Sequenciamento do Genoma Completo para Caracterização Rápida do Vírus Rábico Utilizando Tecnologia de Nanoporos
In This Article
Summary
Aqui, apresentamos um fluxo de trabalho rápido e econômico para caracterizar genomas do vírus da raiva (RABV) usando a tecnologia de nanoporos. O fluxo de trabalho destina-se a apoiar a vigilância genômica informada em nível local, fornecendo informações sobre linhagens RABV circulantes e sua colocação dentro de filogenias regionais para orientar medidas de controle da raiva.
Abstract
Os dados genômicos podem ser usados para rastrear a transmissão e a disseminação geográfica de doenças infecciosas. No entanto, a capacidade de sequenciamento necessária para a vigilância genômica permanece limitada em muitos países de baixa e média renda (PBMRs), onde a raiva mediada por cães e/ou a raiva transmitida por animais selvagens, como morcegos hematófagos, representam grandes preocupações econômicas e de saúde pública. Apresentamos aqui um fluxo de trabalho rápido e acessível de amostra para sequência para interpretação usando a tecnologia de nanoporos. Os protocolos para coleta de amostras e diagnóstico de raiva são brevemente descritos, seguidos por detalhes do fluxo de trabalho otimizado de sequenciamento do genoma inteiro, incluindo o projeto e a otimização de primers para reação em cadeia da polimerase (PCR) multiplex, preparação de uma biblioteca de sequenciamento modificada e de baixo custo, sequenciamento com chamada de base ao vivo e off-line, designação de linhagem genética e análise filogenética. A implementação do fluxo de trabalho é demonstrada, e etapas críticas são destacadas para a implantação local, como validação de pipeline, otimização de primer, inclusão de controles negativos e o uso de dados publicamente disponíveis e ferramentas genômicas (GLUE, MADDOG) para classificação e colocação dentro de filogenias regionais e globais. O tempo de resposta para o fluxo de trabalho é de 2 a 3 dias, e o custo varia de US$ 25 por amostra para uma execução de 96 amostras a US$ 80 por amostra para uma execução de 12 amostras. Concluímos que a criação de vigilância genômica do vírus da raiva em PBMRs é viável e pode apoiar o progresso em direção à meta global de zero mortes por raiva humana mediadas por cães até 2030, bem como um monitoramento aprimorado da disseminação da raiva na vida selvagem. Além disso, a plataforma pode ser adaptada para outros patógenos, ajudando a construir uma capacidade genômica versátil que contribui para a preparação para epidemias e pandemias.
Introduction
O vírus da raiva (RABV) é um lissavírus da família Rhabdoviridae que causa uma doença neurológica fatal em mamíferos1. Embora a raiva seja 100% evitável pela vacinação, continua sendo um grande problema de saúde pública e econômico em países endêmicos. Das 60.000 mortes por raiva humana estimadas para ocorrer a cada ano, mais de 95% estão na África e na Ásia, onde os cães são o principal reservatório2. Em contraste, a vacinação de cães levou à eliminação da raiva mediada por cães em toda a Europa Ocidental, América do Norte e grande parte da América Latina. Nessas regiões, os reservatórios de raiva estão agora restritos à fauna silvestre, como morcegos, guaxinins, gambás e canídeos selvagens3. Em toda a América Latina, o morcego hematófago comum é uma fonte problemática de raiva devido à transmissão regular de morcegos para humanos e animais durante a alimentação sanguínea noturna4. O impacto econômico global anual da raiva é estimado em US$ 8,6 bilhões, sendo as perdas de gado responsáveis por 6%5.
Dados de sequência de patógenos virais combinados com metadados sobre o momento e a fonte das infecções podem fornecer insights epidemiológicos robustos6. Para o RABV, o sequenciamento tem sido utilizado para investigar a origem dos surtos7,8, identificar associações do hospedeiro com animais selvagens ou cães domésticos 8,9,10,11,12 e rastrear fontes de casos humanos 13,14. Investigações de surtos usando análise filogenética indicaram que a raiva surgiu na província anteriormente livre de raiva de Bali, na Indonésia, através de uma única introdução nas áreas endêmicas próximas de Kalimantan ou Sulawesi15. Enquanto isso, nas Filipinas, um surto na ilha de Tablas, província de Romblon, foi comprovadamente introduzido a partir da ilha principal de Luzon16. Dados genômicos virais também têm sido usados para entender melhor a dinâmica de transmissão de patógenos necessária para direcionar geograficamente as medidas de controle. Por exemplo, a caracterização genômica do RABV ilustra o agrupamento geográfico dos clados 17,18,19, a cocirculação de linhagens 20,21,22, o movimento viral mediado por humanos 17,23,24 e a dinâmica metapopulacional 25,26.
O monitoramento da doença é uma função importante da vigilância genômica que foi aprimorada com o aumento global da capacidade de sequenciamento em resposta à pandemia de SARS-CoV-2. A vigilância genômica apoiou o rastreamento em tempo real das variantes de preocupação do SARS-COV-227,28 e contramedidas associadas6. Os avanços na tecnologia de sequenciamento acessível, como a tecnologia de nanoporos, levaram a protocolos aprimorados e mais acessíveis para o sequenciamento rápido de patógenos humanos 29,30,31,32 e animais33,34,35. No entanto, em muitos países endêmicos de raiva, ainda há barreiras para operacionalizar a vigilância genômica de patógenos, como mostrado pelas disparidades globais na capacidade de sequenciamento de SARS-CoV-236. Limitações na infraestrutura laboratorial, cadeias de suprimentos e conhecimento técnico tornam o estabelecimento e a rotinização da vigilância genômica desafiadores. Neste artigo, demonstramos como um fluxo de trabalho otimizado, rápido e acessível de sequenciamento de genoma inteiro pode ser implantado para vigilância RABV em configurações com recursos limitados.
Protocol
O estudo foi aprovado pelo Comitê Coordenador de Pesquisa Médica do National Institute for Medical Research (NIMR/HQ/R.8a/vol. IX/2788), o Ministério da Administração Regional e Governo Local (AB.81/288/01) e o Conselho de Revisão Institucional do Instituto de Saúde Ifakara (IHI/IRB/No:22-2014) na Tanzânia; o Instituto de Doenças Tropicais e Infecciosas da Universidade de Nairóbi (P947/11/2019) e o Instituto de Pesquisa Médica do Quênia (KEMRI-SERU; protocolo nº 3268) no Quênia; e o Instituto de Pesquisa em Medicina Tropical (RITM), Departamento de Saúde (2019-023) nas Filipinas. O sequenciamento de amostras originárias da Nigéria foi realizado em material diagnóstico arquivado coletado como parte da vigilância nacional.
NOTA: Seção 1-4 são pré-requisitos. As seções 5-16 descrevem o fluxo de trabalho amostra-seqüência-para-interpretação para o sequenciamento de nanoporos RABV (Figura 1). Para etapas subsequentes do protocolo que necessitem de centrifugação por pulso, centrifugar a 10-15.000 x g por 5-15 s.
1. Configuração de ambiente computacional para sequenciamento e análise de dados
- Abra o site37 da Oxford Nanopore Technology (ONT) e crie uma conta para acessar recursos específicos de nanoporos.
- Faça login e instale o software de sequenciamento ONT e basecalling38.
- Abra o GitHub39 e crie uma conta.
- Vá para os repositórios artic-rabv40 e MADDOG41 e siga as instruções de instalação.
2. Projetar ou atualizar o esquema de primer multiplex
NOTA: Os esquemas RABV existentes estão disponíveis no repositório artic-rabv40. Ao visar uma nova área geográfica, deve ser concebido um novo regime ou modificado um regime existente para incorporar uma diversidade adicional.
- Escolher um conjunto de referência genômica para representar a diversidade na área de estudo; este é tipicamente um conjunto de sequências disponíveis publicamente (por exemplo, do NCBI GenBank) ou dados internos preliminares. Siga a etapa 2.1.1 para usar o RABV-GLUE42, um recurso de dados de sequência RABV, para filtrar e baixar sequências NCBI e metadados associados.
NOTA: Escolha sequências de referência com genomas completos (ou seja, sem lacunas e bases mascaradas). Recomenda-se escolher até 10 sequências como um conjunto de referência para o design do primer. Se os dados da sequência disponível estiverem incompletos ou não forem representativos da área de estudo, consulte o parecer43,44,45 do Arquivo Suplementar 1.- Navegue até a página NCBI RABV Sequences by Clade no menu suspenso Sequence Data em RABV-GLUE. Clique no link Vírus da raiva (RABV) para acessar todos os dados disponíveis ou selecionar um clado específico de interesse. Use a opção de filtro para Adicionar filtros que se encaixem nos critérios desejados (por exemplo, país de origem, comprimento da sequência). Baixe sequências e metadados.
- Gere um esquema de primers para reação em cadeia da polimerase (PCR) multiplex seguindo as instruções fornecidas pelo Primal Scheme46. Um esquema de 400 pb com sobreposição de 50 pb é recomendado para sequenciar amostras de baixa qualidade. Baixe e salve todas as saídas (não edite os nomes dos arquivos ou cartilhas).
NOTA: O esquema será indexado à primeira sequência no fasta de entrada, doravante denominado 'referência do índice' (Figura 2). Consulte Arquivo Suplementar 1 para obter opções para otimizar o desempenho do primer.
3. Configurar pipelines de bioinformática RAMPART e ARTIC
- Consulte Arquivo Suplementar 2 para configurar uma estrutura de diretórios para gerenciar os arquivos de entrada/saída para RAMPART e o pipeline de bioinformática ARTIC.
4. Biossegurança e configuração laboratorial
- Manipular amostras potencialmente positivas para raiva em condições de nível de biossegurança (BSL) 2 ou 3.
- Garantir que a equipe do laboratório tenha completado a vacinação pré-exposição à raiva e seja submetida ao monitoramento da imunidade de acordo com as recomendações da Organização Mundial da Saúde (OMS)3.
- Garantir que procedimentos operacionais padrão dedicados e avaliações de risco, seguindo diretrizes nacionais ou internacionais, estejam em vigor para o laboratório.
- Configuração de laboratório necessária: Minimize a contaminação mantendo a separação física entre as áreas pré e pós-PCR. Em laboratórios com espaço limitado ou em ambientes de laboratório em campo, use porta-luvas portáteis ou estações de laboratório improvisadas para minimizar a contaminação.
- Neste protocolo, assegure-se de designar áreas separadas para:
- Extração da amostra: Configure um gabinete/porta-luvas BSL2/3 para manusear material biológico e realizar a inativação e extração de RNA.
- Área do modelo: Configure um gabinete/porta-luvas BSL1 para a adição do molde (RNA/cDNA) à mistura mestre de reação pré-preparada.
- Área de mistura mestre: Configure uma área limpa designada (gabinete BSL1 / porta-luvas) para a preparação de misturas mestras de reagentes. Não deve haver nenhum modelo nesta área.
- Área pós-PCR: Estabeleça uma área separada para trabalhar em amplicons e sequenciar a preparação da biblioteca.
NOTA: Todas as áreas devem ser limpas com um descontaminante de superfície e esterilizadas por ultravioleta (UV) antes e após o uso.
5. Coleta e diagnóstico da amostra de campo
OBS: As amostras devem ser coletadas por pessoal treinado e imunizado, utilizando equipamentos de proteção individual e seguindo os procedimentos padrão referenciados47,48,49.
- Coletar a amostra através do forame magno (isto é, a via occipital), conforme descrito em detalhes em Mauti et al.50.
- Diagnosticar a raiva em campo com testes rápidos e confirmar laboratorialmente utilizando procedimentosrecomendados47, como a reação de imunofluorescência direta (IFD), o teste imunoistoquímico rápido direto (DRIT)51,52 ou transcrição reversa (RT)-PCR em temporeal53.
- Use amostras de cérebro positivas confirmadas para extração de RNA ou armazene em um freezer a -20 °C por 2-3 meses ou -80 °C por períodos mais longos. Preservar o RNA para armazenamento e transporte usando um meio de estabilização de DNA/RNA adequado.
6. Preparação da amostra e extração de RNA (3 h)
NOTA: Use um kit de extração de RNA viral baseado em coluna de spin adequado para o tipo de amostra.
- Prepare dois tubos de contas de cerâmica preenchendo um tubo de PCR de 2 mL com um tubo de aproximadamente 200 μL cheio de contas cerâmicas de 1,4 mm e rotule o tubo.
- Adicione o volume recomendado de tampão de lise fornecido no kit de extração de RNA ao tubo de PCR marcado.
- Obter aproximadamente um cubo de 3 mm da amostra de cérebro confirmada com infecção por raiva usando um aplicador de madeira e colocar em um tubo marcado com ID da amostra e 100 μL de água livre de nuclease no tubo rotulado como controle negativo.
NOTA: Use homogeneização à base de esferas de tubo fechado para limitar a exposição da amostra. Se não for possível, use outros disruptores mecânicos adequados (por exemplo, à base de rotor) ou um micropilão manual. No entanto, estes podem ser menos eficazes do que bater contas em superfície dura para interromper o tecido (amostras de tecido podem endurecer em certos meios de armazenamento). - Interrompa o tecido cerebral manualmente usando um bastão aplicador de madeira e, em seguida, vórtice na velocidade máxima até que a homogeneização completa do tecido seja alcançada.
- Centrifugar o lisado de acordo com as instruções do fabricante e usar uma pipeta para transferir o sobrenadante para um novo tubo de microcentrífuga marcado. Use este sobrenadante somente nas etapas subsequentes.
- Siga as instruções da coluna de spin do kit de extração de RNA para obter o RNA purificado.
- Inclua um controle de extração negativo (NEC) aqui e siga até o estágio de sequenciamento.
7. Preparação de cDNA (20 min)
- Na área de master mix, preparar uma master mix para síntese de cDNA de primeira fita de acordo com o número de amostras e controles a serem processados (com um volume excedente de 10% para garantir o reagente adequado; Tabela 1). Um controle sem modelo (NTC) deve ser incluído neste estágio.
- Rotular 0,2 mL de tubos de tiras de PCR e alíquota de 5 μL da mistura mestre nos tubos.
- Leve os tubos preparados para a área do modelo. Adicionar 5 μL de RNA em cada tubo marcado, incluindo o NEC. Adicionar 5 μL de água livre de nucleases (NFW) ao NTC.
- Incubar em termociclador seguindo as condições mencionadas na Tabela 1.
NOTA: Ponto de pausa opcional: o cDNA pode ser armazenado a -20 °C por até 1 mês, se necessário, mas é preferível proceder ao PCR.
8. Preparação do estoque da piscina da cartilha (1 h)
NOTA: Esta etapa só é necessária se forem feitas novas existências a partir de primers individuais, após as quais podem ser utilizadas soluções de existências pré-preparadas.
- Prepare um pool de primers de 100 μM na área master mix.
- Ressuspender os primers liofilizados em tampão 1x tris-EDTA (TE) ou NFW na concentração de 100 μM cada. Vórtice completamente e gire para baixo.
NOTA: Nas etapas a seguir, os primers individuais são separados em dois pools de primers - numerados ímpares (chamados Pool A) e pares numerados (chamados Pool B) - para evitar interações entre primers flanqueando sobreposições de amplicon. Esses pools de primers geram amplicons sobrepostos de 400 pb abrangendo o genoma alvo. - Organize todos os primers ímpares em um rack de tubo. Gere um estoque de pool de primers adicionando 5 μL de cada primer a um tubo de microcentrífuga de 1,5 mL rotulado como "nome do esquema de primers - Pool A (100 μM)".
- Repita o processo para todos os primers de número par e rotule como "nome do esquema de primers - Pool B (100 μM)".
- Diluir cada pool de primers 1:10 em água de grau molecular para gerar estoques de primers de 10 μM.
NOTA: Fazer alíquotas múltiplas de diluições de primers de 10 μM e congelá-las em caso de degradação ou contaminação.
9. PCR multiplex (5 h)
- Prepare duas misturas mestre de PCR, uma para cada pool de primers na área de mistura mestre.
- Utilizar uma concentração final de 0,015 μM por primer. Calcular o volume necessário do pool de primers para a reação de PCR (Tabela 2) usando a seguinte fórmula:
Volume do pool de primers = número de primers x volume de reação x 0,015/concentração (μM) do estoque de primers
- Utilizar uma concentração final de 0,015 μM por primer. Calcular o volume necessário do pool de primers para a reação de PCR (Tabela 2) usando a seguinte fórmula:
- Alíquota 10 μL cada de mistura mestre Pool A e mistura master Pool B para tubos de tira PCR rotulados na área do modelo. Para cada amostra, adicionar 2,5 μL de cDNA (a partir da etapa 3) a cada uma das reações correspondentes do primer marcado Pool A e B. O excesso de cDNA pode ser armazenado a -20 °C.
- Misture agitando suavemente e centrifugando por pulso.
- Incubar as amostras com as condições mencionadas na Tabela 2 em uma máquina de PCR.
NOTA: O programa não inclui uma etapa de extensão específica devido ao longo tempo de recozimento de 5 min (necessário devido ao alto número de primers) e ao curto comprimento dos amplicons (400 pb) que é suficiente para a extensão.
10. Limpeza e quantificação por PCR (3,5 h)
- Realizar todo o trabalho a partir deste ponto na área pós-PCR.
- Alíquotas de imobilização reversível em fase sólida (SPRI) em tubos de microcentrífuga do frasco principal. Conservar a 4 °C.
- Aqueça uma alíquota de contas SPRI à temperatura ambiente (RT; ~20 °C) e despeje completamente até que as contas sejam totalmente ressuspensas na solução.
- Em tubos de 1,5 mL, combinar os produtos de PCR do primer Pool A e do primer Pool B para cada amostra. Se necessário, adicione água para elevar o volume para 25 μL.
- Adicionar 25 μL de contas SPRI a cada amostra (proporção 1:1 talão:amostra). Misture pipetando para cima e para baixo ou batendo suavemente no tubo.
- Incubar em TR por 10 min, ocasionalmente invertendo ou agitando os tubos.
- Coloque em um rack magnético até que as contas e a solução estejam totalmente separadas. Retire e descarte o sobrenadante, tomando cuidado para não atrapalhar o talão.
- Lavar duas vezes com etanol 80% (aquecido a RT).
- Adicionar 200 μL de etanol ao pellet. Aguarde 30 s para garantir que as contas sejam lavadas corretamente.
- Retire e descarte cuidadosamente o sobrenadante, tentando não tocar no talão de pellet.
- Repita as etapas 10.8.1-10.8.2 para lavar o pellet uma segunda vez.
- Remova todos os vestígios de etanol usando uma ponta de 10 μL. Secar ao ar até que o etanol tenha evaporado (com pequenas contas isso acontece rapidamente, ~30 s); Quando isso acontece, a pelota deve passar de brilhante para fosca. Tome cuidado para não secar demais (se o pellet estiver rachando, está muito seco), pois isso afetará a recuperação do DNA.
- Ressuspender as esferas em 15 μL de NFW e incubar em RT (off magnetic rack) por 10 min.
- Retorne ao rack magnético e transfira o sobrenadante (produto limpo) para um tubo fresco de 1,5 mL.
- Preparar uma diluição de 1:10 de cada amostra num tubo separado (2 μL do produto + 18 μL de NFW).
NOTA: Tenha muito cuidado nesta fase para evitar a contaminação cruzada. Tenha apenas um tubo de amplicon aberto por vez. Alíquota 18 μL de água nos tubos primeiro (em uma área de mistura mestre limpa). - Medir a concentração de DNA de cada amostra diluída usando um fluorômetro altamente sensível e específico, conforme descrito na protocols.io54,55.
11. Normalização (30 min)
- Use o molde de normalização (Arquivo Suplementar 3) e a concentração de DNA (ng/μL) de cada amostra para calcular o volume de amostra diluída (ou pura) necessária para 200 fmol de cada amostra em um volume total de 5 μL.
- Rotule novos tubos de PCR e adicione volumes computados de NFW e amostra para obter DNA normalizado.
- Utilizar o volume calculado para amostras não diluídas (puras) se forem necessários mais de 5 μL da amostra diluída para obter 200 fmol.
NOTA: Ponto de pausa opcional: Neste ponto, o produto PCR limpo pode ser armazenado a 4 °C por até 1 semana ou colocado a -20 °C para armazenamento de longo prazo, se necessário.
12. Preparação final e código de barras (1,5 h)
NOTA: As próximas etapas pressupõem o uso de reagentes específicos de kits de sequenciamento de ligadura e código de barras específicos de nanoporos (consulte a Tabela de Materiais para obter detalhes). O protocolo é transferível entre diferentes versões químicas, mas os usuários devem ter o cuidado de usar kits compatíveis, de acordo com as instruções do fabricante.
- Reparo final e dA-tailing
- Configure a reação de preparação final para cada amostra mencionada na Tabela 3. Prepare uma mistura mestre de acordo com o número de amostras (mais 10% de excesso). Tome cuidado ao pipetar, pois os reagentes são viscosos.
- Adicionar 5 μL de mistura mestre em cada tubo de ADN normalizado (5 μL). A mistura total de reação deve ser de 10 μL. Mude as pontas cada vez e tenha apenas um tubo aberto de cada vez.
- Incubar em termociclador nas condições mencionadas na Tabela 3.
- Codificação de barras
- Alíquota os códigos de barras do kit de código de barras para tubos de tira PCR a 1,25 μL/tubo e registre o código de barras atribuído a cada amostra.
- Adicione 0,75 μL da amostra preparada final à alíquota de código de barras atribuída.
- Preparar uma mistura mestre de ligadura de acordo com o número de amostras (mais 10% de excesso) (Tabela 4).
- Adicionar 8 μL de mistura mestra de ligadura à amostra preparada no final + códigos de barras, dando uma reação total de 10 μL.
- Incubar em um termociclador usando as condições mencionadas na Tabela 4.
- Limpeza de contas SPRI e quantificação de DNA
- Descongelar o tampão de fragmento curto (SFB) no RT, misturar por vórtice, centrífuga de pulso e colocar no gelo.
- Junte todas as amostras com código de barras em um tubo de microcentrífuga de 1,5 mL de lobido. Para não tornar o volume de limpeza muito grande para ser usado, agrupe de 12 a 24 amostras (10 μL/amostra), até 48 amostras (5 μL/amostra) ou até 96 amostras (2,5 μL/amostra) de cada reação nativa de código de barras.
- Adicione um volume de 0,4x de contas SPRI ao pool com código de barras. Misture suavemente (mexendo ou pipetando) e incube em RT por 5 min.
- Coloque as amostras em um ímã até que as contas tenham peletizado e o sobrenadante esteja completamente claro (~2 min). Retire e descarte o sobrenadante. Tome cuidado para não atrapalhar as contas.
- Lavar duas vezes com 250 μL de SFB.
- Retire o tubo do ímã e ressuspenda completamente o pellet em 250 μL de SFB. Incubar por 30 s, centrifugar o pulso e retornar ao ímã.
- Retire o sobrenadante e descarte.
- Repita a etapa 12.3.5 para realizar uma segunda lavagem SFB.
- Centrifugar o pulso e remover qualquer SFB residual.
- Adicionar 200 μL de etanol 80% (RT) para banhar o pellet. Retire e descarte o etanol, tomando cuidado para não atrapalhar o grânulo. Secar ao ar por 30 s ou até que o pellet perca o brilho.
- Ressuspender em 22 μL de NFW no TR por 10 min.
- Coloque no ímã, deixe descansar por ~2 min, depois remova cuidadosamente a solução e transfira para um tubo de microcentrífuga limpo de 1,5 mL.
- Utilizar 1 μL para obter a concentração de ADN, conforme descrito anteriormente (passo 10.13).
NOTA: Ponto de pausa opcional: Neste ponto, a biblioteca pode ser armazenada a 4 °C por até 1 semana ou -20 °C para armazenamento de longo prazo, mas é preferível continuar com a ligadura e o sequenciamento do adaptador.
13. Sequenciamento (máximo de 48 h)
- Prepare o computador (consulte também as seções de Pré-requisitos 1-4).
- Verifique se há espaço suficiente para armazenar novos dados (min 150 GB), os dados de execuções antigas são copiados/movidos para um servidor antes de serem excluídos e a versão mais recente do MinKNOW está instalada.
- Retire a célula de fluxo armazenada da geladeira e deixe que ela atinja o RT.
- Ligadura do adaptador (1 h)
- Centrifugar o adaptador, misturar e ligar e colocar no gelo.
- Descongelar tampão de eluição (EB), SFB e tampão de ligadura em RT. Misture por vórtice, centrífuga de pulso e coloque no gelo.
- Prepare a mistura mestre de ligadura do adaptador (Tabela 5), combinando reagentes na ordem especificada em um tubo de baixa ligação.
NOTA: Alternativas para reagentes de mistura mestre de ligadura do adaptador (Tabela 5) podem ser usadas dependendo da disponibilidade no laboratório. Consulte Arquivo Suplementar 3 e Tabela de Materiais para obter uma lista de alternativas. Use a computação na planilha Arquivo Suplementar 3 para obter o volume da biblioteca de DNA equivalente a 200 fmol. Se menos de 20 μL for calculado, adicione NFW para fazer até 20 μL. - Misture por agitação suave e centrífuga de pulso. Incubar em TR por 20 min.
NOTA: Durante a incubação, inicie a preparação da célula de fluxo (secção 13.5).
- Limpe usando contas SPRI (não use etanol como em limpezas anteriores).
- Adicione um volume de 0,4x de contas SPRI (RT) às amostras. Incubar em RT por 10 min, mexer suavemente intermitentemente para ajudar na mistura.
- Coloque sobre o ímã até que as contas e a solução estejam totalmente separadas (~5 min). Retirar e descartar o sobrenadante; Tome cuidado para não perturbar o grânulo.
- Lavar duas vezes com 125 μL de SFB.
- Ressuspenda completamente o pellet com 125 μL de SFB misturando com uma pipeta. Deixe incubar por 30 s.
- Centrífuga de pulso para coletar o líquido na base do tubo e colocar no ímã. Retire o sobrenadante e descarte.
- Repita os passos 13.4.4-13.4.5 para lavar o pellet uma segunda vez.
- Centrifugar o pulso e remover o excesso de SFB.
- Ressuspender em 15 μL de EB e incubar por 10 min no momento da TR.
- Retorne ao ímã por ~2 min e, em seguida, transfira cuidadosamente a solução para um tubo de microcentrífuga limpo de 1,5 mL.
- Quantificar 1 μL da biblioteca eluída, conforme descrito anteriormente no passo 10.13
NOTA: Para obter melhores resultados, prossiga diretamente para o sequenciamento MinION; no entanto, a biblioteca final pode ser armazenada em EB a 4 °C por até 1 semana, se necessário.
- Execute uma verificação de qualidade da célula de fluxo.
- Conecte o dispositivo de sequenciamento a um laptop e abra o software de sequenciamento.
- Selecione o tipo de célula de fluxo e clique em Verificar célula de fluxo e Iniciar teste.
- Uma vez concluído, o número total de poros ativos (ou seja, viáveis) será exibido. Uma nova célula de fluxo deve ter >800 poros ativos; Se isso não acontecer, entre em contato com o fabricante para uma substituição.
- Escorvamento e carregamento da célula de fluxo (20 min)
- Descongele os seguintes reagentes no RT e, em seguida, coloque o buffer de sequenciamento, um cabo de descarga, o buffer de descarga e as contas de carregamento no gelo.
- Vórtice o tampão de sequenciamento e o tampão de descarga, centrífuga de pulso e coloque no gelo.
- Centrifugar o pulso e misturar por pipetagem; em seguida, coloque no gelo.
- Preparar a mistura de priming de células de fluxo adicionando 30 μL de cabo de descarga diretamente ao tubo de tampão de descarga de um kit de escorvamento de célula de fluxo e misturar por pipetagem.
- Misture as contas de carregamento pipetando imediatamente antes de usar, pois elas se acomodam rapidamente.
- Em um tubo fresco, preparar a diluição final da biblioteca para sequenciamento, conforme mencionado na Tabela 5.
Observação : use computação na planilha de arquivo suplementar 3 para obter o volume de biblioteca de DNA equivalente a 50 fmol. Se menos de 12 μL for calculado, adicione EB para completar até 12 μL. - Inverta a tampa do dispositivo de sequenciamento e deslize a tampa da porta de escorvamento no sentido horário para que a porta de escorvamento fique visível (Figura 3)
- Remova as bolhas de ar cuidadosamente ajustando uma pipeta P1000 para 200 μL, insira a ponta na porta de escorvamento e gire a roda até que um pequeno volume que entra na ponta da pipeta seja visto (giro máximo para 230 μL).
- Coloque 800 μL de mistura de priming de célula de fluxo na célula de fluxo através da porta de escorvamento, tomando cuidado para evitar bolhas.
- Deixe agir por 5 min.
- Levante suavemente a tampa da porta de amostra e carregue 200 μL de mistura de escorvamento na célula de fluxo através da porta de escorvamento usando uma pipeta P1000.
- Pipetar a mistura da biblioteca para cima e para baixo antes do carregamento, garantindo que os grânulos de carregamento na mistura mestre sejam ressuspensos antes do carregamento.
- Carregue 75 μL de mistura de bibliotecas na célula de fluxo através da porta de amostra de forma gota a gota. Certifique-se de que cada gotejamento flua para a porta antes de adicionar o próximo.
- Substitua a tampa da porta de amostra suavemente, certificando-se de que o bung entre na porta de amostra.
- Feche a porta de escorvamento e substitua a tampa do dispositivo de sequenciamento.
- Execução sequencial (máximo de 48 h)
- Conecte o dispositivo de sequenciamento ao laptop e abra o software de sequenciamento.
- Clique em Iniciar e, em seguida, clique em Iniciar Sequenciamento.
- Clique em Novo experimento e siga o fluxo de trabalho da interface gráfica do usuário (GUI) do software de sequenciamento para configurar os parâmetros para a execução.
- Digite o nome do experimento e o ID do exemplo (por exemplo, rabv_run1) e escolha o Tipo de célula de fluxo no menu suspenso.
- Continue para a seleção do kit e escolha o kit de sequenciamento de ligadura relevante e o(s) kit(s) de código de barras nativo usado(s).
- Continue para Opções de execução . Mantenha os padrões, a menos que seja desejado que a execução pare automaticamente após um certo número de horas (as execuções podem ser interrompidas manualmente a qualquer momento).
- Continue para Basecalling. Escolha ativar ou desativar Basecalling de acordo com os recursos de computação (consulte a configuração do computador). Escolha Editar Opções em código de barras e verifique se o Código de Barras de Ambas as Extremidades está ativado. Salve e continue na seção de saída.
- Aceite os padrões e continue para a revisão final, verifique as configurações e registre os detalhes na planilha (Arquivo Suplementar 3). Clique em Iniciar.
Observação : se a célula de fluxo está sendo reutilizada, ajuste a tensão inicial (na seção avançada das opções de execução), conforme indicado pelo esquema no arquivo suplementar 3. - Registre os canais ativos iniciais - se isso for significativamente menor do que a verificação de controle de qualidade (QC), reinicie o software de sequenciamento. Se ainda estiver mais baixo, reinicie o computador.
- Registre os canais iniciais em fio versus poro único para dar uma ocupação aproximada dos poros. Esse número vai flutuar, então dê uma aproximação.
- Monitore a execução à medida que ela progride.
14. Basecalling ao vivo e off-line
Observação : essas instruções pressupõem que a estrutura de diretório pré-existente fornecida no repositório artic-rabv e que as seções de pré-requisitos 1 e 3 do protocolo foram seguidas.
- No sistema de arquivos local, crie um novo diretório chamado análise, onde você armazenará todas as saídas de análise. Para organizar mais: crie um subdiretório com o nome do seu projeto e dentro dele um novo diretório para a execução, usando o ID de exemplo fornecido ao MinKNOW como run_name. Faça isso em um comando da seguinte maneira:
Mkdir -p
análise/project_name/run_name
Em seguida, navegue até sua localização:
CD
caminho/análise/project_name/run_name - Basecalling ao vivo
NOTA: Para executar nanopore basecalling em tempo real, os laptops exigem uma unidade de processamento gráfico (GPU) compatível com NVIDIA CUDA. Verifique se as instruções para a configuração de basecalling da GPU foram executadas usando o protocolo guppy56.- Durante a instalação de execução, ative a base ao vivo.
- Use RAMPART para monitorar a cobertura de sequenciamento em tempo real, de acordo com as instruções abaixo.
- No terminal do computador, ative o ambiente artic-rabv conda:
Conda ativar artic-rabv - Crie um novo diretório para a saída rampart dentro do diretório run_name e navegue até ele:
cd /caminho/análise/project_name/run_name
mkdir rampart_output
rampart_output de cd - Crie um arquivo de código de barras.csv para emparelhar os códigos de barras e os nomes de exemplo. Ele deve ter uma linha por código de barras e especificar apenas os códigos de barras que estão presentes na biblioteca, com os títulos "código de barras" e "amostra". Siga o exemplo no diretório artic-rabv:
análise/example_project/example_run/rampart_output/códigos de barras.csv - Inicie o RAMPART fornecendo a pasta de protocolo relevante e o caminho para a pasta fastq_pass na saída MinKNOW para a execução:
rampart --protocol /path/rampart/scheme_name_V1_protocol - basecalledPath - Abra uma janela do navegador e navegue até localhost:3000 na caixa URL. Aguarde até que dados suficientes sejam basecall antes que os resultados apareçam na tela.
- Basecalling offline (executada após a execução)
- Se a basecalling ao vivo não foi definida, a saída do MinKNOW será dados de sinal brutos (arquivos fast5). Não será possível usar o RAMPART durante a execução. Converta os arquivos fast5 em dados basecalled (arquivos fastq) pós-execução usando guppy (consulte a configuração na etapa de pré-requisitos 1.1.1.). Execute RAMPART post-hoc nos dados basecalled.
- Execute o basecaller guppy:
guppy_basecaller -c dna_r9.4.1_450bps_fast.cfg -i /path/to/reads/fast5_* -s /path/analysis/project_name/run_name -x auto -r
-c é o arquivo de configuração para especificar o modelo basecalling, -i é o caminho de entrada, -s é o caminho de salvamento, -x especifica basecalling pelo dispositivo GPU (excluir se estiver usando a versão de computador do guppy) e -r especifica pesquisar arquivos de entrada recursivamente.
Observação : O arquivo de configuração (.cfg) pode ser alterado para um basecaller de alta precisão substituindo _fast por _hac, embora isso levará significativamente mais tempo.
15. Lavagem das células de fluxo
- As células de fluxo podem ser lavadas e reutilizadas para sequenciar novas bibliotecas se os poros ainda forem viáveis. Veja as instruções de lavagem no protocolo de lavagem de células de fluxoONT 57.
16. Análise e interpretação
- Geração de sequência de consenso com pipeline de bioinformática ARTIC
- Siga as instruções detalhadas no repositório artic-rabv GitHub40 na pasta rabv_protocols para gerar sequências de consenso a partir de arquivos fastq raw5 ou basecalled.
NOTA: Consulte Artic pipeline - Core pipeline58 para obter mais orientações.
- Siga as instruções detalhadas no repositório artic-rabv GitHub40 na pasta rabv_protocols para gerar sequências de consenso a partir de arquivos fastq raw5 ou basecalled.
- Opcional: Analise a profundidade média de leitura por amplicon.
- Adaptar os scripts disponíveis no repositório artic-rabv, referentes ao Arquivo Suplementar 1. Resumidamente, estatísticas detalhadas são geradas usando SAMtools59 e cobertura por nucleotídeo plotado em R.
- Análise filogenética utilizando GLUE
- A partir da RABV_GLUE42, selecione Análise > guia Genotipagem e Interpretação > Adicionar arquivos, selecionando o arquivo fasta de sequências de consenso.
- Clique em Enviar e aguarde. Quando as análises estiverem concluídas, o botão Mostrar Análise estará disponível para clicar, mostrando atribuições de clados e subclados, cobertura por gene, variação de sequências de referência e parente mais próximo.
- Sequências contextuais relevantes também podem ser identificadas na seção Dados de Sequência > Sequências NCBI por Clado .
- Selecione o clado identificado ou clique em Vírus da raiva (RABV) para ver todas as sequências disponíveis.
- Filtro para sequências relevantes (por exemplo, país de origem).
- Faça o download dessas sequências e metadados correspondentes para análise e comparação.
- Atribuição de linhagem usando MADDOG41
- Puxe o repositório MADDOG do GitHub para garantir que você esteja trabalhando com a versão mais atualizada.
- Crie uma pasta de atribuição dentro do repositório MADDOG local (criado anteriormente na seção Pré-requisitos) chamada nome de execução.
- Dentro da pasta, adicione o arquivo fasta contendo as sequências de consenso.
- Adicione um arquivo de metadados à pasta.
Observação : esse arquivo deve ser um csv com 4 colunas chamadas 'ID', 'país', 'ano' e 'atribuição', detalhando os IDs de sequência, o país de amostragem e o ano de coleta da amostra, enquanto a coluna 'atribuição' deve estar em branco. - Na interface da linha de comando, ative o ambiente conda: conda activate MADDOG.
- Na interface da linha de comando, navegue até a pasta do repositório MADDOG.
- Inicialmente, realize a atribuição de linhagem em sequências para verificar quaisquer anormalidades potenciais e identificar se a execução da etapa de designação de linhagem mais longa seria apropriada. Para isso, digite isso na linha de comando: sh assignment.sh.
- Quando solicitado, digite Y para indicar que você puxou o repositório e está trabalhando com a versão mais atualizada do MADDOG.
- Quando solicitado, digite o nome da pasta dentro da pasta do repositório MADDOG que contém o arquivo fasta.
- Quando a atribuição de linhagem estiver concluída, verifique o arquivo de saída em sua pasta. Se a saída for conforme o esperado e houver várias sequências atribuídas à mesma linhagem, execute a designação de linhagem.
- Se estiver executando a designação de linhagem, exclua o arquivo de saída de atribuição recém-criado.
- No terminal, dentro da pasta do repositório MADDOG, execute o comando sh designation.sh.
- Quando solicitado, digite Y para indicar que você puxou o repositório e está trabalhando com a versão mais atualizada do MADDOG.
- Quando solicitado, digite o nome da pasta dentro da pasta do repositório MADDOG que contém o arquivo fasta e os metadados. Isso gera informações de linhagem sobre cada sequência, uma filogenia das novas e relevantes sequências anteriores (a partir de 16.3.6), informações hierárquicas sobre as linhagens e detalhes de linhagens potencialmente emergentes e áreas de subamostragem.
NOTA: Detalhes completos do protocolo, uso e saídas podem ser encontrados em Campbell et al.60. - Quando a análise inicial tiver sido concluída, se for solicitado a testar também linhagens emergentes e subamostradas, digite Y, se necessário. Caso contrário, digite N.
- Se solicitado a confirmar linhagens recém-encontradas, digite Y e siga as instruções no arquivo NEXT_STEPS.eml resultante. Caso contrário, digite N.
Representative Results
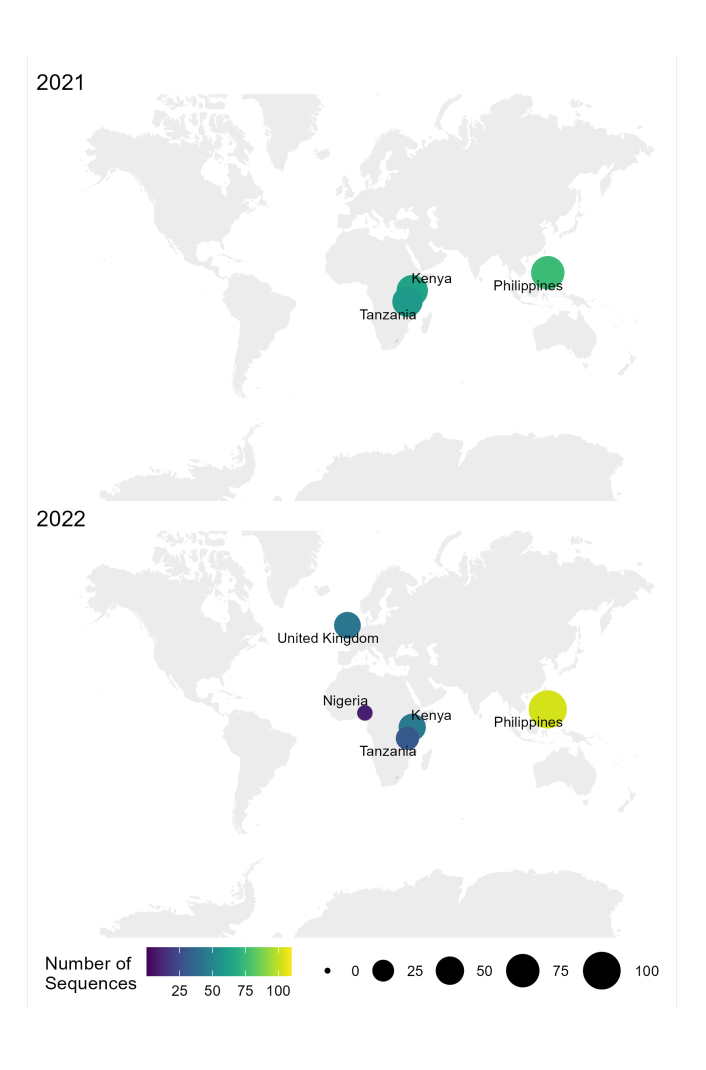
O fluxo de trabalho amostra-seqüência-interpretação para RABV descrito neste protocolo tem sido utilizado com sucesso em diferentes condições laboratoriais em países endêmicos, como Tanzânia, Quênia, Nigéria e Filipinas (Figura 4). O protocolo foi utilizado em diferentes tipos e condições de amostra (Tabela 6): tecido cerebral fresco e congelado, extratos de cDNA e RNA do tecido cerebral transportados sob cadeia de frio por longos períodos e cartões de FTA com esfregaços de tecido cerebral.
Basecalling ao vivo usando RAMPART (Figura 5) mostra a geração quase em tempo real de leituras e a porcentagem de cobertura por amostra. Isso é particularmente útil para decidir quando parar a execução e salvar a célula de fluxo para reutilização. Observou-se variação no tempo de execução, com alguns terminados em 2 h, enquanto outros poderiam levar mais de 12 h para atingir uma profundidade adequada de cobertura (x100). Também podemos ver regiões com pouca amplificação; por exemplo, a Figura 6 mostra um instantâneo de uma execução de sequenciamento onde os perfis de cobertura mostram alguns amplicons com amplificação muito baixa, indicando primers potencialmente problemáticos. Ao investigar mais detalhadamente essas regiões pouco amplificadoras, conseguimos identificar incompatibilidades de primers, o que nos permitirá redesenhar e melhorar os primers individuais. Alguns esquemas de primers mostraram mais incompatibilidades do que outros. Isso é observado no esquema de cartilhas da África Oriental, em comparação com as Filipinas, de acordo com a diversidade direcionada, já que o esquema da África Oriental visa capturar uma diversidade muito mais ampla.
RABV-GLUE42, um recurso de uso geral para o gerenciamento de dados do genoma de RABV, e MADDOG60, um sistema de classificação e nomenclatura de linhagens, foram usados para compilar e interpretar as sequências RABV resultantes. A Tabela 7 mostra os clados maiores e menores que circulam em cada país atribuído usando RABV-GLUE. Também é mostrada uma classificação de maior resolução de linhagens locais após a atribuição MADDOG.

Figura 1: Fluxo de trabalho de amostra para sequência para interpretação para RABV. São apresentadas etapas resumidas para (A) preparação da amostra, (B) PCR e preparação da biblioteca, e (C) sequenciamento e bioinformática até a análise e interpretação. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: Esquema de primers esquemático. Posições de recozimento ao longo do "genoma de referência índice" (roxo escuro) para pares de primers para frente e para trás (meias setas), que são atribuídos em dois pools separados: A (vermelho) e B (verde). Os pares de primers geram amplicons sobrepostos de 400 pb (azul) que são numerados sequencialmente ao longo do genoma de referência do índice no formato 'scheme_name_X_DIRECTION', onde 'X' é um número referente ao amplicon gerado pelo primer e 'DIRECTION' é 'LEFT' ou 'RIGHT', descrevendo o forward ou reverse respectivamente. Valores ímpares ou pares de 'X' determinam o pool (A ou B, respectivamente). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Célula de fluxo de nanoporos48. As etiquetas azuis ilustram as diferentes partes da célula de fluxo, incluindo a tampa da porta de escorvamento que cobre a porta de escorvamento onde a solução de escorvamento é adicionada, a tampa da porta de amostra SpotON que cobre a porta de amostra onde a amostra é adicionada de forma gota a gota, as portas de resíduos 1 e 2 e o ID da célula de fluxo. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Mapa mostrando o local onde o sequenciamento do RABV foi realizado usando o fluxo de trabalho otimizado em 2021 e 2022. O tamanho e a cor da bolha correspondem ao número de sequências por local, onde menor e mais escuro é menor, enquanto maior e mais claro é mais. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Captura de tela da visualização RAMPART no navegador da Web. Os nomes de código de barras são substituídos por nomes de amostra de acordo com a configuração de bioinformática. Os três painéis superiores mostram gráficos de resumo para toda a execução: profundidade de cobertura de leituras mapeadas para cada código de barras por posição de nucleotídeo no genoma de referência do índice (canto superior esquerdo, colorido por código de barras), leituras mapeadas somadas de todos os códigos de barras ao longo do tempo (meio superior) e leituras mapeadas por código de barras (canto superior direito, colorido por código de barras). Os painéis inferiores mostram linhas de gráficos por código de barras. Da esquerda para a direita: a profundidade de cobertura das leituras mapeadas por posição de nucleotídeos no genoma de referência índice (esquerda), distribuição de comprimento das leituras mapeadas (meio) e proporção de posições de nucleotídeos no genoma de referência índice que obtiveram uma cobertura de 10x, 100x e 1.000x de leituras mapeadas ao longo do tempo (direita). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Um exemplo de cobertura de leitura em todo o genoma de uma amostra do vírus da raiva das Filipinas sequenciada usando o protocolo. A cobertura de leitura em cada posição do nucleotídeo no genoma é mostrada, juntamente com a posição dos amplicons sobrepostos (1-41) usados para gerar a biblioteca. Picos na profundidade de cobertura correspondem a áreas de sobreposição de amplicon. Amplicons com baixa profundidade de cobertura correspondem a áreas de sobreposição de amplicon. Amplicons com baixa profundidade de cobertura são destacados em vermelho indicando regiões problemáticas que podem exigir otimização. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Tabela 1: Condições da mistura principal e do termociclador para a preparação de cDNA. Clique aqui para baixar esta tabela.
Tabela 2: Condições da mistura mestre e do termociclador para PCR multiplex. Clique aqui para baixar esta tabela.
Tabela 3: Condições da mistura principal e do termociclador para a reação de preparação final. Clique aqui para baixar esta tabela.
Tabela 4: Condições da mistura mestre e do termociclador para código de barras. Clique aqui para baixar esta tabela.
Tabela 5: Mistura mestre de ligadura do adaptador e diluição da biblioteca final para sequenciamento. Clique aqui para baixar esta tabela.
Tabela 6: O número de sequências do genoma completo do vírus da raiva geradas e o tipo de amostras usadas em diferentes países usando o fluxo de trabalho amostra-a-seqüência-para-interpretação. Clique aqui para baixar esta tabela.
Tabela 7: Atribuições de clados maiores e menores do RABV-GLUE e atribuições de linhagem do MADDOG para sequências geradas usando o fluxo de trabalho. Clique aqui para baixar esta tabela.
Arquivo Suplementar 1: Projeto e otimização do esquema de primers e análise de profundidade de leitura amplicon. Clique aqui para baixar este arquivo.
Arquivo Suplementar 2: Configuração computacional Clique aqui para baixar este arquivo.
Arquivo suplementar 3: Planilha de protocolo RABV WGS Clique aqui para baixar este arquivo.
Discussion
Um fluxo de trabalho acessível de sequenciamento do genoma completo baseado em nanoporos foi desenvolvido por Brunker et al.61, utilizando recursos da rede ARTIC46. Aqui, apresentamos um fluxo de trabalho atualizado, com etapas completas de amostra para sequência para interpretação. O fluxo de trabalho detalha a preparação de amostras de tecido cerebral para sequenciamento do genoma inteiro, apresenta um pipeline de bioinformática para processar leituras e gerar sequências de consenso e destaca duas ferramentas específicas da raiva para automatizar a atribuição de linhagens e determinar o contexto filogenético. O fluxo de trabalho atualizado também fornece instruções abrangentes para a configuração de espaços de trabalho computacionais e laboratoriais apropriados, com considerações para implementação em diferentes contextos (incluindo configurações com poucos recursos). Demonstramos a implementação bem-sucedida do fluxo de trabalho em ambientes acadêmicos e de institutos de pesquisa em quatro PBMRs endêmicos de RABV com nenhuma ou limitada capacidade de vigilância genômica. O fluxo de trabalho provou ser resiliente à aplicação em diversas configurações e compreensível por usuários com experiência variada.
Esse fluxo de trabalho para sequenciamento RABV é o protocolo mais abrangente disponível publicamente (cobrindo etapas de amostra para sequência para interpretação) e especificamente adaptado para reduzir os custos de inicialização e execução. O tempo e o custo necessários para a preparação e sequenciamento de bibliotecas com tecnologia de nanoporos são muito reduzidos em relação a outras plataformas, como o Illumina61, e os desenvolvimentos contínuos da tecnologia estão melhorando a qualidade e a precisão da sequência para serem comparáveis ao Illumina62.
Este protocolo foi projetado para ser resiliente em diversos contextos de poucos recursos. Ao consultar as diretrizes de solução de problemas e modificações fornecidas junto com o protocolo principal, os usuários têm suporte para adaptar o fluxo de trabalho às suas necessidades. A adição de ferramentas de bioinformática amigáveis ao fluxo de trabalho constitui um grande desenvolvimento em relação ao protocolo original, fornecendo métodos rápidos e padronizados que podem ser aplicados por usuários com experiência prévia mínima em bioinformática para interpretar dados de sequência em contextos locais. A capacidade de o fazer in situ é muitas vezes limitada pela necessidade de dispor de competências específicas de programação e filogenéticas, que exigem um investimento intensivo e a longo prazo na formação de competências. Embora esse conjunto de habilidades seja importante para interpretar completamente os dados de sequência, ferramentas de interpretação básicas e acessíveis são igualmente desejáveis para capacitar os "campeões de sequenciamento" locais, cuja experiência principal pode ser baseada em laboratório úmido, permitindo-lhes interpretar e se apropriar de seus dados.
Como o protocolo vem sendo realizado há vários anos em vários países, agora podemos fornecer orientações sobre como otimizar esquemas de primer multiplex para melhorar a cobertura e lidar com a diversidade acumulada. Esforços também têm sido feitos para ajudar os usuários a melhorar a relação custo-efetividade ou para permitir a facilidade de aquisição em uma determinada região, o que é tipicamente um desafio para a sustentabilidade das abordagens moleculares63. Por exemplo, na África (Tanzânia, Quênia e Nigéria), optamos pela mistura mestre de ligase romba/TA na etapa de ligadura do adaptador, que estava mais prontamente disponível em fornecedores locais e uma alternativa mais barata a outros reagentes de ligadura.
A partir da experiência, existem várias maneiras de reduzir o custo por amostra e por execução. Reduzir o número de amostras por corrida (por exemplo, de 24 para 12 amostras) pode estender a vida útil das células de fluxo ao longo de várias corridas, enquanto aumentar o número de amostras por execução maximiza o tempo e os reagentes. Em nossas mãos, conseguimos lavar e reutilizar células de fluxo para uma em cada três sequências, permitindo que mais 55 amostras fossem sequenciadas. Lavar a célula de fluxo imediatamente após o uso, ou se não for possível, remover o fluido residual do canal de resíduos após cada corrida, pareceu preservar o número de poros disponíveis para uma segunda corrida. Levando em consideração o número inicial de poros disponíveis em uma célula de fluxo, uma execução também pode ser otimizada para planejar quantas amostras serão executadas em uma célula de fluxo específica.
Embora o fluxo de trabalho tenha como objetivo ser o mais abrangente possível, com a adição de orientações detalhadas e recursos sinalizados, o procedimento ainda é complexo e pode ser assustador para um novo usuário. O usuário é incentivado a buscar treinamento e suporte presencial, idealmente localmente, ou alternativamente por meio de colaboradores externos. Nas Filipinas, por exemplo, um projeto sobre a capacitação dentro de laboratórios regionais para a vigilância genômica do SARS-CoV-2 usando ONT desenvolveu competências essenciais entre os diagnosticadores de cuidados de saúde que são prontamente transferíveis para o sequenciamento de RABV. Etapas importantes, como a limpeza de contas SPRI, podem ser difíceis de dominar sem treinamento prático, e uma limpeza ineficaz pode danificar a célula de fluxo e comprometer a corrida. A contaminação de amostras é sempre uma grande preocupação quando os amplicons estão sendo processados em laboratório e podem ser difíceis de eliminar. Em particular, a contaminação cruzada entre amostras é extremamente difícil de detectar durante a bioinformática pós-execução. Boas técnicas e práticas de laboratório, como manter superfícies de trabalho limpas, separar áreas pré e pós-PCR e incorporar controles negativos, são imperativas para garantir o controle de qualidade. O ritmo acelerado dos desenvolvimentos de sequenciamento de nanoporos é uma vantagem e uma desvantagem para a vigilância genômica de rotina do RABV. Melhorias contínuas na precisão, acessibilidade e repertório de protocolos dos nanoporos ampliam e melhoram o escopo de sua aplicação. No entanto, os mesmos desenvolvimentos tornam desafiador manter procedimentos operacionais padrão e pipelines de bioinformática. Neste protocolo, fornecemos um documento auxiliando a transição de kits de preparação de bibliotecas de nanoporos mais antigos para os atuais (Tabela de Materiais).
Um obstáculo comum ao sequenciamento em LMICs é a acessibilidade, incluindo não apenas o custo, mas também a capacidade de adquirir consumíveis em tempo hábil (em particular reagentes de sequenciamento, que são relativamente novos para equipes de compras e fornecedores) e recursos computacionais, além de simplesmente ter acesso a energia estável e à internet. Usar a tecnologia de sequenciamento de nanoporos portáteis como base desse fluxo de trabalho ajuda com muitos desses problemas de acessibilidade, e demonstramos o uso de nosso protocolo em uma variedade de configurações, conduzindo o protocolo completo e a análise no país. É certo que a aquisição de equipamento e a sequenciação de consumíveis em tempo útil continua a ser um desafio e, em muitos casos, fomos forçados a transportar ou enviar reagentes do Reino Unido. No entanto, em algumas áreas, pudemos confiar inteiramente em rotas de fornecimento locais para reagentes, beneficiando-nos do investimento no sequenciamento do SARS-CoV-2 (por exemplo, nas Filipinas), que simplificou os processos de aquisição e começou a normalizar a aplicação da genômica do patógeno.
A necessidade de uma conexão de internet estável é minimizada por instalações únicas; por exemplo, repositórios do GitHub, download de software e sequenciamento de nanoporos em si só exigem acesso à internet para iniciar a execução (não por completo) ou podem ser realizados completamente offline com o acordo da empresa. Se os dados móveis estiverem disponíveis, um telefone pode ser usado como um ponto de acesso para o laptop para iniciar a execução de sequenciamento, antes de se desconectar durante a execução. Ao processar amostras rotineiramente, os requisitos de armazenamento de dados podem crescer rapidamente e, idealmente, os dados seriam armazenados em um servidor. Caso contrário, os discos rígidos de unidade de estado sólido (SSD) são relativamente baratos de obter.
Embora reconheçamos que ainda existem barreiras à vigilância genômica em PBMRs, o aumento do investimento na construção de acessibilidade e conhecimento genômico (por exemplo, Africa Pathogen Genomics Initiative [Africa IGP])64 sugere que essa situação irá melhorar. A vigilância genômica é crítica para a preparação para pandemias6, e a capacidade pode ser estabelecida por meio do roteamento da vigilância genômica de patógenos endêmicos, como o RABV. As disparidades globais nas capacidades de sequenciamento destacadas durante a pandemia SARS-CoV-2 devem ser um motor de mudança catalítica para abordar essas desigualdades estruturais.
Este fluxo de trabalho amostra-a-seqüência-para-interpretação para RABV, incluindo ferramentas de bioinformática acessíveis, tem o potencial de ser usado para orientar medidas de controle visando a meta de zero mortes humanas por raiva mediada por cães até 2030 e, finalmente, para a eliminação de variantes RABV. Combinados com metadados relevantes, os dados genômicos gerados a partir desse protocolo facilitam a rápida caracterização do RABV durante investigações de surtos e na identificação de linhagens circulantes em um país ou região60,61,65. Ilustramos nosso pipeline usando principalmente exemplos de raiva mediada por cães; no entanto, o fluxo de trabalho é diretamente aplicável à raiva da vida selvagem. Essa transferibilidade e o baixo custo minimizam os desafios em tornar o sequenciamento de rotina facilmente disponível, não só para a raiva, mas também para outros patógenos46,66,67, para melhorar o manejo e o controle da doença.
Disclosures
Os autores não têm nada a revelar.
Acknowledgements
Este trabalho foi apoiado por Wellcome [207569/Z/17/Z, 224670/Z/21/Z], financiamento Newton do Conselho de Pesquisa Médica [MR/R025649/1] e do Departamento de Ciência e Tecnologia das Filipinas (DOST), o esforço global de pesquisa e inovação do Reino Unido sobre COVID-19 [MR/V035444/1], o Fundo de Apoio Estratégico Institucional da Universidade de Glasgow [204820], Prêmio de Novo Pesquisador do Conselho de Pesquisa Médica (KB) [MR/X002047/1], e Fundo de Desenvolvimento de Parceria Internacional, um DOST British Council-Philippines studentship (CB), um National Institute for Health Research [17/63/82] GemVi scholarship (GJ), e bolsas de estudo da Universidade de Glasgow do MVLS DTP (KC) [125638-06], do EPSRC DTP (RD) [EP/T517896/1] e do Wellcome IIB DTP (HF) [218518/Z/19/Z]. Somos gratos aos colegas e colaboradores que apoiaram este trabalho: Daniel Streicker, Alice Broos, Elizabeth Miranda, DVM, Daria Manalo, DVM, Thumbi Mwangi, Kennedy Lushasi, Charles Kayuki, Jude Karlo Bolivar, Jeromir Bondoc, Esteven Balbin, Ronnel Tongohan, Agatha Ukande, Davis Kuchaka, Mumbua Mutunga, Lwitiko Sikana e Anna Czupryna.
Materials
| Name | Company | Catalog Number | Comments |
| Brand name | |||
| Software | |||
| Sequencing software (MinKnow) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads | |
| Bioinformatics tool kit (Guppy) | Oxford Nanopore Technologies | https://community.nanoporetech.com/docs/prepare/library_prep_protocols/Guppy-protocol/v/gpb_2003_v1_revao_14dec2018 | |
| Equipment | |||
| Thermal cycler (miniPCR™ mini16 thermal cycler) | Cambio | MP-QP-1016-01 | |
| Homogenizer (Precellys Evolution Touch Homogenizer) | Bertin Instruments | EQ02520-300 | |
| Cold Racks (0.2-0.5mL) (PCR Mini-cooler with transparent lid) | BRAND | 781260 | |
| Pipettor | |||
| (Pipetman L Fixed F1000L, 1000 uL) | Gilson | SKU: FA10030 | |
| (Pipetman L Fixed F100L, 100 uL) | Gilson | SKU: FA10024 | |
| (Pipetman L Fixed F10L, 10 uL) | Gilson | SKU: FA10020 | |
| (Pipetman L Fixed F1L, 1 uL) | Gilson | SKU: FA10025 | |
| (Pipetman L Fixed F20L, 20 uL) | Gilson | SKU: FA10021 | |
| (Pipetman L Fixed F250L, 250 uL) | Gilson | SKU: FA10026 | |
| Fluorometer (Qubit 4 Fluorometer) | Thermofisher scientific/Fisher scientific | Q33238 | |
| Laptop (Any brand with ~2 GB of drive space, minimum of 512 GB storage space, msi installer [GPU]) | |||
| Microcentrifuge (Refrigerated centrifuge) | Thermofisher scientific/Fisher scientific | 75004081 | |
| Vortex mixer (Basic vortex mixer) | Thermofisher scientific/Fisher scientific | 88882011 | |
| Magnetic rack (DynaMag -2 Magnet) | Thermofisher scientific/Fisher scientific | 12321D | |
| Sequencing device (MinION) | Oxford Nanopore Technologies | MinION Mk1B | |
| RNA Extraction | |||
| RNA extraction kit (Qiagen RNEasy Mini Kit 250) | Qiagen | 74106 | |
| RNA stabilizing reagents | |||
| (RNA later) | Invitrogen | AM7020 | |
| (DNA/RNA Shield) | Zymo Research | R1100-50 | |
| PCR | |||
| Nuclease-free Water (Nuclease-free Water [not DEPC-treated]) | Thermofisher scientific/Fisher scientific | AM9937 | |
| Master mix for first strand cDNA synthesis (LunaScript RT SuperMix Kit) | New England Biolabs | E3010S | |
| DNA amplification master mix (Q5® Hot Start High-Fidelity 2X Master Mix [NEB]) | New England Biolabs | M0494L | |
| Primer (Scheme) (Custom DNA oligos) | Invitrogen | ||
| SPRI Bead Clean-up | |||
| SPRI beads (Aline Biosciences PCR Clean DX ) | Cambio | AL-AC1003-50 | |
| Ethanol, Pure Absolute, >99.8% (GC) [Riedel-De Haen] | Merck | 818760 | |
| Short Fragment buffer (SFB expansion pack) | Oxford Nanopore Technologies | EXP-SFB001 | |
| DNA Quantification | |||
| DNA quantification kit (Qubit® dsDNA HS Assay Kit) | Thermofisher scientific/Fisher scientific | Q32854 | |
| DNA quantification assay tubes (Qubit™ Assay Tubes) | Thermofisher scientific/Fisher scientific | Q32856 | |
| End Prep and barcoding (Qubit™ Assay Tubes) | |||
| End Prep master mix (NEBNext Ultra End Repair/dA-Tailing Module) | New England Biolabs | E7546L | |
| Barcoding kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 | |
| (Native Barcoding Expansion 1-12) | EXP-NBD104 | ||
| (Native Barcoding Expansion 13-24) | EXP-NBD114 | ||
| (Native Barcoding Expansion 96) | EXP-NBD196 | ||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 | |
| (not compatible) | (not compatible) | ||
| (Native Barcoding Kit 24 V14) | SQK-NBD114.24 | ||
| (Native Barcoding Kit 96 V14) | SQK-NBD114.96 | ||
| Ligation mastermix (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter Ligation | |||
| Adapter ligation master mix | |||
| (NEBNext Quick Ligation Module) | New England Biolabs | E6056S | |
| (NEBNext Ultra II Ligation Module) | New England Biolabs | E7595S | |
| (Blunt/TA Ligase Master Mix) | New England Biolabs | M0367S | |
| Adapter mix | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-AMII001 | |
| (Adapter Mix II [AMII]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-NBA114 | |
| (Native adapter [NA]) | |||
| Sequencing | |||
| Flowcell priming kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 EXP-FLP002 | |
| (Flush Buffer [FB]) | |||
| (Flush Tether [FT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 EXP-FLP004 | |
| (Flow Cell Flush [FCF]) | |||
| (Flow Cell Tether [FCT]) | |||
| Ligation Sequencing Kit | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 SQK-LSK109 | |
| Adapter Mix (Adapter Mix [AMX]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SQB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LB]) | |||
| Sequencing Tether (Sequencing Tether [SQT]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 SQK-LSK114 | |
| Adapter Mix (Ligation Adapter [LA]) | |||
| Ligation Buffer (Ligation buffer [LNB]) | |||
| Short Fragment Buffer (Short Fragment buffer [SFB]) | |||
| Sequencing Buffer (Sequencing Buffer [SB]) | |||
| Elution Buffer (Elution buffer [EB]) | |||
| Loading Beads (Loading Beads [LIB]) | |||
| Sequencing Tether (Flow Cell Tether [FCT]) | |||
| Library solution (Library solution [LIS]) | |||
| Flush buffer (Flow Cell Flush [FCF]) | |||
| Flow Cell | |||
| *Chemistry 9 | Oxford Nanopore Technologies | *Chemistry 9 FLO-MIN106D | |
| (Flow Cell [R9.4.1]) | |||
| *Chemistry 14 | Oxford Nanopore Technologies | *Chemistry 14 FLO-MIN114 | |
| (Flow Cell [R10.4.1]) | |||
| Flow Cell wash | |||
| Flowcell wash kit (Flow cell wash kit) | Oxford Nanopore Technologies | EXP-WSH004 | |
| Consummables | |||
| Surface decontaminant | |||
| (DNA Away Surface Decontaminant, Squeeze Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7010PK | |
| (RNase Away Surface Decontaminant, Bottle [Molecular Bio]) | Thermofisher scientific/Fisher scientific | 7002PK | |
| PCR 8-Tube Strip 0.2ml, individual cap (PCR 8-Tube Strip 0.2ml, with Individual attached Flat Caps, Sterile, DNAse/RNAse, Pyrogen Free,Natural [Greiner]) | Greiner | 608281 | |
| PCR Tube 0.2ml (PCR Tube 0.2ml, Natural [Domed Cap] Bagged in 500s, Non-Sterile [Greiner]) | Greiner | 671201 | |
| 1000µL Filter Tips (500) (Stacked 1000µL Filter Tips [500]) | Thermofisher scientific/Fisher scientific | 11977724 | |
| 100µL Filter Tips (1000) | Thermofisher scientific/Fisher scientific | 11947724 | |
| 10µL Filter Tips (1000) (Stacked 100µL Filter Tips [1000]) | Thermofisher scientific/Fisher scientific | 11907724 | |
| Reinforced tubes tubes (2ml) with screw caps and o-rings (Fisherbrand™ Bulk tubes) | Thermofisher scientific/Fisher scientific | 15545809 | |
| Microcentrifuge tube (1.5ml) (1.5 ml Eppendorf Tubes [500]) | Eppendorf | 1229888 | |
| DNA LoBind Tubes (1.5ml) (DNA LoBind Tubes) | Thermofisher scientific/Fisher scientific | 10051232 | |
| Cryobabies labels | |||
| Gloves (S/M/L) | |||
| Paper towel |
References
- Rupprecht, C. E. Rhabdoviruses: rabies virus. Medical Microbiology. , (1996).
- Rabies. World Health Organization Available from: https://www.who.int/news-room/fact-sheets/detail/rabies (2023)
- WHO Expert Consultation on Rabies: WHO TRS N°1012. World Health Organization Available from: https://www.who.int/publications-detail-redirect/WHO-TRS-1012 (2018)
- Benavides, J. A., et al. Defining new pathways to manage the ongoing emergence of bat rabies in Latin America. Viruses. 12 (9), 1002 (2020).
- Hampson, K. Estimating the global burden of endemic canine rabies. PLoS Neglected Tropical Diseases. 9 (4), e0003709 (2015).
- Global genomic surveillance strategy for pathogens with pandemic and epidemic potential, 2022-2032. World Health Organization Available from: https://www.who.int/publications-detail-redirect/978924004679 (2022)
- Tsai, K. J., et al. Emergence of a sylvatic enzootic formosan ferret badger-associated rabies in Taiwan and the geographical separation of two phylogenetic groups of rabies viruses. Veterinary Microbiology. 182, 28-34 (2016).
- Chiou, H. -. Y., et al. Molecular characterization of cryptically circulating rabies virus from ferret badgers, Taiwan. Emerging Infectious Diseases. 20 (5), 790-798 (2014).
- Sabeta, C. T., Mansfield, K. L., McElhinney, L. M., Fooks, A. R., Nel, L. H. Molecular epidemiology of rabies in bat-eared foxes (Otocyon megalotis) in South Africa. Virus Research. 129 (1), 1-10 (2007).
- Scott, T. P. Complete genome and molecular epidemiological data infer the maintenance of rabies among kudu (Tragelaphus strepsiceros) in Namibia. PLoS One. 8 (3), e58739 (2013).
- Lembo, T., et al. Exploring reservoir dynamics: a case study of rabies in the Serengeti ecosystem. The Journal of Applied Ecology. 45 (4), 1246-1257 (2008).
- Coetzee, P., Nel, L. H. Emerging epidemic dog rabies in coastal South Africa: a molecular epidemiological analysis. Virus Research. 126 (1-2), 186-195 (2007).
- Ou de Munnink, B. B. First molecular analysis of rabies virus in Qatar and clinical cases imported into Qatar, a case report. International Journal of Infectious Diseases. 96, 323-326 (2020).
- Smith, J., et al. Case report: Rapid ante-mortem diagnosis of a human case of rabies imported into the UK from the Philippines. Journal of Medical Virology. 69, 150-155 (2003).
- Mahardika, G. N. K., et al. Phylogenetic analysis and victim contact tracing of rabies virus from humans and dogs in Bali, Indonesia. Epidemiology and Infection. 142 (6), 1146-1154 (2014).
- Tohma, K., et al. Molecular and mathematical modeling analyses of inter-island transmission of rabies into a previously rabies-free island in the Philippines. Infection, Genetics and Evolution. 38, 22-28 (2016).
- Tohma, K., et al. Phylogeographic analysis of rabies viruses in the Philippines. Infection, Genetics and Evolution. 23, 86-94 (2014).
- Saito, M., et al. Genetic diversity and geographic distribution of genetically distinct rabies viruses in the Philippines. PLoS Neglected Tropical Diseases. 7 (4), e2144 (2013).
- Biek, R., Henderson, J. C., Waller, L. A., Rupprecht, C. E., Real, L. A. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proceedings of the National Academy of Sciences. 104 (19), 7993-7998 (2007).
- Reddy, G. B., et al. Molecular characterization of Indian rabies virus isolates by partial sequencing of nucleoprotein (N) and phosphoprotein (P) genes. Virus Genes. 43, 13-17 (2011).
- David, D., Dveres, N., Yakobson, B. A., Davidson, I. Emergence of dog rabies in the northern region of Israel. Epidemiology and Infection. 137 (4), 544-548 (2009).
- Benjathummarak, S. Molecular genetic characterization of rabies virus glycoprotein gene sequences from rabid dogs in Bangkok and neighboring provinces in Thailand, 2013-2014. Archives of Virology. 161 (5), 1261-1271 (2016).
- Denduangboripant, J., et al. Transmission dynamics of rabies virus in Thailand: implications for disease control. BMC Infectious Diseases. 5, 52 (2005).
- Talbi, C., et al. Phylodynamics and human-mediated dispersal of a zoonotic virus. PLoS Pathogens. 6 (10), e1001166 (2010).
- Bourhy, H., et al. Revealing the micro-scale signature of endemic zoonotic disease transmission in an African urban setting. PLoS Pathogens. 12 (4), e1005525 (2016).
- Zinsstag, J., et al. Vaccination of dogs in an African city interrupts rabies transmission and reduces human exposure. Science Translational Medicine. 9 (421), (2017).
- Yakovleva, A., et al. Tracking SARS-COV-2 variants using Nanopore sequencing in Ukraine in 2021. Scientific Reports. 12, 15749 (2022).
- Mannsverk, S., et al. SARS-CoV-2 variants of concern and spike protein mutational dynamics in a Swedish cohort during 2021, studied by Nanopore sequencing. Virology Journal. 19, 164 (2022).
- Soufi, M., et al. Fast and easy nanopore sequencing workflow for rapid genetic testing of familial Hypercholesterolemia. Frontiers in Genetics. 13, 836231 (2022).
- Cabibbe, A. M. Application of targeted next-generation sequencing assay on a portable sequencing platform for culture-free detection of drug-resistant tuberculosis from clinical samples. Journal of Clinical Microbiology. 58 (10), 00632 (2020).
- Xu, Y., et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Euro Surveillance. 26 (27), 2000004 (2021).
- Stubbs, S. C. B., et al. Assessment of a multiplex PCR and Nanopore-based method for dengue virus sequencing in Indonesia. Virology Journal. 17, 24 (2020).
- Croville, G., et al. Rapid whole-genome based typing and surveillance of avipoxviruses using nanopore sequencing. Journal of Virological Methods. 261, 34-39 (2018).
- Theuns, S., et al. Nanopore sequencing as a revolutionary diagnostic tool for porcine viral enteric disease complexes identifies porcine kobuvirus as an important enteric virus. Scientific Reports. 8, 9830 (2018).
- O'Donnell, V. K., et al. Rapid sequence-based characterization of African swine fever virus by use of the Oxford Nanopore MinION sequence sensing device and a companion analysis software tool. Journal of Clinical Microbiology. 58, 01104-01119 (2019).
- Brito, A. F. Global disparities in SARS-CoV-2 genomic surveillance. Nature Communications. 13, 7003 (2022).
- ONT login/register. Oxford Nanopore Technology Available from: https://nanoporetech.com/login-register (2023)
- Software Downloads. Oxford Nanopore Technology Available from: https://community.nanoporetech.com/downloads (2023)
- . GitHub Available from: https://github.com/ (2023)
- . Artic-rabv Available from: https://github.com/kirstyn/artic-rabv (2022)
- . MADDOG: Method for Assignment, Definition and Designation of Global Lineages Available from: https://github.com/KathrynCampbell/MADDOG (2022)
- RABV-GLUE. Centre for Virus Research Available from: https://github.com/KathrynCampbell/MADDOG (2022)
- Itokawa, K., Sekizuka, T., Hashino, M., Tanaka, R., Kuroda, M. Disentangling primer interactions improves SARS-CoV-2 genome sequencing by multiplex tiling PCR. PLoS ONE. 15 (9), e0239403 (2020).
- Davis, M. W., Jorgensen, E. M. ApE, A plasmid editor: A freely available DNA manipulation and visualization program. Frontiers in Bioinformatics. 2, 818619 (2022).
- Döring, M., Pfeifer, N. . openPrimeR: Multiplex PCR primer design and analysis. , (2023).
- Quick, J. Multiplex PCR method for MiniON and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nature Protocols. 12 (6), 1261-1276 (2017).
- Laboratory Techniques in Rabies. World Health Organization Available from: https://apps.who.int/iris/bitstream/handle/10665/310836/9789241515153-eng.pdf (2018)
- Lembo, T. Partners for Rabies Prevention. The blueprint for rabies prevention and control: a novel operational toolkit for rabies elimination. PLoS Neglected Tropical Diseases. 6 (2), e1388 (2012).
- Terrestrial Manual Online Access. World Organization for Animal Health Available from: https://www.woah.org/en/what-we-do/standards/codes-and-manuals/terrestrial-manual-online-access/ (2023)
- Mauti, S. Field postmortem rabies rapid immunochromatographic diagnostic test for resource-limited settings with further molecular applications. Journal of Visualized Experiments. (160), e60008 (2020).
- Patrick, E., et al. Enhanced rabies surveillance using a direct rapid immunohistochemical test. Journal of Visualized Experiments. (146), e59416 (2019).
- Lembo, T., et al. Evaluation of a direct, rapid immunohistochemical test for rabies diagnosis. Emerging Infectious Diseases. 12 (2), 310-313 (2006).
- Marston, D. A., et al. Pan-lyssavirus real time RT-PCR for rabies diagnosis. Journal of Visualized Experiments. (149), e59709 (2019).
- . DNA quantification using the Qubit fluorometer Available from: https://www.protocols.io/view/dna-quantification-using-the-qubit-fluorometer-bc6vize6 (2020)
- . DNA quantification using the Quantus fluorometer Available from: https://www.protocols.io/view/dna-quantification-using-the-quantus-fluorometer-7pzhmp6 (2020)
- Guppy protocol. Nanopore Community Available from: https://community.nanoporetech.com/protocols/Guppy-protocol/v/gpb_2003_v1_revaq_14dec2018 (2023)
- Flow Cell Wash Kit (EXP-WSH004). Nanopore Community Available from: https://community.nanoporetech.com/protocols/flow-cell-wash-kit-exp-wsh004/v/wfc_9120_v1_revh_08dec2020 (2023)
- . Core Pipeline - arctic pipeline Available from: https://artic.readthedocs.io/en/latest/minion/ (2023)
- . Samtools Available from: https://www.htslib.org (2023)
- Campbell, K., et al. Making genomic surveillance deliver: A lineage classification and nomenclature system to inform rabies elimination. PLoS Pathogens. 18 (5), e1010023 (2022).
- Brunker, K., et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Research. 5, 3 (2020).
- Bull, R. A., et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nature Communications. 11, 6272 (2020).
- Okeke, I. N., Ihekweazu, C. The importance of molecular diagnostics for infectious diseases in low-resource settings. Nature Reviews. Microbiology. 19 (9), 547-548 (2021).
- Inzaule, S. C., Tessema, S. K., Kebede, Y., Ouma, A. E. O., Nkengasong, J. N. Genomic-informed pathogen surveillance in Africa: opportunities and challenges. The Lancet Infectious Diseases. 21 (9), 281-289 (2021).
- Kennedy, L. Integrating contact tracing and whole-genome sequencing to track the elimination of dog-mediated rabies: an observational and genomic study. eLife. , (2023).
- Pallerla, S. R. Diagnosis of pathogens causing bacterial meningitis using Nanopore sequencing in a resource-limited setting. Annals of Clinical Microbiology and Antimicrobials. 21, 39 (2022).
- Quick, J. Real-time, portable genome sequencing for Ebola surveillance. Nature. 530 (7589), 228-232 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionExplore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved