Functional Characterization and Visualization of Esophageal Fibroblasts Using Organoid Co-Cultures
In This Article
Summary
Organoid-fibroblast co-cultures provide a model to study the in vivo stem cell niche. Here, a protocol for esophageal organoid-fibroblast co-cultures is described. Additionally, whole mount imaging is used to visualize the fibroblast-organoid interaction.
Abstract
Epithelial stem and progenitor cells contribute to the formation and maintenance of the epithelial barrier throughout life. Most stem and progenitor cell populations are tucked away in anatomically distinct locations, enabling exclusive interactions with niche signals that maintain stemness. While the development of epithelial organoid cultures provides a powerful tool for understanding the role of stem and progenitor cells in homeostasis and disease, the interaction within the niche environment is largely absent, thereby hindering the identification of factors influencing stem cell behavior. Fibroblasts play a key role in directing epithelial stem and progenitor fate. Here, a comprehensive organoid-fibroblast co-culture protocol enabling the delineation of fibroblast subpopulations in esophageal progenitor cell renewal and differentiation is presented. In this protocol, a method to isolate both epithelial cells and fibroblasts in parallel from the esophagus is described. Distinct fluorescence-activated cell sorting strategies to isolate both the esophageal progenitor cells as well as the fibroblast subpopulations from either transgenic reporter or wild-type mice are outlined. This protocol provides a versatile approach that can be adapted to accommodate the isolation of specific fibroblast subpopulations. Establishing and passaging esophageal epithelial organoid mono-cultures is included in this protocol, enabling a direct comparison with the co-culture system. In addition, a 3D clearing approach allowing for detailed image analysis of epithelial-fibroblast interactions is described. Collectively, this protocol describes a comparative and relatively high-throughput method for identifying and understanding esophageal stem cell niche components in vitro.
Introduction
Organoids are used as 3D in vitro assays to characterize stem and progenitor cells, as well as to understand the signaling cues derived from the cellular components of the stem cell niche1,2,3,4. Mouse esophageal organoids were first described in 2014 and several papers have identified growth factors, like R-Spondin (RSPO), NOGGIN, and epidermal growth factor (EGF), needed to maintain and passage esophageal organoids5,6,7, arguing that similar signaling cues are required for in vivo progenitor cell renewal. However, growth factors are commonly added in non-physiological concentrations, resulting in organoid growth conditions which do not necessarily reflect the in vivo signaling environment.
Fibroblasts are heterogeneous stromal cell populations that support progenitor cell properties in many stem cell niches8. Combining epithelial progenitor cells and fibroblasts in the same organoid culture enables organoid formation in reduced concentrations of exogenously supplemented growth factors. Organoid co-culture systems from intestinal and hepatic epithelia are described, but a protocol to establish esophageal organoid-fibroblast co-cultures is still outstanding9,10,11.
In this protocol, two fluorescence-activated cell sorting (FACS) strategies for fibroblasts from the esophagus, using either transgenic PdgfrαH2BeGFP mice12 or wild-type mice with classical antibody staining are outlined. Different subpopulations of fibroblasts can be isolated using cell surface markers of choice, thereby providing flexibility to the protocol. In addition, a 3D fluorescence imaging technique preserving organoid morphology is used to characterize fibroblast-organoid interactions. Organoid clearing provides a quick method to increase the light penetration depth in the organoids, improving the visualization of organoid-fibroblast connections and enabling the recapitulation of the organoid structure in its entirety. This protocol combines esophageal organoid co-culture with a whole mount imaging strategy, enabling functional characterization of the fibroblast-organoid interaction.
Protocol
Animal experiments for this study were approved by Stockholms Norra djurförsöksetiska nämnd (ethical permit no 14051-2019). Animals were housed in pathogen-free conditions according to the recommendations of the Federation of European Laboratory Animal Science Association.
1. Preparation
- Thaw the enzymatic stock solutions used for dissociation (see Table of Materials) on ice. Thaw an aliquot of growth factor reduced (GFR) basement membrane matrix (matrix) at 4 °C.
- Prepare the culture medium. Use the medium described in Table 1 for organoid cultures and co-cultures and prepare it before starting the protocol.
2. Dissection and separation of the esophageal epithelium and stroma
NOTE: Ensure that all instruments used for dissection and tissue processing are sterile. Prepare 2 mL of dissociation solution (see Table of Materials) in Hanks' balanced salt solution (HBSS) per three esophagi.
- Use a mouse strain of choice, such as C57BL/6J mice. Esophageal development in mice finishes after post-natal day (p) 70, so it is recommended to use mice that are older than p7013. Use four or five mice as they provide sufficient material to establish 8-10 organoid co-cultures.
NOTE: Organoid-forming efficiency decreases with the age of the mice. If specific fibroblast subpopulations are of interest, it is possible that the low fibroblast yield is limiting the number of organoid co-cultures that can be established. - Use genetically modified (e.g., PdgfrαH2BeGFP) or wild-type (WT) mice for the isolation of fibroblast populations. When using WT mice, perform antibody staining to isolate specific fibroblast subpopulations from the stroma via FACS.
- Euthanize the mice by CO2 asphyxiation. Dissect the esophagus using forceps and dissection scissors. To remove the whole esophagus, cut the distal end of the esophagus directly above the stomach and the proximal end at the beginning of the trachea. Submerge the esophagus in PBS and put it on ice.
- Mechanically remove the muscularis externa using a dissection microscope (total magnification range of 8x-40x) and forceps. Hold the distal end of the dissected esophagus using one pair of forceps and use the other forceps to grab and pull the muscle from the distal to the proximal end of the dissected esophagus. Remove and discard the muscle layer.
- Open the esophagus longitudinally. This works best using microdissection spring scissors with a ball tip to prevent tissue damage. Hold one end of the esophagus in order to insert the ball of the spring scissors into the lumen of the esophagus and cut open the esophagus while holding on to the end.
- Place the esophagus in a 1.5 mL micro-centrifuge tube or a 24-well plate. Incubate the opened esophagus in 0.5 mg/mL thermolysin in HBSS at 37 °C on a rocker-shaker for 15 min. Submerge the esophagus completely in thermolysin solution.
NOTE: The volume of thermolysin solution used depends on the well or tube-size. Several esophagi can be placed and submerged in the same well or tube. - Take out the esophagus from the thermolysin solution. Use a dissection microscope to carefully separate the esophageal epithelium from the stroma. Using fine forceps, grab both the epithelial side and the stroma side of the tissue and slowly pull them apart to separate the two layers.
NOTE: The stroma is identified by its white and opaque appearance, in contrast to the transparent epithelium. The stroma contains the lamina propria and submucosal layer. - Transfer the epithelial and stromal layer to two separate 1.5 mL micro-centrifuge tubes with 200 µL of dissociation solution in HBSS. Put on ice.
3. Isolation of esophageal progenitor cells
NOTE: The isolation of esophageal progenitor cells (step 2) and fibroblasts (step 3) can be performed simultaneously. Prepare a 50 mL tube of 1% FBS in HBSS (1% FBS).
- Transfer the separated esophageal epithelium from the 1.5 mL micro-centrifuge tube (step 2.8) to a clean Petri dish and use a sharp scalpel to mince. Collect the minced tissue from the Petri dish with 200 µL of dissociation solution and transfer it back to the 1.5 mL micro-centrifuge tube.
NOTE: The epithelium is minced properly when the pieces can be placed back into the 1.5 mL micro-centrifuge tube using a 200 µL pipette tip. - Add 800 µL of fresh dissociation solution to the 1.5 mL micro-centrifuge tube to a total volume of 1 mL.
NOTE: It is important to add a sufficient volume of dissociation solution. Per one to three esophagi, 1 mL of solution is recommended. Scale up when more esophagi are processed at once. - Place the tube with the minced epithelial layer on a rocker-shaker at 37 °C for 60 min. Pipette the solution up and down approximately 20 times using a 200 µL pipette tip every 15 min to enhance digestion.
- After 60 min, pipette up and down another 20 times using a 200 µL pipette tip. Pass the solution through a 40 µm cell strainer into a new 1.5 mL micro-centrifuge tube.Centrifuge at 300 x g for 10 min at 4 °C.
NOTE: Wet the cell strainer with 1% FBS before straining the cells to minimize adherence of the cells to the filter. The epithelium will not be completely digested, and pieces of tissue will still be visible. Longer incubation will, however, decrease cell survival and not result in a higher yield of viable cells. - Discard the supernatant by removing excess liquid with a 1 mL pipette and resuspend the pellet in 1 mL of 1% FBS. Centrifuge at 300 x g for 10 min at 4 °C.
- During centrifugation, prepare the conjugated antibody mix for FACS of esophageal progenitor cells.
- Mix 1 μL of CD324-PE-Cy7 (ECADHERIN) and CD104-A647 (INTEGRIN-β4) in 200 μL of 1% FBS per one million cells.
NOTE: 1 µL of antibody (200 µL final volume) is sufficient for one or two esophagi. When processing more esophagi at once, increase the volume of the antibody staining solution.
- Resuspend the pellet in 200 µL of antibody mix and transfer to a flow cytometry tube. After adding fluorescent antibodies, keep the cell suspensions in the dark to avoid bleaching of signal. Incubate the cells for 30 min at 4 °C. Add 3 mL of 1% FBS and centrifuge at 300 x g for 10 min at 4 °C, and then resuspend the cells in a minimum of 200 µL of 1% of FBS.
NOTE: 500 µL of 1% FBS is used for up to four or five esophagi. Increase the volume when more esophagi are processed at once, so a FACS flow rate of 100-300 events/s can be achieved. More events/s will decrease sorting efficiency, and an increase in the flow rate will decrease cell survival. - Add dead cell stain marker to a final concentration of 1:10,000, 5 minutes prior to FACS sorting to isolate live cells. Sort the progenitor cells using a FACS machine (see Figure 1 for gating strategy). Collect the cells in 1.5 mL micro-centrifuge tubes filled with 200 µL of basic organoid medium (Table 1).
4. Isolation of fibroblasts from the stromal layer
- Cut the stomal layer in fine pieces in a 1.5 mL tube containing 200 µL of dissociation solution (step 2.8) using dissection scissors. The tissue is properly minced once the solution can be pipetted up and down using a 200 µL pipette tip.
- Add 800 µL of fresh dissociation solution to the 1.5 mL micro-centrifuge tube to a total volume of 1 mL.
NOTE: It is important to add a sufficient volume of dissociation solution. Per one to three esophagi, 1 mL of solution is recommended. Scale up when more esophagi are processed at once. - Place the tube on a rocker-shaker at 37 °C for 30 min. After 15 min, pipette the solution up and down approximately 20 times using a 200 µL pipette tip to enhance digestion.
- After 30 min of enzymatic digestion, pipette up and down another 20 times using a 200 µL pipette tip. Strain the solution through a 70 µm cell strainer into a new 1.5 mL micro-centrifuge tube. Centrifuge at 300 x g for 10 min at 4 °C.
NOTE: Wet the cell strainer with 1% FBS before straining the cells to minimize adherence of cells to the filter. - Discard the supernatant by removing excess liquid with a 1 mL pipette and resuspend the pellet in 1 mL of 1% FBS. Centrifuge at 300 x g for 10 min at 4 °C.
NOTE: When using a genetically modified mouse strain containing flourescent-labeled fibroblasts antibody stainings are optional. If antibody stainings are not required, continue with step 3.7 and transfer the sample to a flow cytometry tube.- During centrifugation, prepare the conjugated antibody mix for the FACS isolation of fibroblasts. Mix 1 µL of CD26-APC (DPP4) in 200 µL of 1% FBS per one million cells.
NOTE: 1 µL of antibody is sufficient for one or two esophagi. When processing more esophagi at once, increase the volume of the antibody staining solution.
- During centrifugation, prepare the conjugated antibody mix for the FACS isolation of fibroblasts. Mix 1 µL of CD26-APC (DPP4) in 200 µL of 1% FBS per one million cells.
- Resuspend the pellet in 200 µL of conjugated antibody mix in 1% FBS and transfer to a flow cytometry tube. Incubate the cells for 30 min at 4 °C.
- Add 3 mL of 1% FBS and centrifuge at 300 x g for 10 min at 4 °C. Resuspend the cells in a minimum of 200 µL of 1% of FBS.
NOTE: 500 µL of 1% FBS is used for up to four or five esophagi. Increase the volume when more esophagi are processed at once, so a flow rate of 100-300 events/s can be achieved. More events/s will decrease sorting efficiency, and increasing the flow rate will decrease cell survival. After adding fluorescent antibodies, cell suspensions should be kept dark to avoid bleaching of signal. - Add dead cell stain marker to a final concentration of 1:10,000, 5 minutes prior to FACS sorting to isolate live cells. Sort the cells using a FACS machine (see Figure 1 for gating strategy). Collect the cells in 1.5 mL micro-centrifuge tubes filled with 200 µL of basic organoid medium (Table 1).
5. Establishment and culture of esophageal organoids
NOTE: Prewarm ERlow (organoid co-culture), ENR (organoid) medium (see Table 1 for description), and a 48-well plate at 37 °C. Place the thawed matrix (prepared in step 1) aliquot on ice. It is recommended to use the matrix provided here (see Table of Materials) for mouse esophageal organoid culture, since other brands of matrix negatively affect organoid forming efficiency.
- For the organoid co-culture, mix the sorted epithelial cells and fibroblasts at a ratio of 1:2 in a tube. For each matrix dome, use 5,000 epithelial cells and 10,000 fibroblasts. When preparing for more domes, add a multiple of 5,000 epithelial cells and 10,000 fibroblasts to one tube. For organoid cultures without fibroblasts, use 5,000 epithelial cells per matrix dome.
- Centrifuge the mixed cell population at 300 x g for 5 min. Discard the supernatant by carefully removing it with a 200 µL pipette.
- Wash the cells once by resuspending the pellet in cold basic organoid medium and centrifuge at 300 x g for 5 min. Place the cells on ice.
- Prepare a mix consisting of 80% matrix and 20% cold basic organoid medium. Place everything on ice as matrix solidifies at room temperature (RT).
- Discard the supernatant after centrifugation by carefully removing it with a 200 µL pipette. Resuspend the cells in 10 µL of matrix mix/dome and put back on ice.
- Take the prewarmed 48-well plate from the 37 °C incubator and make one matrix dome per well using a 20 µL pipette. Each dome contains 10 µL of 80% matrix, 5,000 epithelial cell,s and 10,000 fibroblasts. Transfer the plate upside down to an incubator and let the domes solidify for another 20-30 min at 37 °C.
NOTE: A decrease in matrix dome volume and/or an increase in cell number will affect the organoid forming efficiency. - Add 200 µL of prewarmed ERlow medium (Table 1) to the matrix domes containing organoid co-cultures and ENR medium (Table 1) to the respective matrix domes containing only epithelial organoids and place the plate in the incubator.
- Grow the organoids at 37 °C and 5% CO2. For the first 2 days, supplement the medium with 10 µM Rock-inhibitor (Y-27623). Rock inhibitor prevents stress induced cell death and increases the chance of a successful establishment of organoid cultures.
- Refresh the medium every 2-3 days. Make sure that the medium is warm to prevent the dissociation of the temperature sensitive matrix.
- Perform analyses of the experiment 6 to 8 days after plating. The organoids can be kept in culture for up to 14 days. Around day 14, loss of dome integrity is observed.
6. Passaging of organoids
NOTE: Passaging of organoids grown in co-culture results in the loss of fibroblasts. Therefore, it is recommended to use ENR medium for all organoids when passaged. Prewarm ENR, PBS, and a 48-well plate at 37 °C.
- Remove medium and wash the well containing the matrix dome with pre-warmed PBS. Add 200 µL of cold 0.25% trypsin solution and pipette up and down to break the matrix dome.
NOTE: The use of cold 0.25% trypsin is recommended, as the matrix is temperature sensitive and this aids in breaking down the matrix domes. - Incubate the organoids with trypsin at 37 °C for ~20 min. Pipette up and down after 10 min to increase the dissociation of the organoids. Monitor the dissociation process with a microscope every 5-10 min. Since trypsin decreases cell viability, this can help to identify the ideal dissociation time.
- After 20 min, pipette the organoids up and down with a 200 µL pipette to dissociate the organoids. Collect the cells in a 1.5 mL micro-centrifuge tube, add 1 mL of basic organoid medium, and centrifuge at 300 x g for 5 min at RT.
- Optional: To ensure best organoid growth conditions and that new organoids are derived from single cells, strain the cells through a pre-wetted 40 µm cell strainer. Filtering the cell suspension results in the removal of organoid cores and cell clumps that are hard to dissociate.
- Prepare a matrix mix consisting of 80% matrix and 20% cold basic organoid medium. Place everything on ice, as the matrix solidifies at RT.
- Discard the supernatant by carefully removing it with a 200 µL pipette, resuspend the cells in 10 µL of matrix mix/dome, and put the mixture back on ice.
NOTE: Organoids can be split at a 1:5 to 1:10 ratio, depending on dome density. Epithelial cells can also be counted and re-plated at 5,000 cells/dome. - Take the prewarmed 48-well plate from the 37 °C incubator and make one dome per well. Transfer the plate upside down to an incubator and let the domes solidify for another 20-30 min at 37 °C.
- Add 200 µL of prewarmed ENR medium to the respective organoid domes. Supplement the medium with 10 µM Rock-inhibitor (Y-27623) for the first 2 days.
- Refresh the medium every 2-3 days. Make sure that the medium is warm to prevent dissociation of the temperature sensitive matrix.
7. Organoid processing for whole mount staining
NOTE: Coat the tips and tubes with 10% FBS in PBS before use to avoid organoids adhering to plastics. For pipette tips, it is sufficient to pipette once or twice up and down in 10% FBS/PBS solution before using the tip. For tubes, fill the tube with 10% FBS/PBS and then remove the solution.
- Remove the organoid medium and add 200 µL of ice-cold PBS to the matrix domes. Place the plate on ice for 5-10 min.
- Pipette up and down and transfer the solution to 0.6 mL micro-centrifuge non-adherent tubes. Centrifuge shortly for 30-60 s at 100 x g to let the organoids settle in the bottom.
NOTE: Excess pipetting and long centrifugation breaks the organoids and disrupts the fibroblast-organoid interaction. - Remove excess liquid and add ice cold PBS. Centrifuge shortly for 30-60 s at 100 x g to let the organoids settle in the bottom.
- Remove excess liquid and fix the organoid with 200 µL of cold 4% formaldehyde in PBS solution for 30 min on ice.
CAUTION: Formaldehyde is toxic and must always be used in a chemical fume hood. Nitril gloves, safety glasses, and lab coats must always be worn. - Fixation of the organoid causes them to sink, and centrifugation is not needed anymore. Let the organoids sink by placing the tube upright and wait 2-3 min, remove the formaldehyde, and add 500 µL of cold PBS to wash away the formaldehyde.
- Let the organoids sink, remove the excess PBS, and add 500 µL of fresh cold PBS. Let the organoids sink, remove the excess PBS, and add 500 µL of blocking buffer (5% normal donkey serum, 1% BSA, and 0.5% Triton X-100 in PBS). Put the tube on a rocker-shaker for 60 min at RT.
NOTE: Blocking can also be done overnight (O/N) at 4 °C. - Let the organoids sink, remove the blocking buffer, and resuspend the organoids in 200 µL of blocking buffer with primary antibodies (see Table of Materials). Keep the organoids on a rocker-shaker O/N at 4 °C.
NOTE: To improve the staining of nuclear proteins, low expressed proteins, or antibodies that show unspecific staining, the incubation time can be increased for 1 or 2 days at 4 °C. Placing the organoids on a rocker-shaker is essential as it prevents organoids from clumping together and increases staining efficiency. - Let the organoids sink, remove the primary antibody mix, and wash the organoids using 500 µL of 0.02% Triton X-100 in PBS (0.02% Tx) for 60 min at RT. Repeat three times.
NOTE: When longer primary antibody incubation is needed, add a washing step with 0.02% Tx in PBS O/N at 4 °C. - Let the organoids sink, remove the washing buffer, and add 200 µL of fluorescence-conjugated secondary antibody in blocking buffer O/N at 4 °C. After adding fluorescent secondary antibodies, keep the organoids in the dark to avoid bleaching of signal.
- Let the organoids sink, remove the secondary antibody mix, and wash the organoids using 500 µL of 0.02% Tx for 60 min at RT. Repeat three times.
- Counterstain the organoids with 200 µL of DAPI (0.25 µg/mL) solution for nuclear staining if needed. Incubate the samples for 60 min at RT on a rocker-shaker.
- Let the organoids sink, remove DAPI solution, and wash the organoids in 500 µL of PBS for 15 min at RT. Repeat three times.
- Let the organoids sink and remove all excess liquid. Add 10 µL of clearing solution to the organoids and incubate for 15 min at RT.
NOTE: The clearing solution is viscous, so cut off the outer part of a 20 µL pipette tip before pipetting the clearing solution. More clearing solution can be added if a bigger spacer is used for imaging. Mounting solution can be used instead of the clearing solution when not clearing the organoids. The clearing solution reduces the background when imaging and increases the imaging depth. - Place a 0.05 mm double-sided sticky 4-well spacer on a microscope slide. Add the 10 µL of clearing solution with the organoids in one well and place a cover slip on top of the spacer.
NOTE: Organoids can also be mounted without the use of a spacer. The spacer keeps the organoid shape intact and prevents the organoids from being flattened. - Keep the slide at RT O/N to clear the organoids. For long term storage, keep the slides at 4 °C. Acquire images using a confocal microscopy system.
Representative Results
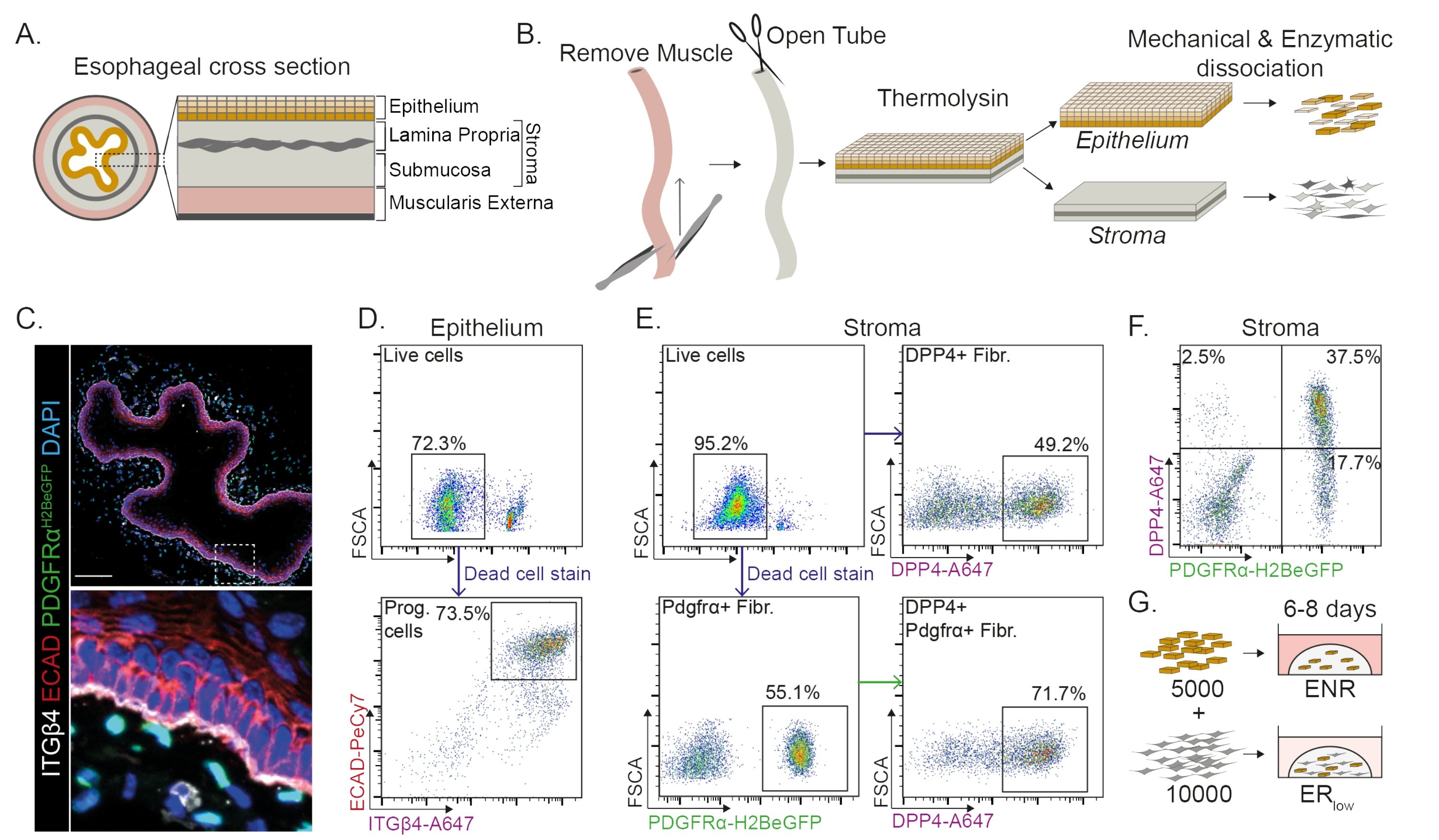
The esophagus is divided into different layers: epithelium, lamina propria, submucosa, and muscularis externa (Figure 1A). Fibroblasts reside within the submucosa and lamina propria, referred to as the stroma. In this protocol, the muscularis externa is mechanically removed (Figure 1B), which does not lead to a loss of fibroblasts (PdgfrαH2BeGFP+) residing in the stroma (Figure 1C). Before dissociation, the epithelium is separated from the stroma resulting in two tissue segments (Figure 1B). Separating the two layers provides the opportunity to increase dissociation time for the more robust epithelial layer compared to the fragile stromal layer. In this way, an efficient isolation protocol yielding both viable epithelial progenitor cells as well as stromal fibroblasts is established (Figure 1B). Esophageal progenitor cells are sorted based on their high INTEGRIN-β4 and E-CADHERIN expression (Figure 1C,D).
Subpopulations of fibroblasts can be isolated by using distinct markers. In this protocol, a strategy for fibroblast isolation based on commonly used fibroblast markers PDGFRα and DPP4 (CD26) is provided. Isolation by either the PdgfrαH2BeGFP reporter expression or DPP4 antibody shows that around 50% of the isolated cells are fibroblasts (Figure 1E,F). Additionally, 70% of the PDGFRα+ fibroblasts are DPP4+, indicating that a largely overlapping, but not identical, fibroblast population is obtained. After isolating both epithelial and stromal cell populations, esophageal progenitor cells are either cultured alone or together with fibroblasts in a matrix dome. To study the contribution of fibroblasts to organoid formation, the co-culture is maintained in a growth factor reduced medium (Figure 1G).
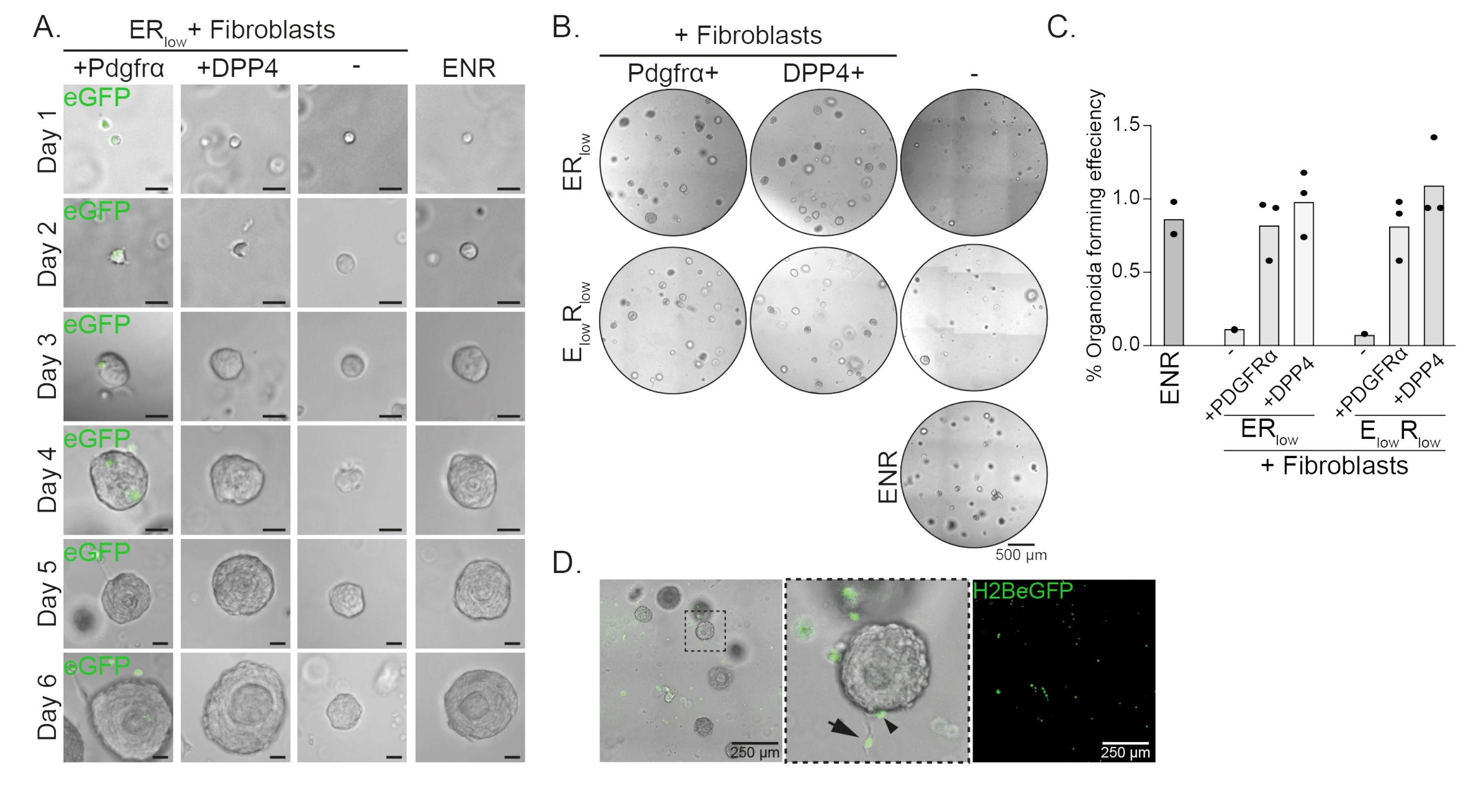
Esophageal progenitor cells form organoids in the presence of EGF, NOGGIN and RSPO (ENR). Removing NOGGIN and reducing the amount of RSPO (25 ng/µL; ERlow) is sufficient to prevent organoid formation (Figure 2A). Interestingly, adding either DPP4+ or PDGFRα+ fibroblasts to the esophageal progenitor cells in the ERlow medium restores the organoid forming ability, demonstrating a supportive function for both fibroblast populations (Figure 2A-D). Visualization of the PdgfrαH2BeGFP transgene shows that fibroblasts are in close contact with the epithelial progenitor cells during organoid formation (Figure 2A). At day 6, PdgfrαH2BeGFP+ fibroblasts are still abundantly present in the co-culture. Fibroblasts are present throughout the dome, near and touching the organoids (full arrow), or attached to the organoids (arrowhead; Figure 2D).
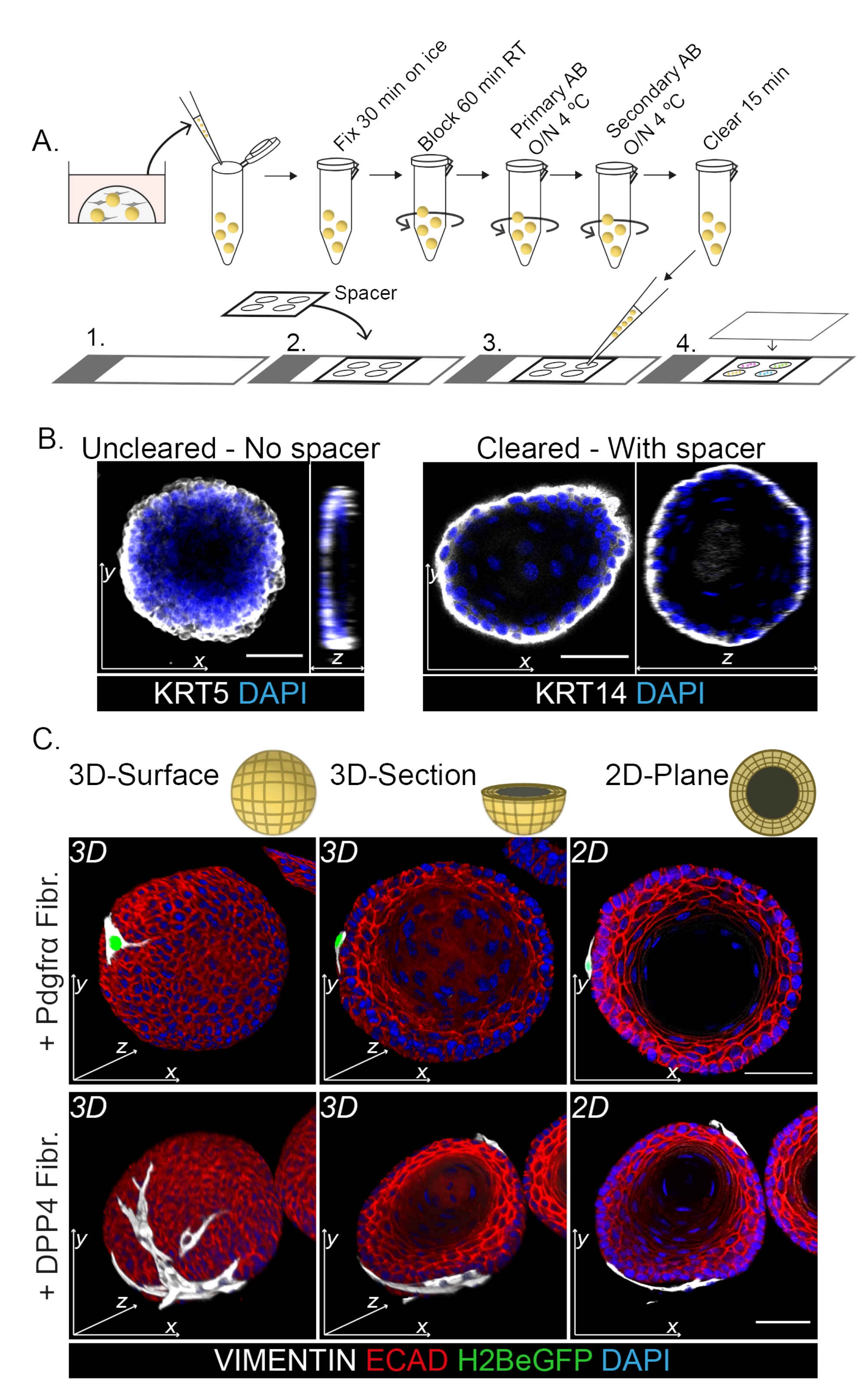
Whole mount staining shows a 3D representation of the interaction of the fibroblasts with the organoids (Figure 3). While it is possible to perform the whole mount protocol without the use of a clearing solution, it decreases transparency and laser penetration of the organoid (Figure 3B, z-view). When mounting organoids, the spacer helps to maintain organoid morphology. In contrast, plating the coverslip directly on the organoids (without a spacer) flattens the organoids and results in loss of organoid structure (Figure 3A,B).
Both the DPP4+ and PDGFRα+ fibroblasts are found to be wrapped around the organoids (Figure 3C, Video1, and Video 2). Differentiation of esophageal organoids can be assessed using different markers. Figure 4 shows that the staining protocol provided is suitable for easier-to-stain keratins (KRT14/13) as well as more-difficult-to-stain transcription factors (TRP63/KLF4). The co-culture protocol generates organoids with a similar differentiation pattern, as demonstrated in vivo13,14 and as seen in organoids grown in ENR medium; KRT14+ or TRP63+ progenitor cells form the outer layer and KRT13+ or KLF4+ differentiated cells oriente inwards.
This protocol provides a tool to study the esophageal stem cell niche in vitro and visualizes the interaction between organoids and fibroblasts. By implementing a protocol for the isolation of fibroblasts using antibodies, the method is adaptable and can be used to study fibroblast subpopulations without the need of transgenic mice.

Figure 1: Isolation of progenitor cells and fibroblast subpopulations from the esophagus. (A) Schematic overview of the different layers in the esophagus. The stroma contains the lamina propria and submucosa. (B) Schematic overview of the isolation protocol. The muscle (muscularis externa) is mechanically removed using forceps; the remaining esophagus is cut open and incubated in thermolysin to separate the epithelial layer from the stroma. The epithelium and stroma are separated, mechanically minced, and enzymatically digested to single cell suspensions. Dissociated cells are then stained and prepared for FACS. (C) Cross section of the esophagus stripped from the muscularis externa showing PdgfrαH2BeGFP+ fibroblasts in the stroma. INTEGRIN-β4 (ITGβ4) and E-CADHERIN (ECAD) double positive cells are the epithelial progenitor cells of the esophagus. Scale bar = 100 µm. (D) Representative flow cytometry plot of epithelial cell isolation showing the percentage of live cells (upper panel) from all single cells. The lower panel shows the percentage of isolated ITGβ4+ ECAD+ progenitor (Prog.) cells from all live cells. (E) Representative flow cytometry plot of stromal cell isolation showing the percentage of live cells (upper left panel). Representative flow cytometry plots showing the percentage of isolated DPP4+ fibroblasts (Fibr.; upper right panel) and Pdgfrα+ fibroblasts (lower left panel) of all live cells. 70% of the Pdgfrα+ fibroblasts are also DPP4+ (lower right panel). (F) Representative flow cytometry plot of the stroma showing DPP4+ only cells (2.5%), DPP4+ PDGFRα+ cells (37.5%), and PDGFRα+ only cells (17.7%). The percentages are of all live cells. (G) Epithelial only cells are plated in a matrix dome in the presence of 50 ng/µL EGF, 100 ng/µL NOGGIN, and 250 ng/µL RSPO (ENR), or together with fibroblasts in the presence of EGF and a low concentration of RSPO (25 ng/µL). Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative results of organoid co-cultures. (A) Brightfield images showing growth of the organoids from day 1 to day 6. The brightfield images with the organoids co-cultured with PdgfrαH2BeGFP+ fibroblasts also show the nuclear eGFP signal. Scale bar = 25 µm. (B) Brightfield images of the whole matrix dome at day 6. The left column shows organoid co-cultures grown in the presence of Pdgfrα+ fibroblasts in ERlow or ElowRlow medium. The middle column shows organoid co-cultures grown in the presence of DPP4+ fibroblasts in ERlow or ElowRlow medium. The right column shows organoid mono-cultures grown in ENR medium. ENR medium = EGF (50 ng/µL), NOGGIN (100 ng/µL), and RSPO (250 ng/µL). ERlow = EGF and 25 ng/µL RSPO. ElowRlow = 5 ng/µL EGF and 25 ng/µL RSPO. Scale bar = 500 µm. (C) Graph showing the organoid forming efficiency (%) (i.e., the percentage of cells forming organoids in different culture conditions). Each dot represents a matrix dome and the bar represents the mean of all dots per condition. (D) Brightfield and fluorescent image of day 6 organoids co-cultured with PdgfrαH2BeGFP+ fibroblasts. PdgfrαH2BeGFP+ fibroblasts are present throughout the dome, attached to the organoids (arrowhead), and unattached but in contact with the organoids (full arrow). Scale bar = 250 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Whole mount staining protocol for the study of fibroblast-organoid interactions. (A) Schematic overview of the whole mount immunofluorescence protocol. AB = antibody. (B) Immunofluorescence picture of uncleared whole mount staining showing a decreased transparency and penetration of the laser light compared to the cleared organoids. The absence of a spacer results in flattening of the organoid and loss in organoid morphology. (C) Whole mount images of the co-cultured organoids show 3D surfaces of the organoids with VIMENTIN+ fibroblasts (Fibr.) wrapped around and in close contact with the organoid. 3D cross sections and 2D plane images show the lumen of the organoid. Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Whole mount images reveal distinct basal and suprabasal cell populations. (A) Whole mount staining of mono- and co-cultured organoids with PdgfrαH2BeGFP+ fibroblasts showing KRT14+ basal cells in the outer layer and KRT13+ differentiated suprabasal cells. Scale bar = 50 μm. (B) Whole mount staining of mono- and co-cultured organoids with PdgfrαH2BeGFP+ fibroblasts showing TRP63+ basal cells in the outer layer and KLF4+ differentiated suprabasal cells. Scale bar = 50 µm. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Table describing the organoid culture media components. Please click here to download this Table.
Video 1: PdgfrαH2BeGFP+ fibroblast wrapped around and in close contact with the organoid. The video accompanies the upper panel of Figure 3C. The scale bar in Figure 3C is 50 µm, and the organoid is ~120 µm in diameter. VIMENTIN is shown in white, E-CADHERIN in red, PdgfrαH2BeGFP in green, and DAPI in blue. Please click here to download this Video.
Video 2: DPP4+ fibroblast wrapped around and in close contact with the organoid. The video accompanies the lower panel of Figure 3C. The scale bar in Figure 3C is 50 µm, and the organoid is ~120 µm in diameter. VIMENTIN is shown in white, E-CADHERIN in red, and DAPI in blue. Please click here to download this Video.
Discussion
The protocol presented here establishes an in vitro model for investigating functional esophageal epithelial-fibroblast interactions.
The epithelial layer is separated from the stroma, allowing for an optimized dissociation protocol for both the epithelial and stromal compartment. Despite optimization of the epithelial dissociation protocol, tissue clumps remain apparent. Pipetting up and down vigorously every 15 min decreases the number and size of clumps substantially. Other protocols use trypsin to further dissociate the epithelial layer5,6. Here, the use of trypsin, or increasing the dissociation time further, is not recommend, as this tends to result in decreased epithelial cell viability and organoid forming efficiency. In contrast to the epithelium, the stroma is easily dissociated, and 30 min in dissociation solution results in a single-cell suspension with ~90% fibroblast viability (Figure 1E). Excluding the epithelial-stomal separation step in the protocol increases the dissociation time substantially, resulting in a decreased fibroblast viability and a lower yield of epithelial cells. Additionally, separating the epithelium from the stroma provides an opportunity to determine cell numbers of each population and mix epithelial cells and fibroblasts from different mouse lines when setting up the co-cultures.
Studying fibroblast function on organoid growth is a commonly used method in stem cell biology 9,10,11,15,16. Established co-culture media are either DMEM supplemented with 10% fetal calf serum (FCS)9,15 or growth factor reduced medium10,16. In this protocol, the growth factor reduced medium is used to mimic the conditions in the in vivo stem cell niche, where fibroblasts are largely quiescent. FCS is a growth factor rich serum which results in activation and proliferation of fibroblasts in the co-cultures, likely corresponding to a fibroblast cell state distinct from the in vivo state. By excluding FCS and reducing growth factors, so that the medium alone (ERlow) does not support organoid growth and does not stimulate fibroblast proliferation, it is possible to isolate the effect of the fibroblasts on the organoid growth. In this medium, NOGGIN is removed and RSPO reduced to a minimum (10% RPSO). Both NOGGIN and RSPO have been demonstrated to be essential for esophageal organoid growth6. EGF was retained in the co-culture medium, as it does not support organoid growth by itself. However, fibroblasts are also capable of supporting organoid growth in an EGF-reduced medium (ElowRlow; Figure 2B,D).
Organoid co-cultures cannot be sustained through passaging as fibroblasts are lost during trypsinization. However, organoid passaging was included in the protocol since esophageal organoids can be maintained, expanded, and used for further experiment as mono-cultures. Passaged organoids from mono-cultures can be used to set up co-cultures with freshly isolated fibroblasts. A disadvantage of using primary cells is the number of mice needed to set up multiple organoid co-cultures. When focusing on small subpopulations of fibroblasts, the number of co-cultures obtained is limited. In other protocols, fibroblasts are first expanded in culture before using them to set up organoid co-cultures10. However, fibroblasts change morphology and identity during passaging, shown by using primary skin and cardiac fibroblasts17,18. Conventional 2D passaging of esophageal fibroblasts results in both morphology and phenotype changes, demonstrating that in vitro enrichment of fibroblasts is not suitable for co-cultures aiming to phenocopy the endogenous stem cell niche.
Whole mount staining provides a tool to maintain and visualize fibroblast-organoid interaction. It should be noted that, while not all organoids will have fibroblasts directly attached to them, most organoids are in contact with fibroblasts (see Figure 2C). To maintain epithelial-fibroblast interactions, it is important to handle the organoids with care and avoid vigorous pipetting, vortexing, and high-speed spinning. Optimal fixation is important to maintain 3D tissue architecture, as well as keep endogenous fluorescence. A 30 min fixation suffices to retain the H2BeGFP signal and is optimal for the antibodies used in this protocol, however this might vary between the fluorophores and antibodies used. Clearing of the organoids reduces light scattering and improves visualization of the whole 3D structure substantially. As the organoids are small, clearing is easy and fast; however, imaging whole organoids using laser-scanning confocal microscopy can be time-consuming, as multiple Z-stacks need to be made. Confocal microscopes, like the spinning disc, can be used to reduce imaging time.
Overall, esophageal organoids grown in the presence of fibroblasts provide a valuable tool to understand aspects of the esophageal stem cell niche. In addition, whole mount clearing provides an accessible method to visualize the interaction between fibroblasts and organoids.
Acknowledgements
This study was supported by ERC StG TroyCAN (851241). E.E. is a Cancerfonden Postdoctoral Associate. M.G. is a Ragnar Söderberg Fellow and Cancerfonden Junior Investigator. We are grateful for the technical assistance from Karolinska Institutet core facilities, including the Biomedicum Flow Cytometry Core Facility, the Biomedicum Imaging Core (BIC), and the Comparative Medicine Biomedicum (KMB) animal facility. We thank members of the Genander lab for carefully reading and commenting on the protocol.
Materials
| Name | Company | Catalog Number | Comments |
| B-27 Supplement (50X), serum free | Thermo Fisher (Gibco) | 17504001 | |
| Corning Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix | fisher scientific | 356231 | |
| Dimethyl sulfoxide | Sigma-Aldrich | 276855-100ML | |
| DMEM/F-12 | Thermo Fisher (Gibco) | 11320074 | |
| DPBS | Thermo Fisher (Gibco) | 14190250 | |
| Fetal Bovine Serum | Sigma-Aldrich | F7524 | |
| GlutaMAX Supplement | Thermo Fisher (Gibco) | 35050061 | |
| HBSS, no calcium, no magnesium, no phenol red | Thermo Fisher (Gibco) | 14175-129 | |
| Normal Donkey Serum | Jackson Immuno | 017-000-121 | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher (Gibco) | 15140122 | |
| Triton X-100 solution | Merck | 93443-100ML | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher (Gibco) | 25200-056 | |
| Chemicals, Peptides, and recombinant proteins | |||
| DAPI Solution | Thermo Fisher | 62248 | |
| Dissociation solution: 0.25 mg/ml Liberase TM, 0.25 mg/ml Dnase in HBSS | |||
| Dnase I | Sigma-Aldrich | 11284932001 | |
| Formaldehyde, 37%, with 10-15% methanol | Sigma-Aldrich | 252549-1L | |
| Liberase | Sigma-Aldrich | 5401127001 | |
| N-Acetyl-cysteine | Sigma-Aldrich | A9165-25G | |
| Noggin murine | Peprotech | 250-38 | |
| RapiClear 1.47 | SunJin Lab | #RC147001 | |
| Recombinant Mouse EGF Protein, CF | R&D systems | 2028-EG-200 | |
| R-spondin-1 murine | Peprotech | 315-32 | |
| SYTOX Blue Dead Cell Stain | Thermo Fisher | S34857 | |
| Thermolysin | Sigma-Aldrich | T7902-25MG | |
| Y-27632 dihydrochloride | Sigma-Aldrich | Y0503-5MG | |
| Plastic & Glassware | |||
| Corning Sterile Cell Strainers 40um | VWR | 15360801 | |
| Corning Sterile Cell Strainers 70um | VWR | 431751 | |
| Menzel Deckgläser/ cover slips | Thermo Fisher | Q10143263NR15 | |
| SafeSeal reaction tube, 1.5 mL, PP | Sarstedt | 72.706 | |
| Snap Cap Low Retention Microcentrifuge Tubes 0.6 mL | Thermo Fisher | 3446 | |
| SuperFrost Slides | VWR | 631-9483 | |
| Tools | |||
| 0.05 mm 4 circular well iSpacer | SunJin Lab | #IS204 | |
| Dumont #5 forceps, biology tip | F.S.T | 11252-20 | |
| ImmEdge Pen | VectorLaboratories | H-4000 | |
| Spring Scissors Angled to Side Ball Tip 8mm Cutting Edge | F.S.T | 15033-09 | |
| Instruments | |||
| Confocal microscope Stellaris 5 | Leica | ||
| Dissection microscope ZEISS Stemi 305 | Zeiss | ||
| FACS ARIA III | BD Biosciences | ||
| Conjugated Antibodies for FACS | |||
| Alexa Fluor 647 anti-mouse CD104 Antibody Clone: 346-11A | 123608 | 123608 | |
| APC anti-mouse CD26 (DPP-4) Antibody | H194-112 | H194-112 | |
| PE/Cy7 anti-mouse/human CD324 (E-Cadherin) Antibody | 147310 | 147310 | |
| Antibodies for Immunofluorescence | |||
| CD104 (ß-integrin 4) Clone: 346-11A | BioLegend | 553745 | |
| Cytokeratin 14 | Acris Antibodies (AbD Serotec) | BP5009 | |
| Cytokeratin13 Clone: EPR3671 | abcam | ab92551 | |
| E-cadherin (CD324) Clone: 2.40E+11 | Cell Signaling Technology | 3195 | |
| Keratin 5 Polyclonal Chicken Antibody, Purified Clone: Poly9059 | BioLegend | 905901 | |
| p63 Clone: 4a4 | abcam | ab735 | |
| Recombinant Anti-KLF4 antibod Clone: EPR20753-25 | abcam | ab214666 | |
| Vimentin | Sigma-Aldrich | AB5733 | |
| Secondary antibodies | |||
| Donkey anti-species* antibodies with fluorophore of choice | Jackson Immuno |
References
- Sachs, N., et al. Long-term expanding human airway organoids for disease modeling. The EMBO Journal. 38 (4), 100300 (2019).
- Lohmussaar, K., et al. Patient-derived organoids model cervical tissue dynamics and viral oncogenesis in cervical cancer. Cell Stem Cell. 28 (8), 1380-1396 (2021).
- Sato, T., et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 459 (7244), 262-265 (2009).
- Smukler, S. R., et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell. 8 (3), 281-293 (2011).
- Zheng, B., et al. A new murine esophageal organoid culture method and organoid-based model of esophageal squamous cell neoplasia. iScience. 24 (12), 103440 (2021).
- DeWard, A. D., Cramer, J., Lagasse, E. Cellular heterogeneity in the mouse esophagus implicates the presence of a nonquiescent epithelial stem cell population. Cell Reports. 9 (2), 701-711 (2014).
- Kasagi, Y., et al. The esophageal organoid system reveals functional interplay between notch and cytokines in reactive epithelial changes. Cellular and Molecular Gastroenterology and Hepatology. 5 (3), 333-352 (2018).
- Plikus, M. V., et al. Fibroblasts: Origins, definitions, and functions in health and disease. Cell. 184 (15), 3852-3872 (2021).
- McCarthy, N., et al. Distinct mesenchymal cell populations generate the essential intestinal BMP signaling gradient. Cell Stem Cell. 26 (3), 391-402 (2020).
- Cordero-Espinoza, L., et al. Dynamic cell contacts between periportal mesenchyme and ductal epithelium act as a rheostat for liver cell proliferation. Cell Stem Cell. 28 (11), 1907-1921 (2021).
- Pastula, A., et al. Three-dimensional gastrointestinal organoid culture in combination with nerves or fibroblasts: a method to characterize the gastrointestinal stem cell niche. Stem Cells International. 2016, 3710836 (2016).
- Hamilton, T. G., Klinghoffer, R. A., Corrin, P. D., Soriano, P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Molecular and Cellular Biology. 23 (11), 4013-4025 (2003).
- McGinn, J., et al. A biomechanical switch regulates the transition towards homeostasis in oesophageal epithelium. Nature Cell Biology. 23 (5), 511-525 (2021).
- Zhang, Y., Bailey, D., Yang, P., Kim, E., Que, J. The development and stem cells of the esophagus. Development. 148 (6), (2021).
- Ohlund, D., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. The Journal of Experimental Medicine. 214 (3), 579-596 (2017).
- Pentinmikko, N., et al. Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature. 571 (7765), 398-402 (2019).
- Janson, D., Rietveld, M., Willemze, R., El Ghalbzouri, A. Effects of serially passaged fibroblasts on dermal and epidermal morphogenesis in human skin equivalents. Biogerontology. 14 (2), 131-140 (2013).
- Landry, N. M., Rattan, S. G., Dixon, I. M. C. An improved method of maintaining primary murine cardiac fibroblasts in two-dimensional cell culture. Scientific Reports. 9 (1), 12889 (2019).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved