Super-Resolution Microscopy of the Synaptonemal Complex Within the Caenorhabditis elegans Germline
In This Article
Summary
Super-resolution microscopy can provide a detailed insight into the organization of components within the synaptonemal complex in meiosis. Here, we demonstrate a protocol to resolve individual proteins of the Caenorhabditis elegans synaptonemal complex.
Abstract
During meiosis, homologous chromosomes must recognize and adhere to one another to allow for their correct segregation. One of the key events that secures the interaction of homologous chromosomes is the assembly of the synaptonemal complex (SC) in meiotic prophase I. Even though there is little sequence homology between protein components within the SC among different species, the general structure of the SC has been highly conserved during evolution. In electron micrographs, the SC appears as a tripartite, ladder-like structure composed of lateral elements or axes, transverse filaments, and a central element.
However, precisely identifying the localization of individual components within the complex by electron microscopy to determine the molecular structure of the SC remains challenging. By contrast, fluorescence microscopy allows for the identification of individual protein components within the complex. However, since the SC is only ~100 nm wide, its substructure cannot be resolved by diffraction-limited conventional fluorescence microscopy. Thus, determining the molecular architecture of the SC requires super-resolution light microscopy techniques such as structured illumination microscopy (SIM), stimulated-emission depletion (STED) microscopy, or single-molecule localization microscopy (SMLM).
To maintain the structure and interactions of individual components within the SC, it is important to observe the complex in an environment that is close to its native environment in the germ cells. Therefore, we demonstrate an immunohistochemistry and imaging protocol that enables the study of the substructure of the SC in intact, extruded Caenorhabditis elegans germline tissue with SMLM and STED microscopy. Directly fixing the tissue to the coverslip reduces the movement of the samples during imaging and minimizes aberrations in the sample to achieve the high resolution necessary to visualize the substructure of the SC in its biological context.
Introduction
Reducing the number of chromosomes by half during meiosis is key to generating healthy progeny in sexually reproducing organisms. To achieve this reduction in chromosome number, homologous chromosomes must pair and segregate during meiosis I. To ensure the accurate segregation of homologous chromosomes, germ cells undergo an extended prophase I, during which homologous chromosomes pair, synapse, and recombine to generate physical links between homologs1. The SC has emerged as the central structure that is key in regulating the correct progression through meiotic prophase2.
The SC is a complex whose general structure is evolutionarily conserved, even though there is little homology between its protein components. The SC was first identified in electron micrographs as a tripartite, ladder-like structure consisting of two lateral elements or axes, a central region formed by transverse filaments, and a central element3,4. Determining the organization of individual components within the complex is key to advancing our understanding of the SC's role during meiotic prophase.
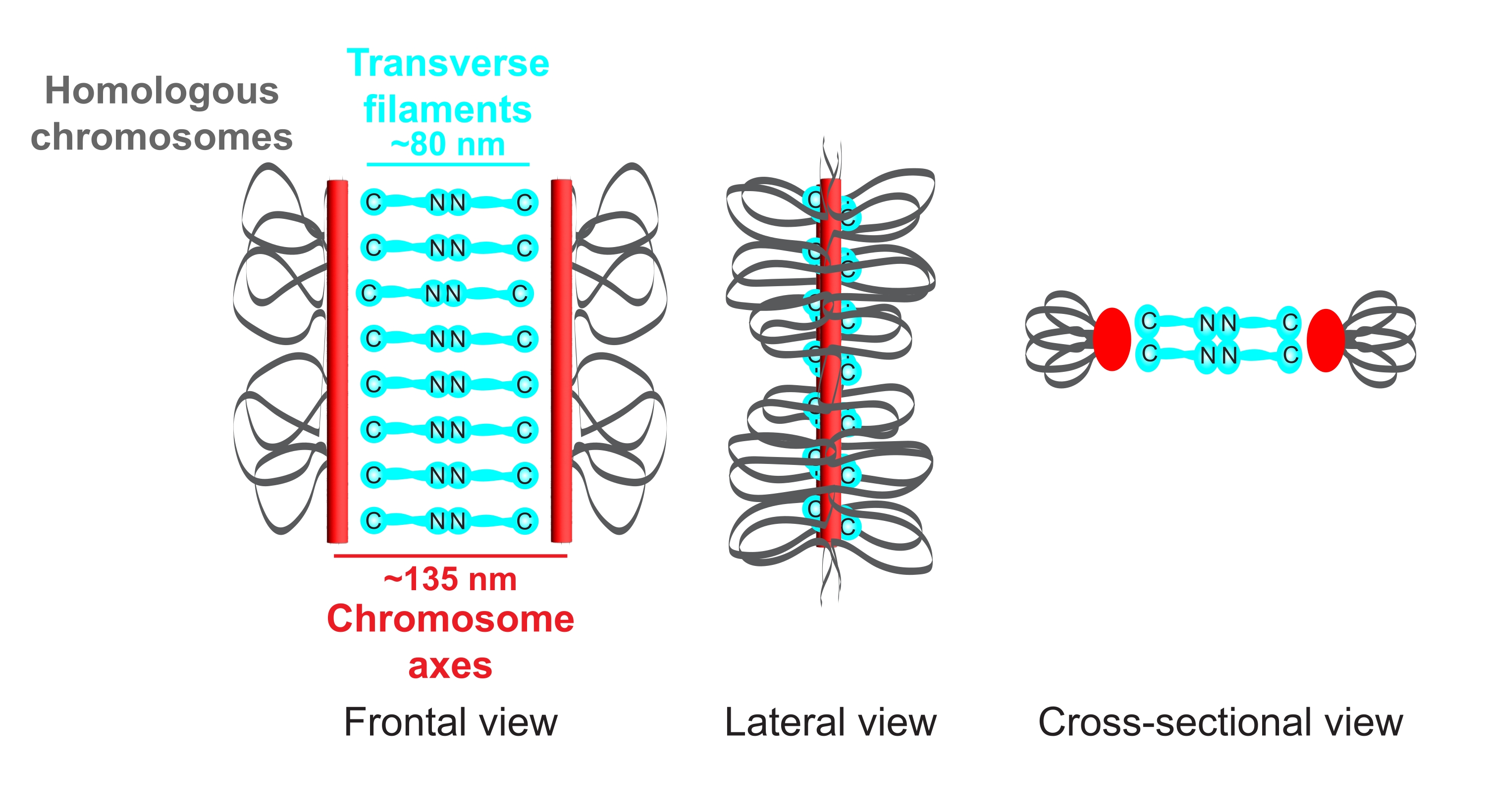
The model organism C. elegans is ideally suited to study the structure and function of the SC since its germlines contain a large number of meiotic nuclei with fully assembled SCs5. Genetic and biochemical studies have revealed that the chromosome axes are formed by three distinct cohesin complexes6,7 and four HORMA domain proteins called HTP-1/2/3 and HIM-37,8,9,10,11 in C. elegans. In the central region of the SC, six proteins containing coiled-coil domains have been identified to date12,13,14,15,16,17. To bridge the distance between the two axes, SYP-1, -5, and -6 dimerize in a head-to-head manner (Figure 1), while three additional proteins stabilize their interaction in the central element16,17,18,19.
Obtaining detailed insight into the organization of these proteins is essential in understanding the SC's many functions during meiosis. Since the width of the central region of the SC is only ~100 nm, its substructure cannot be resolved by diffraction-limited fluorescence microscopy. However, visualizing components within a structure of this size is readily achievable by super-resolution microscopy. Indeed, structured illumination microscopy (SIM), expansion microscopy20, stimulated-emission depletion (STED) microscopy21, and single-molecule localization microscopy (SMLM)22,23 have emerged as essential tools to study the molecular architecture of the SC across species16,24,25,26,27,28,29,30.
To overcome the resolution limit, STED microscopy relies on overlaying the diffraction-limited spot of the emission light with a donut-shaped beam from the STED laser, which theoretically constricts the point spread function down to molecular dimensions31,32. However, the resolution that is practically achievable by STED within biological samples remains in the range of a few tens of nanometers in xy33.
Even higher resolution in biological samples can be obtained with SMLM techniques. SMLM harnesses the blinking properties of specific fluorophores to resolve objects at the sub-diffraction level by separating spatially overlapping fluorophores in time. The sample is then imaged repeatedly to capture different subsets of fluorophores. The position of the fluorophores within the sample is then determined by fitting the point spread function (PSF) to the obtained signals across all images, which can resolve structures down to 15 nm23,34.
Taken together, the localized images encode the positions of all the fluorophores. The resolution of SMLM is determined by the labeling density and the blinking characteristics of the fluorophore. According to the Nyquist-Shannon criterion, it is impossible to reliably resolve objects that are less than twice the average label-to-label distance. Thus, a high labeling density is needed for high-resolution imaging. For the SC in C. elegans, a high labeling density can be achieved by using epitope tags attached to specific sites of endogenous proteins using genome editing. The epitope tags can then be stained at a high density using specific monoclonal antibodies with high affinities19,30. At the same time, the on-cycle of individual fluorophores must be short enough to ensure that spatially overlapping fluorophores are not captured at the same time35.
Due to these two requirements, resolving the structure of big macromolecular complexes such as the SC requires imaging a sufficiently large number of images, and can thus take several hours. The pitfall of long imaging times is that samples tend to drift due to movement of the stage or small currents within the sample buffer; even small movements in the order of 10 nm are detrimental at nm resolution and must be corrected for. However, the drift correction methods commonly used are not robust enough to accurately overlay images of two channels imaged sequentially36. This is problematic because biological questions often ask for precise detection and localization of multiple targets within the same sample. To circumvent these issues, methods such as ratiometric imaging have been developed. Ratiometric imaging allows for the simultaneous imaging of multiple fluorophores with overlapping excitation and emission spectra, with a subsequent assignment of each detected signal to its respective fluorophore based on the ratio of intensities in spectrally distinct channels37,38.
Additionally, studying the organization of macromolecular complexes such as the SC calls for three-dimensional (3D) information. To achieve super-resolution in three dimensions (3D-SMLM), a cylindrical lens is incorporated in the optical path of the emitted light that distorts the shape of the PSF of a fluorophore depending on its distance from the focal plane. Hence, the precise position of a fluorophore in the z-plane can be extrapolated by analyzing the shape of its emission signal35,39. Combining these advances in SMLM allows for imaging of the 3D organization of macromolecular complexes, including the SC.
Protocol
1. Preparation of solutions and coverslips
NOTE: See the Table of Materials for details related to all materials and reagents and Table 1 for the composition of solutions used in this protocol.
- Poly-L-lysine-coated coverslips
- Prepare 0.01% (w/v) poly-L-lysine (see Table 1).
- Wash a precision coverslip (24 mm diameter; 0.17 ± 0.005 mm, No. 1.5) in ethanol for 10-30 min. Rinse the coverslip with ddH2O to remove ethanol, and leave the coverslip to dry at room temperature.
- Plasma clean the coverslip using a plasma cleaner.
NOTE: Plasma cleaning increases the hydrophilicity of the coverslip and facilitates the following steps. If a plasma cleaner is not available, this step can be skipped, although this may require adjusting the volume and/or concentration of the poly-L-lysine solution. This modification has not been tested. - Place one drop (120 µL) of 0.01% (w/v) poly-L-lysine on the coverslip. Incubate for 10 min at room temperature.
- After incubation, rinse the coverslip in ddH2O and dry at room temperature. Store at 4 °C up to 1 month.
- F(ab')2 fragments conjugated with fluorescent organic dyes
- Add in the following order to a PCR tube: 10 µL of 0.6-0.7 mg/mL F(ab')2 fragment in PBS, 1 µL of 0.1 M NaHCO3 (pH 8.3) and 1 µL of 1 mM succinimidyl (NHS) ester reactive fluorophore in DMSO (molar ratio of F(ab')2:dye is ~1:17). Mix well by pipetting up and down.
- Incubate for 1 h at room temperature.
- Separate the F(ab')2 fragment from the remaining free reactive dye using a desalting column (7K MWCO) following the manufacturer's specifications. Use 1x PBS for equilibration of the column and elution of the labeled F(ab')2 fragment.
- Store the labeled F(ab')2 fragment at 4 °C for up to 3 months.
NOTE: Storage times of longer than 3 months have not been tested.
2. Dissection and fixation
NOTE: The dissection and fixation procedures are modified from previously recommended procedures16,40 to obtain optimal samples for super-resolution microscopy.
- Dissection
- Pick age-matched C. elegans worms (grown at 20 °C for this study) into a 30 µL drop of EBTT (1x Egg buffer41 with 0.2% nonionic detergent, Table 1) on a coverslip (22 mm x 22 mm, No. 1). Place the cover slip on a glass slide for easier manipulation. Wash with 30 µL of EBTT by pipetting up and down several times. Remove 30 µL of the solution to leave a 30 µL drop on the coverslip.
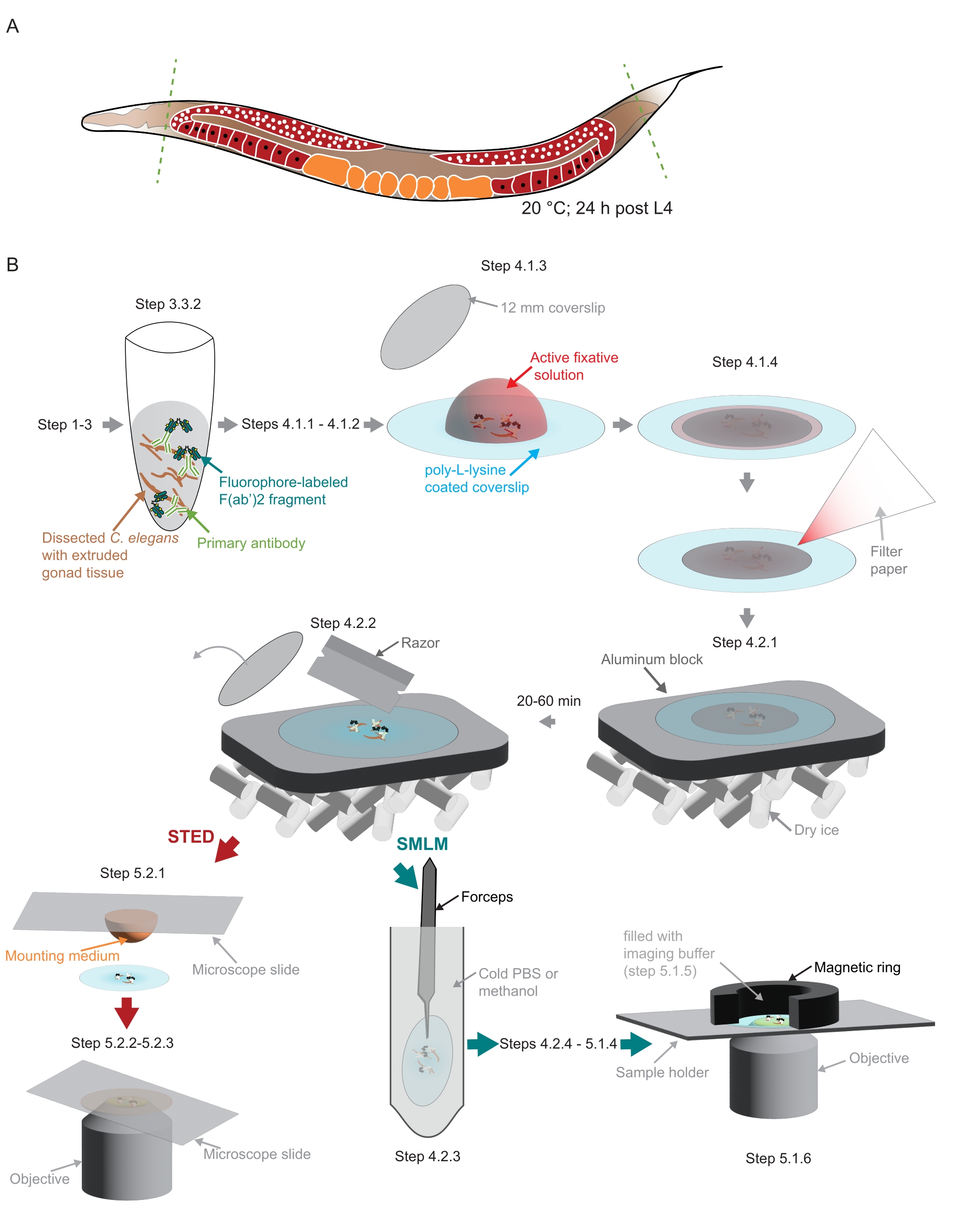
NOTE: Small amounts of nonionic detergent must be added to all solutions in which worms are pipetted to prevent the worms from sticking to the plastic tips. - Use a scalpel blade to cut off the heads and/or the tails of the worms to extrude the gonad (Figure 2A).
- Pick age-matched C. elegans worms (grown at 20 °C for this study) into a 30 µL drop of EBTT (1x Egg buffer41 with 0.2% nonionic detergent, Table 1) on a coverslip (22 mm x 22 mm, No. 1). Place the cover slip on a glass slide for easier manipulation. Wash with 30 µL of EBTT by pipetting up and down several times. Remove 30 µL of the solution to leave a 30 µL drop on the coverslip.
- Fixation
- Pipet 30 µL of fixative solution (Table 1) into the drop of the dissected worms and pipet up and down to mix.
NOTE: Pipetting up and down a few times may help to release more gonads. - Fix for exactly 1 min after adding the fixative solution.
- Stop the fixation by transferring the worms into a PCR tube filled with TBST (Table 1). Transfer the worms in as little volume as possible (~15 µL).
- Spin down the PCR tube on a mini benchtop centrifuge (2,000 × g, 10 s). Remove the supernatant and wash 2x with 200 µL of TBST each.
- Wash with 200 µL of PBST (Table 1) for 5-10 min. Repeat steps 2.1.1 to 2.2.5 for up to four samples while keeping the dissected samples on ice.
NOTE: If processing more than four samples, proceed with steps 2.2.6 to 2.2.7 after every four samples to ensure sample fixation remains consistent across all samples. - Spin the dissected samples on a mini benchtop centrifuge (2,000 × g, 10 s), remove the PBST, and add 50-100 µL of cold methanol (-20 °C).
CAUTION: Methanol is toxic. Wear protective equipment and avoid inhalation. - Mix by pipetting up and down and leave the samples in methanol for 30-60 s. Wash the samples 2x in 200 µL of PBST.
NOTE: If processing more than four samples, proceed with the dissection of the remaining samples (steps 2.1.1 to 2.2.7). - Wash the samples a third time with 200 µL of PBST.
- Pipet 30 µL of fixative solution (Table 1) into the drop of the dissected worms and pipet up and down to mix.
3. Antibody incubations
- Blocking
- Block the samples in 1x Blocking Solution (Table 1) for 45-60 min at room temperature.
NOTE: The incubation time can range from 30 min at room temperature to several days at 4 °C (testing was done up to 3 days).
- Block the samples in 1x Blocking Solution (Table 1) for 45-60 min at room temperature.
- Primary antibody solution
- Dilute anti-HTP-3 (chicken42) and anti-HA (mouse) antibodies (or the antibodies of choice) to the working solutions (1:250 for SMLM and 1:1,000 for STED microscopy samples) in 1x Blocking Solution.
NOTE: Antibodies used to label SMLM samples are more concentrated than for STED samples since a higher labeling density is recommended for SMLM microscopy. - Spin the samples on a mini benchtop centrifuge (2,000 × g, 10 s), remove the blocking buffer, and add 30-50 µL of the primary antibody solution. Incubate overnight at 4 °C (preferred) or for 1-2 h at room temperature.
- After incubation, wash 3 x 5-15 min with PBST.
- Dilute anti-HTP-3 (chicken42) and anti-HA (mouse) antibodies (or the antibodies of choice) to the working solutions (1:250 for SMLM and 1:1,000 for STED microscopy samples) in 1x Blocking Solution.
- Working solution of F(ab')2 fragments conjugated to fluorescent dye
- Dilute the labeled F(ab')2 fragments (step 1.2.4) to the working solutions (1:100 for SMLM and 1:1,000 for STED microscopy samples) in 1x Blocking Solution.
NOTE: For both super-resolution techniques, previously reported fluorophore pairs were used, namely AlexaFluor647/CF680 for SMLM and AlexaFluor594/Abberior STAR635P for STED. AlexaFluor647 and STAR645P were used to label anti-mouse (Fab')2 fragments to target the C-terminus of SYP-5, and CF680/AlexaFluor594-labeled anti-chicken (Fab')2 fragments to target HTP-3. - Spin the samples on a mini benchtop centrifuge (2,000 × g, 10 s), remove the PBST, and add 30-50 µL of secondary antibody solution. Incubate for 30 min to 2 h at room temperature (preferred) or overnight at 4 °C. Wash 3 x 5-15 min with PBST.
- Dilute the labeled F(ab')2 fragments (step 1.2.4) to the working solutions (1:100 for SMLM and 1:1,000 for STED microscopy samples) in 1x Blocking Solution.
4. Mounting samples on a coverslip
- Postfixation
NOTE: Process samples individually through steps 4.1.1-4.2.1.- Spin down the stained samples and remove the supernatant. Add 50 µL of PBST0.2 and transfer the stained worms onto a 22 mm x 22 mm No. 1 coverslip.
NOTE: Use fresh PBST0.2 with 0.2% nonionic detergent (Table 1) for this step to prevent the worms from adhering to the coverslip. - Pipet 5.7-6.3 µL of postfixative solution onto a poly-L-lysine coverslip.
NOTE: Poly-L-lysine coverslips stored at 4 °C should be brought to room temperature first. - Pipet off the dissected worms in the same volume (5.7-6.3 µL) and transfer into the drop of fixative on the poly-L-lysine coverslip (Figure 2B).
NOTE: In this and the following step, it is very important to retain the dissected tissue in the center of the poly-L-lysine-coated coverslip. This is especially important if mounting the samples in a custom holder to fit the custom-built SMLM microscope used here (see step 5.1, Figure 2B). - Cover the sample with a small coverslip (12 mm diameter, Figure 2B). Remove excess liquid using a small piece of filter paper (Figure 2B). Fix for 3-5 min in a dark chamber.
- Spin down the stained samples and remove the supernatant. Add 50 µL of PBST0.2 and transfer the stained worms onto a 22 mm x 22 mm No. 1 coverslip.
- "Freeze-cracking"
- Freeze the samples by placing them on an aluminum block in dry ice (Figure 2B).
NOTE: The aluminum block must be well cooled in the dry ice before placing the samples on it. Proceed with postfixation of the remaining samples (steps 4.1.1 to 4.2.1). The sample needs to be on dry ice for at least 20 min or up to 1 h prior to the next step (4.2.2). - Remove the smaller coverslip using a razor (Figure 2B).
NOTE: For STED, proceed to step 5.2.1. For SMLM, continue with step 4.2.3. - Dip the coverslip into a 50 mL conical tube containing ice-cold PBS (preferred) or -20 °C methanol for approximately 10 s.
NOTE: Temperature is a very important factor for this step. Therefore, use PBS that is freshly thawed or kept in an ice/ethanol bath. - Place the coverslip into a well of a six-well plate filled with PBST buffer. Remove the PBST from the wells and add fresh PBS. Leave the samples in PBS for 5 min.
NOTE: Pipet the PBS onto the side of the well to avoid damaging and detaching the samples. - Wash with fresh PBS and leave the samples at 4 °C until imaging.
NOTE: Samples are stable for up to 2 weeks, but the best results are achieved if the samples are imaged within 2 days. - Before imaging, assess the quality of the sample mounting under a stereo microscope.
NOTE: Successfully mounted germlines are stably attached with no discernable movement relative to the coverslip. Poorly attached germlines will flap in the buffer solution.
- Freeze the samples by placing them on an aluminum block in dry ice (Figure 2B).
5. Imaging
- Single-molecule localization microscopy
NOTE: Images were acquired at the EMBL Imaging Centre using a custom-built single-molecule localization microscope that was constructed around a custom body, as previously reported38,43, with the unique features specified in the Table of Materials; refer to https://www.embl.org/about/info/imaging-centre- Acquiring 3D bead calibration
- Prepare a precision coverslip (24 mm diameter; 0.17 ± 0.005 mm, No. 1.5) with adherent 100 nm fluorescent beads as described previously38,44.
- Place the calibration sample from step 5.1.1.1 onto a sample holder.
- Add a drop of immersion oil on the clean 100x/1.5 oil objective and mount the calibration sample on the microscope.
- Within MicroManager 245,46, specify 15-20 positions in the calibration sample.
- Within the EMU plugin window47, set up the acquisition of a z-stack image for each of the positions from step 5.1.1.5.
NOTE: Here, a compound cylindrical lens provides the astigmatism required for 3D imaging, and 201 z-slices were acquired for each position spanning the range between -1 µm to 1 µm, with an increment of 10 nm. A 2 kW/cm2 640 nm laser illumination was used for 25 ms for each z-slice. - Acquire the z-stack images of the 100 nm fluorescent beads through an identical optical path that will be used to acquire sample images in step 5.1.11.
- Using the super-resolution microscopy analysis platform (SMAP48), generate a cspline model of the experimental point spread function (PSF) that will be used to fit the 3D-SMLM data in step 5.1.13.
- Prepare the sample holder. For the custom-built holder used here that uses a magnetic ring to create the imaging chamber (Figure 2B), wrap the magnetic ring with parafilm.
NOTE: Alternatively, a microscope slide with a concave depression cavity can be used to mount samples for microscopes with slide holders. - Prepare 1 mL of imaging buffer44 (Table 1).
- Take one coverslip from step 4.2.6 and place it in the custom-made holder. Fix the coverslip in the holder with the parafilm-wrapped magnetic ring (step 5.1.2).
- Gently pipet the imaging buffer (step 5.1.3) in the chamber created by the magnetic ring on top of the sample (Figure 2B). Seal the chamber with a piece of parafilm.
- To mount the sample, add one drop of immersion oil on the clean 100x/1.5 oil objective. Without introducing any air into the immersion oil, gently place the sample holder with the mounted sample (step 5.1.5) onto the microscope stage.
NOTE: Before placing the sample on the microscope, clean the bottom of the coverslip with tissue and 70% ethanol. - Using the EMU plugin window47 within MicroManager 245,46, move the piezo stage until the signal from the focus lock laser is detected at the quadrant photodiode (QPD).
NOTE: To maintain a fixed focus across the imaging time, focus locking is achieved by total internal reflection of a near-infrared fiber-coupled laser from the coverslip and subsequent height sensitive detection on a quadrant photodiode (QPD). The QPD signal provided closed-loop control of the objective lens piezo mount. - Acquire a back focal plane image with a 640 nm excitation laser at low power (i.e., 1-5%) to confirm the absence of air bubbles in the immersion oil.
NOTE: Remove the sample from the stage if an air bubble is detected. Clean the bottom of the coverslip and the objective and repeat steps 5.1.6-5.1.8. Otherwise, proceed to lock the focus within the EMU software47. - Localize the gonad tissue using the brightfield illumination. Using a low-intensity 640 nm illumination, focus on the section of the tissue that contains many SC stretches.
NOTE: Do not focus on structures that are more than 2 µm from the coverslip. Do not use a higher laser power to locate the sample, as this may turn some fluorophores into a blinking state prematurely. Here, 1 kW/cm2 was used in rising mode with a pulse set to 1,000. - Proceed to expose the sample with 640 nm illumination at high irradiance (27 kW/cm2) for ~30 s until an appropriate blinking rate is achieved (Supplementary Video 1).
- Acquire 200,000 frames with a 20 ms exposure time using the multidimensional acquisition tool in MicroManager 245,46.
- Meanwhile, set up the UV activation using the activation option of the EMU plugin38,47 to maintain the desired blinking rate.
NOTE: Use the UV laser at a 3 kW/cm2 irradiance in rising mode with maximum pulse length set to 10,000. - Perform SMLM image reconstruction and postprocessing.
NOTE: To reconstruct images from raw SMLM data, refer to published methods. The data presented here was processed using the SMAP48,49 software. Super-resolved image reconstruction, channel assignment, drift correction, and filtering of localizations with poor localization precision and a maximum likelihood filter were performed in the SMAP48 software.
- Acquiring 3D bead calibration
- Stimulated emission depletion microscopy
NOTE: Images were acquired on the integrated STED microscopic system equipped with a white light laser, a 775 nm pulsed STED laser, and the FALCON Fluorescence Lifetime IMaging module (Table of Materials) at the EMBL Imaging Centre (https://www.embl.org/about/info/imaging-centre).- Place a 20 µL drop of mounting medium (Table of Materials) onto a microscope slide. Take one coverslip from step 4.2.2 and place the sample gently onto the slide facing the mounting medium (Figure 2B).
NOTE: Avoid introducing air pockets within the mounting medium. - Let the mounting medium cure overnight.
NOTE: Image the samples on the following day or keep them at 4 °C until imaging. - To mount the sample, add one drop of immersion oil on the coverslip of the sample from the step 5.2.2. Gently place the sample onto the microscope stage using a 100x/1.40 oil objective.
- Focus on the sample and locate the germline tissue using the bright-field illumination.
- Using the microscope software, specify the region of interest for which the TauSTED image will be acquired.
- Select the excitation lasers and their appropriate power used to excite the fluorophores used in the sample.
NOTE: Here, the 580 nm laser at 4% power was used to image AlexaFluor 594-conjugated F(ab')2 secondary antibody fragments, and 635 nm at 3% power to image STAR 635P-conjugated F(ab')2 fragments. - Using the microscope software, select an appropriate STED depletion laser power and set up the image detection.
NOTE: Here, the 775 nm STED depletion laser power was set to 40%. The detector was used in counting mode with a gain value of 10 for photon detection, with a scan rate of 100 Hz, and at a pixel size of 17 nm. Four-line accumulation was used for TauSTED acquisition.
- Place a 20 µL drop of mounting medium (Table of Materials) onto a microscope slide. Take one coverslip from step 4.2.2 and place the sample gently onto the slide facing the mounting medium (Figure 2B).
Representative Results
To image the SC within the C. elegans germline tissue by SMLM, we have employed 2-color ratiometric 3D-SMLM to localize HTP-3, a component of the chromosome axes, and the C-terminus of the transverse filament SYP-5 endogenously tagged with a hemagglutinin (HA) tag. The location of both proteins within the SC of C. elegans was previously determined by other studies16,30.
To minimize light scattering and optical aberrations inherent in thick biological samples, we imaged the bottom-most z-section of meiotic nuclei that contain the SCs (Figure 3, yellow lines). For each acquired image, the piezo stage position of the imaging plane was marked relative to the piezo stage position when the objective was focused on the coverslip. This allowed the calculation of the piezo distance from the coverslip. Successfully mounted samples are stably attached close to the coverslip and retain the gonad shape (i.e., the tissue is not crushed between the two coverslips during the postfixation step). The quality of the sample mounting can readily be assessed under a stereo microscope since well-attached gonads do not show any movement in solution (step 4.2.6). Nevertheless, due to the stochasticity of the mounting process, the gonad tissue will not necessarily be laid out completely flat on the coverslip. Therefore, the bottom plane of the nuclei containing SCs may be found at varying distances relative to the coverslip within the same gonad.
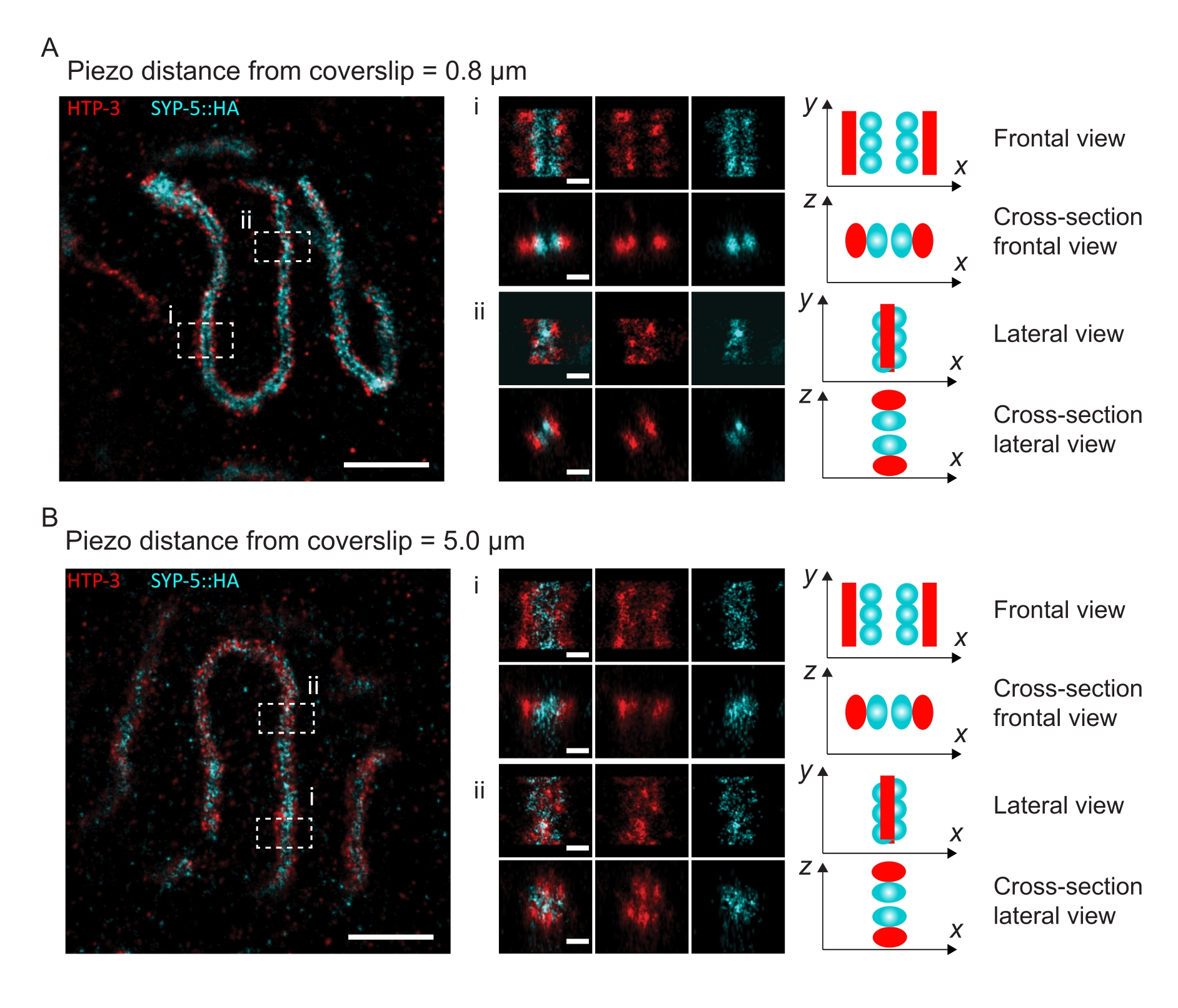
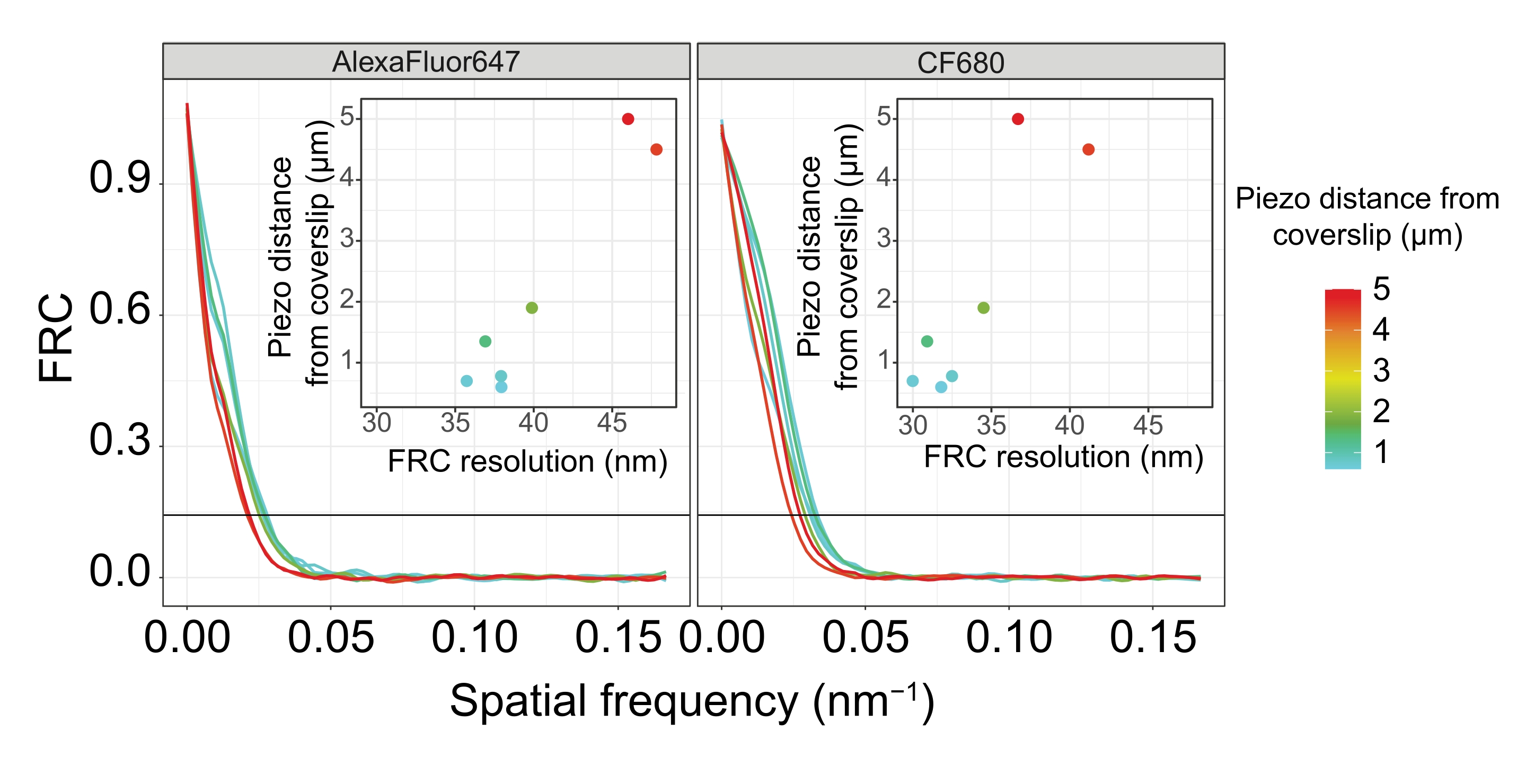
To illustrate how the resolution changes depending on the attachment of the tissue to the coverslip, we acquired images at different piezo distances to the coverslip. To assess the quality of an individual image, Fourier ring correlation (FRC) curves50,51 were calculated and the resolution was determined using the FRCResolution plugin within the SMAP software48. Two representative nuclei extracted from two separate 3D-SMLM images taken at different distances to the coverslip are displayed in Figure 4. In SCs located close to the coverslip, the chromosome axes and the C-terminus of SYP-5::HA are well resolved in all three dimensions (Figure 4A, 0.8 µm from the coverslip). To resolve two structures separated by a given distance, the achieved FRC resolution generally must be smaller than half this distance in the axial resolution.
To separate the same structures laterally, even smaller FRC resolution values need to be achieved. Indeed, in samples that are located in close proximity to the coverslip, the FRC resolution is 38 nm for the AlexaFluor 647 channel and 34 nm for the CF680 channel, and thus well below the expected distance of 84 nm between the C-termini of SYP-516. This resolution therefore readily resolves the organization of the SC not only in frontal but also in lateral views (Figure 4B i,ii). By contrast, the resolution deteriorates in SCs located at a 5 µm distance from the coverslip due to light scattering and spherical aberrations (Figure 4B). The FRC resolutions at this distance drop to 47 nm (AlexaFluor 647) and 41 nm (CF680), which cannot fully resolve the C-termini of SYP-5. Since the optical aberrations impair the lateral resolution more severely than the axial resolution, HTP-3 and SYP-5 bands are no longer clearly resolved in the cross-section of the lateral view in samples located at a 5 µm distance from the coverslip (Figure 4B ii). Comparing the FRC resolution of images acquired at different piezo distances from the coverslip revealed that the imaged tissue should be no further than 2 µm from the coverslip (Figure 5). This result highlights the importance of correct execution of the postfixation step, during which the tissue must be successfully crosslinked to the poly-L-lysine coating of the coverslip.
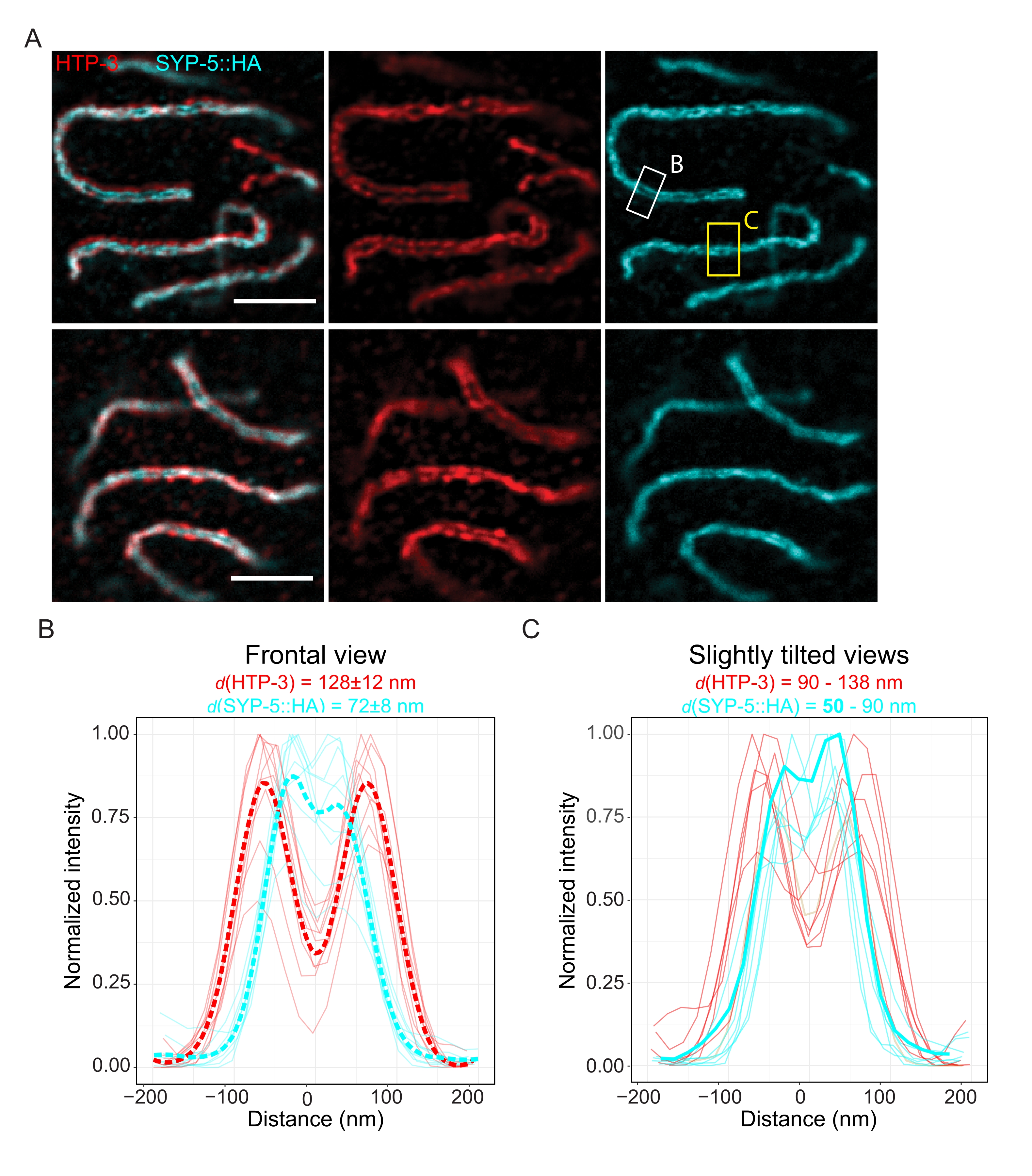
To demonstrate the achievable resolution with another super-resolution technique, we also imaged SCs in fixed intact germline tissue with TauSTED microscopy. Figure 6A shows TauSTED images with the highest and lowest resolution achieved within this study as estimated from line profiles of the SC in frontal view (Figure 6B). In both nuclei, we could resolve the two localization bands of HTP-3 in the chromosome axes and the C-termini of SYP-5 in the central region, demonstrating that the resolution achievable in TauSTED using this optimized protocol is below 84 nm. Under optimal conditions (Figure 6A, top), we could resolve the C-termini in slightly tilted views of the SC that were separated by only 50 nm (Figure 6A, yellow rectangle and 6C).

Figure 1: Schematic of the organization of the synaptonemal complex in Caenorhabditis elegans. The cartoon shows a simplified structure of the SC in C. elegans bridging two homologous chromosomes (gray). The structure is shown in frontal, lateral, and cross-sectional views. Chromosome axes are displayed as red bars while transverse filaments are shown in cyan. Transverse filament proteins (SYP-1, 5, 6 in C. elegans) are oriented in a head-to-head manner (cyan ball-stick graphics) in the central region to bridge the distance between the two axes. The expected distances between axes and the C-termini of transverse filaments are indicated. Abbreviation: SC = synaptonemal complex. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Illustration of the sample preparation used in the study. (A) Young C. elegans adults are dissected at their head or tail (green, dashed lines) and processed as described in the protocol. (B) Individual steps of the method are indicated with graphics that are connected with gray arrows. Abbreviations: STED = stimulated-emission depletion; SMLM = single-molecule localization microscopy; PBS = phosphate-buffered saline. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Location of the tissue section that can be observed by single molecule localization microscopy. MIP of a spinning disk confocal image of a whole mount C. elegans gonad. The tissue was stained for HTP-3 and the C-terminus of SYP-5 (SYP-5::HA), and the combined signal is shown in gray. Individual confocal images were stitched using the Grid/Collection stitching Fiji plugin52 to create an image of the whole gonad. The inset shows an xy view of the bottommost z-plane containing the SCs. The localization of this plane is shown in orthogonal views of the tissue section indicated by a rectangle in the MIP image of the gonad (yellow lines). Scale bars = 10 µm. Abbreviations: MIP = maximum intensity projection; SCs = synaptonemal complexes. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Single-molecule localization microscopy of HTP-3 and the C-termini of the SYP-5. (A,B) Left: SMLM images showing pachytene nuclei stained for HTP-3 (red) and the C-terminus of SYP-5 (SYP-5::HA, cyan) (scale bar = 1 µm). Center: Zoomed-in images of regions of interest that are indicated in A and B with corresponding cross-sectional views displayed below each image (i, ii; scale bar = 100 nm). The stretches of the SC within zoomed-in images are rotated to orient the chromosome axes parallel to the y-axis. Right: Graphical representation of the localization of the proteins of interest within the SC portraying the orientation of the SC in the zoomed-in regions displayed in the center of the figure. Abbreviations: SMLM = single-molecule localization microscopy; SC = synaptonemal complex. Raw data to reconstruct SMLM images are available through the BioStudies database60 (Accession ID: S-BIAD504). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: The Fourier ring correlation resolution of single-molecule localization microscopy images depends on the distance of the imaged z-plane from the plane of the coverslip. Colored lines show FRC curves of images acquired at different distances (as depicted by the color bar) from the coverslip. The 1/7 threshold used to determine the FRC resolution is indicated by a black horizontal line. Insets show the dependence of the FRC resolution on the piezo distance from the coverslip. Plotting was performed by a custom-written R script (version 4.1.2, Supplementary File 1) in which original curves were smoothed with functions from the "ggplot2" package. Abbreviations: FRC = Fourier ring correlation; SMLM = single-molecule localization microscopy; SC = synaptonemal complex. Data for FRC curves and SMLM data are available through the BioStudies database60 (Accession ID: S-BIAD504). Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Stimulated emission depletion microscopy enhanced by fluorescence lifetime-based information (TauSTED) resolves two localization bands for both HTP-3 and the C-terminus of SYP-5. (A) Two representative TauSTED images show pachytene nuclei stained for HTP-3 (red) and the C-terminus of SYP-5 (SYP-5::HA, cyan) with higher (top) and lower (bottom) structural definition (scale bar = 1 µm). The rectangles mark regions with the resolved C-termini of SYP-5 in frontal (white) and a slightly tilted view (yellow) of the SC. (B,C) Distribution of the HTP-3 (red) and the C-terminus of SYP-5 (cyan) signal resolved by TauSTED. Line profiles of regions of interest that contain the SC in frontal (B) or slightly tilted (C) views are shown as full lines with intensity normalized to the maximum value. Line profiles were generated using Fiji ImageJ. Dashed lines in B show the averaged data for each protein. The thick cyan line in C corresponds to the line profile with the shortest resolved distance between the C-termini of SYP-5. To determine the distances between the antibodies targeting specific proteins, the line profiles (n = 9 (B), n = 7 (C)) were fitted with double gaussians using a custom-written R script (version 4.1.2, Supplementary File 1). Mean distance ± standard deviation (B) and the range with minimum value highlighted in bold (C) are indicated on top of each graph, respectively. Abbreviations: STED = stimulated emission depletion microscopy; SC = synaptonemal complex. Displayed images and data points of plotted line profiles are available through the BioStudies database60 (Accession ID: S-BIAD504). Please click here to view a larger version of this figure.
{kind=link}
Table 1: Composition of buffers and solutions used in this protocol. Please click here to download this Table.
Supplementary Video 1: Single molecule localization microscopy acquisition. Video showing fluorophores blinking at an appropriate rate (50 frames are shown, scale bar = 5 µm, 20 ms/frame). Please click here to download this Video.
Supplementary File 1: Data analysis script. Please click here to download this File.
Discussion
The ladder-like organization of the SC, which is essential for the correct recombination and segregation of homologous chromosomes, was first observed almost 70 years ago in electron microscopy3,4. While the overall organization of the SC is readily resolved in electron microscopy, the localization of individual components within this complex requires a more targeted approach. With its width of only ~100 nm, the substructure of the SC cannot be resolved by conventional fluorescence microscopy. However, super-resolution microscopy has become a major driver for novel discoveries on the structure and function of the synaptonemal complex16,19,24,25,26,27,28,29,30. To facilitate this research, we have demonstrated a mounting procedure that allows studying the architecture of the SC within C. elegans gonad tissue with SMLM and STED microscopy.
A critical step to optimize the resolution in SMLM imaging is directly cross-linking the germline tissue to a poly-L-lysine coated coverslip (step 4). The covalent attachment of the tissue to the coverslip is essential to reduce movements within the sample that would result in large drifts and make imaging over long periods of time for SMLM impossible. Additionally, even a suboptimal attachment that leaves the nuclei containing SCs at a distance from the coverslip leads to a significant drop in the achievable resolution resulting from spherical aberrations (Figure 4). Alternatively to the covalent attachment used here, stained germline tissue can also be immobilized between two sealed coverslips in a small drop of imaging buffer19,30. However, this immobilization method severely reduces the volume of imaging buffer in the sample from 1 mL used in the optimized protocol here to just a few µL, which will result in an acidification of the imaging buffer and severely reduce the time for which the sample can be imaged38,53,54.
Long acquisition times for both SMLM and STED microscopy limit the use of these methods to imaging of chemically fixed samples. Here, paraformaldehyde fixation ensures that the structure of the SC is preserved during sample preparation and imaging. However, despite the precautions taken here to image the SC within intact tissue, the resulting structure of the SC after fixation is not necessarily identical to the structure in its native state within a living organism. Moreover, since a single image of the fixed SC represents a single "snapshot" of the biological structure, this approach remains blind to the dynamics of the native structure in vivo.
However, information on the dynamics and variability of macromolecular structures can also be obtained by acquiring not just a single but many "snapshots". While this approach can resolve changes in the structure of the SC during pachytene19, there are several factors that limit the number of images that can be acquired from a single sample prepared using this protocol. First, the high laser powers used during image acquisition lead to permanent bleaching of the fluorophores and preclude imaging of adjacent regions of interest or multiple z-planes, thereby significantly reducing the number of images that can be acquired from a single sample. Second, the sample/tissue density on the coverslip prepared by this method is low, which significantly limits the number of images that can be acquired from a single coverslip. The low sample density also prohibits the use of automated image acquisition pipelines that helped shed light on other biological questions34,55,56,57,58,59. However, the sample density can be increased slightly by an experienced user.
The protocol presented here is optimized to obtain a high labeling density that is necessary to achieve optimal resolution in SMLM35. While previous protocols covalently attach the tissue to the coverslip before immunostaining16, this new protocol cross-links the tissue to the coverslip only after the samples were stained in solution. This modification allows the antibodies used for immunolabeling to freely access the tissue from all sides, while the covalent attachment of the tissue to the coverslip may restrict antibodies from reaching the nuclei closest to the coverslip, thereby reducing the degree of labeling. Together, the modifications described here improve the resolution from 40-50 nm (FRC resolution)16 to 30-40 nm (this protocol).
Importantly, while a high labeling density and a high concentration of antibodies is essential for SMLM, we found that better STED microscopy images are obtained using lower antibody concentrations (step 3). At a resolution of tens of nanometers, the size of the molecules used to label the protein of interest becomes increasingly important. We therefore employed F(ab')2 fragments that are half the size of full-length antibodies. The improvement in local contrast due to a smaller signal source, and therefore resolution gained by this modification compared to using full-length secondary antibodies, allowed the resolution of the two C-termini of SYP-5 within the central region by TauSTED, which are not resolved by conventional STED using full-length antibodies (16 and data not shown). We anticipate that this optimized protocol for imaging SCs in intact C. elegans germlines will facilitate investigating the structure-function relationship of the SC during meiosis.
Acknowledgements
We would like to thank Jonas Ries and the Ries lab for sharing imaging buffers for SMLM imaging. We also thank Yumi Kim for the C. elegans strain used in this protocol and Abby F. Dernburg for the chicken-anti-HTP-3 antibody. We thank Marko Lampe and Stefan Terjung from the Advanced Light Microscopy Facility at EMBL Heidelberg for their support in using the Olympus iXplore SPIN SR confocal microscope. This work was supported by the European Molecular Biology Laboratory and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation - 452616889, SK). We acknowledge the access and services provided by the Imaging Centre at the European Molecular Biology Laboratory (EMBL IC), generously supported by the Boehringer Ingelheim Foundation.
Materials
| Name | Company | Catalog Number | Comments |
| 100x/1.5 oil objective | Olympus | UPLAPO100XOHR | UPLAPO100XOHR |
| 2-mercaptoethylamine (MEA) | Sigma-Aldrich | 30070-10G | Dissolved in MilliQ water to 5 M solution, pH 8.7 adjusted with HCl. Aliquoted to a single-use volume, frozen, and kept at -80 °C. |
| Additional 640 nm booster laser | Toptica | IBEAM-SMART-640-S-HP | |
| AlexaFluor 594, NHS ester | ThermoFischer Scientific | A37572 | Dissolved in DMSO to 1 mM solution, aliquoted to single use volume, frozen and kept at -80 °C |
| AlexaFluor 647, NHS ester | ThermoFischer Scientific | A37573 | Dissolved in DMSO to 1 mM solution, aliquoted to single use volume, frozen and kept at -80 °C |
| anti-HA | Thermo Fisher Scientific | 2-2.2.14 | Mouse monoclonal, 1:250 (SMLM), 1:1,000 (STED Microscopy) |
| anti-HTP-3 | a gift from Abby F. Dernburg | MacQueen et al., 2005 | Chicken polyclonal, 1:250 (SMLM), 1:1,000 (STED Microscopy) |
| Caenorhabditis elegans strain YKM349 | a gift from Yumi Kim | Hurlock et al., 2020 | syp-5(kim9[syp-5::HA]) I; meIs8[pie-1p::GFP::cosa-1, unc-119(+)] II |
| CF6680, NHS ester | Biotium | 92139 | Dissolved in DMSO to 1mM solution, aliquoted to single use volume, frozen and kept at -80 °C |
| Circular cover glass 12 mm No. 1 | Menzel-Gläser; VWR | 631-0713 | |
| Circular cover glass 24 mm No. 1.5 | Carl Roth | PK26.1 | |
| Cylindrical lenses | Thorlabs | LJ1516RM-A, LK1002RM-A | |
| Egg Buffer (10x) | Edgar 1995 | 250 mM HEPES, 1.18 M NaCl, 480 mM KCl, 20 mM EDTA, 5 mM EGTA, pH 7.4 | |
| Ethanol (absolute for analysis) | Merck | 64-17-5 | |
| F(ab’)2 fragment anti-chicken IgY | Jackson Immunoresearch | AB_2340347 | Donkey polyclonal, 1:100 (SMLM), 1:1,000 (STED Microscopy) |
| F(ab’)2 fragment anti-mouse IgG | Jackson Immunoresearch | AB_2340761 | Donkey polyclonal, 1:100 (SMLM), 1:1,000 (STED Microscopy) |
| Fisherbrand Microscope slides T/F Ground 0.8-1.0 mm thick | Fisher scientific | 7107 | |
| Gauge Worm Pick 30 diameter 0.254 mm - Iridium 10% | Kisker | 789265 | |
| Glucose oxidase/Catalase enzyme mix (GlOX/Cat ) | a gift from Jonas Ries | Hoess, Mund, Reitberger, & Ries, 2018 | 20x, 1916 U/mL glucose oxidase (Sigma G7141), 42350 U/mL catalase (Sigma C3155), 50 mM Tris-HCl pH 8.0, 51% glycerol, MilliQ water. Stored at -20 °C. |
| Imaging buffer base | a gift from Jonas Ries | Hoess, Mund, Reitberger, & Ries, 2018 | 50 mM Tris-HCl, pH 8.0, 10 mM NaCl, 10% D-Glucose. Aliquoted to a single-use volume (950 μL), frozen, and kept at -80 °C. |
| Invitrogen ProLong Glass Antifade Mountant | ThermoFischer Scientific | P36982 | |
| Leica Stellaris 8 STED FALCON | Leica | N/A | The microscope is equiped with the latest generation white light laser, a 775nm pulsed STED laser, the FALCON Fluorescence Lifetime IMaging module, HC PL APO CS2 100x/1.40 oil objective, and Leica HyD X detector. The system is capable of FLIM module enhanced Tau-STED which measures the specific fluorescence lifetime of a dye and is therefore capable of removing background signal based on differences in fluorescence lifetimes of the dyes, and dye conditions in the sample. Additionally, the resolution is increased by accounting for the variation of fluorescence lifetimes in different areas of the depletion donut. |
| Longwave channel emission filter | AHF Analysentechnik | F47-702 | 700/100 nm bandpass |
| Methanol (absolute for analysis) | Merck | 67-56-1 | |
| NaHCO3 | Sigma-Aldrich/Merck | S5761-500G | 100 mM NaHCO3, pH 8.3 |
| Near-infrared fiber-coupled laser | Toptica | IBEAM-SMART-PT-CD | Custom Design, 808 nm - 75mW |
| Objective lens piezo mount (PIFOC ) | Physik Instrumente | P-726.1.CD | 100 µm travel range |
| Orca Fusion BT sCMOS camera | Hamamatsu | C15440-20UP | |
| PCR tubes | Greiner Bio-One | 673283 | 0.2 mL |
| Phosphate Saline Buffer (PBS 10x) | N/A | 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4 | |
| Pierce 16% formaldehyde (w/v), methanol free | ThermoFischer Scientific | 28906 | 16% formaldehyde is transferred from the original glass ampule into 1.5 mL tube and kept at room temperature. |
| PlasmaPrep2 plasma cleaner | GaLa Instrumente GmbH | N/A | |
| Poly-L-lysine hydrobromide | Sigma-Aldrich/Merck | P2636-25MG | 0.1% w/v solution was prepared in Milli-Q water, and stored in aliquots at -20 °C. |
| Primary dichroic (illumination reflecting) | AHF Analysentechnik | F73-866S | quad bandpass @ 405, 488, 561, 640 |
| Quadnotch filter | AHF Analysentechnik | F40-072 | 405/488/561/640 nm |
| Quadrant photodiode (QPD) | Laser Components | SD197-23-21-041, LC301DQD-PV | |
| Razor blades | Apollo Herkenrath Solinger | N/A | |
| Refractive beam shaper | AdlOptica | PiShaper 6_6_VIS | |
| Roche Blocking Reagent | Roche | 11096176001 | 10x solution was prepared according to recommendation. Frozen aliquots were stored at -20 °C. |
| Scalpel blade (Feather brand #11, No. 3) | Heinz Herenz Medizinalbedarf GmbH | 1110911 | |
| Scalpel removal box | Fisher scientific | 10002-50 | |
| Secondary dichroic (emission reflecting) | AHF Analysentechnik | F38-785S | 750 nm longpass |
| Shortpass filter | Semrock | BSP01-785R-25 | 750 nm |
| Shortwave channel emission filter | AHF Analysentechnik | F37-677 | 676/37 nm bandpass |
| Single molecule localization microscope | EMBL Imaging Centre | Diekmann et al., 2020 with modifications | The microscope provides widefield epi-illumination via a single-mode fiber-coupled laser engine, additional booster laser, and refractive beam shaper to provide a uniform illumination field (Stehr et al, 2019). Widefield images are captured on a sCMOS camera and appropriate relay optics for a system magnification of 61x and a pixel size of 106 nm. For ratiometric imaging of spectrally overlapping far-red dyes, an image splitter produces two spectrally distinct images on the camera (splitting dichroic: 665 nm long pass, shortwave channel emission filter: 676/37 nm bandpass, longwave emission filter: 700/100 nm bandpass. An additional 405/488/561/640 nm quadnotch filter and 750 nm shortpass filter are common to the two paths and provide additional laser blocking). A compound cylindrical lens provides the astigmatism required for 3D imaging. To maintain a fixed focus across acquisitions exceeding 2 hours in time (comprising 200 000 - 250 000 images), focus locking is achieved by total internal reflection of a near-infrared fiber-coupled laser from the coverslip and subsequent height sensitive detection on a quadrant photodiode (QPD). The QPD signal provided closed-loop control of the objective lens piezo mount. For access to this microscope, refer to https://www.embl.org/about/info/imaging-centre or contact ic-contact@embl.de |
| Single-mode fiber-coupled multi-laser engine | Toptica | iCHROME MLE-LFA-HP | Provides widefield epi-illumination of 100 mW at 405, 488, 561, 640 nm |
| Splitting dichroic | AHF Analysentechnik | F48-665SG | 665 nm long pass |
| Square cover glass 22 x 22 mm No.1 | Menzel-Gläser; VWR | 630-2882 | |
| STAR 635P, NHS ester | Abberior | ST635P-0002-1MG | Dissolved in DMSO to 1 mM solution, aliquoted to single use volume, frozen and kept at -80 °C |
| Stereo microscope Stemi 305 Stand K LAB | Zeiss | N/A | |
| Tetramisole hydrochloride | Sigma-Aldrich/Merck | T1512-2G | 1% (w/v) solution was prepared in Milli-Q water. Frozen aliquots were stored at -20 °C. Thawed aliquot was kept at 4 °C and used for several months. |
| TetraSpeck Microspheres | ThermoFischer Scientific | T7279 | 0.1 µm, fluorescent blue/green/orange/dark red |
| Tris Saline Buffer (TBS 10x) | N/A | 200 mM Tris-HCl, 1.5 M NaCl, pH 7.5 | |
| TWEEN 20 | Sigma-Aldrich/Merck | P9416-50ML | Kept at room temperature in original packaging. |
| WormStuff worm pick | Kisker | 789277 | |
| XY microscope stage | Smaract | N/A | Custom Design |
| Zeba Micro Spin Desalting Column | ThermoFischer Scientific | 89877 | 7K MWCO, 75 µL |
References
- Zickler, D., Kleckner, N. Meiotic chromosomes: integrating structure and function. Annual Review of Genetics. 33, 603 (1999).
- Ur, S. N., Corbett, K. D. Architecture and dynamics of meiotic chromosomes. Annual Review of Genetics. 55, 497-526 (2021).
- Fawcett, D. W. The fine structure ot chromosomes in the meiotic prophase of vertebrate spermatocytes. The Journal of Biophysical and Biochemical Cytology. 2 (4), 403-406 (1956).
- Moses, M. J. Chromosomal structures in crayfish spermatocytes. The Journal of Biophysical and Biochemical Cytology. 2 (2), 215-218 (1956).
- Hillers, K. J., Jantsch, V., Martinez-Perez, E., Yanowitz, J. L. Meiosis. WormBook. , 433-434 (2017).
- Pasierbek, P., et al. A Caenorhabditis elegans cohesion protein with functions in meiotic chromosome pairing and disjunction. Genes & Development. 15 (11), 1349-1360 (2001).
- Severson, A. F., Ling, L., Van Zuylen, V., Meyer, B. J. The axial element protein HTP-3 promotes cohesin loading and meiotic axis assembly in C. elegans to implement the meiotic program of chromosome segregation. Genes & Development. 23 (15), 1763-1778 (2009).
- Zetka, M. C., Kawasaki, I., Strome, S., Müller, F. Synapsis and chiasma formation in Caenorhabditis elegans require HIM-3, a meiotic chromosome core component that functions in chromosome segregation. Genes & Development. 13 (17), 2258-2270 (1999).
- Martinez-Perez, E. HTP-1-dependent constraints coordinate homolog pairing and synapsis and promote chiasma formation during C. elegans meiosis. Genes & Development. 19 (22), 2727-2743 (2005).
- Couteau, F., Zetka, M. HTP-1 coordinates synaptonemal complex assembly with homolog alignment during meiosis in C. elegans. Genes & Development. 19 (22), 2744-2756 (2005).
- Goodyer, W., et al. HTP-3 Links DSB Formation with Homolog Pairing and Crossing Over during C. elegans Meiosis. Developmental Cell. 14 (2), 263-274 (2008).
- Colaiácovo, M. P., et al. Synaptonemal complex assembly in C. elegans is dispensable for loading strand-exchange proteins but critical for proper completion of recombination. Developmental Cell. 5 (3), 463-474 (2003).
- MacQueen, A. J., Colaiácovo, M. P., McDonald, K., Villeneuve, A. M. Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes & Development. 16 (18), 2428-2442 (2002).
- Smolikov, S., et al. Synapsis-defective mutants reveal a correlation between chromosome conformation and the mode of double-strand break repair during Caenorhabditis elegans meiosis. Genetics. 176 (4), 2027-2033 (2007).
- Smolikov, S., Schild-Prüfert, K., Colaiácovo, M. P. A yeast two-hybrid screen for SYP-3 interactors identifies SYP-4, a component required for synaptonemal complex assembly and chiasma formation in Caenorhabditis elegans meiosis. PLoS Genetics. 5 (10), 1000669 (2009).
- Hurlock, M. E., et al. Identification of novel synaptonemal complex components in C. Elegants. The Journal of Cell Biology. 219 (5), (2020).
- Zhang, Z., et al. Multivalent weak interactions between assembly units drive synaptonemal complex formation. The Journal of Cell Biology. 219 (5), (2020).
- Schild-Prüfert, K., et al. Organization of the synaptonemal complex during meiosis in Caenorhabditis elegans. Genetics. 189 (2), 411-421 (2011).
- Köhler, S., Wojcik, M., Xu, K., Dernburg, A. F. The interaction of crossover formation and the dynamic architecture of the synaptonemal complex during meiosis. bioRxiv. , (2020).
- Chen, F., Tillberg, P. W., Boyden, E. S. Expansion microscopy. Science. 347 (6621), 543-548 (2015).
- Klar, T. A., Jakobs, S., Dyba, M., Egner, A., Hell, S. W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences. 97 (15), 8206-8210 (2000).
- Rust, M. J., Bates, M., Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nature Methods. 3 (10), 793-796 (2006).
- Betzig, E., et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642-1645 (2006).
- Schücker, K., Holm, T., Franke, C., Sauer, M., Benavente, R. Elucidation of synaptonemal complex organization by super-resolution imaging with isotropic resolution. Proceedings of the National Academy of Sciences. 112 (7), 2029-2033 (2015).
- Cahoon, C. K., et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proceedings of the National Academy of Sciences. 114 (33), 6857-6866 (2017).
- Zwettler, F. U., et al. Tracking down the molecular architecture of the synaptonemal complex by expansion microscopy. Nature Communications. 11 (1), 1-11 (2020).
- Yoon, S., Choi, E. H., Kim, J. W., Kim, K. P. Structured illumination microscopy imaging reveals localization of replication protein A between chromosome lateral elements during mammalian meiosis. Experimental & Molecular Medicine. 50 (8), 1-12 (2018).
- Prakash, K., et al. Superresolution imaging reveals structurally distinct periodic patterns of chromatin along pachytene chromosomes. Proceedings of the National Academy of Sciences. 112 (47), 14635-14640 (2015).
- Xu, H., et al. Molecular organization of mammalian meiotic chromosome axis revealed by expansion STORM microscopy. Proceedings of the National Academy of Sciences. 116 (37), 18423-18428 (2019).
- Köhler, S., Wojcik, M., Xu, K., Dernburg, A. F. Superresolution microscopy reveals the three-dimensional organization of meiotic chromosome axes in intact Caenorhabditis elegans tissue. Proceedings of the National Academy of Sciences. 114 (24), 4734-4743 (2017).
- Hell, S. W. Far-field optical nanoscopy. Science. 316 (5828), 1153-1158 (2007).
- Hein, B., Willig, K. I., Hell, S. W. Stimulated emission depletion (STED) nanoscopy of a fluorescent protein-labeled organelle inside a living cell. Proceedings of the National Academy of Sciences. 105 (38), 14271-14276 (2008).
- Jahr, W., Velicky, P., Danzl, J. G. Strategies to maximize performance in STimulated Emission Depletion (STED) nanoscopy of biological specimens. Methods. 174, 27-41 (2019).
- Thevathasan, J. V., et al. Nuclear pores as versatile reference standards for quantitative superresolution microscopy. Nature Methods. 16 (10), 1045-1053 (2019).
- Xu, K., Shim, S. -. H., Zhuang, X. Super-resolution imaging through stochastic switching and localization of single molecules: an overview. Far-Field Optical Nanoscopy. , 27-64 (2013).
- Wang, Y., et al. Localization events-based sample drift correction for localization microscopy with redundant cross-correlation algorithm. Optics Express. 22 (13), 15982 (2014).
- Winterflood, C. M., Platonova, E., Albrecht, D., Ewers, H. Dual-color 3D superresolution microscopy by combined spectral-demixing and biplane imaging. Biophysical Journal. 109 (1), 3-6 (2015).
- Diekmann, R., et al. Optimizing imaging speed and excitation intensity for single molecule localization microscopy. Nature Methods. 17 (9), 909 (2020).
- Huang, B., Wang, W., Bates, M., Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 319 (5864), 810-813 (2008).
- Phillips, C. M., McDonald, K. L., Dernburg, A. F. Cytological analysis of meiosis in Caenorhabditis elegans. Methods in Molecular Biology. 558, 171-195 (2009).
- Edgar, L. G. Blastomere culture and analysis. Methods in Cell Biology. 48, 303-321 (1995).
- MacQueen, A. J., et al. Chromosome sites play dual roles to establish homologous synapsis during meiosis in C. elegans. Cell. 123 (6), 1037-1050 (2005).
- Stehr, F., Stein, J., Schueder, F., Schwille, P., Jungmann, R. Flat-top TIRF illumination boosts DNA-PAINT imaging and quantification. Nature Communications. 10 (1), 1-8 (2019).
- Hoess, P., Mund, M., Reitberger, M., Ries, J. Dual-color and 3D super-resolution microscopy of multi-protein assemblies. Methods in Molecular Biology. 1764, 237-251 (2018).
- Edelstein, A., Amodaj, N., Hoover, K., Vale, R., Stuurman, N. Computer Control of microscopes using µManager. Current Protocols in Molecular Biology. 92 (1), 14-20 (2010).
- Edelstein, A. D., et al. Advanced methods of microscope control using µManager software. Journal of Biological Methods. 1 (2), 10 (2014).
- Deschamps, J., Ries, J. EMU: reconfigurable graphical user interfaces for Micro-Manager. BMC Bioinformatics. 21 (1), 1-13 (2020).
- Ries, J. SMAP: a modular super-resolution microscopy analysis platform for SMLM data. Nature Methods. 17 (9), 870-872 (2020).
- Li, Y., et al. Global fitting for high-accuracy multi-channel single-molecule localization. Nature Communications. 13 (1), 1-11 (2022).
- Nieuwenhuizen, R. P. J., et al. Measuring image resolution in optical nanoscopy. Nature Methods. 10 (6), 557-562 (2013).
- Banterle, N., Bui, K. H., Lemke, E. A., Beck, M. Fourier ring correlation as a resolution criterion for super-resolution microscopy. Journal of Structural Biology. 183 (3), 363-367 (2013).
- Preibisch, S., Saalfeld, S., Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics. 25 (11), 1463-1465 (2009).
- Shi, X., Lim, J., Ha, T. Acidification of the oxygen scavenging system in single-molecule fluorescence studies: in situ sensing with a ratiometric dual-emission probe. Analytical Chemistry. 82 (14), 6132-6138 (2010).
- Olivier, N., Keller, D., Rajan, V. S., Gönczy, P., Manley, S. Simple buffers for 3D STORM microscopy. Biomedical Optics Express. 4 (6), 885-899 (2013).
- Mund, M., et al. Superresolution microscopy reveals partial preassembly and subsequent bending of the clathrin coat during endocytosis. bioRxiv. , (2022).
- Mund, M., et al. Systematic nanoscale analysis of endocytosis links efficient vesicle formation to patterned actin nucleation. Cell. 174 (4), 884-896 (2018).
- Sabinina, V. J., et al. Three-dimensional superresolution fluorescence microscopy maps the variable molecular architecture of the nuclear pore complex. Molecular Biology of the Cell. 32 (17), 1523-1533 (2021).
- Cieslinski, K., et al. Nanoscale structural organization and stoichiometry of the budding yeast kinetochore. bioRxiv. , (2021).
- Sieben, C., Banterle, N., Douglass, K. M., Gönczy, P., Manley, S. Multicolor single-particle reconstruction of protein complexes. Nature Methods. 15 (10), 777-780 (2018).
- Sarkans, U., et al. The BioStudies database—one stop shop for all data supporting a life sciences study. Nucleic Acids Research. 46, (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved