Immunofluorescenza multipla combinata con analisi di immagini spaziali per la valutazione clinica e biologica del microambiente tumorale
In This Article
Summary
In questo articolo viene descritto un protocollo per l'immunofluorescenza multipla (mIF) manuale di amplificazione del segnale di tiramido (TSA) combinato con l'analisi delle immagini e l'analisi spaziale. Questo protocollo può essere utilizzato con sezioni FFPE (paraffina-embedded fissate in formalina) per la colorazione di due o sei antigeni per vetrino a seconda dello scanner di vetrini disponibile in laboratorio.
Abstract
Il microambiente tumorale (TME) è composto da una pletora di diversi tipi di cellule, come le cellule immunitarie citotossiche e le cellule immunomodulatorie. A seconda della sua composizione e delle interazioni tra cellule tumorali e cellule peritumorali, la TME può influenzare la progressione del cancro. La caratterizzazione dei tumori e del loro complesso microambiente potrebbe migliorare la comprensione delle malattie tumorali e potrebbe aiutare scienziati e medici a scoprire nuovi biomarcatori.
Recentemente abbiamo sviluppato diversi pannelli di immunofluorescenza multiplex (mIF) basati sull'amplificazione del segnale del tiramido (TSA) per la caratterizzazione della TME nel cancro del colon-retto, nel carcinoma a cellule squamose della testa e del collo, nel melanoma e nel cancro del polmone. Una volta completata la colorazione e la scansione dei pannelli corrispondenti, i campioni vengono analizzati su un software di analisi delle immagini. La posizione spaziale e la colorazione di ogni cella vengono quindi esportate da questo software di quantificazione in R. Abbiamo sviluppato script R che ci permettono non solo di analizzare la densità di ciascun tipo di cellula in diversi compartimenti tumorali (ad esempio il centro del tumore, il margine del tumore e lo stroma), ma anche di eseguire analisi basate sulla distanza tra diversi tipi di cellule.
Questo particolare flusso di lavoro aggiunge una dimensione spaziale alla classica analisi della densità già eseguita di routine per diversi marcatori. L'analisi mIF potrebbe consentire agli scienziati di avere una migliore comprensione della complessa interazione tra le cellule tumorali e la TME e di scoprire nuovi biomarcatori predittivi di risposta ai trattamenti, come gli inibitori del checkpoint immunitario e terapie mirate.
Introduction
Con lo sviluppo di terapie mirate e inibitori del checkpoint immunitario, è diventato della massima importanza caratterizzare meglio le interazioni tra le cellule tumorali e il loro microambiente tumorale, e questo è attualmente un importante campo di ricerca traslazionale. La TME è composta da una pletora di diversi tipi cellulari, con un equilibrio di cellule immunocitotossiche che colpiscono le cellule tumorali e cellule immunomodulatrici che potrebbero favorire la crescita e l'invasività tumorale 1,2,3,4. La caratterizzazione di questo ambiente complesso potrebbe migliorare la comprensione delle malattie tumorali e potrebbe aiutare scienziati e clinici a scoprire nuovi biomarcatori predittivi e prognostici al fine di selezionare meglio i pazienti per il trattamento futuro 5,6. Ad esempio, Galon e il suo team hanno sviluppato l'Immunoscore, che è un metodo di punteggio riproducibile che può essere utilizzato come biomarcatore predittivo. L'Immunoscore viene calcolato utilizzando la densità delle cellule T CD3+ e CD8+ nel margine invasivo e al centro del tumore 7,8.
Negli ultimi decenni sono state sviluppate soluzioni commerciali per mIF, ma queste sono spesso costose e progettate per specifici pannelli di antigeni. Per superare la necessità di pannelli specifici di antigeni nella ricerca accademica e traslazionale, abbiamo sviluppato un metodo economico per eseguire mIF su sezioni tumorali FFPE, consentendo la colorazione di due o sei antigeni aggiunti alla controcolorazione dei nuclei cellulari su campioni umani e murini.
Una volta che le intere sezioni di tessuto sono state colorate e scansionate con uno scanner a vetrini a fluorescenza, i campioni possono essere analizzati da diversi software di analisi delle immagini che supportano grandi set di dati piramidali. Infine, i dati grezzi possono essere utilizzati in un ambiente per il calcolo statistico e grafico come il software R (v.4.0.2) al fine di eseguire analisi di densità e spaziali.
Un protocollo ottimizzato per la colorazione a cinque marcatori, nonché trucchi e suggerimenti per ottimizzare i nuovi pannelli, è presentato in questo manoscritto. Inoltre, vengono spiegati i passaggi dettagliati dell'analisi dell'immagine e le funzioni R utilizzate per l'analisi statistica e spaziale.
Protocol
Tutti i campioni utilizzati nel presente protocollo provengono da uno studio approvato dai comitati etici locali e autorizzato dall'autorità competente. Tutti i partecipanti allo studio hanno fornito il consenso informato scritto. Lo studio è registrato presso ClinicalTrials.gov (NCT03608046).
1. Immunofluorescenza multiplex
- Sezionamento FFPE
- Fissare il tessuto in paraformaldeide al 4% e incorporare il tessuto fisso in paraffina.
- Tagliare sezioni da 5 μm e posizionarle su vetrini adesivi per microscopio.
- Asciugare i vetrini per una notte a temperatura ambiente (RT).
- Deparaffinizzazione e inibizione delle perossidasi endogene
- Decerare i tessuti immergendo i vetrini in toluene (3x per 5 min ciascuno) e metanolo (3x per 5 min ciascuno) sotto una cappa aspirante.
- Inibiscono le perossidasi endogene immergendo i vetrini in perossido di idrogeno al 3% diluito in metanolo per 20 minuti sotto una cappa aspirante.
- Risciacquare i vetrini in distillato (d) H2O (1x per 3 min).
- Colorazione con immunofluorescenza multiplex
- Immergere i vetrini in un barattolo colorante da 300 ml contenente 10 mM di citrato (pH 6) o tampone EDTA (pH 9) integrato con TritonX-100 allo 0,1%.
NOTA: Il tampone utilizzato (pH 6 o pH 9) dipende dall'antigene colorato (vedere Tabella 1). - Posizionare il barattolo di colorazione con il coperchio chiuso in un forno a microonde per 3-5 minuti alla massima potenza (ad esempio, 900 W) fino a quando il tampone inizia a bollire.
NOTA: Il momento ottimale per l'ebollizione dipende dal microonde e dal volume del tampone. Potrebbero essere necessari aggiustamenti per trovare il tempismo perfetto. Per alcuni antigeni fragili o campioni fragili e meno aderenti (ad esempio, organoidi e sferoidi), l'ebollizione a microonde può essere troppo dura. In questo caso, è possibile utilizzare una pentola a pressione. - Mantenere il tampone a una temperatura vicina all'ebollizione mettendo il barattolo di colorazione chiuso nel microonde a bassa potenza (ad esempio, 90 W) per 15 minuti.
- Eseguire l'ultima fase di riscaldamento mettendo il microonde alla massima potenza per 90 s.
- Rimuovere il barattolo dal microonde e lasciare raffreddare il buffer per 15 minuti a RT.
- Risciacquare i vetrini 3 volte per 5 minuti ciascuno in dH2O e 1x per 5 minuti in soluzione salina tris-tamponata contenente lo 0,1% Tween 20 (TBS-T).
- Rimuovere il TBS-T asciugando le diapositive su un tovagliolo di carta
- Posizionare i vetrini (piatti) su un vassoio della camera di colorazione o su una scatola di vetrini per microscopio (vedere Tabella dei materiali).
- Circondare il tessuto con una penna idrofoba.
- Bloccare i siti di legame non specifici coprendo il tessuto con albumina sierica bovina al 5% (BSA) sciolta in TBS-T per 30 minuti.
- Rimuovere il buffer di blocco tamponando le diapositive su un tovagliolo di carta.
NOTA: non risciacquare i vetrini dopo la fase di blocco. - Incubare il tessuto per 60 minuti con l'anticorpo primario (vedere Tabella 1) diluito in TBS-T all'1% di BSA coprendo il tessuto con circa 300 μL della soluzione.
- Risciacquare i vetrini 3 volte per 3 minuti ciascuno con TBS-T.
- Incubare il tessuto per 40 minuti con anticorpi secondari poli-HRP (vedere Tabella 1) coprendo il tessuto con circa 300 μL della soluzione.
- Risciacquare i vetrini 3 volte per 3 minuti con TBS-T.
- Incubare il tessuto per 10 minuti con reagente fluorocromo-tiramide (vedere Tabella 1) diluito 200 volte in tampone borato (0,1 M borato, pH 7,8, 3 M NaCl) estemporaneamente integrato con 0,003% H 2 O2coprendo il tessuto con circa 300 μL della soluzione.
- Risciacquare i vetrini 3 volte per 3 minuti con TBS-T.
- Ripetere i passaggi 1.3.1-1.3.16 fino a quando non è stata eseguita tutta la colorazione TSA.
- Incubare il tessuto per una notte a 4 °C con l'ultimo anticorpo primario (vedere Tabella 1) diluito in TBS-T all'1% di BSA.
NOTA: Poiché l'incubazione avviene durante la notte, è importante coprire il vassoio della camera di colorazione o la scatola del vetrino del microscopio e aggiungere dH2O su un tovagliolo di carta sul fondo della scatola (sotto i vetrini) per assicurarsi che i tessuti non si asciughino durante l'incubazione. - Risciacquare il fazzoletto 3 volte per 5 minuti ciascuno con TBS-T.
- Incubare il tessuto per 120 minuti con l'anticorpo secondario (direttamente accoppiato con fluorocromo) diluito 200 volte in 1% BSA TBS-T.
- Risciacquare il fazzoletto 3 volte per 5 minuti ciascuno con TBS-T.
- Colorare i nuclei incubando il tessuto per 5 minuti in bisbenzimmide (20 mM) diluito 1.000 volte in 10% BSA TBS-T.
NOTA: La bisbenzimmide può essere sostituita dal DAPI, ma quest'ultimo è più tossico e deve essere maneggiato con cura sotto una cappa aspirante. - Risciacquare il fazzoletto 3 volte per 3 minuti ciascuno in dH2O.
- Montare le guide utilizzando un mezzo di montaggio a fluorescenza e occhiali di copertura in borosilicato.
- Immergere i vetrini in un barattolo colorante da 300 ml contenente 10 mM di citrato (pH 6) o tampone EDTA (pH 9) integrato con TritonX-100 allo 0,1%.
2. Scansione dei vetrini
- Digitalizzare i vetrini scansionandoli su uno scanner per vetrini a fluorescenza con ingrandimento 20x (i dettagli dello scanner per vetrini sono forniti nella tabella dei materiali).
NOTA: nella Figura 1 è illustrata una scansione rappresentativa di un multiplex ottimale.
3. Analisi delle immagini
- Importare le scansioni in un software di analisi delle immagini (File > Open Image).
- Vai alla scheda Classificatori e seleziona il plug-in DenseNet AI V2 .
- Addestra il plugin DenseNet AI V2 a riconoscere i nuclei circondando circa 500 nuclei in un'immagine.
- Addestra l'IA su diversi altri vetrini dello stesso lotto e diversi lotti di colorazione mIF circondando diversi nuclei (50) su diversi vetrini (circa 10).
NOTA: istruzioni dettagliate su come utilizzare il plug-in AI sono disponibili nel manuale del software. L'utilizzo dell'intelligenza artificiale per il rilevamento dei nuclei è facoltativo. Altri metodi per rilevare i nuclei sono disponibili a seconda del software di analisi delle immagini utilizzato. - Salva l'IA addestrata (Azioni classificatore > Salva).
- Passare alla scheda Annotazioni e creare un'annotazione per ogni area di interesse (ROI), ad esempio il centro del tumore e il margine del tumore, utilizzando lo strumento di annotazione penna.
- Se necessario, rimuovete le aree con pieghe e le aree che appaiono sfocate con lo strumento di annotazione di esclusione.
NOTA: La colorazione dell'ematossilina-eosina di una sezione adiacente a quella utilizzata per mIF può essere eseguita prima della colorazione mIF per garantire che le cellule tumorali siano presenti nel campione e per aiutare gli anatomopatologi a determinare i ROI. - Vai alla scheda Analisi e seleziona l'algoritmo HighPlex FL (Impostazioni azioni > Carica > HighPlex FL).
- Selezionare la scheda Selezione colorante e selezionare il colorante di interesse.
- Nella scheda Rilevamento nucleare, vai a Tipo di segmentazione nucleare e seleziona AI personalizzato.
- In Nuclear Segmentation Classifier, selezionare l'IA salvata al passaggio 3.5.
- Nella scheda Rilevamento membrana e citoplasma, scegliere il raggio massimo del citoplasma (in questo studio è stato utilizzato 1,5) e il numero di coloranti di membrana.
- Per ogni colorante, selezionare la soglia positiva del nucleo, la soglia positiva del citoplasma e la soglia positiva della membrana.
NOTA: La soglia è diversa per ogni colorazione e deve essere regolata per ogni lotto di vetrini e ogni antigene colorato. L'uso dello strumento Impostazioni di visualizzazione (Visualizza > Impostazioni di visualizzazione) può aiutare a selezionare una soglia adeguata utilizzando il valore di intensità alla fine del picco di intensità (a destra). - Per ogni colorante, selezionare la percentuale di nucleo, membrana e citoplasma dei valori di completezza.
NOTA: questo parametro è importante per evitare il rilevamento di falsi positivi quando due celle con colorazione diversa sono vicine l'una all'altra (Figura 2). - Salvare l'algoritmo (Impostazioni azioni > Salva).
- Analizzare i ROI (Analyze > Annotation Layer).
- Vai alla scheda Risultati e seleziona tutti i dati in Dati oggetto (ctrl + A).
- Esportare i dati in formato .csv (fare clic con il pulsante destro del mouse > Esporta > dati oggetto . CSV).
Nota : questa tabella contiene la posizione (Xmin, Xmax; Ymin, Ymax) e la positività di ciascun marcatore di ogni cellula analizzata.
4. Bioinformatica con R
NOTA: uno script R che fornisce maggiori dettagli sui seguenti passaggi è disponibile su GitHub (benidovskaya/Ring: Pipeline for the analysis of multiplex immunofluorescence stainings. [github.com])
- Utilizzando la tabella esportata, definire prima i diversi tipi di celle in base alle colorazioni di colocalizzazione. Ad esempio, definire le cellule T citotossiche mediante cellule CD3+/CD8+ doppie positive.
- Quindi, ricostruire un'immagine semplificata della diapositiva su un dot plot utilizzando le coordinate esportate dal software di analisi delle immagini e ggplot2 (Figura 3). Utilizzando questi dati, è possibile eseguire diversi tipi di analisi:
- Analisi della densità
NOTA: l'analisi più semplice è un'analisi della densità.- Eseguire un'analisi della densità per tutti i tipi di cellule utilizzando l'intero vetrino per biopsie o alcune aree specifiche del tessuto. Ad esempio, calcolare la densità delle cellule T CD3 + e CD8 + al centro del tumore e il margine del tumore (Figura 4A-C).
- Per calcolare tali densità, utilizzare il software di analisi delle immagini per produrre un frame di dati specifico per campione con il fenotipo e le coordinate di ciascuna cellula. Attraverso una funzione di clustering (k-nearest neighbor) su R, creare un oggetto poligonale utilizzando i bordi della biopsia studiata e calcolare la densità dei tipi di cellule di interesse al suo interno.
NOTA: Ciò consente di confrontare le densità di diversi tipi di cellule tra diverse condizioni (come diversi punti temporali, tipi di trattamento, tipi di tessuto e risposta al trattamento) e localizzazioni (centro del tumore, margine del tumore, fibrosi stroma e area di necrosi) a seconda dell'ipotesi biologica. A causa dell'elevata vicinanza tra cellule tumorali, cellule peritumorali e cellule infiltranti il tumore, il software di analisi delle immagini può rilevare cellule doppie positive come cellule immunitarie e tumorali allo stesso tempo. In tal caso, è necessario correggere bioinformaticamente questo problema menzionando quali sono queste cellule doppie positive. In questo caso, le cellule CD3 + CD8 + citocheratina + sono state etichettate come cellule citotossiche perché la positività della citocheratina era dovuta alle cellule tumorali che circondano i linfociti infiltranti.
- Mappe di calore
- Utilizzando la densità di ciascun tipo di cella da diversi pannelli e applicando una normalizzazione (ad esempio, centratura del ridimensionamento), disegnare mappe di calore (Figura 5) che rappresentano l'abbondanza di cellule nella popolazione di campioni.
- Utilizzando il clustering gerarchico non supervisionato basato sulla densità cellulare, raggruppare i pazienti con composizioni simili di TME e correlare questi cluster con parametri clinici come la risposta al trattamento e la sopravvivenza.
NOTA: le mappe di calore e la clusterizzazione possono essere facilmente eseguite con il pacchetto R ComplexHeatmap9.
- Distribuzione spaziale delle cellule
- Calcolare bioinformaticamente le distanze tra le cellule (ad esempio, cellule immunitarie e tumorali; Figura 6A, B) basata sulle coordinate di cella fornite dall'analisi dell'immagine. Utilizzare le distanze mediane e medie tra i tipi di cellule di interesse per confrontare la vicinanza cellulare tra tutti i campioni di una coorte.
- Funzioni descrittive spaziali
- Utilizzare la funzione G-cross del vicino più vicino di tipo incrociato, disponibile attraverso il pacchetto R spatstat10, per determinare la probabilità che una cellula di interesse, X (ad esempio, una cellula tumorale), incontri la cellula più vicina, Y (ad esempio, una cellula T), entro un certo raggio attorno alla cella X.
- Calcolare l'area sotto la curva empirica per ottenere un valore numerico che rappresenti l'infiltrazione tumorale delle cellule T CD3+ attorno alle cellule tumorali11 (Figura 6C). Utilizzare altre funzioni descrittive spaziali come la funzione F o la funzione J12.
- Analisi dell'immunoscore
- Calcola l'Immunoscore (I), sviluppato dal team di Galon7,8, utilizzando la densità delle cellule T CD3 + e CD8 + al centro del tumore e il margine invasivo del tumore.
NOTA: il punteggio varia da I0 a I4. Una bassa densità di cellule T CD3+ e CD8+ al centro e il margine del tumore sono associati a un punteggio di I0, mentre un'alta densità di cellule T CD3+ e CD8+ in entrambe le regioni è associata a un punteggio di I4. Recentemente, l'impatto prognostico dell'Immunocore è stato convalidato in uno studio con campioni di 2.681 pazienti con cancro del colon in stadio I-III provenienti da 14 centri in 13 paesi7. Tuttavia, per essere calcolato, Immunoscore richiede un campione resecato chirurgicamente contenente sia il centro che il margine del tumore. Per le biopsie, che di solito mancano di margine, è stato recentemente sviluppato un Immunoscore adattato alla biopsia13. - Per calcolare l'Immunoscore adattato alla biopsia, convertire il valore della densità delle cellule T CD3+ e CD8+ in un percentile, quindi utilizzare il percentile medio delle cellule T CD3+ e CD8+ per il punteggio in una delle tre categorie (cioè basso, intermedio e alto)13.
- Calcola l'Immunoscore (I), sviluppato dal team di Galon7,8, utilizzando la densità delle cellule T CD3 + e CD8 + al centro del tumore e il margine invasivo del tumore.
- Analisi degli hotspot
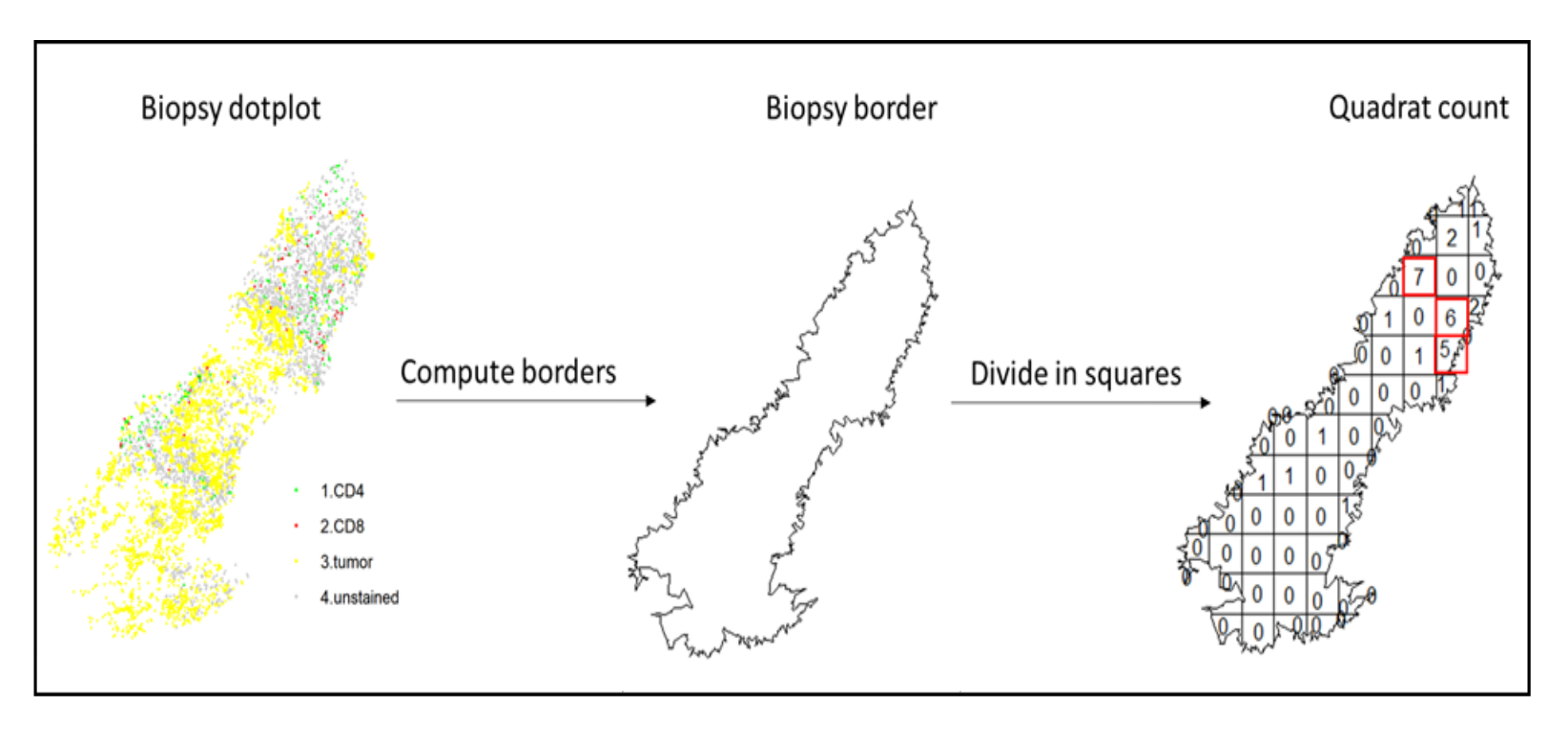
- Utilizzare l'analisi hotspot, utilizzando la funzione quadratcount (spatstat)10, per confrontare le densità di diversi tipi di cellule nell'area più infiltrata del tessuto. Ad esempio, è possibile calcolare un punteggio "Immunoscore-like" utilizzando il valore della densità delle cellule T CD3 e CD8 dei quadrati più infiltrati del tessuto (Figura 7). Applicare questo metodo per l'analisi di qualsiasi tipo di cellula con una distribuzione non omogenea attraverso il tessuto.
- Analisi della densità
Representative Results
Seguendo questo protocollo, è necessario studiare diversi parametri per garantire che il tessuto sia colorato correttamente. In primo luogo, la colorazione TSA dovrebbe mostrare una buona gamma dinamica quando si utilizzano tempi di esposizione ridotti (in genere 2-100 ms) durante il processo di scansione. Un basso tempo di esposizione implica che l'amplificazione sia stata eseguita correttamente durante la reazione con HRP. Per gli antigeni colorati con l'anticorpo secondario direttamente accoppiato con il fluorocromo, il tempo di esposizione potrebbe essere molto più lungo, il che potrebbe portare al fotosbiancamento (una diminuzione dell'intensità del segnale dovuta a un lungo tempo di esposizione). In secondo luogo, è importante verificare che ogni colorazione mostri un SNR elevato. Un segnale di fondo alto con un segnale di antigene basso può essere un'indicazione che l'anticorpo primario non è abbastanza specifico, che le perossidasi endogene non sono state inattivate correttamente o che un passaggio del protocollo non è stato eseguito adeguatamente. In terzo luogo, a seconda dello scanner di vetrini e dei set di filtri utilizzati per la scansione, è possibile vedere sovrapposizioni tra due colori (ad esempio, AF555, AF594 e AF647). La scelta dei giusti set di filtri sullo scanner e la giusta diluizione degli anticorpi primari sono fondamentali per evitare possibili rilevamenti incrociati. Il controllo di qualità consiste nel rilevamento di singole celle colorate per ciascun marcatore sul file scansionato. Infine, è anche importante aggiungere un controllo positivo e negativo per ogni lotto di colorazione. Per le cellule immunitarie, la tonsilla è un buon controllo positivo. Un risultato rappresentativo della colorazione ottimale è mostrato nella Figura 1.

Figura 1: Carcinoma rettale localmente avanzato colorato mediante immunofluorescenza multiplex. Abbreviazioni: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = recettore orfano gamma correlato a RAR; CD3 = cluster di differenziazione 3; hPanCK = pan-citocheratina umana. Ogni colorazione dell'antigene viene scansionata in scala di grigi e i colori presentati nella figura sono pseudocolori. Basso ingrandimento della barra della scala: 200 μm; Barra di scala ad alto ingrandimento: 100 μm. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 2: Rilevamento di nuclei e colorazione di un carcinoma rettale localmente avanzato utilizzando un software di analisi delle immagini. Senza il parametro percentuale di completezza impostato correttamente, il software rileva due cellule CD8+ (cerchio verde) perché sono vicine l'una all'altra, ma solo una cella è macchiata. L'utilizzo del 70% di completezza consente di evitare questo rilevamento di falsi positivi. Verde = hPanCK; Giallo = CD3; Arancione = CD8. Barra scala: 100 μm Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

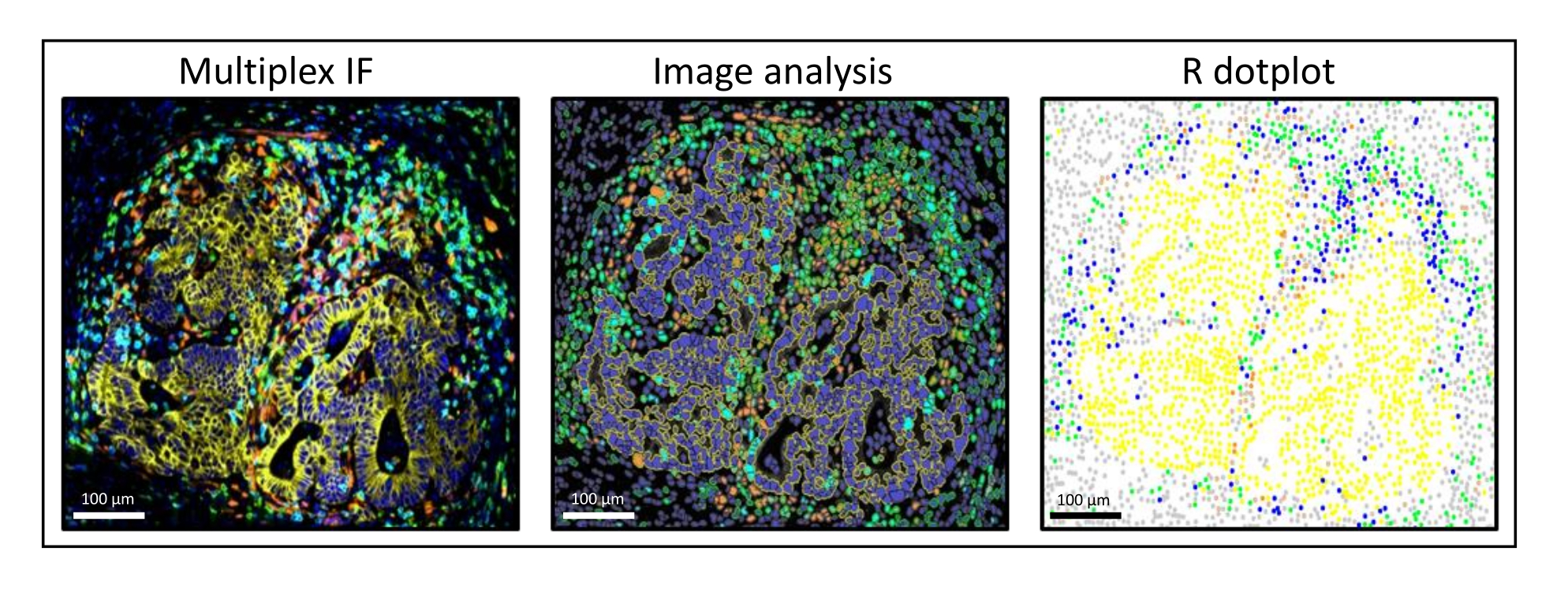
Figura 3: Analisi delle immagini e ricostituzione R dot-plot di una metastasi del cancro del colon-retto del fegato. Sulla colorazione multiplex (a sinistra), la pan-citocheratina umana è in giallo, CD3 è in verde, CD8 è in azzurro e IDO è in arancione. Sul dot-plot (a destra), le cellule pan-citocheratine + umane sono in giallo, le cellule CD3 + CD8 sono in verde, le cellule CD3 + CD8 + sono in blu e le cellule IDO + sono in arancione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

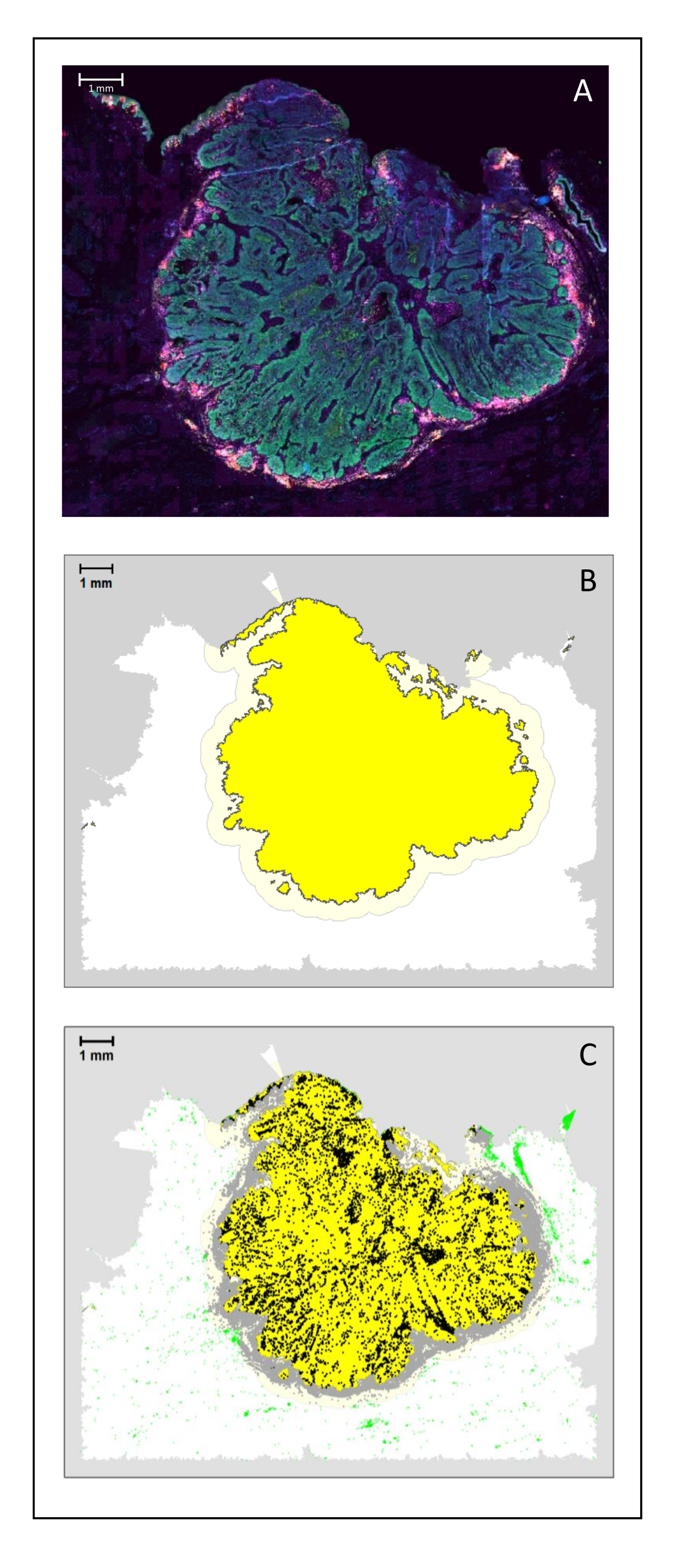
Figura 4: Analisi di una sezione chirurgica di un HNSCC. (A) Una sezione chirurgica di un HNSCC. Le cellule tumorali sono visibili in verde. Le cellule peritumorali sono visualizzate intorno alle isole tumorali (CD3 in giallo e CD8 in viola). (B) Il centro del tumore (in giallo con un bordo nero) è calcolato bioinformaticamente dall'algoritmo k-nearest-neighbor basato sulla distanza tra le isole tumorali da una singola area. Intorno a quest'area, un margine invasivo (giallo chiaro con un bordo grigio) viene calcolato su una base arbitraria di 500 μm. (C) Le cellule T invasive sono evidenziate con punti neri al centro del tumore e punti grigi nel margine invasivo. Altre cellule T sono evidenziate in punti verde chiaro. Barra scala: 1 mm. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

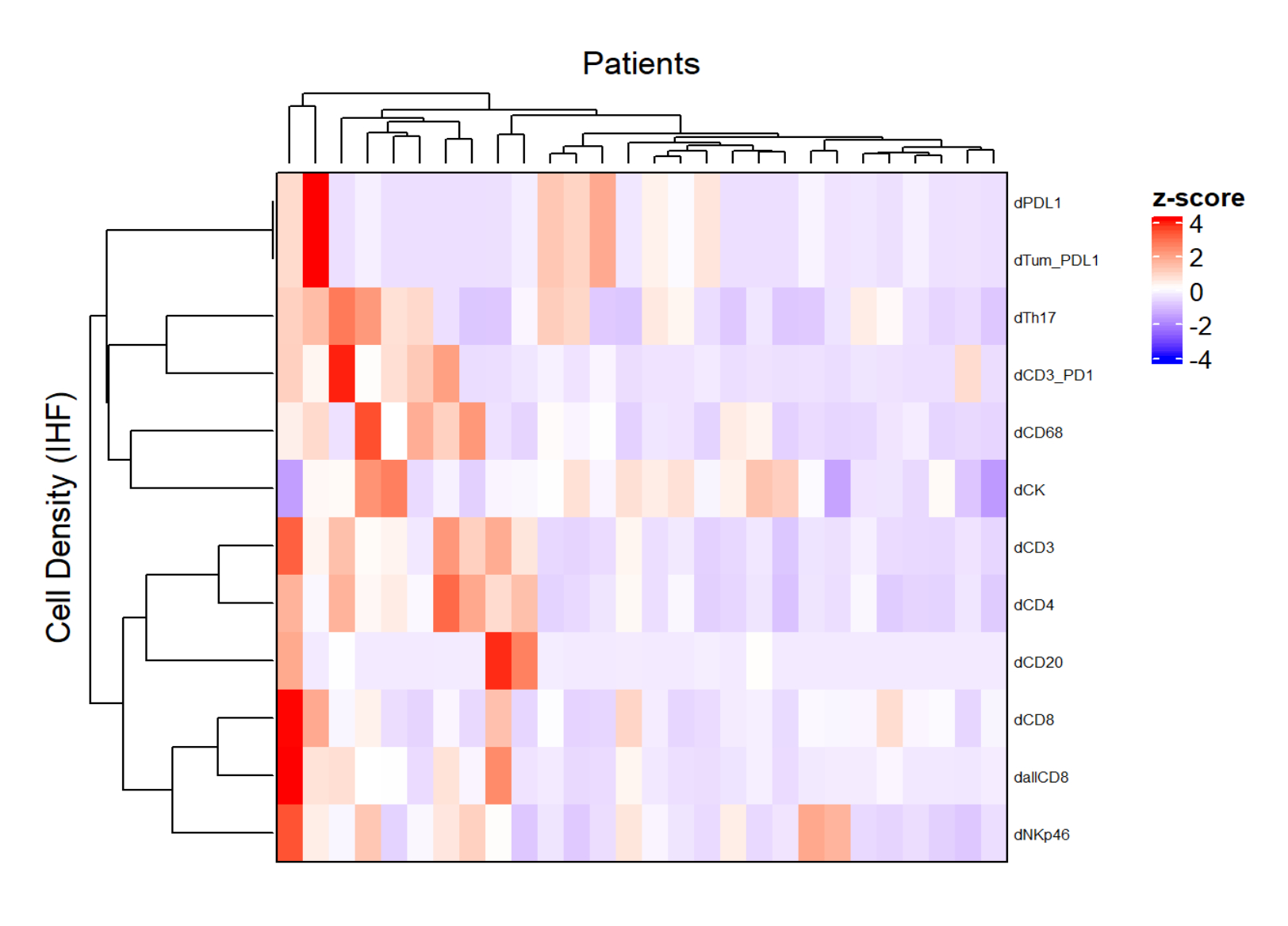
Figura 5: Mappa termica della densità di diversi tipi cellulari di biopsie localmente avanzate del cancro del retto. La mappa di calore è stata disegnata utilizzando il clustering non supervisionato delle densità di diversi tipi di celle da diversi pannelli multiplex con il pacchetto ComplexHeatmap. Il ridimensionamento e la centratura sono stati utilizzati per la normalizzazione. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

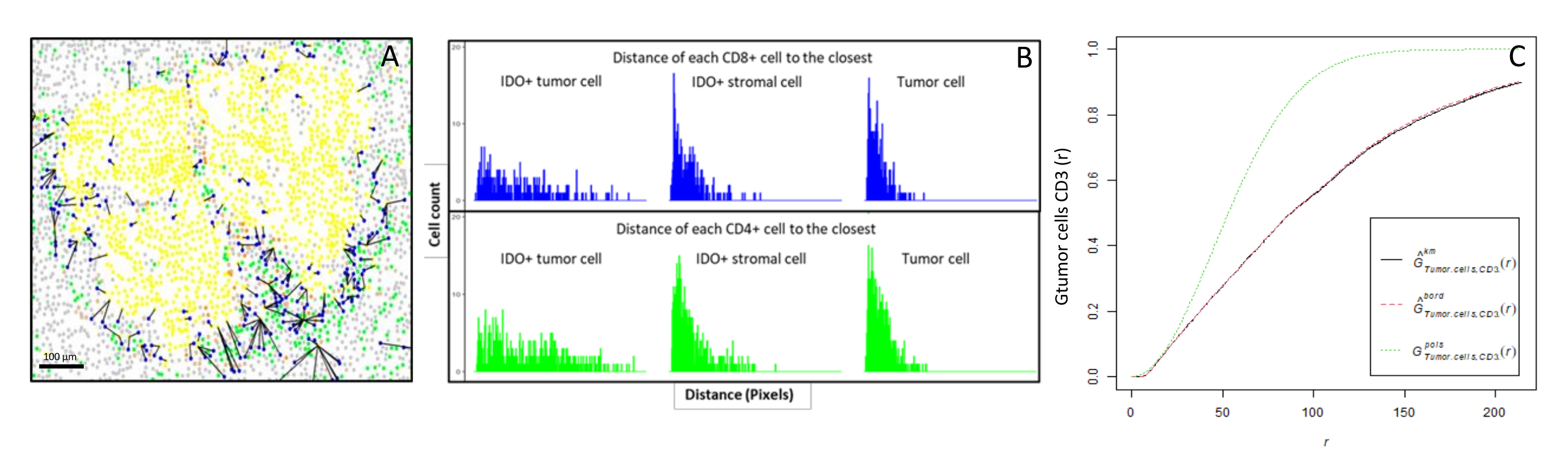
Figura 6: Distanze delle cellule CD4+ e CD8+ da ciascuna cellula IDO+ o tumorale. Le cellule umane pan-citocheratina+ sono in giallo, le cellule CD3 + CD8 sono in verde, le cellule CD3 + CD8 + sono in blu e le cellule IDO + sono in arancione. (A) La distanza minima tra le cellule tumorali e ciascuna cellula T CD8 +. (B) Grafici a barre delle distanze tra le cellule IDO+ e ciascuna cellula T CD8+ (blu) o cellula T CD4+ (verde). (C) Esempio di un campione analizzato dalla funzione G-cross. L'asse y mostra la probabilità che una cellula tumorale incontri un linfocita CD3+ in un raggio compreso tra 0 e 200 μm attorno alla cellula tumorale. Vengono mostrate tre curve; la curva teorica è in verde punteggiato (distribuzione di Poisson), la curva empirica corretta con correzione km è in nero e la curva empirica corretta con correzione del bordo è in rosso punteggiato. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

Figura 7: Illustrazione di un Quadratcount. Il calcolo del bordo e il quadratcount sono stati eseguiti utilizzando il pacchetto spatstats. I quadrati più infiltrati (hotspot) possono essere utilizzati per le statistiche a valle. CD4 è in verde, CD8 è in rosso e le cellule tumorali sono in giallo. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}

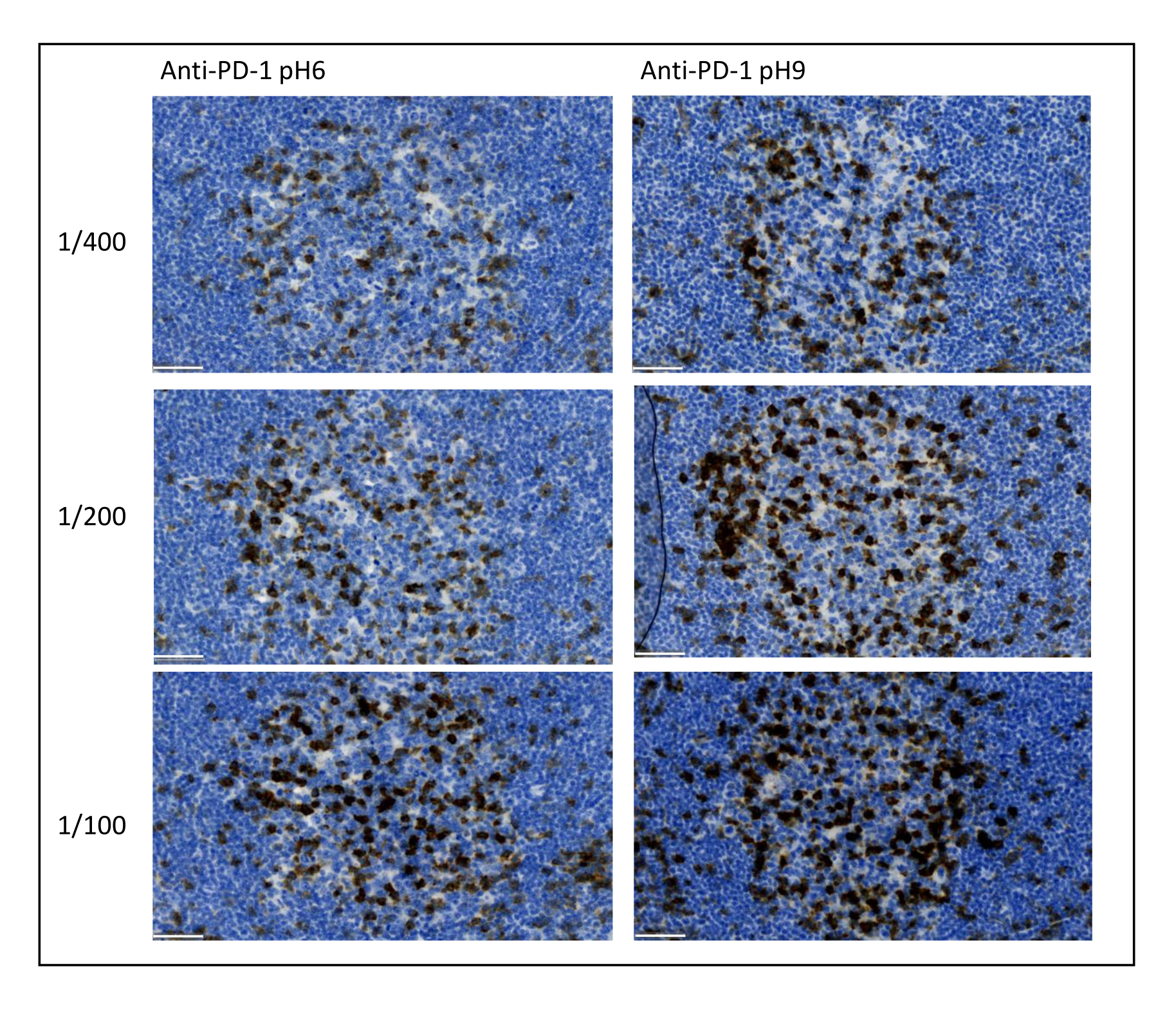
Figura 8: Diluizione degli anticorpi e ottimizzazione del recupero dell'antigene. Rilevazione cromogenica di PD-1 utilizzando tre diverse diluizioni e due diverse soluzioni di recupero dell'antigene dell'anticorpo primario (citrato pH 6 e EDTA pH 9). Barra scala: 50 μm. Fare clic qui per visualizzare una versione ingrandita di questa figura.
{kind=link}
| Anticorpo primario | Diluizione | Recupero dell'antigene | Anticorpo secondario | Fluorocromo | Posizione |
| PD-1 · | 1/100 | EDTA (pH 9) | Anti-coniglio | AF647 · | 1 |
| PD-L1 | 1/1000 | EDTA (pH 9) | Anti-coniglio | AF488 · | 2 |
| ROR-γ | 1/200 | EDTA (pH 9) | Anti-mouse | ATT0-425 | 3 |
| CD3 | 1/100 | Citrato (pH 6) | Anti-coniglio | AF555 | 4 |
| hPanCK | 1/50 | Citrato (pH 6) | Anti-mouse accoppiato con AF750 | 5 | |
Tabella 1: Esempio di pannello multiplex ottimizzato. Abbreviazioni: PD-1 = programmed cell death protein 1; PD-L1 = Programmed death-ligand 1; ROR-γ = recettore orfano gamma correlato a RAR; CD3 = cluster di differenziazione 3; hPanCK = pan-citocheratina umana; AF = AlexaFluor; EDTA = acido etilendiamminotetraacetico. CD3 è usato per rilevare i linfociti T; PD-1 è usato per rilevare i linfociti esausti; ROR- γ viene utilizzato per rilevare Th-17; e hPanCK viene utilizzato per rilevare le cellule tumorali. La colonna posizione indica l'ordine in cui deve essere eseguito il multiplex sequenziale.
Discussion
I parametri più importanti da prendere in considerazione per ottimizzare la colorazione multiplex sono la diluizione, la specificità e il recupero dell'antigene utilizzato per ciascun anticorpo primario. Prima di iniziare un protocollo multiplex, la diluizione ottimale di ciascun anticorpo primario e il recupero ottimale dell'epitopo (pH 6 o pH 9) devono essere testati utilizzando la colorazione cromogenica (DAB). Si consiglia di testare tre diluizioni per ciascun tampone di recupero dell'antigene: la diluizione che di solito è specificata dal marchio che commercializza l'anticorpo, la stessa diluizione divisa due volte e la stessa diluizione moltiplicata due volte (Figura 8). La scelta della giusta diluizione è un passo molto importante per verificare la specificità anticorpale e ottimizzare il rapporto segnale-rumore (SNR) della colorazione. Dopo aver scelto la giusta diluizione nel DAB, la stessa diluizione deve essere testata per ciascun anticorpo primario utilizzando uniplex TSA. Una volta selezionato il tampone di diluizione e di recupero dell'epitopo per ogni colorazione dell'antigene, è anche importante impostare correttamente la sequenza del multiplex; In particolare, alcuni antigeni sono meglio colorati nella prima posizione e altri nell'ultima posizione. Consigliamo di testare l'etichettatura multiplex utilizzando tutte le possibili permutazioni dell'ordine per scegliere quale colorazione dell'antigene dovrebbe venire prima, seconda, ecc. Questo è anche un passo molto importante perché alcuni antigeni fragili possono essere degradati dopo diversi cicli di recupero dell'epitopo e alcuni antigeni sono meglio colorati dopo diversi cicli di recupero dell'epitopo. Ad esempio, l'SNR è sempre più alto nell'ultima posizione per CD3 e nella prima posizione per la colorazione PD-1. Inoltre, la colorazione di diversi antigeni co-localizzati può essere ostacolata da un effetto ombrello (la saturazione dei siti reattivi della tiramide). Questo può essere attenuato diminuendo la concentrazione di tiramide. Quando l'espressione di un antigene è condizionata dall'espressione di un altro (CD8 presente solo sulle cellule T che esprimono CD3), si consiglia di colorare l'antigene con l'espressione più ampia (CD3 in questo caso) dopo l'altro. Infine, la scelta del fluorocromo giusto per ogni colorazione dell'antigene in base alle specificità dello scanner è anche un passo importante per evitare il rilevamento incrociato.
I maggiori vantaggi di questa tecnica sono l'amplificazione e il rapporto segnale-rumore ottenuto. Tuttavia, questa tecnica ha una limitazione, ovvero che la colorazione è sequenziale e i fluorocromi sono legati covalentemente al tessuto. Tuttavia, dopo aver eseguito tutti i cicli di amplificazione del segnale della tiramide, è anche possibile aggiungere un'ultima colorazione con un anticorpo secondario direttamente accoppiato con un fluorocromo (senza TSA). In alcuni pannelli, abbiamo usato questo metodo per aggiungere colorazione nel canale 750. Ciò era necessario perché nessun tyramide-AF750 era disponibile in commercio in quel momento. Da notare, il tempo di esposizione (durante la scansione) dell'antigene colorato con AF750 sarà molto più lungo rispetto agli altri antigeni colorati con TSA. In tal caso, si consiglia di colorare una proteina altamente espressa come la citocheratina o di aumentare la concentrazione dell'anticorpo primario. In questo modo, è possibile colorare un massimo di cinque o sei antigeni per vetrino in un lotto a seconda dello scanner a fluorescenza.
In opposizione, diverse tecniche disponibili in commercio utilizzano la colorazione seriale con diversi cicli di colorazione, scansione e stripping o fotosbiancamento per migliorare il numero di antigeni che possono essere colorati su una singola sezione di tessuto. Tuttavia, queste tecniche sono spesso dispendiose in termini di tempo, costose, non hanno amplificazione del segnale, richiedono passaggi computazionali avanzati per unire correttamente le scansioni seriali e, nella nostra esperienza, possono indurre danni irreversibili ai tessuti a causa delle numerose fasi della procedura. Tuttavia, è stato riportato che fino a 30 antigeni potrebbero essere colorati su un singolo tessuto utilizzando questo metodo14.
In conclusione, il nostro metodo è una tecnica di immunoistofluorescenza robusta, riproducibile, facile da usare ed economica che può essere utilizzata in qualsiasi laboratorio dotato di uno scanner a vetrini a fluorescenza. È possibile utilizzare qualsiasi anticorpo primario commercializzato adatto per IHC e i pannelli non sono specifici per nessun kit commerciale. L'analisi delle immagini può essere eseguita su diversi programmi, inclusi programmi open source come QuPath e R. Tuttavia, pensiamo che questo metodo potrebbe anche essere migliorato in futuro per pannelli di antigene di grandi dimensioni, consentendo di eseguire la colorazione / scansione seriale dello stesso vetrino con diversi pannelli di antigeni e con il vantaggio dell'amplificazione del segnale.
Disclosures
Gli autori non hanno conflitti di interesse da dichiarare.
Acknowledgements
Gli autori desiderano ringraziare la dottoressa Derouane F per il suo aiuto e supporto. Nicolas Huyghe è un ricercatore sostenuto da una sovvenzione del Fondo nazionale belga per la ricerca scientifica (Télévie/FNRS 7460918F).
Materials
| Name | Company | Catalog Number | Comments |
| anti-CD3 primary antibody | Abcam | ab16669 | rabbit monocolonal |
| anti-CD8 primary antibody | DAKO | M710301 | mouse monoclonal |
| anti-hPanCK primary antibody | DAKO | M3515 | mouse monoclonal |
| anti-PD-1 primary antibody | Cell Signalling | D4W2J | rabbit monocolonal |
| anti-PD-L1 primary antibody | Cell Signalling | 13684 | rabbit monocolonal |
| anti-RORC primary antibody | Sigma | MABF81 | mouse monoclonal |
| ATTO-425 | ATTOtec | ||
| Axioscan Z1 | Zeiss | Light source: Colibri 7 (385, 430, 475, 555, 590, 630, 735 nm) Filtersets: Excitation 379/34 – beam splitter 409 – emission 440/40; Excitation 438/24 – beam splitter 458 – emission 483/32; Excitation 490/20 – beam splitter 505 – emission 525/20; Excitation 546/10 – beam splitter 556 – emission 572/23; Excitation 592/21 – beam splitter 610 – emission 630/30; Excitation 635/18 – beam splitter 652 – emission 680/42; Excitation 735/40 – beam splitter QBS 405 + 493 + 611 + 762 - emission QBP 425/30 + 524/51 + 634/38 + 785/38; Objective: Plan-Apochromat 20x/0.8; Camera : Orca Flash 4.0 V3 | |
| Borosilicate Cover Glass | VWR | 631-0146 | |
| Envision+ anti-mouse | DAKO | K4001 | |
| Envision+ anti-rabbit | DAKO | K4003 | |
| Fluorescence mounting medium | DAKO | S3023 | |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 750 | ThermoFischer | A-21037 | |
| HALO software | Indicalabs | ||
| Hoescht | Sigma | 14533 | |
| Superfrost plus microscope slides | Fisherscientific/Epredia | 10149870 | |
| Tyramide-AF488 | ThermoFischer | B40953 | |
| Tyramide-AF555 | ThermoFischer | B04955 | |
| Tyramide-AF647 | ThermoFischer | B04958 |
References
- Ge, P., et al. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomedicine & Pharmacotherapy. 118, 109228 (2019).
- Fridman, W. H. The immune microenvironment as a guide for cancer therapies. Oncoimmunology. 1 (3), 261-262 (2012).
- Fridman, W. H., Pages, F., Sautes-Fridman, C., Galon, J. The immune contexture in human tumours: Impact on clinical outcome. in Nature Reviews. Cancer. 12 (4), 298-306 (2012).
- Hanahan, D., Weinberg, R. A. Hallmarks of cancer: The next generation. Cell. 144 (5), 646-674 (2011).
- Calu, V., et al. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Scientific Reports. 11 (1), 7940 (2021).
- Havel, J. J., Chowell, D., Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature Reviews. Cancer. 19 (3), 133-150 (2019).
- Pages, F., et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 391 (10135), 2128-2139 (2018).
- Mlecnik, B., et al. Integrative analyses of colorectal cancer show immunoscore is a stronger predictor of patient survival than microsatellite instability. Immunity. 44 (3), 698-711 (2016).
- Gu, Z., Eils, R., Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 32 (18), 2847-2849 (2016).
- Baddeley, A., Rubak, E., Turner, R. . Spatial Point Patterns: Methodology and Applications with R. , (2022).
- Barua, S., et al. Spatial interaction of tumor cells and regulatory T cells correlates with survival in non-small cell lung cancer. Lung Cancer. 117, 73-79 (2018).
- Parra, E. R. Methods to determine and analyze the cellular spatial distribution extracted from multiplex immunofluorescence data to understand the tumor microenvironment. Frontiers in Molecular Biosciences. 8, 668340 (2021).
- El Sissy, C., et al. A diagnostic biopsy-adapted immunoscore predicts response to neoadjuvant treatment and selects patients with rectal cancer eligible for a watch-and-wait strategy. Clinical Cancer Research. 26 (19), 5198-5207 (2020).
- Bolognesi, M. M., et al. Multiplex staining by sequential immunostaining and antibody removal on routine tissue sections. The Journal of Histochemistry and Cytochemistry. 65 (8), 431-444 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved