Production of Human Neurogenin 2-Inducible Neurons in a Three-Dimensional Suspension Bioreactor
In This Article
Summary
This article describes a protocol for the generation of human induced pluripotent stem cell-derived neurons in a benchtop 3D suspension bioreactor.
Abstract
The derivation of neuronal lineage cells from human induced pluripotent stem cells (hiPSCs) marked a milestone in brain research. Since their first advent, protocols have been continuously optimized and are now widely used in research and drug development. However, the very long duration of these conventional differentiation and maturation protocols and the increasing demand for high-quality hiPSCs and their neural derivatives raise the need for the adoption, optimization, and standardization of these protocols to large-scale production. This work presents a fast and efficient protocol for the differentiation of genetically modified, doxycycline-inducible neurogenin 2 (iNGN2)-expressing hiPSCs into neurons using a benchtop three-dimensional (3D) suspension bioreactor.
In brief, single-cell suspensions of iNGN2-hiPSCs were allowed to form aggregates within 24 h, and neuronal lineage commitment was induced by the addition of doxycycline. Aggregates were dissociated after 2 days of induction and cells were either cryopreserved or replated for terminal maturation. The generated iNGN2 neurons expressed classical neuronal markers early on and formed complex neuritic networks within 1 week after replating, indicating an increasing maturity of neuronal cultures. In summary, a detailed step-by-step protocol for the fast generation of hiPSC-derived neurons in a 3D environment is provided that holds great potential as a starting point for disease modeling, phenotypic high-throughput drug screenings, and large-scale toxicity testing.
Introduction
Neurological disorders are the leading cause of disability worldwide1. One in six people are affected, and the incidence continues to rise. The associated financial burden on societies and their health-care systems is huge. An evaluation of 30 European countries in 2010 estimated annual costs of €800 billion related to mental and neurological disorders2. The increasing socioeconomic burden demands for effective treatment strategies, and although our understanding of disease pathophysiology has increased substantially, the translation into clinics is often insufficient. In general, only 12% of pharmaceuticals enter clinical trials, of which more than 80% fail during later stages, primarily due to inefficacy or unanticipated toxicity3,4. The reasons are varied, but the limited transferability from animal testing in preclinical stages to human trials has increasingly come to the fore5. Human in vitro cell and tissue models may bridge the gap of interspecies translation, and advances in human-induced pluripotent stem cell (hiPSC) technology possess great potential in this regard. hiPSCs are widely used in basic research and share essential characteristics with embryonic stem cells (hESCs), such as an almost unlimited self-renewal capacity and the ability to differentiate into all three germ layers6, while circumventing ethical concerns associated with embryo destruction.
The derivation of neuronal cells from hESCs, and later hiPSCs, marked a milestone in brain research. Initial differentiation protocols were based on the application of growth factors mimicking critical steps during embryogenesis, most of them involving dual SMAD inhibition either in suspension or adherent cultures7,8,9. Mature neurons have been successfully generated, but several drawbacks of these protocols still hamper their wide use in drug development, such as the low yield and high batch-to-batch variability of generated neurons, as well as the extensive workload related to long culture times. Improvements have been achieved by the forced expression of transcription factors critically involved in neurogenesis, and members of the neurogenin (NGN) family, in particular NGN2, have been identified as effective drivers10. The lentiviral-mediated ectopic expression of NGN2 in hiPSCs accelerated early stages of neuronal differentiation significantly, and induced neuronal cell fate within only 1 week11. Subsequent terminal maturation in co-cultures with astrocytes yielded functional neurons in high purity and quantity with reproducible properties. Site-directed gene-editing of the adeno-associated virus integration site 1 (AAVS1) safe-harbor locus was then applied to create hiPSC lines with a stable and doxycycline-inducible expression cassette for NGN212,13, thus minimizing unwanted side effects of lentiviral delivery.
The robust and efficient differentiation of doxycycline-inducible neurogenin 2 (iNGN2) neurons holds great potential for high-throughput phenotypic drug screenings and toxicity assays10,14,15; and substantial progress in bioprocessing has been made during the last decade implementing bioreactors for a scalable cell expansion and differentiation16,17,18,19. However, the majority of differentiation protocols are optimized for adherent cultures, and translation into a three-dimensional (3D) environment often requires essential modifications. Recently, the successful use of a benchtop 3D bioreactor with reduced shear stress features has been reported for the expansion of hiPSCs and for the reproducible differentiation into hepatocytes, cardiomyocytes, and neurons20. Here, a detailed protocol for the generation and characterization of iNGN2 neurons is provided using the identical benchtop 3D bioreactor.
Protocol
NOTE: All cell manipulations, as well as culture dishes and medium preparations, should be performed under sterile conditions. The laminar flow hood should be cleaned thoroughly prior to use and after processing by wiping all the surfaces with 70% ethanol. The described protocol is optimized for neuronal differentiations in a CERO 3D Incubator and Bioreactor (in the following referred to as benchtop bioreactor). This benchtop bioreactor offers four slots for specialized bioreactor tubes, each with a maximum capacity of 50 mL. Temperature and CO2 levels are continuously controlled, and cultivation parameters (e.g., rotational speed and time) are regulated for each tube independently. All aspiration steps have been performed using an aspiration pipette and vacuum pump, if not stated otherwise.
1. Culturing and expansion of hiPSCs
NOTE: The engineered doxycycline-inducible NGN2 hiPSC line BIONi010-C-13 is used in this protocol. The expansion protocol provided here is optimized for 6 cm Petri dishes, but alternative culture formats can be used if preferred.
- Coat 6 cm Petri dishes with basement membrane matrix diluted in cold Dulbecco's modified Eagle's medium (DMEM)/F12 (-/-) at a final concentration of 0.083 mg/mL/10 cm2. Incubate the coated dishes at 37 °C for at least 30 min.
NOTE: A detailed protocol for the preparation of basement membrane matrix solution can be found in the manufacturer's instructions. hiPSCs can be cultured on alternative extracellular matrices for expansion. - Thaw cryopreserved hiPSCs according to the EBiSC Cell line User Protocol21 in feeder-free iPSC maintenance medium + 10 µM ROCK inhibitor (Y-27632). A seeding density of 1 × 106 viable cells per 60 mm dish is recommended.
- Change the medium the next day to feeder-free iPSC maintenance medium without ROCK inhibitor, and perform daily media changes.
- Start passaging when the hiPSC cultures reach a confluence of 60%-80%. Check for differentiated areas and clean the colonies manually if the area exceeds 5%.
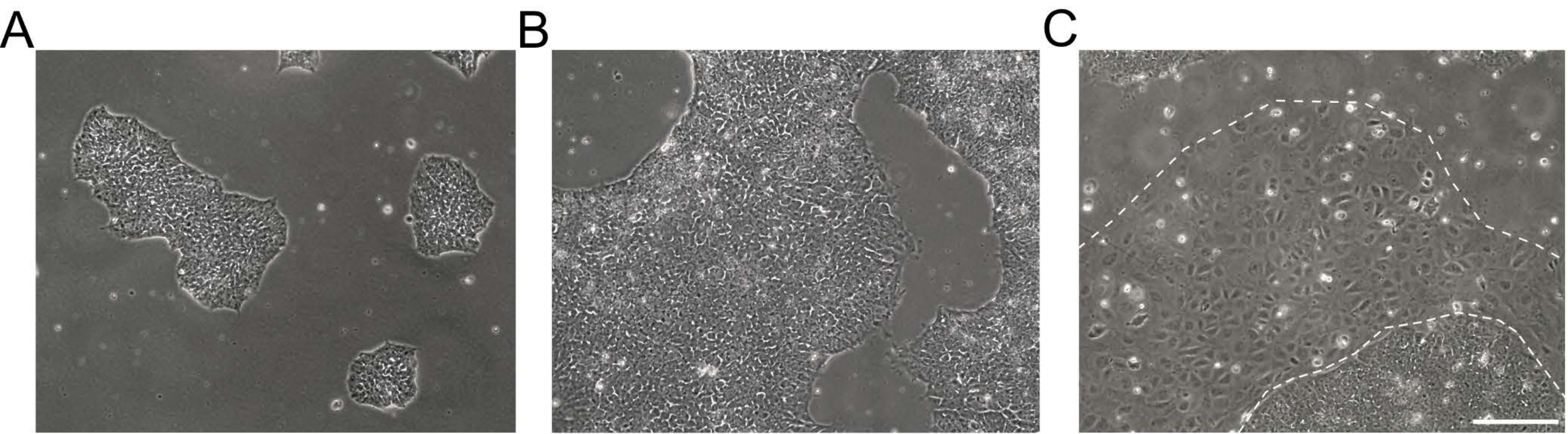
NOTE: Undifferentiated hiPSCs appear as round cells with a prominent nucleolus and less cytoplasm. Flat and tightly packed colonies are formed early after thawing or passaging. Exemplary brightfield images of hiPSC cultures are shown in Figure 1. Further information is provided in the EBiSC Cell line User Protocol21. For passaging, prepare basement membrane matrix-coated dishes and feeder-free iPSC maintenance medium. - Aspirate the medium from hiPSC cultures and rinse the cells 2x with 0.5 mM ethylenediaminetetraacetic acid (EDTA) in 1x Dulbecco's phosphate buffered saline (DPBS) without Ca2+ and Mg2+ (DPS [-/-], see Table of Materials).
- Remove the supernatant and add 2 mL of 0.5 mM EDTA in 1x DPBS (-/-) to the 6 cm Petri dishes. Incubate the cells at 37 °C for 3 min in the incubator.
- Aspirate 1.5 mL of the EDTA solution and continue incubation for 3-5 min.
- Gently tap the dishes to facilitate cell detachment.
NOTE: Check detachment by visual assessment. hiPSC colonies should start to detach after 5 min, but a longer incubation time may be required with higher confluent hiPSC cultures. If the colonies do not detach, increase the incubation time until 10 min, but do not exceed this time frame. - Add 5 mL of feeder-free iPSC maintenance medium to the 6 cm Petri dishes, and gently resuspend the colonies 2x with a 10 mL serological pipette or by using wide-bore pipette tips. Transfer the cells to a 15 mL tube.
NOTE: hiPSCs are highly sensitive to mechanical stress; thus, multiple resuspending should be avoided. The final cell suspension should consist of small colony fragments (50-200 µm). - Aspirate the supernatant from the coated culture dishes, and prepare 4 mL of feeder-free iPSC maintenance medium per 6 cm dish.
- Transfer small colony fragments to the freshly prepared culture dishes in split ratios of 1:10 to 1:40, and cultivate at 37 °C and 5% CO2 with daily media changes.
NOTE: Small colonies should attach within 1 or 2 h after passaging.

Figure 1: Morphology of human induced pluripotent stem cell cultures. (A,B) Good-quality hiPSC cultures of different confluence showing compacted hiPSC colonies with a homogenous morphology and defined edges. (C) hiPSC culture with emerging clusters of differentiated cells around the colony edges (dashed white line). Scale bar = 200 µm. Abbreviation: hiPSC = human induced pluripotent stem cell. Please click here to view a larger version of this figure.
{kind=link}
2. Precultivation of hiPSCs in the benchtop bioreactor system (day-2)
NOTE: Start precultivation when the hiPSC cultures reach a confluence between 60% and 80%. Check the hiPSC colonies for differentiated areas. During this precultivation phase, the hiPSCs are maintained in feeder-free iPSC maintenance medium for 2 days.
- Prepare the culture media, consisting of feeder-free iPSC maintenance medium and 10 µM ROCK inhibitor (Y-27632).
- Aspirate the medium completely from the hiPSCs and gently rinse the cells with 1x DPBS (-/-) twice.
- Add 2.0 mL of prewarmed trypsin-EDTA solution to the 6 cm Petri dishes and incubate the cells for 3 min at 37 °C in the incubator.
- Gently tap the dishes to facilitate cell detachment, or incubate for 1-2 min longer.
- Resuspend the cells in 5 mL of feeder-free iPSC maintenance medium + ROCK inhibitor per dish. Transfer the cell suspension to a 15 mL or 50 mL tube and mix gently by pipetting to ensure cell singularization.
- Determine the cell numbers in 100 µL of cell suspension using an automated cell counter as previously described20. Transfer the corresponding volume for 15 × 106 cells per bioreactor tube into a 50 mL tube.
- Centrifuge the cells at 300 × g for 3 min.
- Aspirate the supernatant and resuspend the cells in 2 mL of feeder-free iPSC maintenance medium + ROCK inhibitor.
- Fill up each 50 mL tube with 18 mL of medium (cell seeding density of 0.75 × 106 cells/mL). Dispense the cell suspension into the bioreactor tubes (20 mL per tube).
- Place the tubes into the bioreactor system. Set the following cultivation parameters: 2 s rotation period, 60 rpm rotation speed, no agitation pause, 37 °C, and 5% CO2 for an unlimited duration20.
- Start the cultivation program via the bioreactor display.
- Change the media the next day. Let the aggregates settle down in the bioreactor tubes for ~5 min. Carefully aspirate the supernatant.
NOTE: Do not let the aggregates settle down for longer than 10 min, as they might stick to each other and build a heterogeneous aggregate suspension. It is recommended to leave ~5 mL of culture medium in the bioreactor tube. - Add 15 mL of fresh feeder-free iPSC maintenance medium without ROCK inhibitor per tube, and continue the cultivation in the benchtop bioreactor for 24 h.
3. Differentiation of hiPSCs into early neurons (day 0)
- Prepare neurobasal medium (NBM): 50% DMEM/F-12 with stabilized L-alanyl-L-glutamine dipeptide, 50% neurobasal medium, 0.5x serum-free supplement (50x), 0.5x serum-free supplement based on Bottenstein's N-1 formulation (100x), 0.5x stabilized L-alanyl-L-glutamine dipeptide, 0.5x MEM non-essential amino acids solution (100x), 500 nM sodium pyruvate (100 mM), 50 nM 2-mercaptoethanol (50 mM), 0.025% human insulin solution, and 5 U/mL penicillin-streptomycin.
NOTE: NBM has to be stored at 4 °C and can be used for up to 2 weeks. - Start neuronal differentiation by adding doxycycline (DOX) to the hiPSC cultures. For this, let the aggregates settle down in the bioreactor tubes. Carefully aspirate the supernatant from the cells, leaving ~5 mL in the tube, and add 35 mL of neural induction medium (NIM) consisting of NBM and 2 µg/mL DOX.
NOTE: As DOX is light-sensitive, it is recommended to turn off the light while working. - Place the tubes back into the benchtop bioreactor and continue cultivation.
- Perform media changes every day for 2 days, as described in step 3.2.
NOTE: After 4 days in suspension culture, the aggregates can be dissociated, and early neurons cryopreserved or directly replated for terminal maturation.
4. Cryopreservation of early neurons (day 2)
NOTE: Cryopreservation is not required and not critical for the differentiation process, but highly recommended as large stocks of early neurons can be produced and stored for subsequent maturation and analyses.
- Let the aggregates settle down in the bioreactor tube. Aspirate the supernatant as previously described.
- Transfer the aggregates to a sterile 15 mL or 50 mL tube and gently rinse the aggregates 2x with 1x DPBS (-/-). Carefully aspirate the supernatant as much as possible without disturbing the aggregates.
- Add 2-5 mL of prewarmed cell dissociation enzyme, depending on the pellet size, and incubate the cells for approximately 10 min at 37 °C in a water bath. Gently resuspend the sedimented aggregates every 2 min until the aggregates dissociate.
NOTE: An almost homogenous cell suspension should be obtained after 7-10 min of incubation. - Add the triple volume of prewarmed NBM medium and resuspend the cells carefully to ensure cell singularization.
- Determine the cell numbers and transfer the corresponding volume for cryopreservation into a 15 mL or 50 mL tube.
NOTE: A cell density of 5-10 × 106 cells/mL of freezing medium is recommended. - Centrifuge the cells at 300 × g for 3 min.
- Aspirate the supernatant and gently resuspend the cell pellet in the corresponding volume of freezing medium containing 10% dimethyl sulfoxide (DMSO).
- Aliquot the cell suspension in suitable vials for cryopreservation (1 mL/vial).
- Transfer the vials immediately to a prechilled, slow-rate freezing container filled with 2-propanol and place the container at -80 °C overnight. Place the vials at -150 °C the next day for long-term storage.
NOTE: The liquid 2-Propanol is highly flammable and may cause eye damage upon contact. Keep away from heat and wear protective gloves as well as glasses.
5. Maturation of hiPSC-derived neurons in monolayer cultures
- Prepare poly-L-ornithine/laminin-coated culture dishes for the long-term cultivation of hiPSC-derived neurons.
- Dilute the poly-L-ornithine stock solution to 0.001% in 1x DPBS (-/-) and coat the dishes overnight at 4 °C, or for 4 h at 37 °C. Aspirate the poly-L-ornithine solution and wash the plates once with 1x DPBS (-/-).
- Dilute the laminin solution in 1x DPBS (-/-) to a final concentration of 10 µg/mL and incubate the dishes overnight at 4 °C, or for 4 h at 37 °C.
NOTE: A volume of 0.1-0.15 mL per cm2 is recommended for the coating procedures, and 0.2 mL per cm2 for all respective washing steps. Alternative coating substrates can be used, but potential effects on cell attachment and maturation should be evaluated.
- Thaw the cryopreserved cells. For this, place the cryovial in a water bath (set to 37 °C) and swirl it for approximately 1 min, until a small clump of frozen cell suspension is left.
- Carefully transfer the cell suspension dropwise to a 15 mL tube prepared with 10 mL of prewarmed NBM. Rinse the cryovial with 1 mL of NBM and transfer the cell suspension to the identical 15 mL tube.
- Centrifuge the cells at 300 × g for 3 min.
- Aspirate the supernatant and add 1-2 mL of NIM supplemented with 10 µM ROCK inhibitor.
- Resuspend the cell pellet carefully and determine the cell numbers.
- Aspirate the remaining laminin solution from the coated cell culture dishes and seed the cells at a seeding density of 1 × 105 cells/cm2 in NIM supplemented with 10 µM ROCK inhibitor.
- Switch the medium after 24 h to NIM without ROCK inhibitor.
- Perform daily medium changes for 4 days.
- After this initial phase of NGN2 induction, omit the DOX and culture the cells in NBM, with half-media changes 2x a week until reaching the desired stage of maturation.
NOTE: Co-culturing of iNGN2 neurons with astrocytes is recommended to increase cell survival, attachment, maturity, and electrical activity13,22.
6. Characterization of hiPSC-derived neurons
NOTE: Differentiation into neuronal derivatives can be evaluated by the following techniques.
- Immunocytochemistry and imaging
- Aspirate the medium from the hiPSC-derived neurons and wash the cells once with 1x DPBS with Ca2+ and Mg2+ (+/+).
NOTE: Pipette carefully, preferentially on the well edges as cells may detach very easily from the surface. A volume of 0.2 mL per cm2 is recommended for all washing steps to ensure a complete coverage of cells with 1x DPBS (+/+). - Fix the neuronal cells using a fixation solution containing 4% paraformaldehyde in DPBS (+/+) for 15 min at room temperature (RT). A volume of 0.1 mL per cm2 is recommended.
NOTE: Paraformaldehyde (4%) in DPBS is a hazardous and skin irritating solution with acute toxicity and potential carcinogenicity. Keep away from heat, wear protective gloves and glasses, avoid inhalation, and use the solution only in a ventilated environment. - Gently rinse the cells 2x in 1x DPBS (+/+) and proceed with staining.
NOTE: Fixed cells can be stored in DPBS (+/+) at 4 °C for up to 1 month until further processing. - Aspirate the DPBS (+/+) from samples. Permeabilize the cells and block non-specific binding sites with 1x DPBS (+/+) containing 1% BSA and 0.2% Triton-X-100 for 60 min at RT.
NOTE: A volume of 0.2 mL per cm2 is recommended. - Remove the supernatant and incubate the cells overnight at 4 °C, with respective primary antibodies diluted in staining buffer (1x DPBS [+/+] containing 1% BSA) (see Table of Materials). Ensure that the cells are completely covered by solution.
NOTE: A volume of 0.1-0.15 mL per cm2 is recommended. - Aspirate the staining buffer and rinse the cells 3x in 1x DPBS (+/+).
- Dilute the corresponding secondary antibodies (see Table of Materials) in staining buffer at a final concentration of 1:1,000.
- Incubate the cells in diluted antibody solution for 1 h at RT in the dark. Ensure that the cells are completely covered by solution.
NOTE: A volume of 0.1-0.15 mL per cm2 is recommended. - Aspirate the secondary antibody solution and rinse the cells 2x in 1x DPBS (+/+).
- Counterstain the nuclei with 4',6-diamidino-2-phenylindole (DAPI), diluted in DPBS (+/+) for 5 min at RT.
NOTE: A working volume of 0.1-0.15 mL per cm2 is recommended. - Rinse the cells 2x in 1x DPBS (+/+).
- Store the cells in DPBS (+/+) at 4 °C until imaging.
NOTE: Brightfield imaging is suitable to track morphological changes and neurite outgrowth. In addition, stereotypic marker expression can be assessed by fluorescence microscopy. While undifferentiated hiPSCs express OCT 3/4 and NANOG, beta-III-tubulin (TUBB3) and microtubule associated protein 2 (MAP2) may serve as neuronal markers for visualizing neurites and axons. The neuritic network can be further assessed by determining the neurite length in brightfield or fluorescent images.
- Aspirate the medium from the hiPSC-derived neurons and wash the cells once with 1x DPBS with Ca2+ and Mg2+ (+/+).
- Gene expression analyses by quantitative real-time polymerase chain reaction (qPCR)
- Aspirate the medium and rinse the cells once in 1x DPBS (-/-).
NOTE: A volume of 0.2 mL per cm2 is recommended. - Add cold RNA lysis buffer and harvest the cells by scratching.
- Isolate the RNA using a suitable column-based kit, and determine the RNA concentrations by UV photospectrometry.
- Generate cDNA using 250 ng of total RNA and a suitable kit for reverse transcription.
- Prepare molecular beacon qPCR reactions containing 2.5 ng of cDNA in duplicate. Use an appropriate master mix and corresponding primer assays20 (see Table of Materials).
- Run the qPCR by applying 45 cycles with primer annealing at 60 °C for 20 s.
- Normalize relative gene expression levels to the mean of the housekeeping genes GAPDH, HPRT1, and GUSB. Apply the ΔΔCt method23, using undifferentiated hiPSCs as the calibrator.
- Aspirate the medium and rinse the cells once in 1x DPBS (-/-).
Representative Results
During the initial steps, adherent cultures of hiPSCs are detached, singularized, and transferred into suspension (Figure 2). Aggregates are formed within 24 h and continuously grow in size. After 2 days of transgene induction, early neurons can be cryopreserved for subsequent experiments.
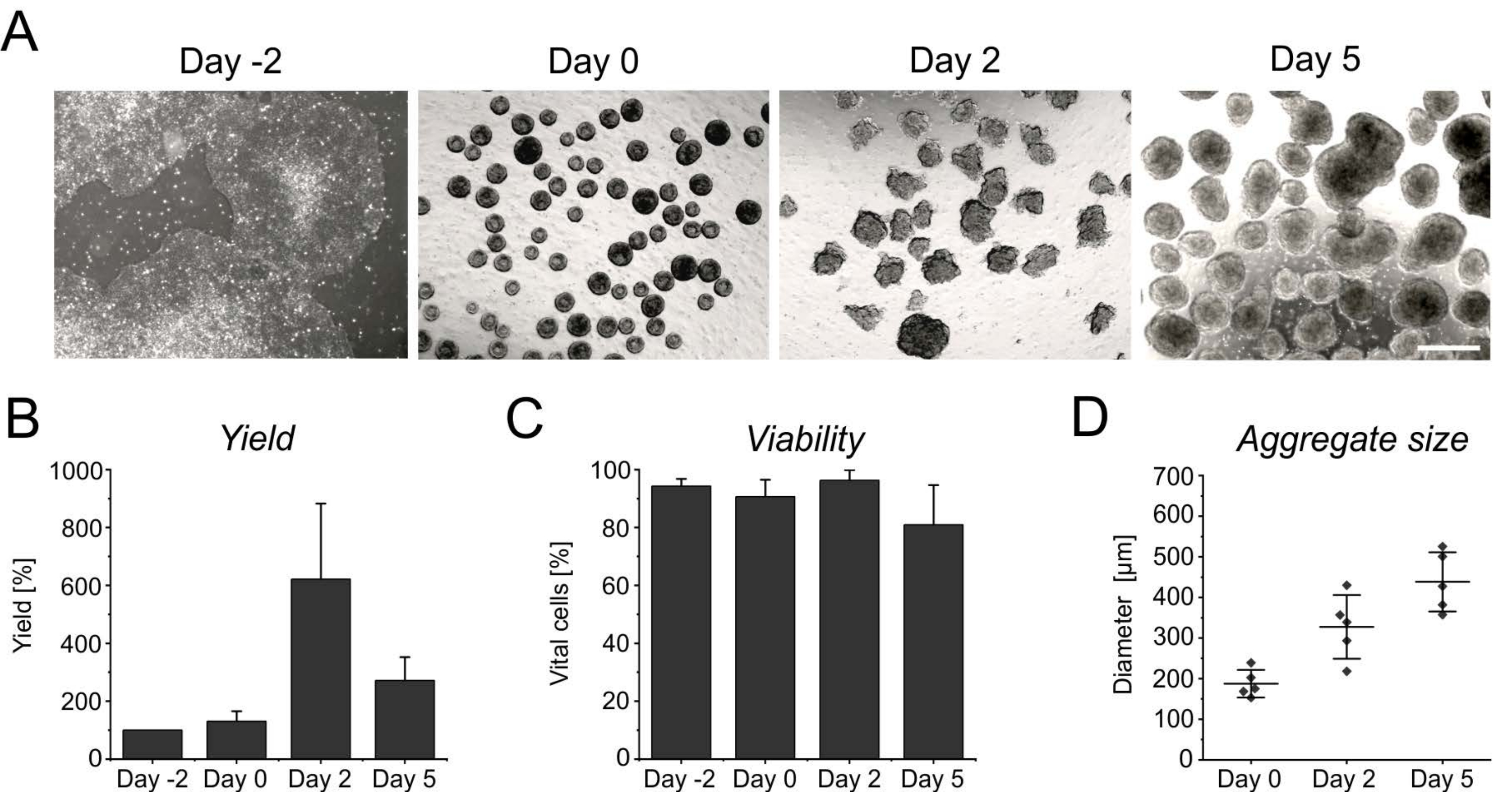
The persistent proliferation during the initial days in suspension yields an increase in cell number, reaching its peak after 2 days of induction (Figure 3A,B). Along with the proliferation, aggregates start to grow. Compared to day 0, the diameter shows an increase of 50% at day 2, and is almost doubled at day 5 (Figure 3D). Although an increasing diameter limits nutrient supply inside the aggregates, the viability of cells is not affected at day 2 or at day 5 of differentiation (Figure 3C). However, a prolonged cultivation for more than 4 days in suspension does not further improve cell yield, as the aggregates become increasingly resistant to enzymatic singularization (Figure 3B).
Upon cryopreservation, day 2 cells are thawed and plated onto poly-L-ornithine (PLO)-laminin-coated dishes for terminal maturation. In general, cells attach very well after thawing, and start to extend neurites early on. The temporal gene expression profile, as well as immunocytochemical staining for the neuronal markers TUBB3 and MAP2, confirms neuronal cell identity (Figure 4A,B). In addition to rising levels of TUBB3 and MAP2, neuronal cultures are enriched in microtubule-associated protein tau transcripts (MAPT), encoding a neuronal protein involved in axon stabilization, and show a concomitant decline in the expression of the pluripotency-regulating transcription factor POU5F1. Moreover, a dense neuritic network is formed within the first week after thawing (Figure 4C). These morphological changes, in conjunction with the transcriptional profile, suggest an increasing maturation of the neuronal cultures.

Figure 2: Generation of hiPSC-derived iNGN2 neurons using a benchtop bioreactor. (A) Schematic overview of the differentiation paradigm, highlighting the key steps of cell culture. (B-F) Brightfield images visualize (B) hiPSCs and (C,D) the formation of aggregates in the benchtop bioreactor. (E,F) iNGN2 neurons extend neurites and undergo morphological changes during differentiation. Scale bar = 100 µm. Abbreviations: BMM = basement membrane matrix; iPSC-MM = iPSC maintenance medium; PLO = poly-L-ornithine. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Cell yield and viability during aggregate formation and growth. (A) Brightfield images depict the formation of aggregates within 2 days of culture in the benchtop bioreactor, as well as the increase in aggregate size over time. Scale bar = 500 µm. (B) Quantification of the cell yield and (C) viability over the differentiation course. Bar graphs represent the mean + SD from four independent experiments. (D) The diameter, indicative of the size of aggregates, was determined semi-automatically using the open-source software ImageJ (version 1.53). First, brightfield images were converted into binary images, and then further assessed using the analyze particles tool, applying the following parameters: size > 2,500 µm2, circularity 0.45-1, exclude edges, and include holes. Horizontal lines indicate the mean ± SD of five independent differentiations. Single dots represent the mean of individual differentiation experiments with at least 20 aggregates per time point. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Characterization of hiPSC-derived iNGN2 neurons. Temporal gene expression profile for the neuronal genes TUBB3, MAP2, and MAPT, as well as the pluripotent stem cell-associated gene POU5F1. Relative expression levels were normalized to the reference genes GAPDH, HPRT1, and GUSB. Undifferentiated hiPSCs (day-2) were chosen as the calibrator. The geometrical symbols indicate the mean ± SD of four independent differentiations. (B) Representative images of iNGN2 neurons at day 7 after thawing (day-2 + 7), stained for beta-III-tubulin (TUBB3, magenta) and microtubule-associated protein 2 (MAP2, cyan). Cell nuclei were counterstained with DAPI. Scale bars = 100 µm. (C) Evaluation of the neuritic network in brightfield images of neuronal cultures after thawing. The total length of neurites was determined in an area of 930.82 × 698.11 µm2 using ImageJ (version 1.53). After adjusting the brightness and contrast, the images were converted into 8-bit images and the colors were inverted. Neurites were subsequently skeletonized and then analyzed using the ImageJ plugins 'Skeletonize' and 'Analyze Skeleton', respectively. The total length of neurites was divided by the number of somas in the region of interest to yield the average neurite length/cell. Bar graphs represent the mean + SD from three independent experiments. Abbreviations: DAPI = 4',6-diamidino-2-phenylindole. Please click here to view a larger version of this figure.
{kind=link}
Discussion
The ectopic expression of the neuronal transcription factor NGN2 has been previously shown to accelerate early stages of neuronal differentiation and induce neuronal lineage commitment in hiPSCs within 1 week of cultivation12,13,20. This work describes a detailed protocol for the differentiation of gene-edited BIONi010-C-13 hiPSCs into iNGN2 neurons using a benchtop bioreactor.
The CERO 3D bioreactor is a low-volume bioreactor with four slots for specialized tubes, each with a maximum capacity of 50 mL. Temperature and CO2 levels are continuously controlled and cultivation parameters (e.g., rotational speed and time) are regulated for each tube, independently enabling users to run several differentiation approaches in parallel. Integrated wavebreakers on the inner tube wall allow for a perturbation of the medium with low shear stress and maintain 3D aggregates in suspension during controlled rotation. Limited access to nutrients, as well as enforced 3D cell-cell and cell-extracellular matrix (ECM) interactions, may significantly influence cellular differentiation and behavior24,25,26,27.
Culturing cells as 3D aggregates in suspension increases these cell interactions, thereby mimicking the in vivo environment of tissue or organs more closely28. The concomitant perturbation of the medium regulates aggregate size due to mechanical friction, improves gas exchange, and reduces the gradient of nutrients and waste in the tube, while preserving physiological relevant gradients inside the aggregates29,30,31,32,33,34. Although medium changes are performed manually, the benchtop bioreactor reduces labor and operating costs compared to static culture flasks. The bioreactor also offers several advantages over fully automated stir-tank bioreactors, as the impeller free design reduces hydrodynamic shear stress at the aggregate surface, thereby not only improving cell survival, but also limiting the effects of shear stress on sensitive iPSCs, cell differentiation, and function35,36,37,38.
Vertical wheel bioreactors, in which cells are agitated by a large vertical impeller, as well as rocking motion bioreactors using inflated culture bags attached to a motorized platform, represent alternative 3D suspension platforms. Although excessively tested for the cultivation of hiPSCs17,18,19,39,40, little is known about their applicability in neuronal differentiation approaches, and reports are limited to the successful expansion of mammalian and human neural precursor cells in stir-tank bioreactors41,42,43,44, with a rare number of studies focusing on maturation44,45. In general, automated 3D suspension platforms offer the advantage of fully automated and computer-controlled medium changes, thereby reducing variations in handling and minimizing the risk of contamination. Furthermore, nutrient parameters such as lactate and glucose concentration can be continuously monitored. However, the establishment of these systems often requires a significant initial investment, laboratory space, and the training of qualified personnel. The benchtop bioreactor represents a compact and easy-to-use alternative for culturing cells in suspension, but does not provide concurrent monitoring of nutrients.
As in the majority of protocols used for the cultivation of cells in 3D suspension platforms, the initial formation of aggregates represents a critical step. hiPSCs are cultured as adherent monolayers and subsequently transferred as single-cell suspensions to a 3D environment. To minimize cell loss at that stage, several aspects need to be considered. The design of the tubes with the integrated wavebreakers means that a minimal culture volume of 10 mL per tube is required to ensure optimal medium perturbation. The critical volume should therefore not be undercut over the long term. Furthermore, a starting seeding density of 7.5 million cells per 10 mL of culture volume, and a rotational speed of 60 rpm, is highly recommended to form stable aggregates in the short term, although adaptations might be necessary dependent on the cell line.
After transferring hiPSCs into suspension, aggregates should be formed within 24 h. The first medium change is critical, as the aggregates are small and may not settle down properly. The time for the sedimentation of aggregates can be extended but should not exceed more than 10 min, as aggregates may start to clump and grow in a heterogeneous manner. During aspiration, a minimal culture volume of 5 mL should be left in the tube, and any perturbations of the medium should be avoided. Nevertheless, a loss of cells and aggregates is to be expected during the initial phases. Cell counts indicate a constant cell yield over the first 2 days in culture, indicating that the high proliferative capacity of hiPSCs compensates for the initial cell loss. Once in suspension, the gentle cultivation and perturbation leads to homogenously sized aggregates that start growing over time.
Neuronal differentiation was performed according to a previously published protocol based on doxycycline-induced NGN2 overexpression13, and the individual steps were optimized for a 3D cultivation. The neuronal transcription factor NGN2 is a well-described driver in neuronal differentiation and accelerates neuronal induction significantly11,13,46. While conventional patterning with defined growth factors takes several weeks to months7,8,9, NGN2 expression induces neuronal cell fate within days. In addition to the shortened cultivation time, the protocol depends neither on expensive growth factors nor coating matrices for the initial suspension culture, thus reducing cultivation costs additionally.
Moreover, a direct comparison of the NGN2-driven generation of immature neurons in adherent and suspension cultures revealed a 1.36-fold increase in cells after 4 days in culture using the benchtop bioreactor20, while the volume of medium required for the generation of 1 million cells is expected to be halved. Employing the benchtop bioreactor for the generation of iNGN2 neurons may therefore be beneficial, as a higher number of cells can be produced with a lower handling time and cost. In line with previous reports, neuronal lineage commitment was observed early on. After 2 days of NGN2 induction, a profound expression of classical neuronal markers such as TUBB3, MAP2, and MAPT was evident, supporting the applicability of the benchtop bioreactor for neuronal induction. Following this protocol, approximately 6.2 million immature iNGN2 neurons can be obtained from 1 million hiPSCs within 4 days of culture. However, the output capacity may vary across batches and depend on the volume of suspension culture at seeding.
To increase the yield further, the cultivation time was extended for 3 additional days; however, although the aggregates became larger, they also became highly compact and could not properly be dissociated for cryopreservation or replating. Despite advances in 3D culture approaches, adherent 2D neuronal cultures are still the gold standard for functional analyses like microelectrode arrays or calcium imaging. Cell singularization is thus an important prerequisite for a homogenously distributed neuronal monolayer and neuritic network. Stronger mechanical detachment of the cells or harsh dissociation reagents may be used to ensure singularization, yet with potential effects on cell viability, physiology, and re-attachment capacity47,48. With regard to the need for single cells for further analyses and maturation, aggregates were dissociated after 4 days in suspension, and the (day 2) iNGN2 neurons cryopreserved. Post-thaw differentiation and characterization of iNGN2 neurons confirmed an increasing maturity of the neuronal cultures and the formation of a dense neuritic network.
The provided protocol yields a high number of iNGN2 neurons. However, several limitations need to be considered. As for most suspension culture platforms, the transfer of hiPSCs from a 2D adherent culture to a 3D environment is critical and often associated with a profound cell loss. Further significant cell and aggregate loss is expected during media changes. Compared to automated platforms, the handling time and contamination risk using the benchtop bioreactor is increased, as medium changes must be performed manually. Apart from the lack of automation, the benchtop bioreactor does not offer the possibility to monitor the nutrients, and the maximum capacity of 50 mL per tube limits the upscaling potential of the protocol. Finally, it is important to note that, although NGN2 accelerates neuronal lineage commitment, the duration of terminal maturation is not shortened and still requires long-term culture over several weeks. Given the importance of astrocytic support for neuronal function, attachment, and survival49,50,51, co-culture approaches should be considered, and may be even essential for long-term cultivation.
In summary, a 2D differentiation protocol was successfully translated to a 3D environment for the fast and reproducible generation of iNGN2 neurons by implementing a benchtop bioreactor. The described protocol yields high quantities of good-quality, iPSC-derived neurons, which may serve as a starting point for complex model testing, high-throughput drug screenings, and large-scale toxicity assays.
Acknowledgements
The EBiSC2 project has received funding from the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No 821362. The JU receives support from the European Union's Horizon 2020 research and innovation programme and EFPIA. We thank Nadine J. Smandzich, Helene D. M. Hemmer, Johnn-Majd Balsters, and Vanessa M. Nalewaja for their assistance in immunocytochemical and gene expression analyses, as well as Heike Arthen for her excellent technical support. Moreover, we thank Stephanie Bur for the establishment of the bioreactor program. Figure 2A was created with BioRender.com.
Materials
| Name | Company | Catalog Number | Comments |
| 2-Mercaptoethanol 50 mM | Gibco | 11528926 | |
| 60 mm Nunclon Delta Surface | Nunc | 734-2040 | |
| Anti-beta III Tubulin antibody [TU-20] (dilution 1:1,000) | Abcam | ab7751 | |
| Applied Biosystems High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 10400745 | |
| B-27 Supplement (50x) | Gibco | 11530536 | serum-free Supplement (50x) |
| BIONi010-C-13 hiPSC line | European Bank for induced pluripotent Stem Cell (EBiSC), BIONEER as Depositor | SAMEA103988285 | Bioneer is depositor in EBiSC and owner of the hiPSC line. |
| Biorender | Biorender.com | ||
| BSA Cell Culture grade | Thermo Fisher Scientific | 12330023 | |
| CERO 3D Incubator & Bioreactor | OLS OMNI Life Science | 2800000 | |
| CEROtubes Cell culture Tubes 50 mL | OLS OMNI Life Science | 2800005 | |
| Citavi 6 | Swiss Academic Software | ||
| CryoStor CS10 | Stemcell | 7930 | freezing medium containing 10% DMSO |
| Cytofix Fixation Solution | BD Biosciences | 554655 | fixation solution containing 4% paraformaldehyde |
| DAPI (NUCBLUE FIXED CELL STAIN) | Thermo Fisher Scientific | 12333553 | |
| Doxycycline hydrochloride (DOX) | Sigma-Aldrich | D3447 | |
| DPBS without Calcium and Magnesium (DPBS -/-) | Gibco | 14190250 | |
| DPBS with Calcium and Magnesium (DPBS +/+) | Gibco | 11580456 | |
| DMEM/F12 (-/-) | Gibco | 21331-020 | |
| DMEM/F-12, GlutaMAX Supplement | Gibco | 31331-028 | |
| EVOS XL Core Cell Imaging System | Thermo Fisher Scientific | ||
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | A21424 | |
| GlutaMAX supplement | Gibco | 35050-038 | stabilized L-alanyl-L-glutamine dipeptid |
| ImageJ 1.51v | National Institute of Health | ||
| Insulin solution human | Sigma-Aldrich | I9278l | |
| Laminin | Merck | L2020 | |
| MACSQuant Analyzer | Miltenyi | ||
| MAP2 Monoclonal Antibody (dilution 1: 300) | Thermo Fisher Scientific | 13-1500 | |
| Matrigel Growth factor reduced, phenol-red free | Corning Life Science | 356231 | basement membrane matrix |
| MEM Non-Essential Amino Acids Solution (100x) | Gibco | 11140035 | |
| MicroAmp Optical Adhesive Film Kit | Thermo Fisher Scientific | 10095714 | |
| mTeSR1 | Stemcell | 85850 | feeder-free iPSC maintenance medium |
| N-2 Supplement (100x) | Gibco | 17502-048 | serum-free Supplement based on Bottenstein’s N-1 formulation |
| Neurobasal Medium | Gibco | 11570556 | |
| Nikon Eclipse TS2 | Nikon Instruments Europe B.V. | ||
| NucleoCounter-NC200 | ChemoMetec A/S | ||
| Origin 2021 | OriginLab | ||
| Penicillin-Streptomycin | Gibco | 11548876 | |
| Perm/Wash Buffer | BD Biosciences | 554723 | |
| Primer assay TUBB3(Hs00801390_s1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay GAPDH (Hs99999905_m1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay GUSB (Hs99999908_m1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay HPRT1 (Hs99999909_m1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay MAP2 (Hs00258900_m1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay MAPT (Hs00902194_m1) | Thermo Fisher Scientific | 11620099 | |
| Primer assay POU5F1 (Hs04260367_gH) | Thermo Fisher Scientific | 11620099 | |
| Poly-L-ornithine 0.01% | Merck | P4957 | |
| RNeasy Plus Micro Kit (50)-Kit | Qiagen | 74034 | column-based RNA isolation kit |
| RLT Buffer | Qiagen | 79216 | cell lysis buffer |
| Sodium Pyruvat (100 mM) | Gibco | 12539059 | |
| StemPro Accutase | Gibco | 11599686 | cell dissociation enzyme |
| TAQMAN FAST ADVANCED MASTER MIX | Thermo Fisher Scientific | 11380912 | qPCR master mix |
| Triton-X-100 | Sigma-Aldrich | T8787 | |
| TrypLE Select Enzym | Gibco | 12563-011 | Trypsin-EDTA solution |
| UltraPure 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | AM9260G | |
| Via1-Cassette | ChemoMetec A/S | 941-0012 | |
| Wide Bore Filtered Pipette Tips | Thermo Fisher Scientific | 10088880 | |
| X20 OPTICAL 96WELL FAST CLEAR REACTION PLATES | Thermo Fisher Scientific | 15206343 | |
| Y-27632 dihydrochloride, Rho kinase inhibitor (ROCK inhibitor) | Abcam | ab120129 |
References
- Feigin, V. L., et al. The global burden of neurological disorders: translating evidence into policy. The Lancet. Neurology. 19 (3), 255-265 (2020).
- Olesen, J., Gustavsson, A., Svensson, M., Wittchen, H. -. U., Jönsson, B. The economic cost of brain disorders in Europe. European Journal of Neurology. 19 (1), 155-162 (2012).
- Preclinical Development: The Safety Hurdle Prior to Human Trials. American Pharmaceutical Review Available from: https://www.americanpharmaceuticalreview.com/Featured-Articles/187349-Preclinical-Development-The-Safety-Hurdle-Prior-to-Human-Trials (2016)
- van Norman, G. A. Phase II trials in drug development and adaptive trial design. JACC. Basic to Translational Science. 4 (3), 428-437 (2019).
- van Norman, G. A. Limitations of animal studies for predicting toxicity in clinical trials: part 2: potential alternatives to the use of animals in preclinical trials. JACC. Basic to Translational Science. 5 (4), 387-397 (2020).
- Takahashi, K., et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131 (5), 861-872 (2007).
- Chambers, S. M., et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 27 (3), 275-280 (2009).
- Shi, Y., Kirwan, P., Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nature Protocols. 7 (10), 1836-1846 (2012).
- Reinhardt, P., et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PloS One. 8 (3), 59252 (2013).
- Hulme, A. J., Maksour, S., St-Clair Glover, M., Miellet, S., Dottori, M. Making neurons, made easy: the use of Neurogenin-2 in neuronal differentiation. Stem Cell Reports. 17 (1), 14-34 (2022).
- Zhang, Y., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 78 (5), 785-798 (2013).
- Schmid, B., et al. Generation of two gene edited iPSC-lines carrying a DOX-inducible NGN2 expression cassette with and without GFP in the AAVS1 locus. Stem Cell Research. 52, 102240 (2021).
- Shih, P. -. Y., et al. Development of a fully human assay combining NGN2-inducible neurons co-cultured with iPSC-derived astrocytes amenable for electrophysiological studies. Stem Cell Research. 54, 102386 (2021).
- Chang, C. -. Y., et al. Induced pluripotent stem cell (iPSC)-based neurodegenerative disease models for phenotype recapitulation and drug screening. Molecules. 25 (8), 2000 (2000).
- Silva, M. C., Haggarty, S. J. Human pluripotent stem cell-derived models and drug screening in CNS precision medicine. Annals of the New York Academy of Sciences. 1471 (1), 18-56 (2020).
- Elanzew, A., Sommer, A., Pusch-Klein, A., Brüstle, O., Haupt, S. A reproducible and versatile system for the dynamic expansion of human pluripotent stem cells in suspension. Biotechnology Journal. 10 (10), 1589-1599 (2015).
- Davis, B. M., Loghin, E. R., Conway, K. R., Zhang, X. Automated closed-system expansion of pluripotent stem cell aggregates in a rocking-motion bioreactor. SLAS Technology. 23 (4), 364-373 (2018).
- Kropp, C., et al. Impact of feeding strategies on the scalable expansion of human pluripotent stem cells in single-use stirred tank bioreactors. Stem Cells Translational Medicine. 5 (10), 1289-1301 (2016).
- Borys, B. S., et al. Overcoming bioprocess bottlenecks in the large-scale expansion of high-quality hiPSC aggregates in vertical-wheel stirred suspension bioreactors. Stem Cell Research & Therapy. 12 (1), 55 (2021).
- Kwok, C. K., et al. Scalable expansion of iPSC and their derivatives across multiple lineages. Reproductive Toxicology. 112, 23-35 (2022).
- User Protocol for Human induced Pluripotent Stem Cells. EBiSC Available from: https://ebisc.org/docs/ebisc/EBiSC_User_Protocol_for_Human_induced_Pluripotent_Stem_Cells.pdf (2023)
- Neyrinck, K., et al. SOX9-induced generation of functional astrocytes supporting neuronal maturation in an all-human system. Stem Cell Reviews and Reports. 17 (5), 1855-1873 (2021).
- Livak, K. J., Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25 (4), 402-408 (2001).
- Griffith, L. G., Swartz, M. A. Capturing complex 3D tissue physiology in vitro. Nature Reviews. Molecular Cell Biology. 7 (3), 211-224 (2006).
- Burdick, J. A., Vunjak-Novakovic, G. Engineered microenvironments for controlled stem cell differentiation. Tissue Engineering. Part A. 15 (2), 205-219 (2009).
- Baker, B. M., Chen, C. S. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. Journal of Cell Science. 125, 3015-3024 (2012).
- Duval, K., et al. Modeling physiological events in 2D vs. 3D cell culture. Physiology. 32 (4), 266-277 (2017).
- Haycock, J. W. 3D cell culture: a review of current approaches and techniques. Methods in Molecular Biology. 695, 1-15 (2011).
- Sen, A., Kallos, M. S., Behie, L. A. Effects of hydrodynamics on cultures of mammalian neural stem cell aggregates in suspension bioreactors. Industrial and Engineering Chemical Research. 40 (23), 5350-5357 (2001).
- Gerecht-Nir, S., Cohen, S., Itskovitz-Eldor, J. Bioreactor cultivation enhances the efficiency of human embryoid body (hEB) formation and differentiation. Biotechnology and Bioengineering. 86 (5), 493-502 (2004).
- Zhao, F., et al. Effects of oxygen transport on 3-d human mesenchymal stem cell metabolic activity in perfusion and static cultures: experiments and mathematical model. Biotechnology Progress. 21 (4), 1269-1280 (2005).
- Cimetta, E., Figallo, E., Cannizzaro, C., Elvassore, N., Vunjak-Novakovic, G. Micro-bioreactor arrays for controlling cellular environments: design principles for human embryonic stem cell applications. Methods. 47 (2), 81-89 (2009).
- Kehoe, D. E., Jing, D., Lock, L. T., Tzanakakis, E. S. Scalable stirred-suspension bioreactor culture of human pluripotent stem cells. Tissue Engineering. Part A. 16 (2), 405-421 (2010).
- Sargent, C. Y., et al. Hydrodynamic modulation of embryonic stem cell differentiation by rotary orbital suspension culture. Biotechnology and Bioengineering. 105 (3), 611-626 (2010).
- Liu, N., Zang, R., Yang, S. -. T., Li, Y. Stem cell engineering in bioreactors for large-scale bioprocessing. Engineering in Life Sciences. 14 (1), 4-15 (2014).
- Wolfe, R. P., Leleux, J., Nerem, R. M., Ahsan, T. Effects of shear stress on germ lineage specification of embryonic stem cells. Integrative Biology: Quantitative Biosciences from Nano to Macro. 4 (10), 1263-1273 (2012).
- Wolfe, R. P., Ahsan, T. Shear stress during early embryonic stem cell differentiation promotes hematopoietic and endothelial phenotypes. Biotechnology and Bioengineering. 110 (4), 1231-1242 (2013).
- Shafa, M., et al. Impact of stirred suspension bioreactor culture on the differentiation of murine embryonic stem cells into cardiomyocytes. BMC Cell Biology. 12 (1), 53 (2011).
- Kwok, C. K., et al. Scalable stirred suspension culture for the generation of billions of human induced pluripotent stem cells using single-use bioreactors. Journal of Tissue Engineering and Regenerative Medicine. 12 (2), 1076-1087 (2018).
- Rodrigues, C. A. V., et al. Scalable culture of human induced pluripotent cells on microcarriers under xeno-free conditions using single-use vertical-wheel™ bioreactors. Journal of Chemical Technology & Biotechnology. 93 (12), 3597-3606 (2018).
- Gilbertson, J. A., Sen, A., Behie, L. A., Kallos, M. S. Scaled-up production of mammalian neural precursor cell aggregates in computer-controlled suspension bioreactors. Biotechnology and Bioengineering. 94 (4), 783-792 (2006).
- Baghbaderani, B. A., et al. Expansion of human neural precursor cells in large-scale bioreactors for the treatment of neurodegenerative disorders. Biotechnology Progress. 24 (4), 859-870 (2008).
- Baghbaderani, B. A., et al. Bioreactor expansion of human neural precursor cells in serum-free media retains neurogenic potential. Biotechnology and Bioengineering. 105 (4), 823-833 (2010).
- Serra, M., Brito, C., Costa, E. M., Sousa, M. F. Q., Alves, P. M. Integrating human stem cell expansion and neuronal differentiation in bioreactors. BMC Biotechnology. , 82 (2009).
- Zhao, S., et al. Generation of cortical neurons through large-scale expanding neuroepithelial stem cell from human pluripotent stem cells. Stem Cell Research & Therapy. 11 (1), 431 (2020).
- Pawlowski, M., et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports. 8 (4), 803-812 (2017).
- Bajpai, R., Lesperance, J., Kim, M., Terskikh, A. V. Efficient propagation of single cells Accutase-dissociated human embryonic stem cells. Molecular Reproduction and Development. 75 (5), 818-827 (2008).
- Jager, L. D., et al. Effect of enzymatic and mechanical methods of dissociation on neural progenitor cells derived from induced pluripotent stem cells. Advances in Medical Sciences. 61 (1), 78-84 (2016).
- Allen, N. J. Astrocyte regulation of synaptic behavior. Annual Review of Cell and Developmental Biology. 30, 439-463 (2014).
- Verkhratsky, A., Nedergaard, M., Hertz, L. Why are astrocytes important. Neurochemical Research. 40 (2), 389-401 (2015).
- Jäkel, S., Dimou, L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Frontiers in Cellular Neuroscience. 11, 24 (2017).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved