm. A 1024 x 1024 frame (line average 4) was acquired every 30 s. Please click here to view a larger version of this figure.
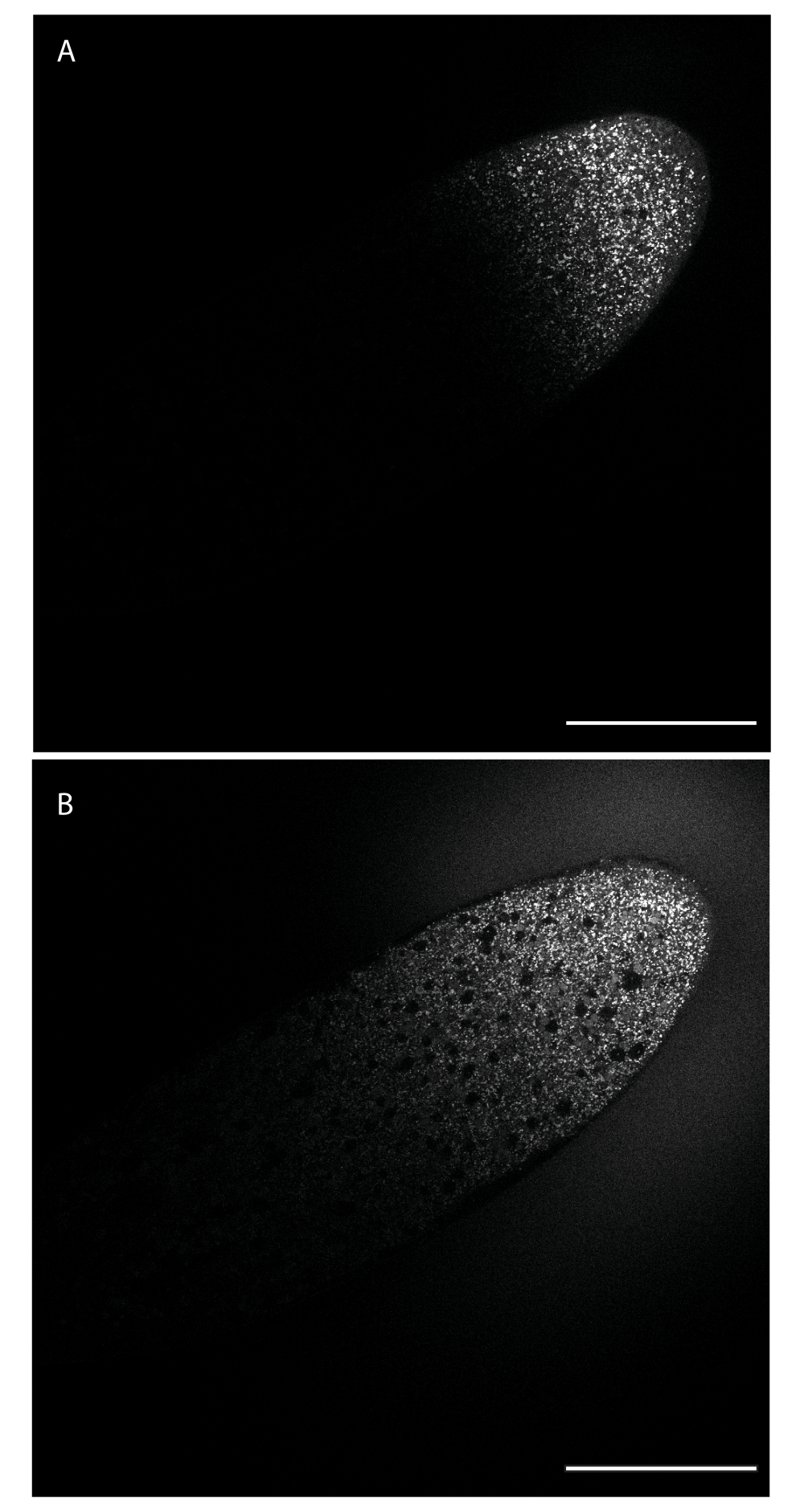{kind=link}

Figure 2: Particle image velocimetry (PIV) for LDs and acidic organelles. An embryo was injected with both BODIPY 493/503 and LysoTracker Red. (A) BODIPY channel. (B) LysoTracker Red channel. (A',B') Streamline diagrams generated by PIV analysis of the two channels generated from 10 sequential frames, including those shown in A and B. A' corresponds to the flow of LDs, and B' corresponds to the flow of acidic organelles. Note that both A' and B' show a left-of-center confluence where embryonic contents are flowing out of the plane of view, into the center of the embryo. Also, note that BODIPY has diffused more than LysoTracker as different dyes have different diffusive properties. Scale bar: 100 µm. A 1024 x 1024 frame (line average 4) was acquired every 30 s. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: ER tracker labels syncytial nuclear divisions. A syncytial blastoderm embryo was microinjected with ER tracker and a portion of its surface was imaged over time. (A) Spindle assembly during a nuclear division. (B) Abscission of the nuclear envelope during the same division. (C) Subsequent interphase. (D) The onset of the next division is indicated by centrosome appearance (occurrence of circular ER-free regions, marked by arrowheads). Note the gradual dye bleaching. Excitation wavelength: 488 nm. Scale bar: 5 µm. A: initial frame, B: 3 min elapsed, C: 10 min elapsed, D: 13 min elapsed. A 1024 x 1024 frame (line average 4) was acquired every 30 s. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Mitoview 633 labeling of mitochondria. An embryo was injected with Mitoview 633 during the syncytial blastoderm stage and imaged 4 h later, after cellularization. The image shows a neuroectodermal cell of an embryo in germ-band extension. Scale bar: 5 µm. A 1024 x 1024 frame (line average 4) was acquired every 30 s. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Co-labeling of LDs and microtubules. A cellularizing embryo was injected with a mixture of BODIPY 493/503 (yellow A,B,C) and SiR Tubulin (magenta A',B',C'). A'', B'', C'' show the merged channels. Panels A, A', and A'' show the initial frame, panels B, B' and B'' show the frame after 5 s, and panels C, C' and C'' show the frame after 10 s. Scale bar: 5 µm. A 512 x 512 frame (line average 3) was acquired every 2.5 s. Please click here to view a larger version of this figure.
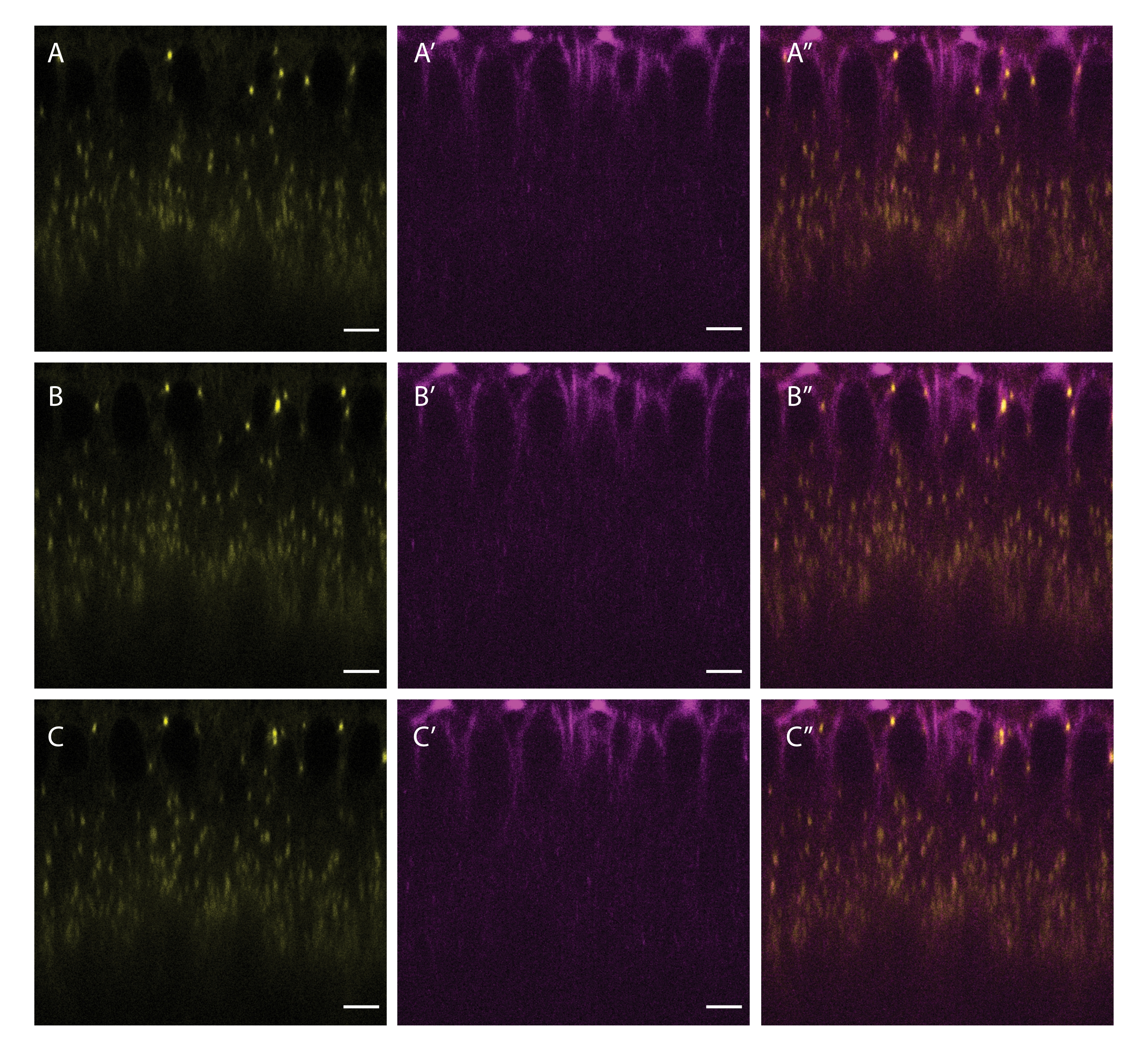{kind=link}

Figure 6: Imaging yolk vesicle autofluorescence during syncytial cleavage stages. (A) At the start of the acquisition. (B) After 8 min. (C) After 16 min. Excitation wavelength: 405 nm. Low excitation intensity was used to keep the embryo alive. Scale bar: 100 µm. A 1024 x 1024 frame (line average 4) was acquired every 30 s. Please click here to view a larger version of this figure.
{kind=link}
Supplemental data. Please click here to download this File.
Discussion
The Drosophila embryo is a powerful and convenient model to study fundamental questions in cellular and organismal biology. Its relative simplicity, powerful genetics, and small size make it an excellent system for imaging both cellular processes and development. Here, a standard microinjection protocol is adapted to enable FP usage in embryos. This approach allows for fluorescent imaging of specific cellular structures without the need for genetically encoded fluorophores, opening many genetic backgrounds to imaging. Combining multiple dyes plus strategically chosen fluorescently tagged proteins can open multichannel live imaging spanning the whole spectrum of visible light.
Critical steps in the protocol:
This protocol uses BODIPY 493/503 to label LDs. This approach can easily be adapted to mark other cellular structures. For subsequent image analysis, one of the most important factors is the signal-to-noise ratio, i.e., the brightness of the dye compared to the background signal. Lysosomes have been successfully imaged (LysoTracker Red, 1 mM), as well as mitochondria (Mitoview 633, 200 µM), the ER (ER tracker Green, 10 µM), and microtubules (SiR tubulin, 200 nM in DMSO), as shown in Figure 2, Figure 3, Figure 4, Figure 5. In addition, yolk vesicles are autofluorescent and give off blue light upon UV excitation (image using 405 nm as the excitation wavelength (Figure 6)). For other dyes, aim for the dye concentration to be 100-1,000x of what would be needed for staining cultured cells live; this is similar to the concentration of a stock solution that would be diluted into cell culture media. As this protocol calls for an injection of 100 fL and the Drosophila embryo is roughly 9 nL in volume18, these dye concentrations will average out to an internal embryonic concentration of under 1/100th of what is present in the cell culture media. Temporarily, the local concentration will be higher at the site of injection, which is most relevant for FPs that do not diffuse well (i.e., mitochondrial dyes and SiR-tubulin). For these FPs, start at the recommended high concentrations; if unexpected death is observed, successively dilute two-fold until an acceptable compromise between survival and signal strength is reached.
When co-injecting multiple dyes, both dyes should either be in the same solvent, or both the solvents and dyes should be compatible with the mixture (alcohol concentrations exceeding ~10% are not recommended).
The quality of the needles is essential for the success of this procedure, as the tip needs to be as fine as possible. Otherwise, damage from the injection wound can compromise the subsequent development of the embryo. As commercial needle pullers differ, it is important to follow the suggestions of the manufacturer and try out multiple pulling parameters until the desired shape is achieved. It is critical to perform the quality control step 1.1.3 as working with a cracked, jagged, or large bore needle tip will make the successful injection more difficult or even impossible.
The embryo needs to be partially desiccated so that additional volumes of liquid can be added during the injection. If the embryo is under-desiccated, the needle will not enter easily, and cytoplasm will squirt out as the needle penetrates or as the solution is injected. If the embryo is over-desiccated, it will look deflated and will not develop properly. The exact drying time depends on local conditions, e.g., air humidity, and can change from day to day. It has to be determined empirically for each session.
The ability of the embryo to survive after microinjection depends critically on the quality of the needle, proper desiccation, and limiting injection volume to less than 1 pL (ideally 100 fL). As long as these parameters are optimized, no significant toxicity is apparent when the described dyes are injected at the recommended concentrations. If embryos survive the desiccation and injection steps, they typically develop successfully well into germ-band extension, an exception being the microtubule and ER probes which caused cellularization defects at high levels (100 fL injection of the stock concentration of each). Testing found no obvious developmental defects when DMSO, water, and mixtures of the two were injected at the recommended volumes, with an embryo survival rate through germ-band extension of ~75% or more. Injection volumes more than over 1 pL caused defects and embryos injected with ~4 pL volumes developed for less than 1 h. Therefore, injection volumes need to be kept low, which means that dye concentrations have to be high.
Generally, injections along the lateral edge of the embryo are recommended as those result in the least damage. However, the injection site may need to be adjusted depending on the diffusive properties of the FPs employed. BODIPY 493/503 and LysoTracker diffuse faster across the entire embryo than Lipid Spot 610 (another dye to mark LDs), while SiR-Tubulin and Mitoview 633 never diffuse fully across the embryo (imaging as late as 7 h post-injection). Thus, injection in or near the site of interest may be necessary. When injecting in the anterior or posterior regions, a particularly fine needle is recommended.
Image acquisition relies on confocal microscopy to optically section and resolve small organelles and all cytoskeletal components. Techniques requiring analysis of many images (e.g., STORM or PALM) will not work because the embryonic contents are in motion and the fluorophores are not optimized for photoswitching. Epifluorescence microscopy lacks the lateral and axial resolution to make out most organelles and smaller cellular structures. For these reasons, it is strongly recommended to use a confocal microscope or employ light sheet technology.
The reproducibility of the image analysis relies greatly on consistent imaging data. For the greatest chance of success, optimization of the injection technique and image acquisition is required. Establishing and practicing a technique where the dye(s) of interest, site of injection, age of the embryo, injection volume, and acquisition setup are all consistent will generate the most robust data for image analysis.
Modifications and troubleshooting of the method
This protocol demonstrates a method for analyzing the bulk flow of LDs in cleavage stage embryos using particle image velocimetry. The same approach can be used for other organelles, other developmental stages, and other analysis methods. For example, Figure 2 shows analysis of LDs and acidic organelles flowing in the syncytial stages of embryogenesis, visualized by co-injecting BODIPY 493/503 and LysoTracker Red. Further successful imaging of LD motion in embryos up to 7 h post-fertilization has been achieved; these embryos do retain the injection wound but are able to develop for several hours.
Data gathered using this protocol has been used for particle image velocimetry, but many other analysis techniques are available. For example, particle tracking programs like those found in ImageJ, Imaris, or manual tracking can be used to obtain velocities and directionalities of moving structures. Note that most such tracking software are built to work with data from planar cell culture systems and do not always adapt well to 3D structures like the Drosophila embryo. Further, for the generation of the best quality particle tracking data, multiple Z planes would need to be imaged; this should be feasible if image stack acquisition times are under ~2 s. This benchmark should be reachable on spinning disc confocal, lattice light sheet, and recent laser scanning confocal systems. However, the feasibility of particle tracking for abundant organelles such as LDs, mitochondria, and lysosomes is low as the amount of positive signal in a field of view is too high for the current tracking methods. Tracking of less abundant structures like nuclei or yolk vesicles may be possible. PIV for flow analysis works well for LDs and acidic organelles in cleavage stages because both organelles move freely. Organelles like nuclei, ER, and mitochondria are tethered to other cellular structures and thus do not move freely and are not suited to software analysis that assumes free motion. The investigator should pick the techniques best suited to the organelle of interest.
During the syncytial and cellular blastoderm stages, LDs (as well as some other organelles) move along radially oriented microtubules5. It is therefore possible to find cross-sectional views (like in Figure 5) where single microtubules are in focus for long distances, allowing particle tracking in 2D. Since these optical planes are deep within the embryo, overall signal strength is diminished, and the signal-to-noise ratio is reduced.
For tracking analysis, imaging as fast as possible can reveal crucial details of the motion and thus of the motile machinery. For example, lipid-droplet motion is a mixture of two motile states, slow-short motion (~200 nm/s; average travel distance ~100 nm) and fast-long motion (~450 nm/s; average travel distance ~1,000 nm)19; thus, if images are taken every second or even less frequently, the slow-short state becomes undetectable. However, frequent imaging also induces fluorophore bleaching and phototoxicity. Imaging conditions, therefore, have to be adjusted depending on the exact question to be addressed.
Limitations of the method
Depending on the dye desired, the method can be limited by the compatibility between the dye solubility and the toxicity of the injection solution. Alcohols like isopropanol and ethanol are difficult to handle within a needle due to their lower viscosity and appear to damage cellular components and kill the embryo.
The method is also not well suited for visualizing the earliest steps in embryogenesis because it takes 30+ min to prepare the embryos for injection. At room temperature, the initial cell cycles of the embryo are just ~10 min long each; so, even if one were to pick a newly fertilized egg in step 5.2.3, the first few cell cycles would already be completed by the time the embryo is ready for imaging.
Illumination with light in the UV/blue range is considerably more phototoxic than for longer wavelengths. Under such conditions (e.g., to follow autofluorescent yolk vesicles; Figure 6), one has to limit imaging time (leading to shorter time series) or use lower laser power (resulting in a reduced signal-to-noise ratio).
After cellularization, dyes injected in a specific location tend to diffuse poorly, as they must traverse many cell membranes. This limits the region of observation in later developmental stages.
The significance of the method with respect to existing/alternative methods
The motion of LDs and other lipid-containing organelles in early embryos can be visualized with genetically encoded fluorophores, label-free techniques, and by the introduction of FPs. The latter can be achieved by permeabilization of the vitelline membrane12 or the microinjection approach discussed here.
Genetically encoded fluorophores are versatile markers whose levels are typically highly reproducible from embryo to embryo. However, they have lower quantum yields and bleach more easily than FPs. Typically, they are only available in one or two tagged version(s) (e.g., GFP or mCherry), limiting the choice of which structures can be imaged simultaneously. FPs, on the other hand, often exist in a large variety; for example, various lipid-droplet specific dyes are available with emission spectra from Autodot in the UV/blue spectrum to Lipidtox and LipidSpot 610 in the far-red spectrum. FPs can also be directly applied to any strain of interest, and thus do not require strain construction to, for example, introduce the desired organelle marker into a mutant strain of interest. This advantage is particularly pronounced when multiple structures are to be labeled simultaneously; instead of time-consuming crosses spanning multiple generations, this can be achieved in a single day by mixing the relevant dyes and introducing them at the same time. Finally, if cellular processes are to be probed with pharmacological inhibition, drugs and dyes can be introduced together.
Label-free methods are very powerful approaches for detecting specific cellular structures. For example, LDs can be specifically detected in early embryos by third-harmonic generation microscopy20 or by femtosecond Stimulated Raman Loss microscopy21. Like FPs, these approaches can be applied in any genetic background, and because they do not cause bleaching, they potentially allow for faster image acquisition. However, they are typically limited to specific organelles and thus do not by themselves support multiplex imaging; they also require specialized microscopes.
There are two general strategies for introducing small molecules into embryos. One is the microinjection approach employed here; the other is chemical (terpene) treatment to permeabilize the vitelline membrane. The latter approach12 is less involved than microinjection, but also more variable from embryo to embryo. In addition, after permeabilization, the protection provided by the vitelline membrane is compromised and the embryo proper is accessible to the external medium, making it more challenging to keep it alive. Microinjection is much less likely to derail embryonic development than permeabilization. However, permeabilization is recommended if many embryos need to be monitored simultaneously, e.g., for drug screening purposes. To follow the movement of cellular structures and obtain reproducible image series suitable for image analysis, microinjection is the method of choice.
Importance and potential applications of the method in specific research areas
The Drosophila embryo is an important model system for studying many cell-biological and developmental processes1,5,6. Tagging organelles with fluorescent proteins has made major contributions to the understanding of how the early embryo develops, how various organelles traffic, and how such trafficking is modulated developmentally and genetically. However, their propensity to bleach and the challenges of generating strains in which multiple organelles are labeled with different colors limit the application of this approach. The use of FPs introduced by microinjection solves many of these challenges and can even be combined with fluorescently tagged proteins. This technique allows for the imaging of multiple organelles, cell structures, and cytoskeletal components in any genetic background. As a result, several genotypes can be compared via live imaging, making it possible to determine the effect of mutations on the trafficking of multiple organelles.
This protocol demonstrates the FP injection approach for embryos of Drosophila melanogaster, but in principle, this approach applies to any insect eggs for which microinjection techniques have been established, including other species of Drosophila, crickets22, and aphids23.
Acknowledgements
We thank Pakinee Phromsiri, Brian Jencik, Jinghong (James) Tang, and Roger White for their comments on the manuscript. We thank Patrick Oakes, Stefano Di Talia, and Victoria Deneke for sharing their expertise on how to perform PIV analysis. This work was supported by National Institutes of Health grants F31 HD100127 (to M. D. K.) and R01 GM102155 (to M. A. W).
Materials
| Name | Company | Catalog Number | Comments |
| AUTODO | abcepta | SM1000a | Stains lipid droplets in violet/blue |
| BODIPY 493/503 | ThermoFisher | D3922 | Stains lipid droplets in green |
| Desiccant Beads –Desiccant-Anhydrous Indicating Drierite | W.A. Hammond Drierite Company | 21001 | |
| Dissecting microscope SteREO DiscoveryV20 with transillumination base | Zeiss | 4350030000000000 | |
| Double sided tape-Permanent Double-Sided Tape | Scotch (3M) | - | Sold by many vendors |
| ER-Tracker Green (BODIPY FL Glibenclamide) | ThermoFisher | E34251 | Stains the ER/nuclear envelope |
| Femptotip II | Eppendorf | H129354N | Ready to use |
| Glass capillaries -Glass Thinw w/fil 1.0mm 4in | World Precision Instruments | TW100F-4 | Must be pulled |
| Glass coverslip | - | - | Buy the appropriate refractive index for your objective lens |
| Glass slides -Double Frosted Microscope Slides precleaned | FisherBrand | 12-550-343 | |
| Halocarbon oil 27 | Sigma-Aldrich | H8773-100ML | |
| Halocarbon oil 700 | Sigma-Aldrich | H8898-50ML | |
| Heptane greener alternative anhydrous, 99% | Sigma-Aldrich | 246654-1L | Heptane is considered toxic by the USA's OSHA |
| Confocal microscope | Leica | Sp5 | Many different types of confocal microscopes will work. |
| LipidSpot 610 | Biotium | #70069 | Stains lipid droplets in far red |
| LysoTracker Red | ThermoFisher | L7528 | Stains lysosomes in red |
| MitoView 633 | Biotium | #70055 | Stains mitochondria |
| Needle loading pipette tips - 20uL microloader tips | Eppendorf | # 5242956.003 | |
| P-1000, Next Generation Micropipette Puller | Sutter Instruments | P-1000 | The settings used are heat-470, pull-70, velocity-60, delay-90, pressure-200, ramp-479 at 1x1. They refer to the intensity and length of the heating, the timing of pulling force and whether the force is applied linearly. Using these conditions, one capillary generates two usable needles with fine openings at the tip. |
| SiR-Tubulin | Cytosketon Inc | CY-SC014 | Made by Spirochrome; Cytoskeleton Inc. is the North American distributor. Stains microtubules in far red |
| TransferMan | Eppendorf | 5178 NK | |
| ViaFluor 405 | Biotium | #70064 | Stains microtubules in violet/blue |
References
- Chowdhary, S., Tomer, D., Dubal, D., Sambre, D., Rikhy, R. Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Scientific Reports. 7 (1), 5502 (2017).
- Mavor, L. M., et al. Rab8 directs furrow ingression and membrane addition during epithelial formation in Drosophila melanogaster. Development. 143 (5), 892-903 (2016).
- Riggs, B., et al. Actin cytoskeleton remodeling during early Drosophila furrow formation requires recycling endosomal components Nuclear-fallout and Rab11. Journal of Cell Biology. 163 (1), 143-154 (2003).
- Welte, M. A., Gross, S. P., Postner, M., Block, S. M., Wieschaus, E. F. Developmental regulation of vesicle transport in Drosophila embryos: forces and kinetics. Cell. 92 (4), 547-557 (1998).
- Welte, M. A. As the fat flies: The dynamic lipid droplets of Drosophila embryos. Biochimica et Biophysica Acta. 1851 (9), 1156-1185 (2015).
- Deneke, V. E., et al. Self-organized nuclear positioning synchronizes the cell cycle in Drosophila embryos. Cell. 177 (4), 925-941 (2019).
- Welte, M. A., et al. Regulation of lipid-droplet transport by the perilipin homolog LSD2. Current Biology. 15 (14), 1266-1275 (2005).
- Bailey, A. P., et al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell. 163 (2), 340-353 (2015).
- Johnson, M. R., et al. H2Av buffering via dynamic sequestration to lipid droplets in Drosophila embryos. Elife. 7, 36021 (2018).
- Lowe, N., et al. Analysis of the expression patterns, subcellular localisations and interaction partners of Drosophila proteins using a pigP protein trap library. Development. 141 (20), 3994-4005 (2014).
- Rambold, A. S., Cohen, S., Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Developmental Cell. 32 (6), 678-692 (2015).
- Rand, M. D., Kearney, A. L., Dao, J., Clason, T. Permeabilization of Drosophila embryos for introduction of small molecules. Insect Biochemistry and Molecular Biology. 40 (11), 792-804 (2010).
- Tran, S. L., Welte, M. A. In-vivo centrifugation of Drosophila embryos. Journal of Visualized Experiments. (40), (2010).
- Rothwell, W. F., Sullivan, W. Drosophila embryo collection. CSH Protocols. 2007, (2007).
- Wieschaus, E., Nüsslein-Volhard, C., Roberts, D. B. . Drosophila: A Practical Approach. , 179-214 (1998).
- . The Interactive Fly: Stages of Development and Mitotic Domains Available from: https://www.sdbonline.org/sites/fly/aimain/2stages.htm (1999)
- Liberzon, A., et al. . OpenPIV/openpiv-python: OpenPIV - Python (v0.22.2) with a new extended search PIV grid option. , (2020).
- Markow, T. A., Beall, S., Matzkin, L. M. Egg size, embryonic development time and ovoviviparity in Drosophila species. Journal of Evolutionary Biology. 22 (2), 430-434 (2009).
- Gross, S. P., Welte, M. A., Block, S. M., Wieschaus, E. F. Dynein-mediated cargo transport in vivo. A switch controls travel distance. Journal of Cell Biology. 148 (5), 945-956 (2000).
- Debarre, D., et al. Imaging lipid bodies in cells and tissues using third-harmonic generation microscopy. Nature Methods. 3 (1), 47-53 (2006).
- Dou, W., Zhang, D., Jung, Y., Cheng, J. X., Umulis, D. M. Label-free imaging of lipid-droplet intracellular motion in early Drosophila embryos using femtosecond-stimulated Raman loss microscopy. Biophysical Journal. 102 (7), 1666-1675 (2012).
- Barry, S. K., et al. Injecting Gryllus bimaculatus Eggs. Journal of Visualized Experiments. (150), (2019).
- Le Trionnaire, G., et al. An integrated protocol for targeted mutagenesis with CRISPR-Cas9 system in the pea aphid. Insect Biochemistry and Molecular Biology. 110, 34-44 (2019).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved