Nettoyage optique rapide pour l’analyse à semi-haut débit des sphéroïdes tumoraux
Dans cet article
Résumé
Les sphéroïdes tumoraux sont de plus en plus utilisés pour évaluer les interactions cellule tumorale-microenvironnement et la réponse thérapeutique. Le présent protocole décrit une méthode robuste mais simple pour l’imagerie à semi-haut débit des sphéroïdes tumoraux 3D en utilisant un nettoyage optique rapide.
Résumé
Les sphéroïdes tumoraux deviennent rapidement monnaie courante dans la recherche fondamentale sur le cancer et le développement de médicaments. L’obtention de données concernant l’expression des protéines dans le sphéroïde au niveau cellulaire est importante pour l’analyse, mais les techniques existantes sont souvent coûteuses, laborieuses, utilisent un équipement non standard, provoquent une distorsion de taille importante ou sont limitées à des sphéroïdes relativement petits. Ce protocole présente une nouvelle méthode de montage et de nettoyage des sphéroïdes qui répondent à ces problèmes tout en permettant une analyse confocale de la structure interne des sphéroïdes. Contrairement aux approches existantes, ce protocole prévoit le montage et le nettoyage rapides d’un grand nombre de sphéroïdes à l’aide d’équipements standard et de fournitures de laboratoire. Le montage de sphéroïdes dans une solution de gel d’agarose-PBS au pH neutre avant d’introduire une solution de nettoyage adaptée à l’indice de réfraction minimise la distorsion de taille commune à d’autres techniques similaires. Cela permet une analyse quantitative et statistique détaillée où la précision des mesures de taille est primordiale. De plus, par rapport aux solutions de nettoyage liquide, la technique du gel d’agarose maintient les sphéroïdes fixés en place, ce qui permet la collecte d’images confocales tridimensionnelles (3D). Le présent article explique comment la méthode produit des images bi- et 3D de haute qualité qui fournissent des informations sur la variabilité intercellulaire et la structure sphéroïde interne.
Introduction
Les cultures cellulaires tridimensionnelles (3D), telles que les sphéroïdes, fournissent des modèles biologiquement réalistes et reproductibles de la croissance cellulaire agrégée 1,2. Ces modèles deviennent rapidement monnaie courante dans la recherche fondamentale et le développement de médicaments, où les différences de taille et de structure des sphéroïdes sont examinées entre les traitements pour déterminer l’efficacité des médicaments 3,4. Dans ces contextes, la capacité de collecter des informations détaillées à partir d’un grand nombre de sphéroïdes est très avantageuse, à la fois du point de vue de la puissance statistique et pour permettre une évaluation rapide du comportement cellulaire à travers plusieurs traitements.
Les techniques couramment utilisées pour obtenir des images microscopiques détaillées de la structure sphéroïde prennent du temps, sont coûteuses ou produisent des images de mauvaise qualité qui ne conservent pas les principales caractéristiques quantitatives telles que la taille des sphéroïdes 4,5. Par exemple, les techniques histologiques basées sur la cryosection peuvent fournir des images de haute qualité, mais prennent souvent beaucoup de temps, nécessitent une main-d’œuvre qualifiée et créent souvent des artefacts de sectionnement 6,7, tandis que les technologies élégantes, telles que la microscopie à éclairage monoplan (SPIM)8 et la microscopie multiphotonique9 nécessitent des microscopes spécialisés qui ne sont pas facilement disponibles. Les technologies modernes de microscopie ont récemment permis ce que l’on appelle la section optique, où les sphéroïdes sont placés dans une solution de nettoyage appariée à l’indice de réfraction et les images sont obtenues à l’aide de la microscopie confocale 4,5. Bien que ces techniques aient le potentiel de produire un rendement élevé, les problèmes courants incluent le mouvement des sphéroïdes lors de l’imagerie, la distorsion de la taille lors du nettoyage et le coût élevé des solutions de nettoyage propriétaires. En outre, de nombreux protocoles existants ne s’appliquent qu’aux sphéroïdes relativement petits de moins de 300 μm de diamètre ou de profondeur jusqu’à 100 μm, limitant la technologie aux premiers stades de la croissance tumorale 5,10,11.
Le présent protocole permet la collecte à semi-haut débit et à haut rendement d’images sphéroïdes détaillées à l’aide d’une solution de compensation à indice de réfraction à faible coût dérivée de procédures de nettoyage d’organes entiers12,13. Pour empêcher le mouvement des sphéroïdes pendant l’imagerie et fournir un support structurel pour réduire la distorsion de taille, les sphéroïdes sont montés dans un gel agarose-PBS dans une plaque inférieure en verre #1.5 de 24 puits. Étant donné que cette technique permet de monter plusieurs sphéroïdes dans chaque puits dans une plaque de 24 puits, jusqu’à 360 sphéroïdes (15 sphéroïdes / puits) peuvent être rapidement montés et imagés dans diverses conditions expérimentales. Une solution de nettoyage adaptée à l’indice de réfraction, construite à partir de consommables facilement disponibles, est utilisée pour éliminer optiquement les sphéroïdes montés et le gel environnant. Après une période de décantation de 24 h, ce protocole fournit des images 2D et 3D de haute qualité de la structure sphéroïde, même pour les sphéroïdes relativement grands (environ 700 μm de diamètre), avec moins de 2% de distorsion de taille.
Protocole
Le protocole décrit la préparation d’une quantité suffisante de sphéroïdes tumoraux pour monter une plaque de 24 puits (environ 240-360 sphéroïdes ou 10-15 sphéroïdes/puits) à 200 μL de gel d’agarose par puits et 500 μL de solution de nettoyage par puits. La procédure complète est illustrée à la figure 1.
1. 2% préparation de gel d’agarose-PBS
- Peser 0,5 g d’agarose à faible fusion (voir tableau des matériaux) dans une bouteille en verre et ajouter 25 mL de solution saline tamponnée au phosphate (PBS).
- Faire fondre l’agarose en faisant bouillir la solution au micro-ondes pendant 30 à 60 s avec le couvercle fermé mais non scellé et avec un tourbillon constant. Assurez-vous que l’agarose est complètement dissoute et que la solution ne bout pas.
REMARQUE: Le gel peut être conservé à température ambiante; vérifier le volume avant utilisation pour tenir compte de l’évaporation. Porter à l’état liquide (micro-ondes; 20-60 s avec tourbillon) avant d’utiliser.
2. Préparation de la solution de compensation
- Peser 9 g de N,N,N′,N′-Tétrakis(2-Hydroxypropyl) éthylènediamine, 22 g d’urée, 44 g de saccharose, 0,1 g de Triton X-100 et 24,9 g d’eau désionisée (voir tableau des matériaux).
REMARQUE: Il est plus facile de peser la quantité approximative de N,N,N′,N′-Tétrakis(2-Hydroxypropyl)éthylènediamine et d’ajuster le poids des autres composants pour obtenir la concentration souhaitée. - Chauffer la solution dans un bain-marie à 56 °C avec un mélange constant et continuer jusqu’à ce que tous les cristaux se dissolvent.
- Reposer la solution à température ambiante pour permettre aux bulles formées lors du mélange de remonter à la surface. La solution est stable à température ambiante jusqu’à 2 mois.
3. Préparation sphéroïde
- Générez des sphéroïdes à l’aide de la méthode de superposition d’agarose décrite précédemment 1,3,14.
REMARQUE: Les sphéroïdes générés par d’autres méthodes sont également compatibles avec ce protocole d’imagerie. - Coupez l’extrémité d’une pipette de 1 mL à l’aide d’un scalpel pour agrandir l’orifice.
REMARQUE: Tous les sphéroïdes ont été manipulés avec des pipettes de 1 mL ou 200 μL avec des pointes de coupe. - À l’aide de la pipette, aspirer les sphéroïdes des puits et les transférer dans un tube conique transparent de 1,5 mL. Plusieurs sphéroïdes des mêmes conditions peuvent être combinés dans le même tube.
REMARQUE: Laissez toujours les sphéroïdes se déposer au fond du tube pour aspirer le milieu. - Laver les sphéroïdes dans 1 mL de PBS deux fois, ajouter une solution neutre de paraformaldéhyde (PFA) préchauffée à 4 % et incuber les sphéroïdes à 37 °C pendant 20 min.
- Retirer la solution de PFA et laver les sphéroïdes deux fois avec 1 mL de PBS.
4. Coloration sphéroïde
- À l’aide d’une pipette, transférez jusqu’à 15 sphéroïdes dans un tube PCR de 200 μL.
REMARQUE: Pour les sphéroïdes fragiles, montez les sphéroïdes dans du gel (étapes 5.1-5.4) avant de continuer. - Perméabiliser les cellules en utilisant 200 μL de Triton X-100 à 0,5 % dans le PBS. Placez les tubes PCR à l’intérieur de tubes à bouchon à vis de 50 mL et incubez les sphéroïdes pendant 2 h à température ambiante avec une légère agitation.
REMARQUE: Toutes les incubations se font par agitation sur un rotor, un rouleau ou un agitateur (voir tableau des matériaux), sauf indication contraire. Ceci est important pour obtenir une coloration uniforme. Les sphéroïdes fixes dans PBS peuvent parfois coller sur les côtés de la pointe de la pipette. Le rinçage de la pointe dans du Triton X-100 à 0,5 % minimise l’adhérence des sphéroïdes aux pointes. - Remplacer la solution (étape 4.2) par 200 μL de tampon de dilution d’anticorps (ABDIL)15 et incuber pendant la nuit à température ambiante.
- Retirer ABDIL, ajouter 75 μL d’anticorps primaire dans ABDIL, et incuber à 4 °C pendant 2,5 jours.
- Retirer le surnageant et laver avec 200 μL de PBS avec 0,1% Tween et 0,1% Triton X-100 (PBS-TT) deux fois et incuber avec 200 μL de PBS-TT pendant 4 h à température ambiante.
- Retirer le PBS-TT, ajouter 100 μL d’anticorps secondaire dans ABDIL et incuber à 4 °C pendant 2,5 jours.
REMARQUE: DAPI ou un autre colorant nucléaire peut être ajouté à ce stade. La plupart des colorants nécessitent moins de temps d’incubation que les anticorps. - Retirer le surnageant et laver avec 200 μL de PBS-TT deux fois et incuber avec 200 μL de PBS-TT pendant 4 h. Les sphéroïdes sont prêts à être montés.
5. Montage
- À l’aide d’une pipette de 200 μL, transférer les sphéroïdes fixes et colorés dans des tubes PCR de 500 μL à un tube par condition (environ 10 à 15 sphéroïdes).
- Remplacer la solution par 200 μL de gel d’agarose liquide à 2 % (p/v) et centrifuger sur la rotation rapide (à la vitesse maximale fixée, voir tableau des matériaux) à température ambiante pendant 30 s.
REMARQUE: Sauf montage immédiat, placer dans un bloc chauffant à 50 ° C pour éviter le durcissement du gel. - Aspirer les sphéroïdes dans environ 50 μL de gel liquide d’agarose et distribuer dans le puits d’une plaque à fond de verre de 24 puits.
- Avant que le gel ne durcisse, séparez les sphéroïdes à l’aide d’une pointe de pipette dans le gel environnant et assurez-vous que les sphéroïdes sont recouverts de gel. En option, placez la plaque sur de la glace pour fixer rapidement le gel.
REMARQUE: Le nombre de sphéroïdes par puits dépend de la taille des sphéroïdes. Les sphéroïdes sont placés loin les uns des autres de sorte qu’un seul sphéroïde pénètre dans le champ de vision lors de l’imagerie. Ceci est important pour le traitement automatisé des images. - Ajouter 500 μL de solution de nettoyage (étape 2) par puits, en veillant à ce que le gel soit immergé. Incuber à RT pendant au moins 24 h et imager les sphéroïdes dans la solution de nettoyage. Les sphéroïdes sont stables dans cette solution jusqu’à un mois à température ambiante et lorsqu’ils sont protégés de l’évaporation.
6. Imagerie
- Choisissez un objectif avec une distance de travail suffisamment longue pour englober l’ensemble du sphéroïde, y compris la hauteur de montage du sphéroïde dans le vaisseau.
REMARQUE: Des objectifs d’une distance de travail de 3 mm ou plus sont nécessaires pour imager le plan équatorial des sphéroïdes. Des objectifs de grossissement plus élevés avec un NA plus élevé peuvent permettre une meilleure résolution, mais limiteront la profondeur maximale à laquelle les sphéroïdes peuvent être imagés. - Pour identifier le plan équatorial, ajustez la mise au point jusqu’à ce que la plus grande surface soit atteinte dans le plan XY et l’image avec le pourcentage de puissance laser, la tension du détecteur, le gain et les paramètres de décalage requis.
REMARQUE: Le pourcentage de puissance laser, la tension du détecteur, le gain et le décalage varient considérablement en fonction du fluorophore, de l’intensité de la coloration, de l’intensité du laser, de la sensibilité du détecteur, etc. - Pour les images 3D, définissez le début et la fin des sphéroïdes et choisissez l’intensité de signal appropriée à différentes profondeurs z à l’aide des paramètres de correction d’intensité z avant l’imagerie.
REMARQUE: Pour une meilleure résolution d’image, utilisez la fréquence d’échantillonnage de Nyquist pour x, y et z (pour plus de détails, voir la référence16).
Résultats
Pour démontrer la capacité de cette méthode de nettoyage à fournir des images bidimensionnelles et tridimensionnelles de haute qualité, des sphéroïdes d’un diamètre de 300 à 600 μm ont été cultivés à partir delignées cellulaires de mélanome transduites FUCCI-WM164et FUCCI-WM983bà base de biquitination F ell Cycle I (FUCCI) à base de C ell C ycle I(FUCCI) , qui expriment la protéine monomère Kusabira Orange2 (mKO2) et la protéine monomère Azami Green (mAG) lorsqu’elles sont dans Gap1, et la phase précoce S/Gap2/mitose du cycle cellulaire, respectivement selon les procédures fournies à l’étape 3 1,3,14. Les sphéroïdes ont ensuite été fixés avec une solution de formaldéhyde à 4 % à 37 °C, perméabilisés et colorés avec de l’anti-p27kip1/anti-lapin Alexa Fluor 647 anti-pimonidazole/anti-souris Alexa Fluor 647, DAPI ou DRAQ7 (voir tableau des matières) (Figure 2). Tous les fichiers de microscopie sont téléchargés dans le référentiel GitHub (https://github.com/ap-browning/SpheroidMounting). Par rapport aux sphéroïdes montés sur PBS, la solution de nettoyage fournit des images de haute clarté avec une distorsion de taille minimale (Figure 2A). Le protocole permet une imagerie à haute résolution des détails au niveau cellulaire plus profondément dans les sphéroïdes sans section histologique, et l’image en coupe transversale a été obtenue à partir d’un objectif d’air 20x (0,7 NA) à une résolution de 4096 x 4096 px sans couture (Figure 2B). En utilisant un grossissement plus faible et un objectif à ouverture numérique plus faible avec une distance de travail plus longue, des images confocales 3D qui fournissent des détails au niveau cellulaire à une profondeur d’au moins 200 μm peuvent être obtenues (Figure 2C). Les sphéroïdes ont également été cryosectionnés et colorés conformément au protocole de Spoerri et al.4 et comparés à la coloration sphéroïde entière (Figure 2D, E). La figure 2D montre la région hypoxique du sphéroïde coloré par le pimonidazole, et la figure 2E montre la coloration p27kip1 qui marque l’arrêt du cycle cellulaire (jaune) et la tache nucléaire DAPI (gris). La localisation des protéines et le schéma de coloration sont similaires entre la cryosection et le nettoyage et ne sont donc pas affectés par cette méthode de nettoyage.
La correction de l’intensité du signal en z permet l’imagerie de l’ensemble du sphéroïde. Cependant, la diffusion de la lumière due au noyau nécrotique limite la capacité d’imager la face cachée du sphéroïde. Une pénétration plus profonde et moins de diffusion d’un fluorophore rouge lointain, tel que la coloration nucléaire DRAQ7, permet d’améliorer encore la représentation de la structure sphéroïde 3D (Figure 3). Le film 1 montre le rendu 3D des sphéroïdes FUCCI colorés avec DRAQ7. Une tranche z plus mince peut permettre une meilleure résolution z, mais cela augmente considérablement le temps d’imagerie et le photoblanchiment des fluorophores.
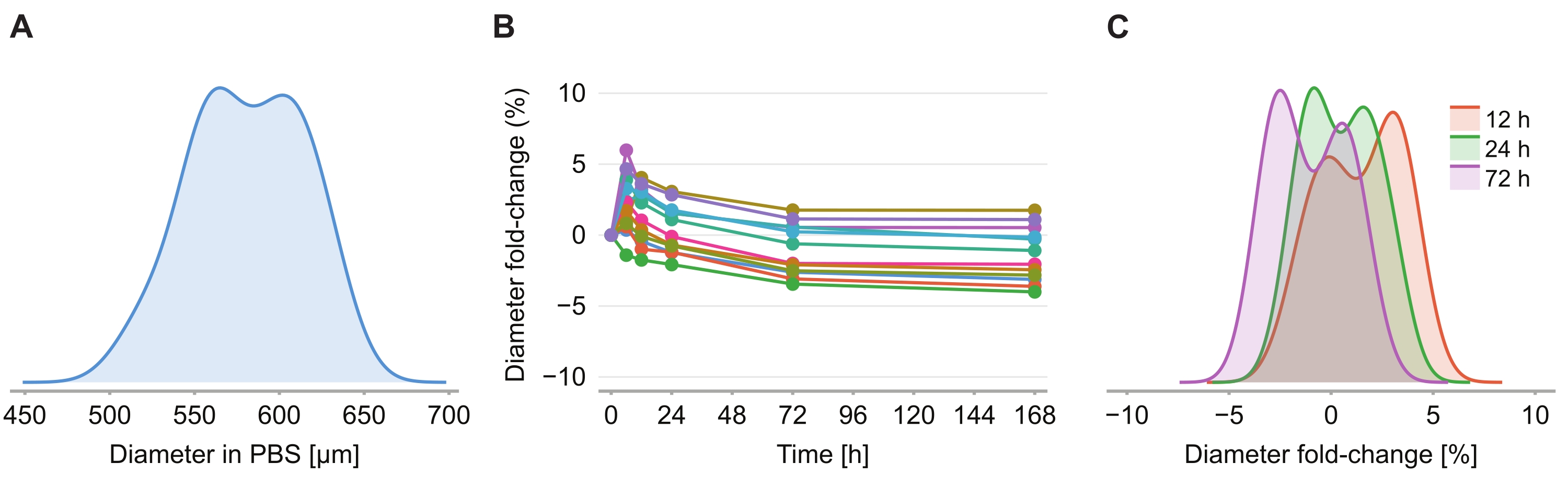
Pour déterminer si la solution de nettoyage provoque une distorsion de taille, douze sphéroïdes dans un gel d’agarose-PBS à 2 % ont été imagés à 6 h, 12 h, 24 h, 72 h et 168 h après l’introduction de la solution de nettoyage. Les images ont été résumées en déterminant le diamètre du sphéroïde, défini sur la base d’une sphère ayant la même section transversale que le sphéroïde (figure 4A). Alors que l’on observe que la taille des sphéroïdes augmente légèrement au cours des 6 premières heures, indiquée par un changement de pliage de diamètre compris entre 2 % et 6 % (figure 4B), après 24 h à 72 h, les sphéroïdes reviennent à une taille approximativement égale à la taille correspondante dans la fixation PBS post-PFA (Figure 4C).

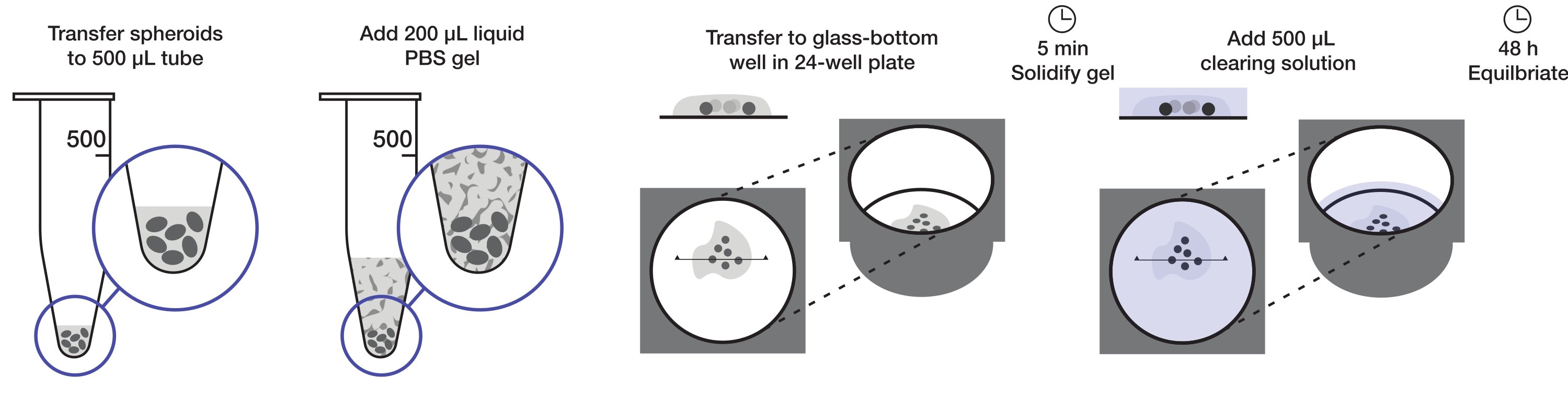
Figure 1 : Illustration du protocole de montage et de nettoyage des sphéroïdes. Les sphéroïdes fixes et colorés sont transférés dans un tube de 500 μL; l’excès de liquide est remplacé par un gel d’agarose-PBS de 200 μL et centrifugé. Les sphéroïdes sont ensuite transférés dans un puits à fond de verre dans une plaque de 24 puits. Une fois que le gel est autorisé à se solidifier, une solution de nettoyage de 500 μL est ajoutée et les sphéroïdes sont autorisés à s’équilibrer. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 2 : Comparaison des sphéroïdes de mélanome humain FUCCI éliminés et non nettoyés. La coloration indique que les noyaux cellulaires sont positifs pour la mKO2 (rouge), ce qui indique les cellules dans l’intervalle 1; et noyaux cellulaires positifs pour mAG (vert), ce qui indique les cellules dans l’intervalle 2. (A) Sphéroïdes cultivés à partir de 5000 cellules FUCCI-WM983b, récoltées au jour 10 et imagées dans un gel d’agarose-PBS et 24 h après l’ajout d’une solution de nettoyage. La comparaison d’images en champ clair et confocales avant et après le nettoyage de la solution montre une distorsion de taille minimale et un gain de clarté important. Les images sont obtenues à l’aide d’un objectif 10x. (B) Sphéroïdes cultivés à partir de cellules FUCCI-WM164 perméabilisées à l’aide de Triton X-100 et colorés avec DRAQ7, colorant tous les noyaux cellulaires. L’image est obtenue à l’aide d’un objectif 20x (0,75 NA), démontrant que la solution de nettoyage permet une imagerie haute résolution des détails au niveau de la cellule. (C) Des images 3D (10x, 0,4 NA) ont été obtenues de sphéroïdes FUCCI-WM164 dans PBS, et 24 h après l’ajout de la solution de nettoyage. Le réglage de la puissance, de la tension et du décalage du laser à différents plans Z permet d’imager plus profondément à l’intérieur du sphéroïde. (D,E) Comparaison entre la cryosection et le sphéroïde entier nettoyé coloré pour le pimonidazole et p27kip1. (D) La coloration au pimonidazole dans le magenta montre la région hypoxique dans les sphéroïdes. Le rouge et le vert indiquent FUCCI. (E) Cryosections et sphéroïdes nettoyés montrant DAPI (gris) et p27kip1 (jaune). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 3 : Le nettoyage permet d’imager plus profondément dans le sphéroïde avec une perte de lumière minimale. Images de microscopie confocale d’un sphéroïde de mélanome humain FUCCI à un grossissement de 10x et à un NA inférieur (0,4), permettant une imagerie à une profondeur z plus élevée avec une perte de signal minimale. (A) Tranches de 3,88 μm de noyaux sphéroïdes colorés avec DRAQ7. (B-D) y/z-résolution dans les canaux 488 (mAG), 568 (mKO2) et 647 nm (DRAQ7), respectivement. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

Figure 4 : La solution de nettoyage a un impact minimal sur la taille des sphéroïdes. (A) Distribution de la taille initiale des sphéroïdes (diamètre équivalent) dans le gel PBS (n = 12 sphéroïdes). (B) Changement de pliage du diamètre au fil du temps depuis l’ajout de la solution de nettoyage. À 0 h, les sphéroïdes sont en gel PBS uniquement. (C) Distribution des changements de pli de diamètre à 12 h, 24 h et 72 h. Veuillez cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Film 1 : Rendu 3D d’un sphéroïde FUCCI teinté avec DRAQ7. Veuillez cliquer ici pour télécharger ce film.
Discussion
Un protocole pour obtenir des images bidimensionnelles et tridimensionnelles de haute qualité des sphéroïdes tumoraux est présenté ici. Les méthodes existantes, telles que CLARITY, See deep brain (SeeDB) et ScaleS, provoquent souvent une distorsion de taille allant jusqu’à 30%, tandis que des techniques telles que l’alcool benzylique / benzoate de benzyle (BABB) et l’imagerie 3D d’organes éliminés par solvant (3DISCO) peuvent éteindre la protéine fluorescente18. Beaucoup de ces méthodes sont conçues pour nettoyer les tissus avec intégrité structurelle et déformer la taille et la structure lorsqu’elles sont appliquées aux sphéroïdes18. Contrairement à d’autres protocoles qui utilisent des solutions de compensation coûteuses disponibles dans le commerce, ce protocole utilise des consommables facilement disponibles tout en maintenant la clarté optique et la fluorescence endogène et en minimisant la distorsion de taille. L’incorporation de sphéroïdes dans le gel agarose-PBS fournit un soutien structurel aux sphéroïdes et minimise le choc osmotique lorsque la solution de nettoyage est ajoutée. Ceci est crucial lors de l’imagerie de sphéroïdes fragiles après un traitement médicamenteux. On suppose que cette méthode de nettoyage optique convient aux sphéroïdes formés par n’importe quelle méthode puisque ce protocole est adapté de la clairance des tissus entiers. L’hypothèse est basée sur la similitude dans les sphéroïdes obtenue par différentes méthodes de formation des sphéroïdes. Le choix du fixateur peut affecter la taille du sphéroïde ainsi que la fluorescence endogène. Cette méthode de nettoyage convient aux sphéroïdes fixés avec une solution neutre de PFA à 4%. Des tests supplémentaires sont nécessaires pour vérifier sa compatibilité avec d’autres fixateurs.
Étant donné que cette technique permet de monter simultanément plusieurs sphéroïdes dans une plaque multi-puits, elle est bien adaptée aux pipelines d’analyse quantitative qui nécessitent des informations sur la structure des sphéroïdes allant jusqu’à 360 sphéroïdes par plaque de 24 puits. Les microscopes dotés de fonctions automatisées de cartographie des scènes et des plaques peuvent rendre l’imagerie moins manuelle. Même si cette méthode est plus rapide et plus facile que le sectionnement, elle est actuellement inadaptée à une automatisation complète. Cependant, les images obtenues par cette méthode conviennent au traitement automatiséd’images 4,19, et la vitesse à laquelle les sphéroïdes peuvent être montés à l’aide de ce protocole permet une analyse quantitative de la structure interne des sphéroïdes 20,21,22.
Pour colorer les sphéroïdes entiers, la concentration, le volume et le temps d’incubation des anticorps doivent être optimisés pour chaque anticorps. À titre indicatif, utilisez 2,5 fois la concentration recommandée d’anticorps d’immunofluorescence 2D et 100 à 200 μL d’anticorps, selon le nombre de sphéroïdes par tube. Assurez-vous que tous les sphéroïdes sont recouverts d’une solution de coloration lorsqu’ils sont sur le rotor. Le temps d’incubation dépend de nombreux facteurs, y compris la taille et la densité des sphéroïdes et de l’anticorps, et peut varier de 16 à 72 h. Malgré la méthode permettant la détection du signal plus profondément dans le sphéroïde, les fluorophores excités par les UV provoquent une diffusion importante de la lumière, conduisant à un faible rapport signal/bruit. Lors du choix des fluorophores, il faut prendre soin de visualiser la protéine cible. Par exemple, la coloration de protéines moins abondantes et structurelles avec des fluorophores de plus grande longueur d’onde et des protéines plus abondantes ou des taches nucléaires avec des fluorophores de longueur d’onde plus courte permettra d’obtenir le meilleur résultat. Enfin, les sphéroïdes nettoyés montrent toujours une perte de lumière due à la diffusion dans le noyau nécrotique, évidente dans la dimension y/z des images obtenues de sphéroïdes de 600 μm (Figure 2C).
Une imagerie à grossissement plus élevé avec des objectifs NA plus élevés est possible avec des sphéroïdes montés et effacés à l’aide de ce protocole, mais la distance de travail de l’objectif limite la profondeur d’imagerie. Pour une lentille d’immersion dans l’huile, il est important d’utiliser de l’huile qui a un RI de 1,51 pour le meilleur résultat.
Pour résumer, la méthode de nettoyage intégré au gel agarose-PBS permet de visualiser les cellules profondément à l’intérieur des sphéroïdes à l’aide de consommables couramment disponibles. Les sphéroïdes montés et éliminés à l’aide de cette méthode subissent une distorsion de taille minimale et maintiennent leur intégrité structurelle, ce qui permet la collecte de données de haute qualité relatives à la structure sphéroïde interne et une quantification automatisée ultérieure.
Déclarations de divulgation
Olympus a contribué aux coûts de publication.
Remerciements
Cette recherche a été réalisée à l’Institut de recherche translationnelle (TRI), Woolloongabba, QLD. TRI est soutenu par une subvention du gouvernement australien. Nous remercions le personnel de l’installation centrale de microscopie de TRI pour son soutien technique exceptionnel. Nous remercions le professeur Atsushi Miyawaki, RIKEN, Wako-city, Japon, pour avoir fourni les constructions FUCCI, le professeur Meenhard Herlyn et Mme Patricia Brafford, The Wistar Institute, Philadelphie, PA, pour avoir fourni les lignées cellulaires. Nous remercions le Dr Loredana Spoerri d’avoir fourni des images de cryosection C8161.
Ce travail a été soutenu par des subventions de projet à N.K.H.: Australian Research Council (DP200100177) et Meehan Project Grant (021174 2017002565).
matériels
| Name | Company | Catalog Number | Comments |
| #1.5 glass bottom 24-well plate | Celvis | P24-1.5H-N | |
| 500 µL clear PCR tubes | Sigma | HS4422 | |
| Alexa Fluor 647 AffiniPure Donkey Anti-Mouse IgG (H+L) | Jackson Immuno research | 715-605-151 | Dilution used 1:500 |
| Bovine serum albumin | Sigma | A7906 | Final concentration 2% w/v |
| DAPI | Sigma | D9542-10MG | Final concentration 5 µg/mL |
| Deionized water | MILLI Q | ||
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermofisher | A-31573 | Dilution used 1:500 |
| DRAQ7 | Thermofisher | D15106 | Dilution used 1:250 |
| Heating block | Ratek | DBH10 | or similar equipment |
| Hypoxyprobe Kits | Hypoxyprobe | HP1-1000Kit | Antibody dilution used 1:500 |
| Low-melting agarose powder | Sigma | A9414 | Final concentration 2% w/v |
| Microwave | Sharp | ||
| N,N,N′,N′-Tetrakis(2-Hydroxypropyl)ethylenediamine | Sigma | 122262 | Final concentration 9% w/w |
| NaCl | Sigma | S9888 | Final concentration 150 mM |
| NaN3 | Sigma | S2002 | Final concentration 0.10% w/v |
| p27 Kip1 (D69C12) XP | Cell Signalling technology | 3686S | Dilution used 1:500 |
| Paraformaldehyde solution | Proscitech | C004 | Final concentration 4% w/v |
| Phosphate Buffered Saline (PBS) | Thermofisher | 18912014 | Final concentration 1x |
| Pipette | Eppendorf | ||
| Quickspin minifuge | or similar equipment | ||
| Roller | Ratek | BTR10-12V | or similar equipment |
| Rotor | Ratek | RSM7DC | or similar equipment |
| Shaker | Ratek | EOM5 | or similar equipment |
| Sucrose | Sigma | S9378 | Final concentration 44% w/w |
| Tris-HCl pH 7.4 | Sigma | T5941 | Final concentration 20 mM |
| Triton X-100 | Sigma | X100 | Final concentration 0.10% v/v |
| Triton X-100 | Sigma | X100 | Final concentration 0.1% v/v |
| Tween 20 | Sigma | P1379 | Final concentration 0.1% v/v |
| Urea | Sigma | U5379 | Final concentration 22% w/w |
Références
- Beaumont, K. A., Anfosso, A., Ahmed, F., Weninger, W., Haass, N. K. Imaging- and flow cytometry-based analysis of cell position and the cell cycle in 3D melanoma spheroids. Journal of Visualized Experiments. (106), e53486 (2015).
- Hirschhaeuser, F., et al. Multicellular tumor spheroids: an underestimated tool is catching up again. Journal of Biotechnology. 148 (1), 3-15 (2010).
- Smalley, K. S., Lioni, M., Noma, K., Haass, N. K., Herlyn, M. In vitro three-dimensional tumor microenvironment models for anticancer drug discovery. Expert Opinion on Drug Discovery. 3 (1), 1-10 (2008).
- Spoerri, L., Gunasingh, G., Haass, N. K. Fluorescence-based quantitative and spatial analysis of tumour spheroids: A proposed tool to predict patient-specific therapy response. Frontiers in Digital Health. 3, 668390 (2021).
- Nürnberg, E., et al. Routine optical clearing of 3D-cell cultures: Simplicity forward. Frontiers in Molecular Biosciences. 7, 20 (2020).
- Kabadi, P. K., et al. Into the depths: Techniques for in vitro three-dimensional microtissue visualization. BioTechniques. 59 (5), 279-286 (2015).
- Ivanov, D. P., Grabowska, A. M. Spheroid arrays for high-throughput single-cell analysis of spatial patterns and biomarker expression in 3D. Scientific Reports. 7, 41160 (2017).
- Spoerri, L., et al. Phenotypic melanoma heterogeneity is regulated through cell-matrix interaction-dependent changes in tumor microarchitecture. bioRxiv. , (2021).
- Haass, N. K., et al. Real-time cell cycle imaging during melanoma growth, invasion, and drug response. Pigment Cell & Melanoma Research. 27 (5), 764-776 (2014).
- Ahmad, A., et al. Clearing spheroids for 3D fluorescent microscopy: combining safe and soft chemicals with deep convolutional neural network. bioRxiv. , 428996 (2021).
- Villani, T., Rossi, A. E., Sherman, H. Image-based characterization of 3-D cell culture models grown in spheroid microplates. American Laboratory. 50 (6), 12 (2018).
- Lloyd-Lewis, B., et al. Imaging the mammary gland and mammary tumours in 3D: optical tissue clearing and immunofluorescence methods. Breast Cancer Research. 18 (1), 127 (2016).
- Tainaka, K., et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 159 (4), 911-924 (2014).
- Spoerri, L., Beaumont, K. A., Anfosso, A., Haass, N. K. Real-time cell cycle imaging in a 3D cell culture model of melanoma. Methods in Molecular Biology. 1612, 401-416 (2017).
- Cold Spring Harbor. Antibody Dilution Buffer (Abdil). Cold Spring Harbor Protocols. , (2018).
- Pawley, J. B., Pawley, J. B. . Handbook Of Biological Confocal Microscopy. , 20-42 (2006).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Tian, T., Yang, Z., Li, X. Tissue clearing technique: Recent progress and biomedical applications. Journal of Anatomy. 238 (2), 489-507 (2021).
- Browning, A. P., Murphy, R. J. . Zenodo. , (2021).
- Browning, A. P., et al. Quantitative analysis of tumour spheroid structure. eLife. 10, 73020 (2021).
- Klowss, J. J., et al. A stochastic mathematical model of 4D tumour spheroids with real-time fluorescent cell cycle labelling. Journal of The Royal Society Interface. 19 (189), 20210903 (2022).
- Murphy, R. J., Browning, A. P., Gunasingh, G., Haass, N. K., Simpson, M. J. Designing and interpreting 4D tumour spheroid experiments. Communications Biology. 5 (1), 91 (2022).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.