Multiplex Immunofluorescence Combined with Spatial Image Analysis for the Clinical and Biological Assessment of the Tumor Microenvironment
In This Article
Summary
In this article, a protocol for manual tyramide signal amplification (TSA) multiplex immunofluorescence (mIF) combined with image analysis and spatial analysis is described. This protocol can be used with formalin-fixed paraffin-embedded (FFPE) sections for the staining of two to six antigens per slide depending on the slide scanner available in the laboratory.
Abstract
The tumor microenvironment (TME) is composed of a plethora of different cell types, such as cytotoxic immune cells and immunomodulatory cells. Depending on its composition and the interactions between cancer cells and peri-tumoral cells, the TME may affect cancer progression. The characterization of tumors and their complex microenvironment could improve the understanding of cancer diseases and may help scientists and clinicians to discover new biomarkers.
We recently developed several multiplex immunofluorescence (mIF) panels based on tyramide signal amplification (TSA) for the characterization of the TME in colorectal cancer, head and neck squamous cell carcinoma, melanoma, and lung cancer. Once the staining and scanning of the corresponding panels are completed, the samples are analyzed on an image analysis software. The spatial position and the staining of each cell are then exported from this quantification software into R. We developed R scripts that allow us not only to analyze the density of each cell type in several tumor compartments (e.g. the center of the tumor, the margin of the tumor, and the stroma) but also to perform distance-based analyses between different cell types.
This particular workflow adds a spatial dimension to the classical density analysis already routinely performed for several markers. mIF analysis could allow scientists to have a better understanding of the complex interaction between cancer cells and the TME and to discover new predictive biomarkers of response to treatments, such as immune checkpoint inhibitors, and targeted therapies.
Introduction
With the development of targeted therapies and immune checkpoint inhibitors, it has become of the utmost importance to better characterize the interactions between cancer cells and their tumor microenvironment, and this is currently an important field of translational research. The TME is composed of a plethora of different cell types, with a balance of immune cytotoxic cells targeting the cancer cells and immunomodulatory cells that could favor tumor growth and invasiveness1,2,3,4. The characterization of this complex environment could improve the understanding of cancer diseases and may help scientists and clinicians to discover new predictive and prognostic biomarkers in order to better select patients for future treatment5,6. For example, Galon and his team developed the Immunoscore, which is a reproducible scoring method that can be used as a predictive biomarker. The Immunoscore is calculated using the density of CD3+ and CD8+ T cells in the invasive margin and in the center of the tumor7,8.
Over the past decades, commercial solutions for mIF have been developed, but these are often expensive and designed for specific panels of antigens. To overcome the need for specific panels of antigens in academic and translational research, we developed a cost-effective method to perform mIF on FFPE tumor sections, allowing the staining of two to six antigens added to the cell nuclei counterstaining on human and mouse samples.
Once the whole tissue sections are stained and scanned with a fluorescence slide scanner, the samples can be analyzed by several image analysis software supporting large pyramidal datasets. Finally, the raw data can be used in an environment for statistical computing and graphics like the R software (v.4.0.2) in order to perform density and spatial-based analyses.
A protocol optimized for five-marker staining, as well as tricks and tips to optimize new panels, is presented in this manuscript. Moreover, detailed steps of the image analysis and the R functions used for the statistical and spatial analysis are explained.
Protocol
All samples used in the present protocol came from a study approved by the local ethics committees and authorized by the competent authority. All the participants in the study provided written informed consent. The trial is registered with ClinicalTrials.gov (NCT03608046).
1. Multiplex immunofluorescence
- FFPE sectioning
- Fix the tissue in 4% paraformaldehyde, and embed the fixed tissue in paraffin.
- Cut 5 µm sections, and place them on adhesive microscope slides.
- Dry the slides overnight at room temperature (RT).
- Deparaffinization and endogenous peroxidases inhibition
- Dewax the tissues by submerging the slides in toluene (3x for 5 min each) and methanol (3x for 5 min each) under a fume hood.
- Inhibit the endogenous peroxidases by submerging the slides in 3% hydrogen peroxide diluted in methanol for 20 min under a fume hood.
- Rinse the slides in distilled (d) H2O (1x for 3 min).
- Multiplex immunofluorescence staining
- Submerge the slides in a 300 mL staining jar containing 10 mM citrate (pH 6) or EDTA (pH 9) buffer complemented with 0.1% TritonX-100.
NOTE: The buffer used (pH 6 or pH 9) depends on the antigen stained (see Table 1). - Place the staining jar with the lid closed in a microwave for 3-5 min at maximum power (e.g., 900 W) until the buffer starts to boil.
NOTE: The optimal time for boiling depends on the microwave and the volume of the buffer. Adjustments may be necessary to find the perfect timing. For some fragile antigens or fragile and less adherent specimens (e.g., organoids and spheroids), microwave boiling can be too harsh. In this case, a pressure cooker can be used instead. - Keep the buffer at near-boiling temperature by putting the closed staining jar in the microwave on low power (e.g., 90 W) for 15 min.
- Perform the last step of heating by putting the microwave at maximum power for 90 s.
- Remove the jar from the microwave, and let the buffer cool down for 15 min at RT.
- Rinse the slides 3x for 5 min each in dH2O and 1x for 5 min in tris-buffered saline containing 0.1% Tween 20 (TBS-T).
- Remove the TBS-T by blotting the slides on a paper towel
- Place the slides (flat) on a staining chamber tray or microscope slide box (see Table of Materials).
- Encircle the tissue with a hydrophobic pen.
- Block the non-specific binding sites by covering the tissue with 5% bovine serum albumin (BSA) dissolved in TBS-T for 30 min.
- Remove the blocking buffer by blotting the slides on a paper towel.
NOTE: Do not rinse the slides after the blocking step. - Incubate the tissue for 60 min with the primary antibody (see Table 1) diluted in 1% BSA TBS-T by covering the tissue with around 300 µL of the solution.
- Rinse the slides 3x for 3 min each with TBS-T.
- Incubate the tissue for 40 min with poly-HRP secondary antibody (see Table 1) by covering the tissue with around 300 µL of the solution.
- Rinse the slides 3x for 3 min with TBS-T.
- Incubate the tissue for 10 min with fluorochrome-tyramide reagent (see Table 1) diluted 200-fold in borate buffer (0.1 M borate, pH 7.8, 3 M NaCl) extemporaneously supplemented with 0.003% H2O2 by covering the tissue with around 300 µL of the solution.
- Rinse the slides 3x for 3 min with TBS-T.
- Repeat steps 1.3.1-1.3.16 until all the TSA staining has been performed.
- Incubate the tissue overnight at 4 °C with the last primary antibody (see Table 1) diluted in 1% BSA TBS-T.
NOTE: Since the incubation is overnight, it is important to cover the staining chamber tray or the microscope slide box and to add dH2O on a paper towel in the bottom of the box (under the slides) to make sure that the tissues do not dry during the incubation. - Rinse the tissue 3x for 5 min each with TBS-T.
- Incubate the tissue for 120 min with the secondary antibody (directly coupled with fluorochrome) diluted 200-fold in 1% BSA TBS-T.
- Rinse the tissue 3x for 5 min each with TBS-T.
- Stain the nuclei by incubating the tissue for 5 min in bisbenzimide (20 mM) diluted 1,000-fold in 10% BSA TBS-T.
NOTE: Bisbenzimide can be replaced by DAPI, but the latter is more toxic and has to be handled carefully under a fume hood. - Rinse the tissue 3x for 3 min each in dH2O.
- Mount the slides by using a fluorescence mounting medium and borosilicate cover glasses.
- Submerge the slides in a 300 mL staining jar containing 10 mM citrate (pH 6) or EDTA (pH 9) buffer complemented with 0.1% TritonX-100.
2. Slide scanning
- Digitalize the slides by scanning them on a fluorescence slide scanner at 20x magnification (slide scanner details are provided in the Table of Materials).
NOTE: A representative scan of an optimal multiplex is shown in Figure 1.
3. Image analysis
- Import the scans into an image analysis software (File > Open Image).
- Go to the Classifiers tab, and select the DenseNet AI V2 plugin.
- Train the DenseNet AI V2 plugin to recognize nuclei by surrounding around 500 nuclei in one image.
- Train the AI on several other slides from the same batch and different batches of mIF staining by surrounding several nuclei (50) on several slides (around 10).
NOTE: Detailed instructions on how to use the AI plugin can be found in the software manual. Using AI for nuclei detection is optional. Other methods to detect nuclei are available depending on the image analysis software used. - Save the trained AI (Classifier Actions > Save).
- Go to the Annotations tab, and create an annotation for each region of interest (ROI), such as the center of the tumor and the margin of the tumor, by using the Pen Annotation Tool.
- If necessary, remove the regions with folds and the regions that appear blurry with the Exclusion Annotation Tool.
NOTE: Hematoxylin-eosin staining of a section adjacent to the one used for mIF can be performed before the mIF staining to ensure that tumor cells are present in the specimen and to help anatomopathologists to determine the ROIs. - Go to the Analysis tab, and select the HighPlex FL algorithm (Settings Actions > Load > HighPlex FL).
- Select the Dye Selection tab, and select the dye of interest.
- In the Nuclear Detection tab, go to Nuclear Segmentation Type, and select AI custom.
- In Nuclear Segmentation Classifier, select the AI saved at step 3.5.
- In the Membrane and Cytoplasm Detection tab, chose the maximum cytoplasm radius (in this study, 1.5 was used) and the number of membrane dyes.
- For each dye, select the nucleus positive threshold, the cytoplasm positive threshold, and the membrane positive threshold.
NOTE: The threshold is different for each staining and should be adjusted for each batch of slides and each antigen stained. Using the View Settings tool (View > View Settings) can help to select an adequate threshold by using the intensity value at the end of the intensity peak (on the right). - For each dye, select the nucleus, membrane, and cytoplasm percentage of completeness values.
NOTE: This parameter is important to avoid false positive detection when two cells with different staining are close to each other (Figure 2). - Save the algorithm (Settings Actions > Save).
- Analyze the ROIs (Analyze > Annotation Layer).
- Go to the Results tab, and select all the data in Object Data (ctrl + A).
- Export the data in .csv format (Right click > Export > Object Data .Csv).
NOTE: This table contains the position (Xmin, Xmax; Ymin, Ymax) and the positivity of each marker of each analyzed cell.
4. Bioinformatics using R
NOTE: An R script providing more details about the following steps is available on GitHub (benidovskaya/Ring: Pipeline for the analysis of multiplex immunofluorescence stainings. [github.com])
- Using the exported table, first define the different cell types based on the colocalization stainings. For example, define cytotoxic T cells by double-positive CD3+/CD8+ cells.
- Then, reconstruct a simplified image of the slide on a dot plot using the coordinates exported from the image analysis software and ggplot2 (Figure 3). Using these data, several types of analysis can be performed:
- Density analysis
NOTE: The simplest analysis is a density analysis.- Perform a density analysis for all the cell types using the entire slide for biopsies or some specific area of the tissue. For example, compute the density of CD3+ and CD8+ T cells at the center of the tumor and the margin of the tumor (Figure 4A-C).
- To compute those densities, use image analysis software to produce a specific data frame per sample with the phenotype and coordinates of each cell. Through a clustering function (k-nearest neighbor) on R, create a polygon object using the borders of the studied biopsy, and calculate the density of the cell types of interest inside of it.
NOTE: This allows for comparing the densities of different cell types between different conditions (such as different time points, treatment types, tissue types, and response to the treatment) and localizations (center of the tumor, margin of the tumor, stroma fibrosis, and necrosis area) depending on the biological hypothesis. Due to the high proximity between cancer cells, peri-tumoral cells, and tumor-infiltrating cells, the image analysis software may detect double-positive cells as immune and cancer cells at the same time. In that case, one needs to bioinformatically correct this issue by mentioning what these double-positive cells are. In this case, CD3+CD8+cytokeratin+ cells were tagged as cytotoxic cells because the cytokeratin positivity was due to the tumor cells surrounding the infiltrating lymphocytes.
- Heatmaps
- Using the density of each cell type from different panels and by applying a normalization (e.g., scaling centering), draw heatmaps (Figure 5) representing the cell abundance in the population of samples.
- Using hierarchical unsupervised clustering based on cell density, cluster patients with similar TME compositions, and correlate these clusters with clinical parameters such as treatment response and survival.
NOTE: Heatmaps and clusterization can be easily performed with the R ComplexHeatmap package9.
- Spatial cell distribution
- Compute bioinformatically the distances between the cells (e.g., immune and tumoral cells; Figure 6A, B) based on the cell coordinates provided by the image analysis. Use the median and mean distances between the cell types of interest to compare the cell proximity across all the samples of a cohort.
- Spatial descriptive functions
- Use the cross-type nearest neighbor G-cross function, available through the R spatstat package10, to determine the probability for a cell of interest, X (e.g., a tumor cell), meeting the nearest cell, Y (e.g., a T cell), within a certain radius around cell X.
- Calculate the area under the empirical curve to obtain a numerical value representing the tumor infiltration of CD3+ T cells around the tumor cells11 (Figure 6C). Use other spatial descriptive functions such as the F-function or J-function12.
- Immunoscore analysis
- Compute the Immunoscore (I), developed by the team of Galon7,8, by using the density of CD3+ and CD8+ T cells in the center of the tumor and the invasive margin of the tumor.
NOTE: The score ranges from I0 to I4. A low density of both CD3+ and CD8+ T cells in the center and the margin of the tumor is associated with a score of I0, while a high density of CD3+ and CD8+ T cells in both regions is associated with a score of I4. Recently, the prognostic impact of the Immunocore was validated in a study with samples from 2,681 stage I-III colon cancer patients from 14 centers in 13 countries7. However, to be computed, Immunoscore requires a surgically resected specimen containing both the center and margin of the tumor. For biopsies, which usually lack a margin, a biopsy-adapted Immunoscore has been recently developed13. - To compute the biopsy-adapted Immunoscore, convert the value of the CD3+ and CD8+ T cell density into a percentile, and then use the mean percentile of CD3+ and CD8+ T cells for scoring into one of three categories (i.e., low, intermediate, and high)13.
- Compute the Immunoscore (I), developed by the team of Galon7,8, by using the density of CD3+ and CD8+ T cells in the center of the tumor and the invasive margin of the tumor.
- Hotspot analysis
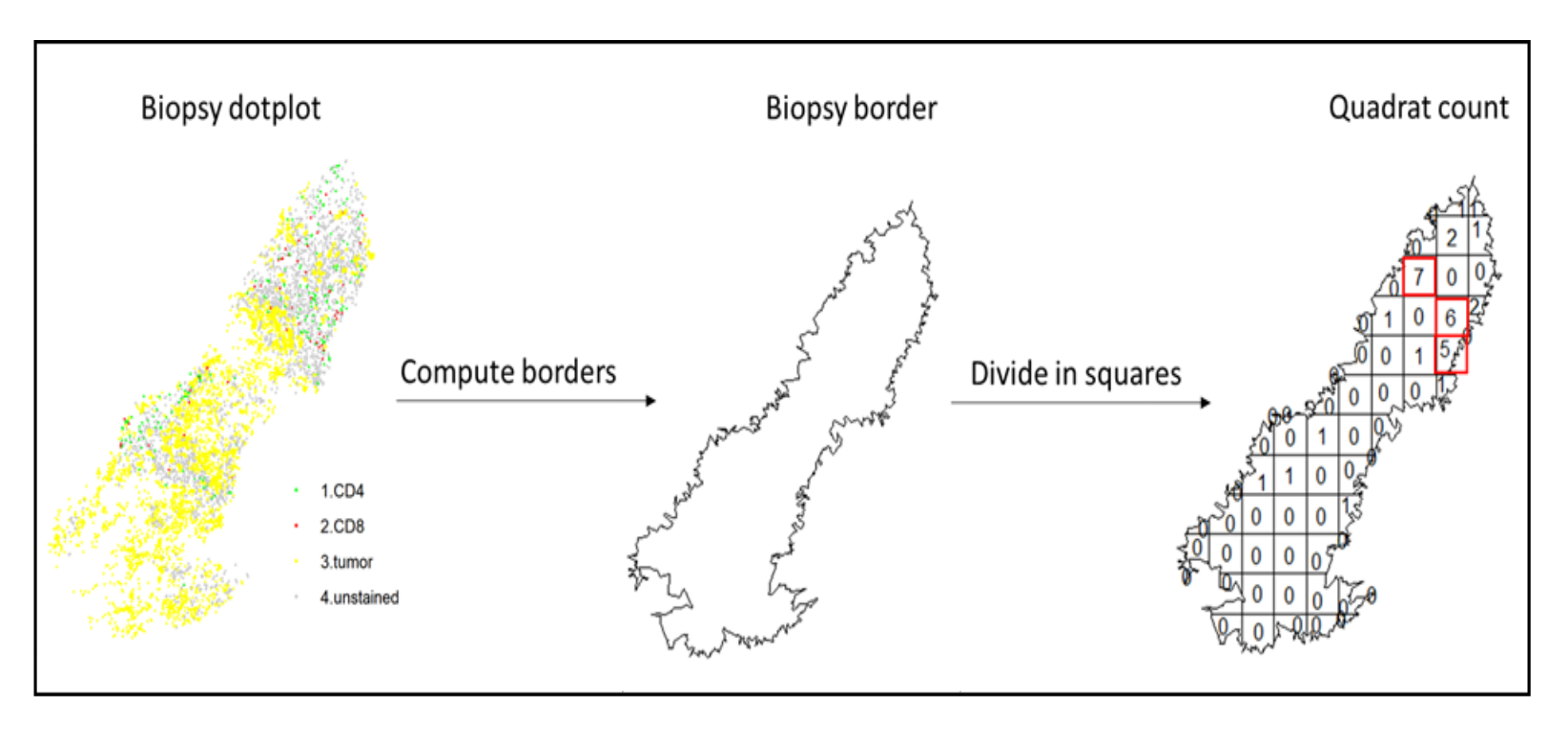
- Use hotspot analysis, by using the quadratcount function (spatstat)10, to compare the densities of different cell types in the most infiltrated area of the tissue. For example, it is possible to compute an "Immunoscore-like" score using the value of the CD3 and CD8 T cell density of the most infiltrated squares of the tissue (Figure 7). Apply this method for the analysis of any cell type with a non-homogeneous distribution across the tissue.
- Density analysis
Representative Results
Following this protocol, several parameters should be investigated to ensure that the tissue is correctly stained. Firstly, the TSA staining should display a good dynamic range when using low exposure times (typically 2-100 ms) during the scanning process. A low exposure time implies that the amplification has been done correctly during the reaction with HRP. For antigens stained with the secondary antibody directly coupled with the fluorochrome, the exposure time could be much longer, which could lead to photobleaching (a decrease in the signal intensity due to a long exposure time). Secondly, it is important to verify that each staining displays a high SNR. A high background signal with a low antigen signal can be an indication that the primary antibody is not specific enough, that the endogenous peroxidases were not inactivated correctly, or that one step of the protocol was not done adequately. Thirdly, depending on the slide scanner and the filter sets used for the scan, it is possible to see overlaps between two colors (e.g., AF555, AF594, and AF647). Choosing the right filter sets on the scanner and the right primary antibody dilution are crucial to avoid possible cross-detections. Quality control consists of the detection of single stained cells for each marker on the scanned file. Finally, it is also important to add a positive and negative control for each batch of staining. For immune cells, the tonsil is a good positive control. A representative result of optimal staining is shown in Figure 1.

Figure 1: Locally advanced rectal cancer stained by multiplex immunofluorescence. Abbreviations: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = RAR-related orphan receptor gamma; CD3 = cluster of differentiation 3; hPanCK = human pan-cytokeratin. Each antigen staining is scanned in grayscale, and the colors presented in the figure are pseudocolors. Scale bar low magnification: 200 µm; scale bar high magnification: 100 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Nuclei and staining detection of a locally advanced rectal cancer using an image analysis software. Without the percentage of completeness parameter correctly set, the software detects two CD8+ cells (green circle) because they are close to each other, but only one cell is stained. Using 70% completeness helps to avoid this false positive detection. Green = hPanCK; Yellow = CD3; Orange = CD8. Scale bar: 100 µm Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Image analysis and R dot-plot reconstitution of a liver colorectal cancer metastasis. On the multiplex staining (left), human pan-cytokeratin is in yellow, CD3 is in green, CD8 is in light blue, and IDO is in orange. On the dot-plot (right), human pan-cytokeratin+ cells are in yellow, CD3+CD8− cells are in green, CD3+CD8+ cells are in blue, and IDO+ cells are in orange. Please click here to view a larger version of this figure.
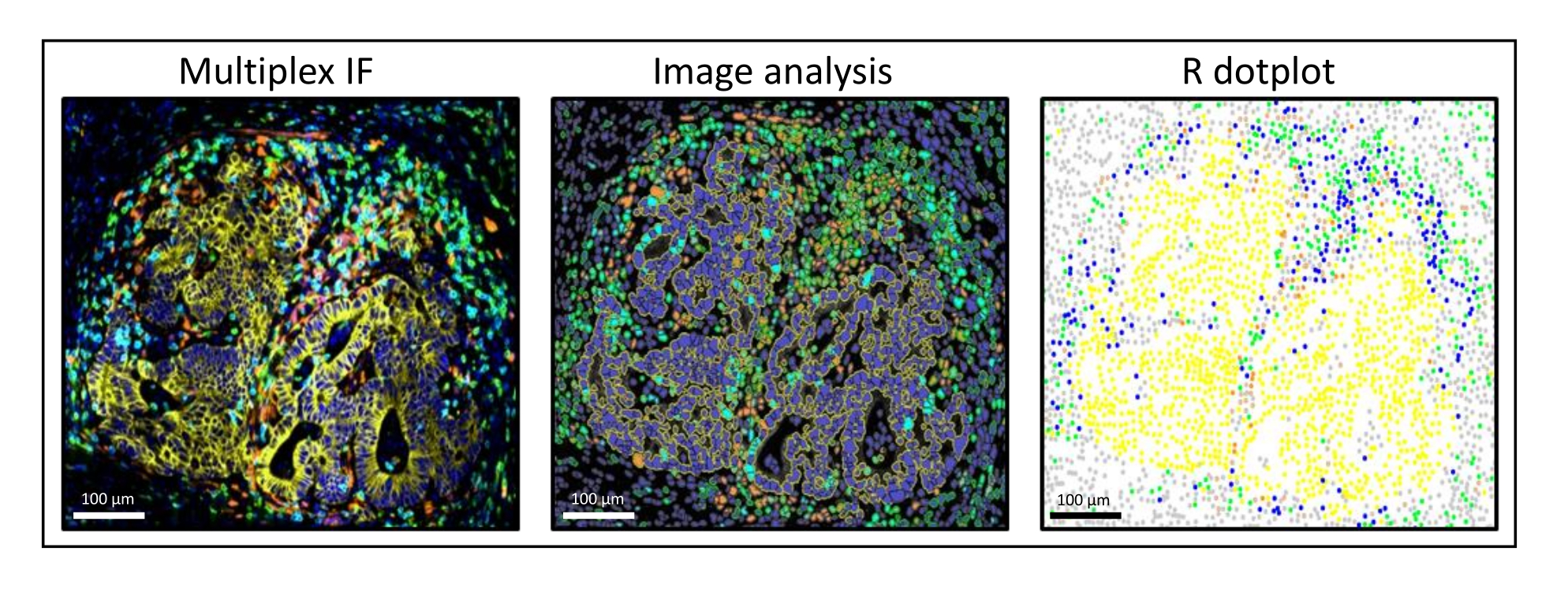{kind=link}

Figure 4: Analysis of a surgical section of an HNSCC. (A) A surgical section of an HNSCC. Cancer cells are visible in green. Peri-tumoral cells are visualized around the tumor islets (CD3 in yellow and CD8 in purple). (B) The center of the tumor (in yellow with a black border) is computed bioinformatically by the k-nearest-neighbor algorithm based on the distance between the tumor islands from a single area. Around this area, an invasive margin (light yellow with a grey border) is computed on an arbitrary 500 µm basis. (C) Invasive T cells are highlighted with black dots in the center of the tumor and grey dots in the invasive margin. Other T cells are highlighted in light green dots. Scale bar: 1 mm. Please click here to view a larger version of this figure.
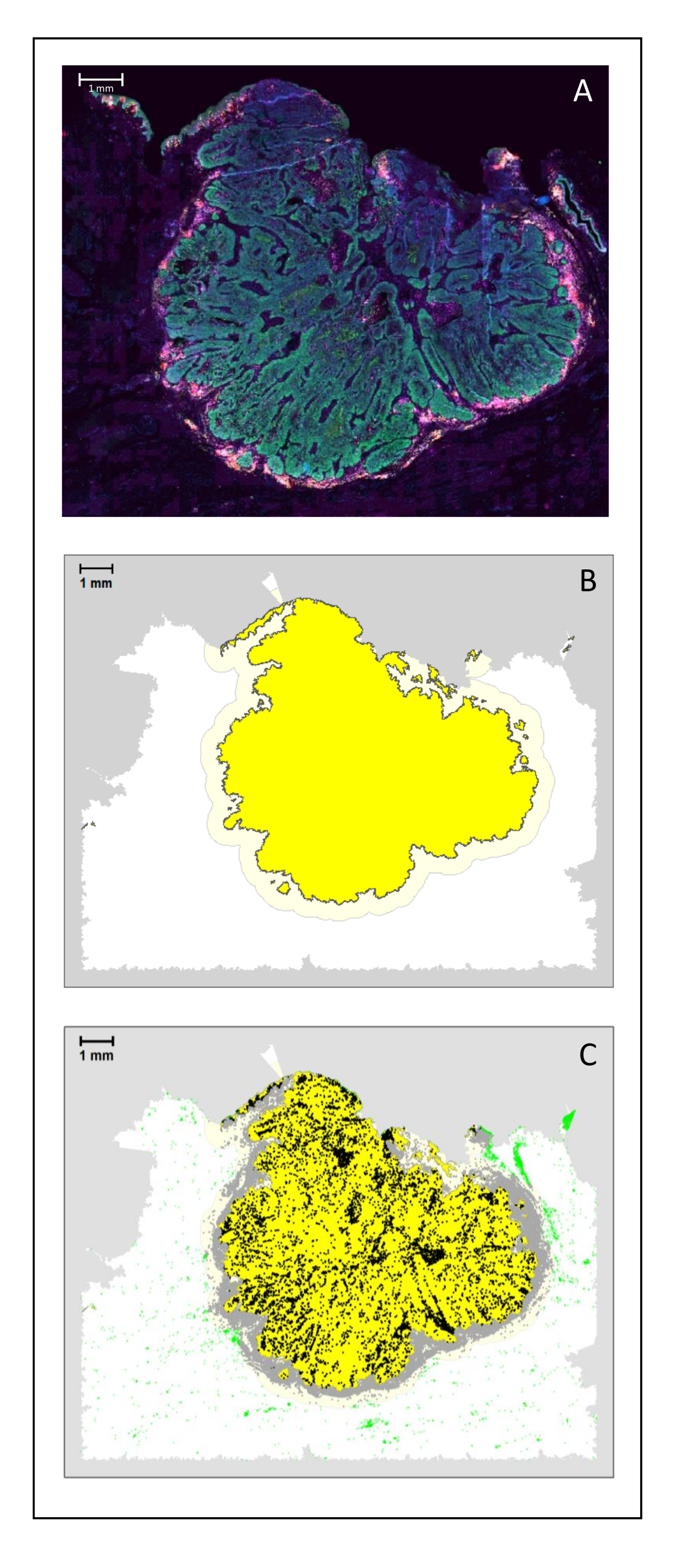{kind=link}

Figure 5: Heatmap of the density of different cell types of locally advanced rectal cancer biopsies. The heatmap was drawn using unsupervised clustering of the densities of different cell types from different multiplex panels with the ComplexHeatmap package. Scaling and centering were used for normalization. Please click here to view a larger version of this figure.
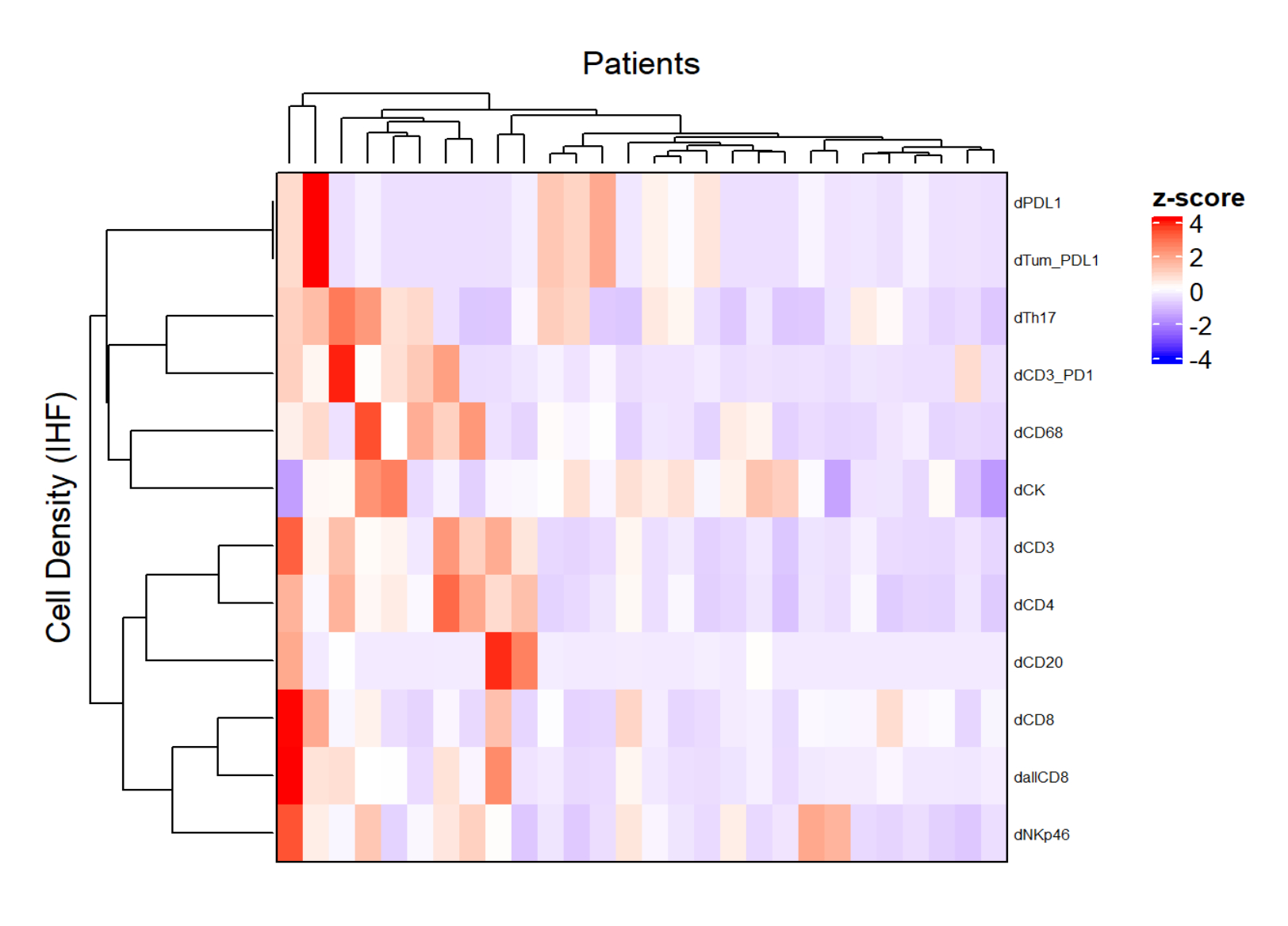{kind=link}

Figure 6: Distances of the CD4+ and CD8+ cells to each IDO+ or tumoral cell. Human pan-cytokeratin+ cells are in yellow, CD3+CD8− cells are in green, CD3+CD8+ cells are in blue, and IDO+ cells are in orange. (A) The closest distance between tumor cells and each CD8+ T cell. (B) Barplots of the distances between IDO+ cells and each CD8+ T cell (blue) or CD4+ T cell (green). (C) Example of a sample analyzed by the G-cross function. The y-axis shows the probability of a tumor cell encountering a CD3+ lymphocyte in a radius ranging from 0-200 µm around the tumor cell. Three curves are shown; the theoretical curve is in dotted green (Poisson distribution), the corrected empirical curve with km correction is in black, and the corrected empirical curve with border correction is in dotted red. Please click here to view a larger version of this figure.
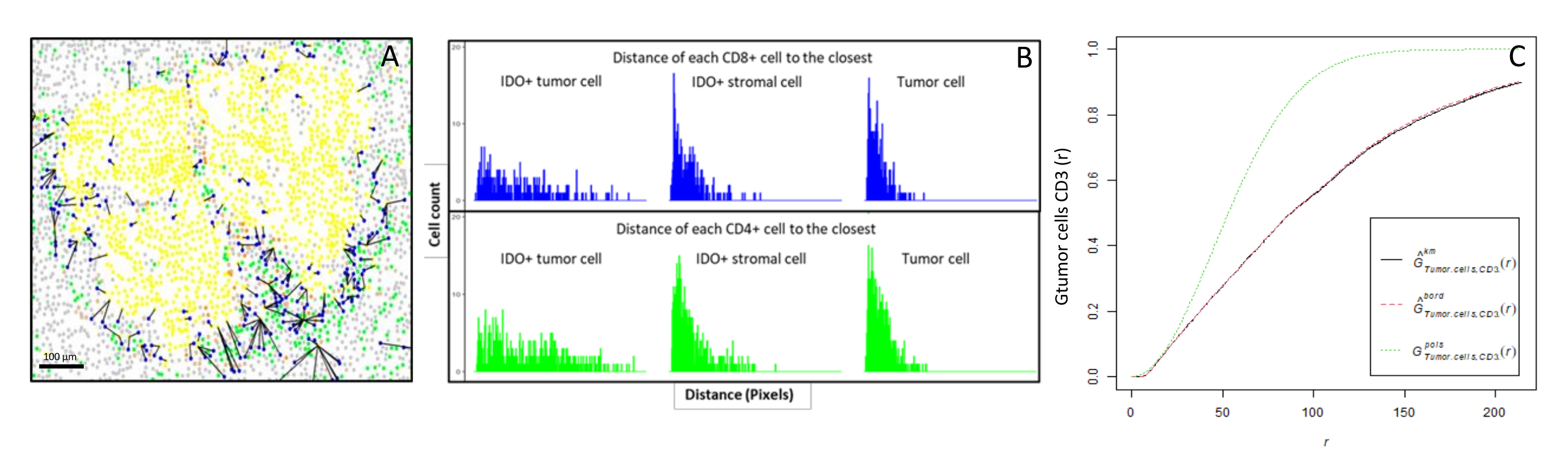{kind=link}

Figure 7: Illustration of a Quadratcount. Border computation and quadratcount were performed using the spatstats package. The most infiltrated squares (hotspots) can be used for downstream statistics. CD4 is in green, CD8 is in red, and tumor cells are in yellow. Please click here to view a larger version of this figure.
{kind=link}

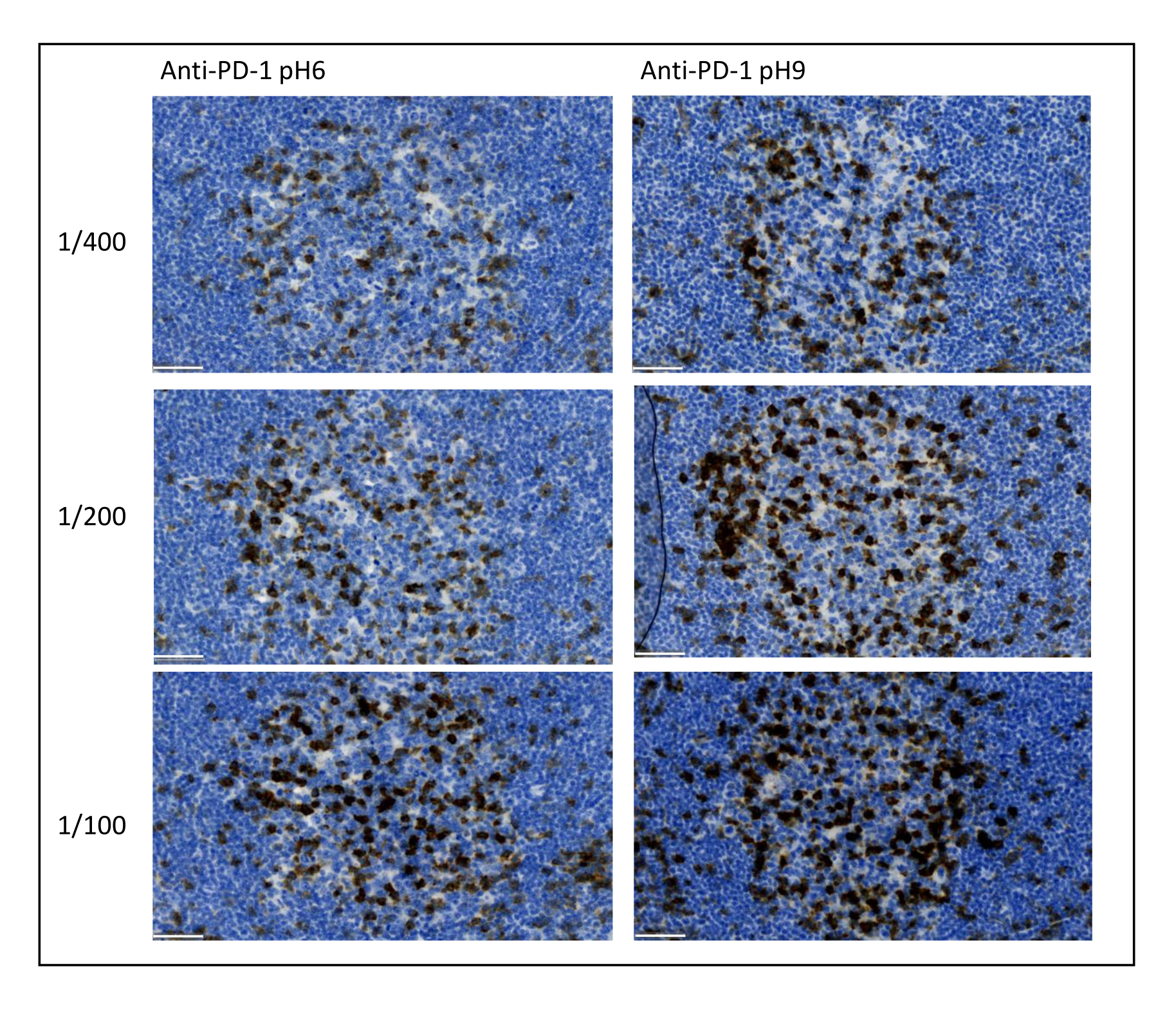
Figure 8: Antibody dilution and antigen retrieval optimization. Chromogenic detection of PD-1 using three different dilutions and two different antigen retrieval solutions of the primary antibody (Citrate pH 6 and EDTA pH 9). Scale bar: 50 µm. Please click here to view a larger version of this figure.
{kind=link}
| Primary antibody | Dilution | Antigen retrieval | Secondary antibody | Fluorochrome | Position |
| PD-1 | 1/100 | EDTA (pH 9) | Anti-rabbit | AF647 | 1 |
| PD-L1 | 1/1000 | EDTA (pH 9) | Anti-rabbit | AF488 | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-mouse | ATT0-425 | 3 |
| CD3 | 1/100 | Citrate (pH 6) | Anti-rabbit | AF555 | 4 |
| hPanCK | 1/50 | Citrate (pH 6) | Anti-mouse coupled with AF750 | 5 | |
Table 1: Example of an optimized multiplex panel. Abbreviations: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = RAR-related orphan receptor gamma; CD3 = cluster of differentiation 3; hPanCK = human pan-cytokeratin; AF = AlexaFluor; EDTA = ethylenediaminetetraacetic acid. CD3 is used to detect T lymphocytes; PD-1 is used to detect exhausted lymphocytes; ROR- γ is used to detect Th-17; and hPanCK is used to detect tumor cells. The position column indicates the order in which the sequential multiplex has to be performed.
Discussion
The most important parameters to take into consideration to optimize multiplex staining are the dilution, the specificity, and the antigen retrieval used for each primary antibody. Before starting a multiplex protocol, each primary antibody's optimal dilution and optimal epitope retrieval (pH 6or pH 9) must be tested using chromogenic staining (DAB). We advise testing three dilutions for each antigen retrieval buffer: the dilution that is usually specified by the brand commercializing the antibody, the same dilution divided two-fold, and the same dilution multiplied two-fold (Figure 8). Choosing the right dilution is a very important step for verifying the antibody specificity and optimizing the signal-to-noise ratio (SNR) of the staining. After choosing the right dilution in the DAB, the same dilution should be tested for each primary antibody using uniplex TSA. Once the dilution and epitope retrieval buffer are selected for each antigen staining, it is also important to correctly set up the sequence of the multiplex; specifically, some antigens are better stained at the first position and others at the last position. We advise testing the multiplex labeling using all the possible order permutations to choose which antigen staining should come first, second, etc. This is also a very important step because some fragile antigens can be degraded after several rounds of epitope retrieval, and some antigens are better stained after several rounds of epitope retrieval. For example, the SNR is always higher in the last position for CD3 and in the first position for PD-1 staining. Moreover, the staining of several co-localized antigens can be hindered by an umbrella effect (the saturation of tyramide reactive sites). This can be attenuated by decreasing the tyramide concentration. When the expression of one antigen is conditioned by the expression of another one (CD8 only present on CD3-expressing T cells), we advise staining the antigen with the broadest expression (CD3 in this case) after the other one. Finally, choosing the right fluorochrome for each antigen staining according to the scanner specificities is also an important step to avoid cross-detection.
The major advantages of this technique are the amplification and the signal-to-noise ratio obtained. However, this technique comes with a limitation, which is that the staining is sequential, and the fluorochromes are covalently bound to the tissue. Nevertheless, after performing all the tyramide signal amplification rounds, it is also possible to add a last staining with a secondary antibody directly coupled with a fluorochrome (no TSA). In some panels, we used this method to add staining in the 750 channel. This was necessary because no tyramide-AF750 was commercially available at that time. Of note, the time of exposure (during the scan) of the antigen stained with AF750 will be much longer than for the other antigens stained with TSA. In that case, we advise staining a highly expressed protein such as cytokeratin or increasing the concentration of the primary antibody. By doing that, it is possible to stain a maximum of five to six antigens per slide in one batch depending on the fluorescence scanner.
In opposition, several commercially available techniques use serial staining with several rounds of staining, scanning, and stripping or photobleaching to improve the number of antigens that can be stained on one single tissue section. However, these techniques are often time-consuming, expensive, have no signal amplification, require advanced computational steps to merge the serial scans correctly, and, in our experience, can induce irreversible tissue damage due to the numerous procedure steps. Nonetheless, it has been reported that up to 30 antigens could be stained on one single tissue using this method14.
In conclusion, our method is a robust, reproducible, easy-to-use, and cost-effective immunohistofluorescence technique that can be used in any lab possessing a fluorescence slide scanner. Any commercialized primary antibody suitable for IHC can be used, and the panels are not specific to any commercial kits. The image analysis can be done on several different programs, including open-source programs such as QuPath and R. However, we think that this method could even be improved in the future for large antigen panels, allowing for performing serial staining/scanning of the same slide with different panels of antigens and with the advantage of signal amplification.
Acknowledgements
The authors would like to thank Dr. Derouane F for her help and support. Nicolas Huyghe is a research fellow supported by a grant from the Belgian National Fund for Scientific Research (Télévie/FNRS 7460918F).
Materials
| Name | Company | Catalog Number | Comments |
| anti-CD3 primary antibody | Abcam | ab16669 | rabbit monocolonal |
| anti-CD8 primary antibody | DAKO | M710301 | mouse monoclonal |
| anti-hPanCK primary antibody | DAKO | M3515 | mouse monoclonal |
| anti-PD-1 primary antibody | Cell Signalling | D4W2J | rabbit monocolonal |
| anti-PD-L1 primary antibody | Cell Signalling | 13684 | rabbit monocolonal |
| anti-RORC primary antibody | Sigma | MABF81 | mouse monoclonal |
| ATTO-425 | ATTOtec | ||
| Axioscan Z1 | Zeiss | Light source: Colibri 7 (385, 430, 475, 555, 590, 630, 735 nm) Filtersets: Excitation 379/34 – beam splitter 409 – emission 440/40; Excitation 438/24 – beam splitter 458 – emission 483/32; Excitation 490/20 – beam splitter 505 – emission 525/20; Excitation 546/10 – beam splitter 556 – emission 572/23; Excitation 592/21 – beam splitter 610 – emission 630/30; Excitation 635/18 – beam splitter 652 – emission 680/42; Excitation 735/40 – beam splitter QBS 405 + 493 + 611 + 762 - emission QBP 425/30 + 524/51 + 634/38 + 785/38; Objective: Plan-Apochromat 20x/0.8; Camera : Orca Flash 4.0 V3 | |
| Borosilicate Cover Glass | VWR | 631-0146 | |
| Envision+ anti-mouse | DAKO | K4001 | |
| Envision+ anti-rabbit | DAKO | K4003 | |
| Fluorescence mounting medium | DAKO | S3023 | |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 750 | ThermoFischer | A-21037 | |
| HALO software | Indicalabs | ||
| Hoescht | Sigma | 14533 | |
| Superfrost plus microscope slides | Fisherscientific/Epredia | 10149870 | |
| Tyramide-AF488 | ThermoFischer | B40953 | |
| Tyramide-AF555 | ThermoFischer | B04955 | |
| Tyramide-AF647 | ThermoFischer | B04958 |
References
- Ge, P., et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomedicine & Pharmacotherapy. 118, 109228 (2019).
- Fridman, W. H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1 (3), 261-262 (2012).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: Impact on clinical outcome. in Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Hanahan, D., Weinberg, R. A. Hallmarks of cancer: The next generation. Cell. 144 (5), 646-674 (2011).
- Calu, V., et al. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Scientific Reports. 11 (1), 7940 (2021).
- Havel, J. J., Chowell, D., Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews. Cancer. 19 (3), 133-150 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Mlecnik, B., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 44 (3), 698-711 (2016).
- Gu, Z., Eils, R., Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32 (18), 2847-2849 (2016).
- Baddeley, A., Rubak, E., Turner, R. . Spatial Point Patterns: Methodology and Applications with R. , (2022).
- Barua, S., et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer. 117, 73-79 (2018).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340 (2021).
- El Sissy, C., et al. A diagnostic biopsy-adapted immunoscore predicts response to neoadjuvant treatment and selects patients with rectal cancer eligible for a watch-and-wait strategy. Clinical Cancer Research. 26 (19), 5198-5207 (2020).
- Bolognesi, M. M., et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. The Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved