Single Extracellular Vesicle Transmembrane Protein Characterization by Nano-Flow Cytometry
* These authors contributed equally
In This Article
Summary
The latest generation of EV characterization tools are capable of single EV analysis across multiple parameters simultaneously. Nano-flow cytometry measures all biological particles larger than 45 nm without labeling and identifies specific characteristics of subpopulations by a variety of fluorescent labeling techniques.
Abstract
Single particle characterization has become increasingly relevant for research into extracellular vesicles, progressing from bulk analysis techniques and first-generation particle analysis to comprehensive multi-parameter measurements such as nano-flow cytometry (nFCM). nFCM is a form of flow cytometry that utilizes instrumentation specifically designed for nano-particle analysis, allowing for thousands of EVs to be characterized per minute both with and without the use of staining techniques. High resolution side scatter (SS) detection allows for size and concentration to be determined for all biological particles larger than 45 nm, while simultaneous fluorescence (FL) detection identifies the presence of labeled markers and targets of interest. Labeled subpopulations can then be described in quantitative units of particles/mL or as a percentage of the total particles identified by side scatter.
Here, EVs derived from conditioned cell culture media (CCM) are labeled with both a lipid dye, to identify particles with a membrane, and antibodies specific for CD9, CD63, and CD81 as common EV markers. Measurements of comparison material, a concentration standard and a size standard of silica nanospheres, as well as labeled sample material are analyzed in a 1-minute analysis. The software is then used to measure the concentration and size distribution profile of all particles, independent of labeling, before determining the particles that are positive for each of the labels.
Simultaneous SS and FL detection can be utilized flexibly with many different EV sources and labeling targets, both external and internal, describing EV samples in a comprehensive and quantitative manner.
Introduction
What are EVs?
Extracellular vesicles (EVs) are the collective term for a range of cell-derived membranous particles integral to many normal cellular and tissue activities. Their impact upon a wide range of scientific fields and their potential clinical relevance have driven a growth in EV research and industrial interest1. Small EV (sEV) research primarily focuses on exosomes, 40-100 nm particles that begin formation in the early endosomes before maturation and release through fusion of multi-vesicular bodies (MVB) to the plasma membrane, as well as microvesicles, which bud directly from the plasma membrane forming 80-1,000 nm particles2. A third EV population are apoptotic bodies, 50-1,500 nm particles formed during cell death meaning their relative proportion to other EVs can be greatly variable3.
Because EV characteristics may represent changes occurring in their cell/tissue of origin, there is potential for their use in diagnostics. Various 'omics' analyses have begun to identify markers of cell origin and disease state, which may allow for non-invasive assessment of patients using EV sources such as blood plasma/serum, urine, saliva and cerebral spinal fluid (CSF)4,5. A driving force behind these EV-related innovations are new characterization techniques that overcome previous limitations.
The need for, and challenges of, single EV characterization
Single EV characterization is becoming increasingly important for both validation and description of EV isolates, as well as elucidating key features of these nanoparticles for progression of EV-based therapies and diagnostics6. Depending on the EV source and intended use, analysis of purity, often described as a ratio of EV to non-EV particles or as EV to free protein, can require significant amounts of data from multiple analytics7.
Particle counting and sizing measurements in EV publications have previously relied heavily on Nanoparticle tracking (NTA), resistive pulse sensing (RPS), and electron microscopy (EM)2. Standard NTA and RPS lack the ability to distinguish EVs from non-EV particles and have their own caveats such as slow throughput8 and unsuitable lower limit of detection seen with NTA9,10,11.
The tetraspanins CD9, CD63, and CD81 have historically been important identifiers for the presence of EVs in EV isolations/preparations. Commonly, western blotting (WB) and dot blotting techniques are used to show the enrichment of these proteins in EV isolates compared to cell lysate12. However, the lack of quantitation to these methods and the heterogeneity of these EV markers, regarding both display within EV subpopulations and cell, tissue or patient related variation, encourages advanced analytical techniques, which unify physical and phenotypic characterization13.
Nano-flow cytometry as a comprehensive EV analysis technique
Determining true EV concentrations requires identification of intact particles and a universal marker, particularly in complex particle isolates, with resolution capable of detecting all EVs while distinguishing them from non-EV particles14.
Nano-flow cytometry (nFCM) is a technique that allows unlabeled analysis of particles sized between 45-1,000 nm while simultaneously utilizing fluorescent labeling and detection to identify particle subpopulations. A key distinction from conventional flow cytometry is the use of equipment dedicated to nanoparticle analysis, allowing for the greatest resolution15. EV analysis, which uses conventional flow instrumentation repurposed for small particle analysis is improving, but still struggles to attain the resolution to detect and analyze <100 nm EVs16,17. Bead-based flow cytometry is a further adaptation often employed for EV analysis, but this removes the possibility of single particle detection and introduces capture based biases18.
Benefitting from a lower limit of detection of ~45 nm in the side scatter (SS) channel for EV analysis, nFCM utilizes SS triggering. This can be thought of as 'particle first' analysis as it means that events must provide a SS signal, surpassing a set threshold before analysis of the fluorescence intensity. This removes false positives such as degraded membrane and fluorophore aggregations, and focuses analysis onto intact EVs15. SS measurements are also used to size individual particles by comparison to a four-modal silica nanosphere standard19. The fluorescence measurements are taken on two further detectors allowing three simultaneous measurements for each particle to describe particle concentration, sizes and presence of markers or other targets of interest in order to identify EV subpopulations20.
In the following experiment, SS and fluorescent (FL) measurements are used to measure >45 nm particles, show the subset of membrane positive EVs, and identify CD9, CD63, CD81 presentation on EV subpopulations. Both the concentration of these subpopulations and their ratio as part of the total particles measured by SS are described, as are their size profiles.
Protocol
1. Setup of the nFCM instrument and measurements of the nFCM standards
NOTE: Three measurements are taken before beginning data acquisition for samples to validate correct alignment of the nFCM instrument and provide strong comparison between instruments. These are the concentration standard/quality control (QC), size standard, and a blank.
- Dilute the QC beads 1:100 in distilled water in an appropriate 0.6 µL tube and place in loading bay.
- From the Sample Flow drop down menu, select Boosting to introduce the QC sample into the system for 45 s to completely replace the previous sample/cleaning solution.
- While boosting, set the laser power to the preset template for QC beads "250 nm FL QC standard", e.g., 10/40 mW for the blue laser, 20/50 mW for the red laser, with 0.2% SS decay.
- Select the Sampling pressure from the same down menu to reduce the system pressure.
- Set the auto sampling pressure to 1.0 kPa to maintain a constant pressure.
- Initiate the 1-minute analysis by selecting Time to Record from the acquisition controls. Data will be plotted on the dot-plot showing a log scale for SS-intensity and a selected FL intensity.
- Insert the file name and sample dilution before saving the file.
- Select unload to remove the tube from the loading bay. Replace with 150 µL of cleaning solution and clean for >30 s by selecting Boosting before removing the cleaning solution by selecting Unload.
- Remove any excess cleaning solution from the capillary tip using a tube containing 150 µL of water.
- Dilute the size standard beads 1:100 in water and load 100 µL into the loading bay, before Boosting the sample for 45 s.
- Set the laser power to the preset template "S16 exo 68-155 nm". This setting is for both the size standard and samples containing EVs < 200 nm. For example, 15/40 mW for the blue laser, 20/50 mW for the red laser, with 0.2% SS decay.
- Select Sampling and proceed to recording the sample for 1 min as before.
- Perform the third measurement with either a water or PBS sample to create a blank measurement of your eluent, identifying false positives for removal by the software. Inputting the standard measurements is described in the following section.
2. Determining the particle concentration of an unlabeled sample and generating a PDF report
- Dilute the unlabeled EV sample in PBS to a suitable particle concentration range for nFCM analysis, 1 x 108- 5 x 108 particles/mL. Load 10-100 µL of the diluted sample into the loading bay.
- When the particle concentration is unknown, begin with a 1:100 dilution of the EV sample. Sample concentration can be quickly approximated by the size of the laser spot on the CCD camera during boosting, or by observation of the Event Burst Trace during sampling.
- Boost the loaded sample for 45 s before selecting Sampling and record for 1 min, saving as previously described.
- To begin analyzing this data, switch from the Acquisition tab to the Analysis tab. Open the saved nfa files.
- To allow for accurate sample measurement, use the two standard measurements taken prior to sample measurement to set values for sample comparison as well as the blank.
- Create the size standard curve, for SS to diameter conversion, by first selecting the size standard file and using the set threshold tool (also known as auto-thresholding). The threshold, visible in the event burst trace, identifies the minimum signal intensity required for an event to be considered significant.
- With the threshold set, check the dot plot x and y parameters are SS-H or SS-A on the x axis and FITC-A on the y axis.
- Open the standard curve generation tool and select S16 exo 68-155 nm as the sizing template. Click on Find Peaks to identify the peak SS intensities as either a 68, 91, 113, or 155 nm diameter particle.
- Check whether the SS to diameter curve has been generated with an r value close to 1 before closing the window.
- Set the concentration standard by selecting the saved file and clicking on Count STD. Input the particle concentration of the standard.
- Having set the standard information, select the EV sample file and set the threshold.
- Select the blank file and click on Set Blank to identify the number of false positives for removal from the sample count. This should be done with the same threshold as your sample. Return to the EV sample file.
- Open the PDF generation tool and select Sizing and Concentration. Input the dilutions of the sample. The concentration of the sample and the size distribution of the particles are shown.
3. Sample labeling
NOTE: Two staining strategies can be used simultaneously for a comprehensive analysis of the particles in suspension. Labeling protocols often require optimization for new antibodies or sample sources.
- Dilute a portion of the EV sample to a concentration of 1.25 x 1010 particles/mL in PBS. Incubate 8 µL of the diluted EV sample with 1 µL of antibody and 1 µL of dye for a particle concentration of 1 x 1010 particles/mL (total particles will be 1 x 108 suspended in 10 µL). The incubation ratio for the antibody is 1:50 (1 µL of 1:5 antibody) in this instance.
- Use more than three different concentrations of antibody or dye during optimization, to provide data indicating the protocol allows for maximum epitope binding without over saturation of the chosen label, which may impair identification of low intensity fluorescence events. The incubation concentration for the membrane dye is 40 nM (1 µL of 400 nM).
- Mix the 10 µL sample by vortexing for 5 s and incubate at RT for 30 min in the dark.
NOTE: Recommendations for controls are included in the discussion. - Following incubation, take 1 µL of the labeled sample and dilute 1:50 in PBS in a 0.6 mL tube.
- Load the sample into the loading bay and apply boosting pressure for 45 s. Ensure laser settings are correct and the appropriate lenses are loaded.
- Switch to sampling pressure and select Time to Record. Following the 1 min acquisition, name the data file and save.
- Unload the sample and replace with cleaning solution. Boost this for >30 s before loading the next labeled sample.
4. nFCM analysis-PDF generation for subpopulation analysis
- For PDF generation, apply the same standards as previously done; only the blank measurement will be set differently.
- Select the sample file and set the threshold. On the dot plot, change the y-axis to show FL measurements from the green or red channel.
- Select the square gating tool and use left click to draw a square around the FL+ population.
- Open the blank measurement file and click on Set Blank before reverting to the sample file.
- Open the PDF generation tool as before and the size distribution, concentration, and percentage (compared to all SS+ events) are identified for the FL+ subpopulation.
- Repeat for each subpopulation of interest identified as "Total, P1, P2 etc."
Representative Results
Tetraspanin presentation on SW620 derived EVs and C2C12 derived EVs
Modern analysis of the abundance of key tetraspanins CD9, CD63, and CD81 on EVs from a variety of sources has highlighted the extreme variability of their presence, both in bulk analyses and when analyzing their presentation on EV subpopulations21. This is likely related to the availability of different biogenesis pathways such as the ESCRT-dependent and independent pathways22.
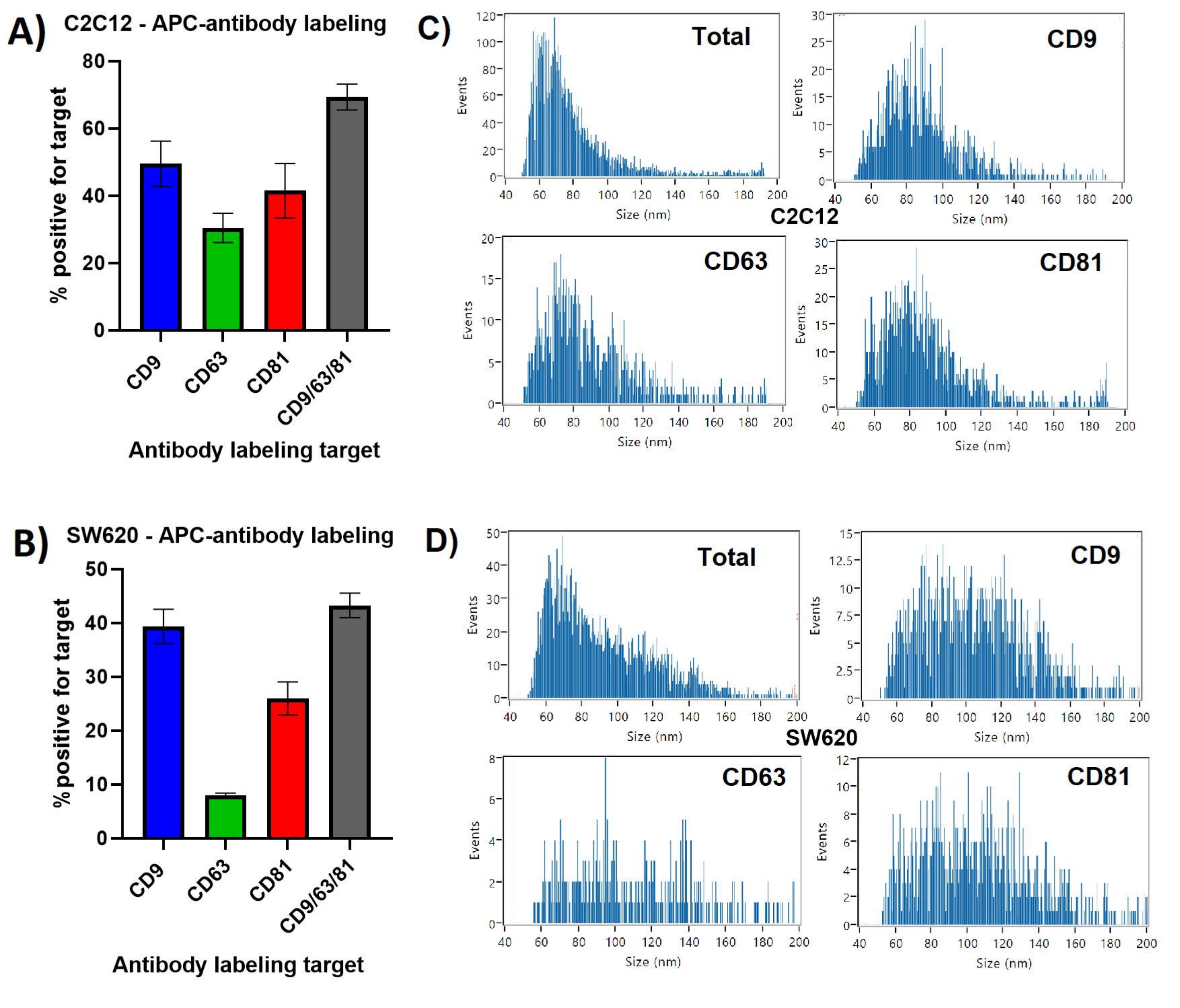
CD9 was presented on the greatest percentage of C2C12 EVs at ~50%, with CD63 presentation on only ~30%, in Figure 1. When labeling was performed with all three anti-tetraspanins in one incubation, ~70% of particles were shown to present at least one of the EV markers. Repeat measurements showed lowest standard deviation for the CD9/63/81 labeling of C2C12 EVs at ~3.8%, while this was ~8.1% for the CD81 labeled C2C12 EVs.
SW620 derived EVs showed a different tetraspanin profile, with a much greater difference between the most presented CD9 at ~40% and the least presented CD63 at ~7%. Similar to the C2C12 EVs, CD9 was present on the majority of the tetraspanin positive population, as the CD9/63/81 combined labeling identified ~42% of particles as having at least one of the three markers.
The majority of C2C12 derived particles, the total SS+ population, ranged in size from 45-120 nm, with a median ~65 nm diameter. The size profiles for each of the single tetraspanin labeled EV-subpopulations, shown in Figure 1, are similar to each other, showing a larger median size of ~75-85 nm and fewer <65 nm EVs compared to the total particle population.
SW620 derived particles ranged between 45-160 nm with a median size of ~65 nm, showing a skewed distribution. The tetraspanin labeled EVs show a more normal distribution and larger median diameter between 90-110 nm, with similar distributions between individual tetraspanin positive subpopulations.
While the most and least presented tetraspanins are the same for these two cell lines, their proportional representation differ greatly and there is an interesting difference between the potential co-presentation of tetraspanins observable from the difference in CD9 positivity and the CD9/63/81 positivity.
Combining EV membrane labeling with antibody labeling
The bilayer membrane of EVs provides a more generic labeling target, which may allow distinction from similar sized non-EV particles such as protein aggregates and some forms of LDL with lipid monolayers23. The specificity of this type of labeling must be validated as some common membrane dyes used in EV research have been shown to bind to non-EV particles as well24.
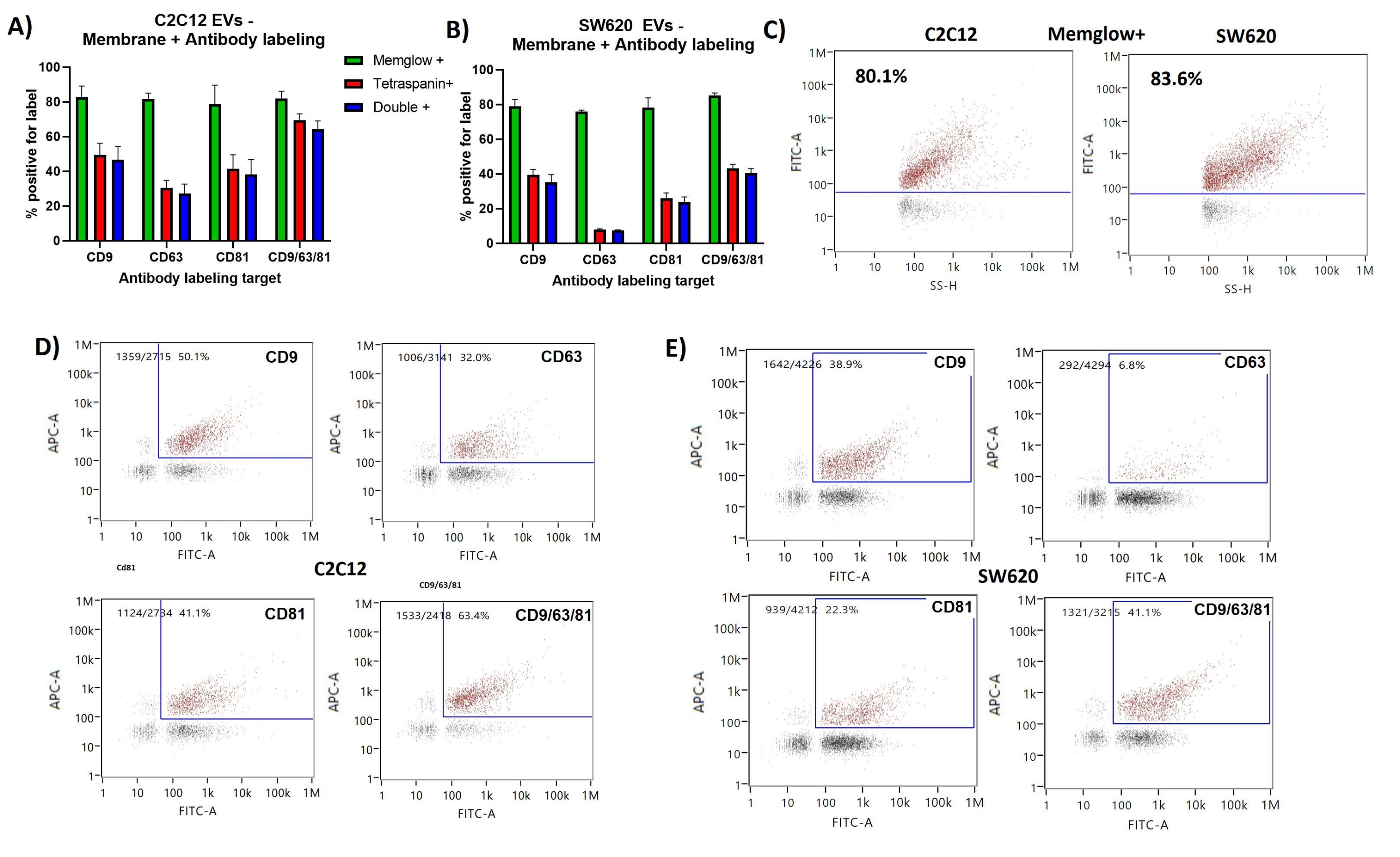
Membrane labeling repeatedly showed ~80% positivity of C2C12 derived EVs and SW620 EVs. The SS intensity (SS-H) shows good correlation with FITC intensity on the dot plots of both C2C12 and SW620 results shown in Figure 2. This is expected as the SS intensity is relative to the particle size and hence the membrane surface area.
The majority of antibody labeled EVs were also positive for the membrane labeling showing double positive percentages (FITC+ & APC+) very similar to the tetraspanin positive percentages. FITC vs APC dot plots show slight correlation between membrane and tetraspanin labeling with CD63 labeling seeming to show the weakest correlation to membrane labeling. Few events were detected that were positive for antibody labeling but negative for membrane labeling.
Reproducibility, dual labeling efficiency, and alternative data outputs
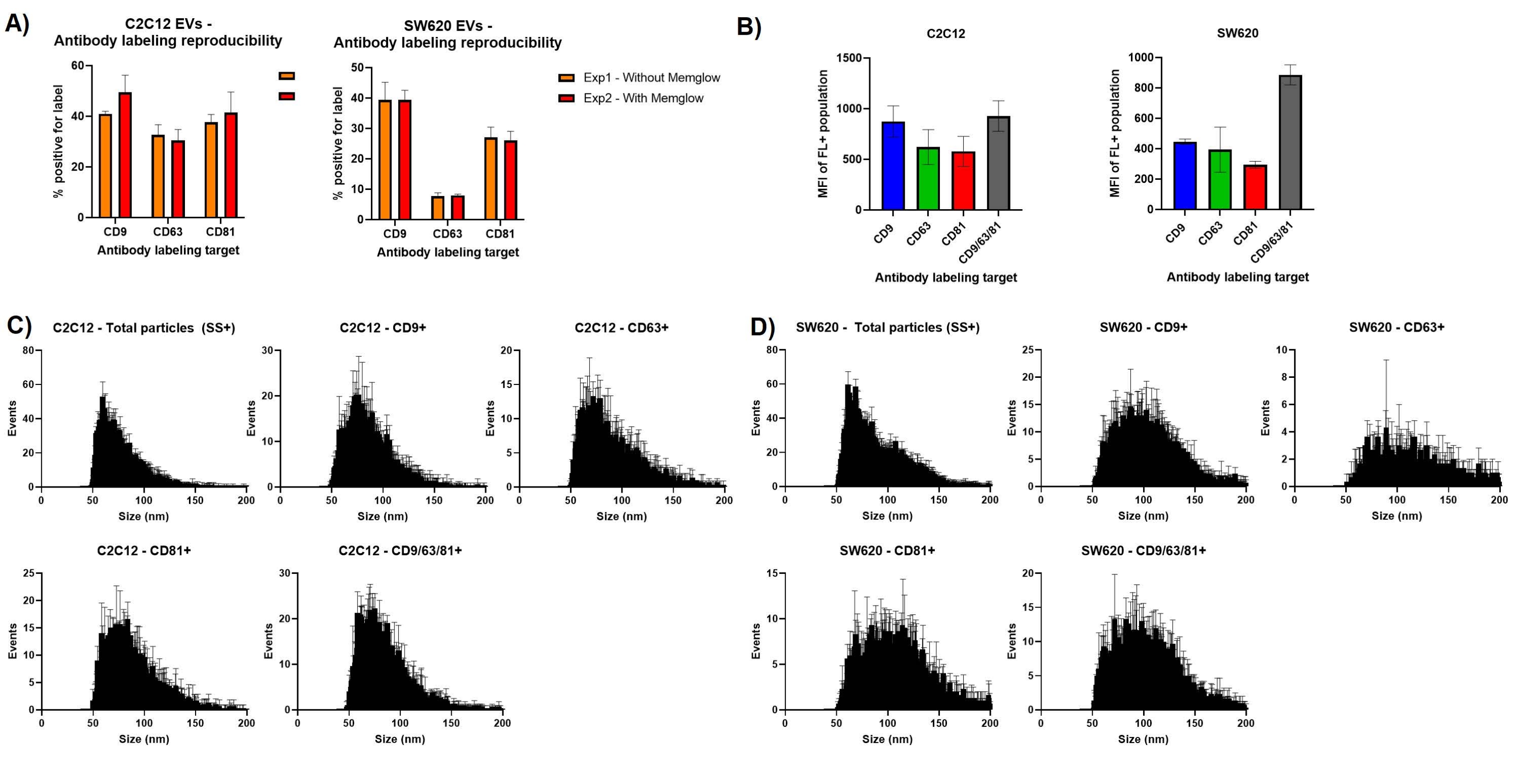
An important consideration in designing nFCM experiments is fluorophore or label interactivity. Figure 3a shows that repeating the antibody labeling with and without additional membrane labeling leads to very similar % positivity measurements. In particular, the SW620 labeling shows very little variation between the two measurement sets. This shows limited interference between the dye and antibody binding and also highlights good reproducibility when repeating EV labeling.
Intensity measurements, both side scatter and fluorescence, can be exported to excel for each individual particle measured. From this we can generate mean fluorescence intensity (MFI) measurements to provide comparative approximations of the abundance of our labeled target. MFI for the fluorescent populations of C2C12 derived EVs suggest slightly lower presentation of CD63 and CD81 with MFI measurements of ~550 and ~600, respectively, compared to the ~850 MFI seen for CD9+ EVs. The units of measurement for MFI in this instance are FL-A and are not standardized against a fluorescent calibration. The use of all three antibodies does not elicit a much greater fluorescence than just CD9 labeling. This is very different to the SW620 MFI measurements, which suggest that using a cocktail of all three antibodies provides a greater fluorescence signal for labeled EVs, than the use of any individual antibody label.
The size distribution histograms produced by the NanoFCM software can also be exported into excel allowing for triplicate data sets to be overlayed. The size distributions profiles of Figure 3 are similar to those of Figure 1 but include error bars. Median EV size of the tetraspanin positive subpopulations in C2C12 derived EVs show a median around 70 nm, slightly larger than the 60 nm average of the total particle populations. SW620 EVs positive for tetraspanin markers are larger, showing peaks at ~100 nm.

Figure 1: CD9, CD63, CD81 positive EV subpopulations identified by antibody labeling. (A) Bar chart showing percentage of labeled EVs compared to total particles observed by SS for C2C12 derived EVs. (B) Bar chart showing percentage of labeled EVs compared to total particles observed by SS for SW620 derived EVs. (C) Representative size distribution profiles for C2C12 derived EVs. Each histogram is taken from the PDFs generated for the first measurement of triplicate datasets. (D) Representative size distribution profiles for SW620 derived EVs. Each histogram is taken from the PDFs generated for the first measurement of triplicate datasets. Error bars represent standard deviation of triplicate measurements of the same labeled samples. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Membrane labeling and antibody labeling of CD9, CD63, CD81 presenting EV subpopulations. (A) Bar chart showing Membrane+ %, tetraspanin+ % and double+ % for C2C12 EVs. (B) Bar chart showing Membrane+ %, tetraspanin+ % and double+ % for SW620 EVs. (C) Representative SS vs FITC dot plots showing gating for the Membrane positive population. (D) Representative FITC vs APC dot plots showing gating for double positive population of EVs from C2C12. (E) Representative FITC vs APC dot plots showing gating for double positive population of EVs from SW620. Error bars represent standard deviation of triplicate measurements of the same labeled samples. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Assessing variation in repeat measurements. (A) Bar charts showing antibody labeling % positivity with and without additional membrane labeling. (B) Mean fluorescence intensity measurements for gated EV subsets. (C) Averaged size distribution histograms for triplicate data sets for C2C12 EVs. (D) Averaged size distribution histograms for triplicate data sets for SW620 EVs. Error bars represent standard deviation of triplicate measurements of the same labeled samples. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Sample and reagent details
Two EV isolates from separate cell lines were selected for demonstration of fluorescent labeling and subsequent nFCM analysis. Both EV sets were suspended in PBS and stored at -80 °C for <3 months but most other conditions were different between isolates. The C2C12 mouse myoblast cell line represents an embryonic precursor to skeletal muscle cells and were grown in 2D culture, conditioning the growth medium over 72 h before EV isolation by ultracentrifugation. SW620 is a human colon adenocarcinoma cell line and was grown in a rudimentary bioreactor, enriching media over 7 weeks, with EV isolation conducted by size exclusion chromatography (SEC) and fractions 7-9 eluted from sepharose CL-2B columns combined into a single sample.
While EVs isolated and concentrated from CCM are the easiest sample types to work with, nFCM is applicable to most EV isolates, including biofluids such as serum, plasma, urine, and CSF. These sample analyses benefit from the nFCM SS detection of all particles, allowing for corroboration with NTA, TRPS, and other particle analyses, while describing the fluorescently labeled EV subpopulations in quantitative terms as well as a proportion of the total, for an unbiased approach of multiparameter particle analysis. Analysis of low processed samples is possible too, such as unclarified urine and EV enriched CCM with the caveat of requiring low contaminate protein.
The membrane dye used here is intended to integrate into the lipid bilayer, with a lipophilic moiety for membrane loading and a hydrophilic dye for remaining in the plasma membrane25. There is currently no perfect EV labeling dye and several criteria must be considered when choosing, including specificity to EVs, suitability with fixation or permeabilization, and efficiency of EV labeling26.
Fluorescent conjugated antibody labeling of surface exposed epitopes has proven to be an effective method for identifying EV subpopulations27. A key aspect of protocol optimization is meeting, and not exceeding, the labeling saturation point to bind to all available epitopes without inhibiting detection of low fluorescence particles by allowing the surrounding buffer to be filled with fluorescent unbound antibody28. Additionally, the availability of exposed binding sites for antibody labeling is potentially influenced by several factors. Storage conditions of EVs has been shown to effect concentration and size profiles of EVs29 with observations also indicating effects on antibody labeling30. The presence of surface corona proteins, protein modifications, and impacts of isolation techniques may also have effects upon antibody labeling in some cases. Ultimately, as the utilization of fluorescent labeling becomes more prevalent for EV studies, experimental design and optimization for multiple analytical techniques will become more refined.
To increase accuracy of results, several controls can be included such as (1) PBS + dye control, to assess micelle or aggregate formation in some dyes, which can appear as SS+ particles in nFCM analysis, (2) PBS + antibody control, aggregates can occur, although often not large enough to scatter enough light for SS detection, (3) EV sample + IgG antibody control, common in flow cytometry and used to identify any non-specific binding, (4) non-EV particle sample + antibody/dye control - particularly important when needing to identify EVs in complex particle samples, controls such as purified low-density lipoprotein (LDL) particles or EV depleted/ablated samples can act as a negative control to validate selective labeling, (5) positive controls are hard to design but validation of an antibody on cells is a useful inclusion.
Presentation of tetraspanins on EVs
The antibody and membrane labeling of this experiment demonstrates the high level of quantitative data that can be obtained in a small timeframe by nFCM analysis. Simultaneous measurement of the key physical attributes of particle diameter/concentration with the phenotypic measurements of membrane and/or protein presence leads to high level descriptions of subpopulations within particle isolation.
Importantly, varying levels of the three 'key' EV-related tetraspanins, CD9, CD63, CD81, were identified in these two EV samples. CD9 was presented on the greatest proportion of EVs for both C2C12 derived EVs and SW620, with CD81 and CD63 being the second and least presented proteins, respectively.
Despite some similarities observed here in the tetraspanin profiles of EVs from two very different cell sources, levels of CD9, CD63, and CD81 can be very different between cell line and patient derived EVs31.
The difference in CD63 expression between the two EV samples is particularly relevant to the ongoing discussion of key identifiers of 'EV-ness'. While presentation of CD63 in only ~8% of the SW620 EVs may be unexpected by some, CD63 has been suggested as poor identifier of the different types of EVs isolated by size or density32, and tetraspanin negative EVs have been identified, even when described as exosome-like33.
Identification of EVs within complex particle isolations
The heterogeneity of EV tetraspanin profiles, in both cell line and patient-derived EVs, cautions against reliance on tetraspanin-based capture of EVs and highlights the future need for new EV identification methods31. EV labeling independent of specific proteins could prove to be highly beneficial to identifying EVs from similar sized non-EV particles if EV specificity can be proven to be very high. This is particularly true for biofluid EV isolates as it has been suggested that the concentration of EVs in human blood plasma is in the range of 1010 particles/mL while lipoproteins are measured at 1016 per mL14,34. Even upon EV enrichment, studies comparing particle positivity for tetraspanin markers and/or LDL marker ApoB suggest ~50-100x greater abundance of LDL in Platelet free plasma (PFP) samples compared to EV35.
The isolation technique used greatly affects the range of co-isolated non-EV particles such as very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and LDL36. There are also descriptions of lipoprotein co-isolates bound to EVs which, while potentially playing an important biological role, makes achieving EV pure samples a challenging goal35.
Therefore, it could be argued that a greater focus be placed on the description of particles which make up a sample, rather than achieving isolation of a pure but limited selection of EVs. Achieving comprehensive description of particles by measuring tetraspanin abundance in bulk and particle counts separately can be insufficient to accurately determine EV concentrations, particularly from biofluid sources36,37. Identifying subpopulations in a tier-based approach, showing total particles, EVs, and EVs presenting certain proteins, as demonstrated in this experiment may provide a robust solution to nanoparticle characterization. This has been the case for projects involving EV-loading with designs for future therapeutic applications20 and identification of CD63+ EVs with luminal cargo such as mitochondria38.
nFCM within the repertoire of EV analytics
A strength of nFCM EV analysis is the way data can corroborate and build upon the most common EV analyses and form bridges between physical and phenotypic data sets. However, this is based on accurate labeling protocols which often need to be optimized for unique labeling reagents such as dyes and antibodies. A key criterion for accurate analysis is having labeled particles suspended in a non-fluorescent buffer, which relies on either removal of excess unbound fluorophore or the refinement of protocols to not exceed epitope saturation.
Comparison studies have shown that nFCM sizing of EVs provides data in line with TRPS and cryo-TEM, techniques which are described as more accurate than NTA for EV size analysis10,39. However, as with any optical-based method, the influence of heterogeneic optical properties seen for EVs and differences between the optical properties of reference material and EVs must be acknowledged when interpreting data10.
Western blotting has been a key method for indicating EV enrichment through identification of EV markers40. But the desire to demonstrate the presence of such markers on particles has driven advances in EV-based flow cytometric analyses17. However, the necessary resolutions to provide robust data are currently best attained through dedicated instrumentation with regards to both scattered and fluorescent light19.
nFCM provides an unbiased approach of initially describing all particles irrelevant of specific markers, by use of side scatter measurement, allowing for corroboration with the most common techniques of NTA, RPS, and TEM1, while simultaneously adding phenotypic measurement similar to WB or Elisa in a quantitative manner.
Disclosures
A.L., B.P., D.A., R.L, R.T are employees of NanoFCM and their contributions to this paper were made as part of their employment.
Acknowledgements
We would like to thank the groups of Owen Davies and Nick Peake for continuing to provide material and expertise.
Materials
| Name | Company | Catalog Number | Comments |
| APC Anti-CD81 antibody (M38) | abcam | ab233259 | Anti-Human CD81 APC conjugated antibody |
| APC Anti-CD9 antibody (EM-04) | Abcam | ab82392 | Anti-mouse CD9 APC conjugated antibody |
| APC Anti-CD9 antibody (MEM-61), prediluted | abcam | ab82389 | Anti-Human CD9 APC conjugated antibody |
| APC anti-mouse CD63 antibody | biolegend | 143905 | Anti-Mouse CD63 APC conjugated antibody |
| APC anti-mouse/rat CD81 antibody | biolegend | 104909 | Anti-Mouse CD81 APC conjugated antibody |
| CD63 monoclonal antibody (MEM-259), APC | invitrogen Via Fisherscientific | A15712 | Anti-Human CD63 APC conjugated antibody |
| Celline AD1000 bioreactor | Merck | Z688037-5EA | |
| Cleaning solution | NanoFCM | 17159 | In house cleaning solution for nanoanalyser |
| EVs from C2C12 | Gifted | Mouse line | |
| EVs from SW620 | Gifted | Human line | |
| Memglow - Fluorogenic Membrane Probe | Cytoskeleton | MG01 | non toxic cell membrane dye |
| NanoAnalyzer | NanoFCM | nano-flow cytometer for measurement of single particles | |
| PBS, pH 7.2 | Gibco Via Fisherscientific | 12549079 | Salt solution for dilution of samples and antibodies |
| Snaplock Microtubes, 0.60mL | Axygen Via Fisherscientific | 11371944 | Tubes required to load sample into nanoanalyser |
References
- Couch, Y., et al. A brief history of nearly EV-erything - The rise and rise of extracellular vesicles. Journal of Extracellular Vesicles. 10 (14), 12144 (2021).
- Doyle, L. M., Wang, M. Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 8 (7), 727 (2019).
- Grant, L. R., Milic, I., Devitt, A. Apoptotic cell-derived extracellular vesicles: structure-function relationships. Biochemical Society Transactions. 47 (2), 509-516 (2019).
- Rontogianni, S., et al. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Communications Biology. 2, 325 (2019).
- Sahoo, S., et al. Therapeutic and diagnostic translation of extracellular vesicles in cardiovascular diseases. Circulation. 143 (14), 1426-1449 (2021).
- Chiang, C. -. Y., Chen, C. Toward characterizing extracellular vesicles at a single-particle level. Journal of Biomedical Science. 26 (1), 9 (2019).
- Karimi, N., et al. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cellular and Molecular Life Sciences. 75 (15), 2873-2886 (2018).
- Akers, J. C., et al. Comparative analysis of technologies for quantifying Extracellular Vesicles (EVs) in Clinical Cerebrospinal Fluids (CSF). PLOS ONE. 11 (2), 0149866 (2016).
- Bachurski, D., et al. Extracellular vesicle measurements with nanoparticle tracking analysis - An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. Journal of Extracellular Vesicles. 8 (1), 1596016 (2019).
- Vogel, R., et al. Measuring particle concentration of multimodal synthetic reference materials and extracellular vesicles with orthogonal techniques: Who is up to the challenge. Journal of Extracellular Vesicles. 10 (3), 12052 (2021).
- Van Der Pol, E., et al. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. Journal of Thrombosis and Haemostasis. 12 (7), 1182-1192 (2014).
- Hartjes, T. A., Mytnyk, S., Jenster, G. W., Van Steijn, V., Van Royen, M. E. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering. 6 (1), 7 (2019).
- Zhao, Z., Wijerathne, H., Godwin, A. K., Soper, S. A. Isolation and analysis methods of extracellular vesicles (EVs). Extracellular Vesicles and Circulating Nucleic Acids. 2, 80-103 (2021).
- Johnsen, K. B., Gudbergsson, J. M., Andresen, T. L., Simonsen, J. B. What is the blood concentration of extracellular vesicles? Implications for the use of extracellular vesicles as blood-borne biomarkers of cancer. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1871 (1), 109-116 (2019).
- Zhu, S., et al. Light-scattering detection below the level of single fluorescent molecules for high-resolution characterization of functional nanoparticles. ACS Nano. 8 (10), 10998-11006 (2014).
- Van Der Pol, E., et al. Standardization of extracellular vesicle measurements by flow cytometry through vesicle diameter approximation. Journal of Thrombosis and Haemostasis. 16 (6), 1236-1245 (2018).
- Lucchetti, D., et al. Measuring extracellular vesicles by conventional flow cytometry: dream or reality. International Journal of Molecular Sciences. 21 (17), 6257 (2020).
- Suárez, H., et al. A bead-assisted flow cytometry method for the semi-quantitative analysis of Extracellular Vesicles. Scientific Reports. 7 (1), 11271 (2017).
- Tian, Y., et al. Protein profiling and sizing of extracellular vesicles from colorectal cancer patients via flow cytometry. ACS Nano. 12 (1), 671-680 (2018).
- Silva, A. M., et al. Quantification of protein cargo loading into engineered extracellular vesicles at single-vesicle and single-molecule resolution. Journal of Extracellular Vesicles. 10 (10), 12130 (2021).
- Dragovic, R. A., Southcombe, J. H., Tannetta, D. S., Redman, C. W. G., Sargent, I. L. Multicolor flow cytometry and nanoparticle tracking analysis of extracellular vesicles in the plasma of normal pregnant and pre-eclamptic women. Biology of Reproduction. 89 (6), 151 (2013).
- Teng, F., Fussenegger, M. Shedding light on extracellular vesicle biogenesis and bioengineering. Advanced Science. 8 (1), 2003505 (2021).
- Laulagnier, K., et al. Mast cell- and dendritic cell-derived exosomes display a specific lipid composition and an unusual membrane organization. Biochemical Journal. 380, 161-171 (2004).
- Simonsen, J. B. Pitfalls associated with lipophilic fluorophore staining of extracellular vesicles for uptake studies. Journal of Extracellular Vesicles. 8 (1), 1582237 (2019).
- Collot, M., et al. MemBright: A family of fluorescent membrane probes for advanced cellular imaging and neuroscience. Cell Chemical Biology. 26 (4), 600-614 (2019).
- Takov, K., Yellon, D. M., Davidson, S. M. Confounding factors in vesicle uptake studies using fluorescent lipophilic membrane dyes. Journal of Extracellular Vesicles. 6 (1), 1388731 (2017).
- Tian, Y., et al. Quality and efficiency assessment of six extracellular vesicle isolation methods by nano-flow cytometry. Journal of Extracellular Vesicles. 9 (1), 1697028 (2020).
- Mondal, A., Ashiq, K. A., Phulpagar, P., Singh, D. K., Shiras, A. Effective visualization and easy tracking of extracellular vesicles in glioma cells. Biological Procedures Online. 21, 4 (2019).
- Gelibter, S., et al. The impact of storage on extracellular vesicles: A systematic study. Journal of Extracellular Vesicles. 11 (2), 12162 (2022).
- Jayachandran, M., Miller, V. M., Heit, J. A., Owen, W. G. Methodology for isolation, identification and characterization of microvesicles in peripheral blood. Journal of Immunological Methods. 375 (1-2), 207-214 (2012).
- Mizenko, R. R., et al. Tetraspanins are unevenly distributed across single extracellular vesicles and bias sensitivity to multiplexed cancer biomarkers. Journal of Nanobiotechnology. 19 (1), 250 (2021).
- Kowal, J., et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America. 113 (8), 968-977 (2016).
- Laulagnier, K., et al. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cellular and Molecular Life Sciences. 75 (4), 757-773 (2018).
- Simonsen, J. B. What are we looking at? Extracellular vesicles, lipoproteins, or both. Circulation Research. 121 (8), 920-922 (2017).
- Sódar, B. W., et al. Low-density lipoprotein mimics blood plasma-derived exosomes and microvesicles during isolation and detection. Scientific Reports. 6, 24316 (2016).
- Brennan, K., et al. A comparison of methods for the isolation and separation of extracellular vesicles from protein and lipid particles in human serum. Scientific Reports. 10 (1), 103 (2020).
- Welton, J. L., Webber, J. P., Botos, L. -. A., Jones, M., Clayton, A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. Journal of Extracellular Vesicles. 4, 27269 (2015).
- Peruzzotti-Jametti, L., et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PLOS Biology. 19 (4), 3001166 (2021).
- Arab, T., et al. Characterization of extracellular vesicles and synthetic nanoparticles with four orthogonal single-particle analysis platforms. Journal of Extracellular Vesicles. 10 (6), 12079 (2021).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750 (2018).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved