Compost Microcosms as Microbially Diverse, Natural-like Environments for Microbiome Research in Caenorhabditis elegans
In This Article
Summary
Compost microcosms bring the microbial diversity found in nature into the laboratory to facilitate microbiome research in Caenorhabditis elegans. Provided here are protocols for setting up microcosm experiments, with the experiments demonstrating the ability to modulate environmental microbial diversity to explore the relationships between environmental microbial diversity and worm gut microbiome composition.
Abstract
The nematode Caenorhabditis elegans is emerging as a useful model for studying the molecular mechanisms underlying interactions between hosts and their gut microbiomes. While experiments with well-characterized bacteria or defined bacterial communities can facilitate the analysis of molecular mechanisms, studying nematodes in their natural microbial context is essential for exploring the diversity of such mechanisms. At the same time, the isolation of worms from the wild is not always feasible, and, even when possible, sampling from the wild restricts the use of the genetic toolkit otherwise available for C. elegans research. The following protocol describes a method for microbiome studies utilizing compost microcosms for the in-lab growth in microbially diverse and natural-like environments.
Locally sourced soil can be enriched with produce to diversify the microbial communities in which worms are raised and from which they are harvested, washed, and surface-sterilized for subsequent analyses. Representative experiments demonstrate the ability to modulate the microbial community in a common soil by enriching it with different produce and further demonstrate that worms raised in these distinct environments assemble similar gut microbiomes distinct from their respective environments, supporting the notion of a species-specific core gut microbiome. Overall, compost microcosms provide natural-like in-lab environments for microbiome research as an alternative to synthetic microbial communities or to the isolation of wild nematodes.
Introduction
The nematode Caenorhabditis elegans is emerging as a useful model for studying interactions between hosts and their gut microbiomes1,2. As a model, it offers several advantages. First, germ-free or gnotobiotic animals are easy to obtain and maintain; bleach can be used to kill gravid worms and associated microbes, leaving their bleach-resistant eggs unharmed to grow as age-synchronized populations that can be colonized by bacteria of interest3,4. In addition, when grown in the presence of bacteria, C. elegans, a bacterivore, ingest the encountered bacteria, with susceptible species digested or excreted, while resistant and persistent species stably colonize the worm gut. Furthermore, C. elegans are mostly hermaphroditic, producing populations of genetically identical progeny, which reduces confounding genetic variation. Coupled with the availability of mutant and transgenic worm strains, working with C. elegans offers researchers a gnotobiotic and genetically tractable model to investigate the molecular underpinnings of host-microbe interactions5,6,7,8.
While experiments with well-characterized bacteria can facilitate the analysis of molecular mechanisms, identifying and studying the bacteria that worms interact with in nature are essential for exploring the diversity of such mechanisms, unraveling the natural context for their function, and understanding the selective forces that have shaped their evolution. Outside the laboratory, C. elegans is found globally in humid temperate climates, where populations are thought to undergo a "boom-and-bust" lifecycle, characterized by rapid population growth when resources are abundant, followed by a developmental shift to pioneering, stress-tolerant dauers when resources are depleted9. Though considered a soil nematode, wild-proliferating C. elegans populations are most commonly found feeding on decomposing organic material such as rotting flowers or fruits, where bacterial populations are abundant and diverse.
Studies of the gut microbiome in nematodes isolated from the wild have identified diverse, yet characteristic, bacterial communities10,11, the composition of which was further supported by studies carried out with worms raised in natural-like microcosm environments12,13. Together, such studies enabled the delineation of a core worm gut microbiome2. Whereas the sampling of C. elegans populations in the wild represents the most direct examination of natural worm-microbe interactions, it is not feasible everywhere and anytime, as it is limited to regions and seasons with ample precipitation10,11. Alternatively, instead of isolating worms from their natural habitat, experiments using microcosms bring the natural habitat into the laboratory6,8,12,13,14,15. Microcosm environments are prepared from soil composted with various fruit or vegetables, which enables further diversification of the starting soil community. They offer tractable experimental methods that combine the microbial diversity and the three-dimensional wild soil environment with the experimental advantages of a controlled laboratory facility and genetically defined worm strains. The protocol below details the steps involved in working with compost microcosms, demonstrating their use in understanding the assembly of a characteristic worm gut microbiome from diverse environments.
Protocol
1. Preparation of compost
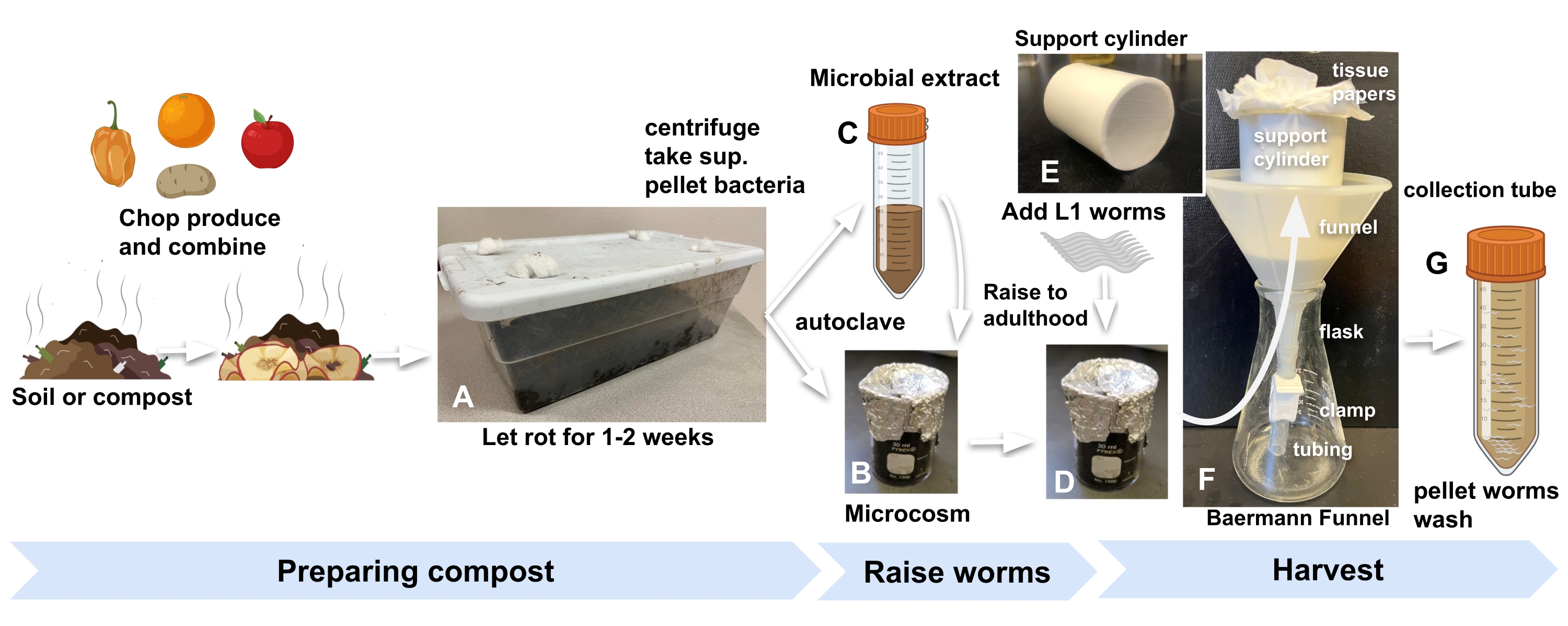
- Obtain compost or garden soil from any convenient source and store inside the laboratory in a standard kitchen plastic container with holes cut in the lid to let air in. Plug the holes with cotton wool to keep fruit flies and other invertebrates out (Figure 1A).
NOTE: Five hundred grams of soil (fitting in a 1.5 gallon container, dimensions: 30 cm x 20 cm x 10 cm) will provide enough material for 12 microcosms. - Enrich the compost or soil with chopped produce or a mixture of different produce in a 1:2 mass ratio of produce to soil.
- Incubate for 7-14 days at 20-25 °C, mixing once a day and adding M9 medium as necessary to maintain moisture without making it muddy.
NOTE: Soils not enriched with produce usually do not support C. elegans growth, but which specific produce to use is up to the researcher, with many types and mixes capable of supporting worm growth. Enrichment with different produce will promote bacterial community diversification in different ways, enabling the study of gut microbiome assembly from different starting points (see the discussion section).
2. Preparation of compost microcosms
- For each microcosm, add 10 g of enriched compost to a 30 mL glass beaker covered with tin foil and autoclave (Figure 1B).
- To prepare microbial extract to replenish the autoclaved compost, start by adding 30 g of the same compost used in step 2.1 to each of three 50 mL tubes and fill with M9. Vortex for 1 min (Figure 1C).
NOTE: This should provide enough bacteria for nine microcosms, each supporting the development of hundreds of nematodes. - Centrifuge the tubes at 560 × g for 5 min at room temperature (RT).
- Paying attention not to disturb the pellet, remove the supernatants with a serological pipette and combine in a new 50 mL tube.
- Concentrate the bacterial extract by centrifuging at maximum speed (2,000 × g) for 15 min at RT. Resuspend the pellets in enough M9 to have 200 µL for each microcosm and 200 µL more to add to a plate that will serve as a visible proxy of worm development inside microcosms.
NOTE: For example, for nine microcosms, resuspend the microbial pellet in 2 mL of M9. - Add 200 µL of the concentrated microbial extract to each beaker of autoclaved compost, as well as to an NGM plate that will serve as the visible proxy plate.
- Incubate microcosms and proxy plate for 24 h at 20-25 °C before addition of worms.
3. Raising worms in compost microcosms
- Add 500-1000 eggs or L1 larvae3 to each microcosm and to the proxy plate (step 2.7) to begin the experiment (Figure 1D).
NOTE: The experiments described here utilize N2 wild-type worms. However, other C. elegans strains (and potentially other nematodes) can be used. - Raise the worms to adulthood at 20 °C (typically 3 days).

Figure 1: Preparing compost microcosms, raising worms, and harvesting. (A) Enrich local soil or compost with produce and incubate for 2 weeks. Combine (B) autoclaved enriched soil with (C) microbial extract and (D) incubate for at least 24 h before adding synchronized L1s worms to microcosms to begin the experiment. (E, F) When ready to harvest, add compost from the microcosm into a Baermann funnel support cylinder and cover with M9. (G) After 15 min, release the filtrate into a 50 mL tube. Abbreviation: sup. = supernatant. Please click here to view a larger version of this figure.
{kind=link}
4. Preparing a Baermann funnel for harvesting worms
- Assemble a Baermann funnel by attaching 5-8 cm of rigid rubber tubing to the end of a plastic funnel.
- Slide a clamp onto the tubing and clamp it shut.
- Place in the funnel a 7 cm long, 5 cm diameter, cylindrical PVC pipe with a 1 mm nylon mesh glued at its bottom (Figure 1E). Line the cylinder with two sheets of tissue paper.
- Place the Baermann funnel into a flask (Figure 1F).
5. Harvesting worms from microcosms and collecting the respective soil samples
- Add 20 mL of M9 to the microcosm in which the worms were raised, agitate the mixture, and then pour the mixture from the beaker into the tissue paper-lined cylinder in the Baermann funnel setup. Add more M9 to completely submerge the compost in the funnel.
NOTE: C. elegans (N2) populations reach adulthood in compost microcosms at a similar rate as on standard agar plates seeded with Escherichia coli. For further accuracy in harvesting worms at a specific developmental stage, refer to the proxy plate from step 3.3. - After 30 min, unfasten the clamp to release the filtrate containing the harvested worms into a 50 mL tube (Figure 1G).
- Add more M9 to the cylinder and repeat for a second round to harvest more worms, and once more if additional worms are required.
NOTE: When repeating harvesting rounds, be careful not to compromise the tissue paper's integrity, which could allow soil particles to pass through. A maximum of four harvest rounds should isolate at least 50% of the worms originally added to the microcosm without compromising tissue paper integrity. - Concentrate the worms by centrifuging at 560 × g for 2 min (RT). Remove 35 mL of the supernatant with a serological pipette.
- Transfer the remaining 15 mL to a 15 mL tube and centrifuge again at 560 × g for 1 min to further concentrate the worms. Remove 14 mL of the supernatant with a serological pipette.
- In parallel, collect 1 g of the remaining microcosm soil into a 1.5 mL tube. Process the soil samples containing the environmental bacterial community immediately or store them at −20 °C for later extraction of nucleic acids, as described below for worm samples.
6. Washing and surface sterilization of harvested worms
- Transfer 1 mL of the concentrated, harvested worms from step 5.5 to a 1.5 mL tube using a glass pipette. Incubate for 2 min to allow the worms to settle at the bottom of the tube. Remove the supernatant, leaving the bottom 100 µL undisturbed.
- Wash 6x with 1.5 mL of M9+T (0.025% Triton-X in M9), allowing the worms to settle at the bottom each time.
- Transfer the washed worms in a volume of 100 µL to a new 1.5 mL tube using a glass pipette.
NOTE: At this point in the protocol, roughly 30 min should have passed since the first wash (step 6.2). Allowing the worms to remain without food for at least 1 h before the addition of levamisole is recommended to allow the excretion of transient bacteria and the full digestion of food bacteria. - Add 100 µL of 25 mM levamisole hydrochloride to paralyze the worms. Incubate for 5 min at RT.
- Add 200 µL of 4% bleach solution. Incubate for 2 min.
- Remove the supernatant, leaving the bottom-most 150 µL undisturbed, and wash 3x with M9+T, as above.
NOTE: To minimize contamination of the samples, it is recommended to use filtered M9+T (through a 0.2 µm filter) and to wear gloves from this point onward. - Following the washes, take 50 µL of the remaining 150 µL from the last wash and plate on an LB plate, incubating at 25 °C for 48 h to confirm the effective removal of external bacteria.
NOTE: Observing up to 30 colonies in the last wash (representing 60 external bacterial cells total remaining in the sample) is permitted and expected to have only a marginal contribution to the analyzed microbiome composition, given the thousands of bacteria that typically colonize each adult worm15. When more are observed, sample integrity might be compromised. - Use the surface-sterilized worms immediately (e.g., to extract live bacteria for culturing [CFU counts]) or store at −20 °C for the subsequent extraction of nucleic acids.
7. DNA extraction
NOTE: The following steps describe DNA extraction of the harvested worms using a commercial kit designed for the extraction of microbial DNA from soil (see Table of Materials), with modifications described below to facilitate the extraction of microbial DNA from worms.
- Transfer the sample containing surface-sterilized worms (defrost at RT if necessary) to the tubes provided by the kit, replacing the provided glass beads with roughly 30-50 1 mm diameter zirconia beads (see Table of Materials).
NOTE: The observed DNA yields are higher with zirconia beads than with the glass beads provided by the kit. Although the reason behind this observation remains undetermined, zirconia beads may break open the nematode cuticle more efficiently, releasing more bacteria. - For compost samples, add roughly 250 mg of the collected soil to the kit's tubes without replacing the glass beads.
- After the addition of the buffer solution provided by the kit to all the samples, homogenize with a power homogenizer at RT (see Table of Materials) for two rounds of 2,000 rpm for 30 s each, pausing for 30 s in between.
- Complete the remaining DNA purification steps according to the kit protocol. Elute the worm samples in no more than 50 µL of elution buffer to ensure DNA concentrations that are high enough for sequencing.
Representative Results
To explore the ability to diversify the community of soil microcosms, we compared the microbial communities in compost microcosms prepared by enriching the same initial soil, an industrial-grade compost available from the city of Berkeley, California, with different produce: apples, bell peppers, oranges, or potatoes (each set in triplicate). We further compared the microbial communities of each compost environment with the gut microbiome of wild-type C. elegans raised in the respective microcosm. Analysis was performed with DNA samples extracted from roughly 500 surface-sterilized adults per microcosm and from 250 mg compost samples of the respective microcosms.
Characterization of the environmental soil and worm gut microbiomes relied on next-generation sequencing of the V4 region of the bacterial 16S rRNA gene. Sequencing library preparation was achieved using the standard kits and performed according to the manufacturers' instructions, with sequencing performed on a commercial sequencer (see Table of Materials). Demultiplexed sequences were processed using DADA2, assigned taxonomy based on the SILVA v132 reference database, and analyzed with phyloseq16,17,18 (see Supplementary File 1, Supplementary Figure S1, Supplementary Figure S2, Supplementary Figure S3, Supplementary Table S1, and Supplementary Table S2 for a detailed description of the sequencing and analysis; the full computational pipeline is available in GitHub [https://github.com/kennytrang/CompostMicrocosms]). Raw data are available at the NCBI Sequence Read Archive (Bioproject ID PRJNA856419).
On average, 73,220 sequences were obtained per sample. These sequences represent 15,027 amplicon sequence variants (ASVs), spanning 27 phyla and 216 families, including families considered part of the core C. elegans gut microbiome13, such as Rhizobiaceae, Burkholderiaceae, and Bacillaceae. Enterobacteriaceae, and Pseudomonadaceae, which were previously found to be dominant members, were a minority this time, but were still enriched (2-10-fold) compared to their respective soil environments. Comparisons based on both unweighted and weighted UniFrac19,20 distances demonstrated good reproducibility among microcosm triplicates enriched with the same produce, as indicated by close clustering. In contrast, environmental soil microbiomes enriched with different produce clustered away from each other, demonstrating the ability to diversify an initial microbial community through the addition of different produce (Figure 2).
In comparisons of worm gut microbiomes and environmental communities, principal coordinate analysis (PCoA) with either unweighted or weighted UniFrac distances showed distinct clustering of worm gut microbiomes away from that of their respective environments for each microcosm type (Figure 2). While PCoA based on unweighted UniFrac distances did not distinguish between soil and worm microbiomes (Figure 2A), clustering based on weighted distances revealed a clear separation of worm gut and compost microbiomes (Figure 2B). These results support a process in which host filtering operates on environmental availability to shape a gut microbiome that is not completely distinct from its environmental source with regard to the presence of taxa but modulates their abundance by enriching for a subset of the available taxa, ultimately resulting in a core worm gut microbiome shared between worms raised in different environments.

Figure 2: Worm gut microbiomes clustering away from their respective produce-diversified microbial environments. Microbiome composition was determined with 16S sequencing, and communities from microcosms enriched with the designated produce or from worms raised in them were clustered using PCoA based on (A) unweighted or (B) weighted UniFrac distances. Axes shown are those that explain the greatest variation in community composition between samples (N = 3 for each microcosm type). Please click here to view a larger version of this figure.
{kind=link}
Supplementary File 1: Next-generation sequencing and data analysis. Presented here are the steps for library preparation, in-lab sequencing, and data analysis. Please click here to download this File.
Supplementary Figure S1: An example of a quality control graph for the reverse reads from one sample. The X-axis (cycle) shows the nucleotide position along the sequence read. The left Y-axis shows the quality score. The greyscale heatmap represents the frequency of the quality score at each nucleotide position; the green line depicts the median quality score at each nucleotide position; the top orange line depicts the quartiles of the quality score distribution; the bottom red line depicts the percent of sequence reads that extended that nucleotide position (right Y-axis, here 100%). Please click here to download this File.
Supplementary Figure S2: Error rates for different samples. The error frequency in the different samples (black dots) should decrease with increasing quality score for each possible base pair substitution depicted, reflecting the expected trend. Please click here to download this File.
Supplementary Figure S3: An example of PCoA based on weighted UniFrac distances. The group names shown in the legend represent the produce used to enrich the compost used in the different microcosms. Please click here to download this File.
Supplementary Table S1: Sequential sequence filtering. aNumber of sequence reads before filtering. b-dEach column represents the number of sequence reads remaining after a filtration step: filtering out low quality reads (step 2.5), denoising algorithm performed by dada() (step 2.8), merging forward and reverse reads (step 2.9), and removing chimeras (step 2.11). Please click here to download this File.
Supplementary Table S2: Metadata table. Please click here to download this File.
Discussion
The protocol presented here describes a method to study the gut microbiome of nematodes raised in natural-like environments, offering an alternative approach to the isolation of worms from nature or to raising them on synthetic communities.
The thousands of potential bacterial species captured in the representative microcosm experiment reflect the microbial diversity that the worms have evolved with and demonstrate the ability of the microcosm pipeline to combine the advantages of working with a model host organism and those of working with natural, diverse microbial communities.
The representative results demonstrate that enriching a common soil with different produce types modulates environmental microbial diversity, highlighting the range of microbial diversity available for exploration using this pipeline. Produce choice is not particularly important. Previous work has used bananas, apples, oranges, strawberries, green tea leaves, and potatoes to enrich soil, resulting in similar worm raising efficiency. Mixed produce has also been used effectively. The key feature is that different produce will diversify a given soil in distinct ways.
Despite the broad variation in environmental microbial diversity, worm gut microbiomes vary considerably less, recapitulating the importance of the worm gut niche and host filtering for the assembly of a core gut microbiome that is distinct from that of its soil environment13. This pattern is observed best with weighted UniFrac PCoA analysis, demonstrating that the differences between environmental soil and worm gut microbiomes stem mostly from differences in the relative abundance of key taxa.
Although the protocol described here focuses on harvesting worms for 16S sequencing, microcosms can be used to explore additional questions of interest. For example, worms harvested from microcosms can be subsequently examined for the effect of a diverse gut microbiome on host resistance to various adverse conditions, including pathogens or toxins. Alternatively, novel bacterial species and strains may be isolated and cultured from ground-harvested worms, expanding the taxonomic and functional diversity of bacteria available to perform experiments with.
While research methods in laboratory settings seek consistency and reproducibility, work with microcosms takes advantage of the natural variation to explore host-microbe interactions in a natural-like context. Nevertheless, this variation also poses some challenges. Some soils that include high levels of endogenous invertebrates may require additional centrifugation, filtration, and examination steps to effectively eliminate undesired organisms from the microcosm preparation. Additionally, low microbial abundance in soil may induce undesired dauer formation in worm populations, requiring an increase in microbial extract or enriching soil with a greater amount of produce. With each microcosm experiment performed, researchers continue to explore the full taxonomic and functional diversity provided by nature, enabling the discovery of new microbial taxa and functional abilities ranging from infection resistance to protection against environmental xenobiotics.
Acknowledgements
The work described in this manuscript was supported by NIH grants R01OD024780 and R01AG061302. K.T. was further supported by a Summer Undergraduate Research Fellowship from the University of California, Berkeley, funded by the Rose Hills Foundation. Cartoon designs in Figure 1 were obtained from BioRender.com.
Materials
| Name | Company | Catalog Number | Comments |
| AMPure XP Reagent, 60 mL | Beckman Coulter | A63881 | Supplementary File Step 1.3 |
| Bleach (Sodium Hypochlorite) | Sigma-Aldrich | 7681-52-9 | Step 6.5 |
| DNeasy PowerSoil Pro Kit | Qiagen | 47016 | Step 7 DNA extractions |
| dNTP set 10 mM | Invitrogen | 18427013 | Supplementary Step 1.2 |
| Easypet 3 Serological Pipette Controller | Eppendorf | 4430000018 | Used to remove supernatant when specified |
| Greiner Bio-One 25 mL Sterile Serological Pipets | Fisher Scientific | 07-000-368 | Used to remove supernatant when specified |
| KH2PO4 | Fisher Scientific | P285-500 | Used to make M9 |
| Levamisole Hydrochloride | Fisher Scientific | AC187870100 | Step 6.4 |
| M9 Minimal Media Solution | Prepared in-house | N/A | Recipe in wormbook.org |
| MgSO4 | Fisher Scientific | M63-500 | Used to make M9 |
| MiniSeq High Output Reagent Kit (150 cycles) | Illumina | FC-420-1002 | Supplementary Step 1.7 |
| MiniSeq System | Illumina | SY-420-1001 | Commercial sequencer used; Supplementary Step 1.7 |
| Na2HPO4 | Fisher Scientific | S374-500 | Used to make M9 |
| NaCl | Fisher Scientific | S271-3 | Used to make M9 |
| Nematode Growth Media (NGM) | Prepared in-house | N/A | Recipe in wormbook.org |
| Nextera XT DNA Library Preparation Kit (96 samples) | Illumina | FC-131-1096 | Library prep kit used; Supplementary Step 1.4 |
| PhiX Control v3 | Illumina | FC-110-3001 | Supplementary Step 1.7 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | M0530L | Supplementary Step 1.2 |
| PowerLyzer 24 Homogenizer (110/220 V) | Qiagen | 13155 | Step 7.3 |
| Qubit dsDNA HS Assay Kit | Invitrogen | Q32851 | Supplementary Steps 1.1 & 1.6 |
| Qubit Fluorometer | Invitrogen | Q33238 | Supplementary Steps 1.1 & 1.6 |
| Triton X-100 | Fisher Scientific | BP-151 | Used to prepare M9+T |
| Zirconia/Silica Beads 1.0 mm diameter | Fisher Scientific | NC9847287 | Step 7.1 |
References
- Shapira, M. Host-microbiota interactions in Caenorhabditis elegans and their significance. Current Opinion in Microbiology. 38, 142-147 (2017).
- Zhang, F., et al. Caenorhabditis elegans as a model for microbiome research. Frontiers in Microbiology. 8, 285 (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook. , 7-8 (2006).
- Dirksen, P., et al. CeMbio - The Caenorhabditis elegans microbiome resource. G3: Genes, Genomes, Genetics. 10 (9), 3025-3039 (2020).
- Ortiz, A., Vega, N. M., Ratzke, C., Gore, J. Interspecies bacterial competition regulates community assembly in the C. elegans intestine. ISME Journal. 15 (7), 2131-2145 (2021).
- Berg, M., et al. TGFβ/BMP immune signaling affects abundance and function of C. elegans gut commensals. Nature Communications. 10, 604 (2019).
- Zhang, F., et al. Natural genetic variation drives microbiome selection in the Caenorhabditis elegans gut. Current Biology. 31 (12), 2603-2618 (2021).
- Montalvo-Katz, S., Huang, H., Appel, M. D., Berg, M., Shapira, M. Association with soil bacteria enhances p38-dependent infection resistance in Caenorhabditis elegans. Infection and Immunity. 81 (2), 514-520 (2013).
- Frézal, L., Félix, M. A. C. elegans outside the Petri dish. eLife. 4, 05849 (2015).
- Dirksen, P., et al. The native microbiome of the nematode Caenorhabditis elegans: Gateway to a new host-microbiome model. BMC Biology. 14 (1), 38 (2016).
- Samuel, B. S., Rowedder, H., Braendle, C., Félix, M. A., Ruvkun, G. Caenorhabditis elegans responses to bacteria from its natural habitats. Proceedings of the National Academy of Sciences of the United States of America. 113 (27), 3941-3949 (2016).
- Berg, M., Zhou, X. Y., Shapira, M. Host-specific functional significance of Caenorhabditis gut commensals. Frontiers in Microbiology. 7, 1622 (2016).
- Berg, M., et al. Assembly of the Caenorhabditis elegans gut microbiota from diverse soil microbial environments. ISME Journal. 10 (8), 1998-2009 (2016).
- Slowinski, S., et al. Interactions with a complex microbiota mediate a trade-off between the host development rate and heat stress resistance. Microorganisms. 8 (11), 1-9 (2020).
- Pérez-Carrascal, O. M., et al. Host preference of beneficial commensals in a microbially-diverse environment. Frontiers in Cellular and Infection Microbiology. 12, 795343 (2022).
- Callahan, B. J., et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods. 13 (7), 581-583 (2016).
- Quast, C., et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research. 41, 590-596 (2013).
- McMurdie, P. J., Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 8 (4), 61217 (2013).
- Lozupone, C., Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Applied and Environmental Microbiology. 71 (12), 8228-8235 (2005).
- Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME Journal. 5 (2), 169-172 (2011).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved