Um ensaio de reação em cadeia da polimerase digital validável em gotículas para a detecção de vetores virais adenoassociados em estudos de biosshedding de lágrimas
In This Article
Summary
Aqui, apresentamos um protocolo para o desenvolvimento e validação de boas práticas laboratoriais na detecção aderente de vetores virais adenoassociados em lágrimas humanas por reação em cadeia da polimerase digital em gotículas em apoio ao desenvolvimento clínico de vetores de terapia gênica.
Abstract
O uso de vetores virais para tratar doenças genéticas aumentou substancialmente nos últimos anos, com mais de 2.000 estudos registrados até o momento. Os vetores virais adenoassociados (AAV) têm encontrado particular sucesso no tratamento de doenças oculares, como exemplificado pela aprovação do voretigene neparvovec-rzyl. Para trazer novas terapias ao mercado, as agências reguladoras normalmente solicitam estudos de bioshedding qualificados ou validados para avaliar a liberação do vetor no ambiente. No entanto, nenhuma diretriz oficial para o desenvolvimento de ensaios de base molecular para apoiar tais estudos de eliminação foi divulgada pela Food and Drug Administration dos Estados Unidos, deixando os desenvolvedores para determinar as melhores práticas por si mesmos. O objetivo deste protocolo é apresentar um protocolo válido para a detecção de vetores de AAV em lágrimas humanas por reação em cadeia da polimerase digital em gotículas (ddPCR) em apoio a estudos clínicos de bioshedding. Este artigo discute as abordagens atuais da indústria para validação de ensaios moleculares e demonstra que o método excede os critérios de aceitação de ensaios alvo atualmente propostos em white papers. Finalmente, etapas críticas na realização de qualquer ensaio ddPCR, independentemente da aplicação, são discutidas.
Introduction
As definições de terapia gênica variam, mas geralmente induzem uma alternância permanente intencional e frequentemente esperada de uma sequência específica de DNA do genoma celular para modificar ou manipular a expressão de um gene ou alterar as propriedades biológicas de uma célula viva para fins clínicos 1,2. Os vetores virais estão sendo cada vez mais utilizados como veículos para a terapia gênica devido à sua eficiência de transdução, com um relato sugerindo que mais de 70% dos ensaios clínicos atuais de terapia gênica utilizam vetores virais3. O interesse em vetores virais para terapia gênica vem ganhando cada vez mais. O Relatório de Dados Trimestrais do 4º Trimestre de 2022 sobre o cenário de terapia de genes, células e RNA da Sociedade Americana de Terapia Gênica e Celular relatou que, em 2022, o pipeline de terapia de genes, células e RNA do pré-registro para o pré-registro cresceu 7%, elevando o número total de terapias em desenvolvimento para 3.726, das quais 2.053 (55%) eram terapias gênicas4. Atualmente, a Food and Drug Administration (FDA) dos Estados Unidos aprovou 27 terapias celulares e gênicas para uso clínico em humanos, cinco das quais utilizam especificamente vetores virais5.
Os vírus adenoassociados (AAVs) têm ganhado interesse específico como veículos para terapia gênica. Uma metanálise recente revelou que houve aproximadamente 136 ensaios clínicos investigando o uso de AAVs nas últimas duas décadas6. Além disso, três das cinco terapias gênicas aprovadas pelo FDA nos EUA são baseadas em AAV. Isso se deve à sua natureza altamente editável, ampla gama de hospedeiros que podem ser ajustados com base no uso de vetores específicos de ocorrência natural ou artificialmente modificados, baixa patogenicidade e toxicidade em humanos e, geralmente, baixa imunogenicidade 7,8. Os VAA também têm sido usados com sucesso para tratar doenças oculares em um ambiente clínico aprovado. O voretigene neparvovec-rzyl é uma terapia baseada em AAV2 que foi aprovada pelo FDA dos EUA em 2017 e pela Agência Europeia de Medicamentos (EMA) em 2018 para tratar a distrofia retiniana associada à mutação bialélica RPE65 9.
Com o crescente interesse no desenvolvimento de terapias baseadas em AAV surge a necessidade de orientação regulatória sobre ensaios. A detecção e quantificação precisas de qualquer vetor viral são parte integrante das fases de descoberta, fabricação e testes pré-clínicos/clínicos do desenvolvimento de produtos. O FDA dos EUA começou a emitir algumas orientações para terapias gênicas, incluindo sobre química, fabricação e controle para aplicações de novos fármacos investigacionais de terapia gênicahumana 10, acompanhamento de longo prazo após a administração de terapia gênica11, testes de retrovírus competentes em replicação 12 e recomendações para vetores microbianos usados em terapias gênicas13. A EMA também divulgou uma série de diretrizes sobre o desenvolvimento de produtos de terapia gênica que geralmente se alinham com as recomendações da FDA, embora existam algumas diferenças14. É importante notar que, embora essas orientações não estabeleçam responsabilidades legalmente exigíveis, exceto quando regulamentações específicas são referenciadas, elas fornecem clareza sobre o pensamento atual das agências reguladoras sobre o tema e suas expectativas para os ensaios necessários para registros de medicamentos e aprovação regulatória.
O FDA recomenda especificamente que estudos sejam realizados para avaliar a distribuição, persistência e depuração de um vetor do local de administração para atingir tecidos oculares e não oculares, fluidos intraoculares e sangue15. Estes assumem a forma de estudos de biodistribuição e eliminação. Os estudos de biodistribuição avaliam a exposição investigando como um produto é espalhado por todo o corpo do paciente a partir do local de administração. O excredimento, especificamente, avalia a liberação do produto do paciente para o ambiente e levanta a possibilidade de transmissão do vetor para indivíduos não tratados16. O FDA faz recomendações para o desenho de estudos de biodistribuição e excreção com relação à frequência de coleta de amostras, duração da coleta de amostras, tipos de amostras coletadas e condições de armazenamento.
Além disso, o FDA recomenda o uso da reação em cadeia da polimerase quantitativa (qPCR, ou PCR em tempo real) para a detecção quantitativa de genomas vetoriais devido à sua facilidade de desempenho, formato de alto rendimento, tempos de resposta rápidos e sensibilidade do ensaio. No entanto, há uma relativa falta de recomendações para o planejamento e avaliação de desempenho de métodos moleculares em comparação com aqueles que existem para moléculas pequenas e grandes. Muitas das diretrizes para tais estudos são difíceis de aplicar a métodos moleculares devido ao design único e complexo tanto dos produtos quanto dos próprios ensaios, levantando questões quanto à adequação das plataformas disponíveis para as avaliações recomendadas e métodos apropriados para validação de ensaios. Até o momento, a FDA não exigiu a validação formal de ensaios baseados em PCR, embora a EMA tenha imposto essa exigência17. À luz desse vazio, diferentes grupos e oficinas emitiram white papers e recomendações que fabricantes e organizações de pesquisa contratadas procuraram seguir 18,19,20,21,22,23,24,25. A maioria dessas recomendações é escrita especificamente com ensaios de qPCR em mente, com sugestões ou alterações para plataformas emergentes, como a PCR digital de gotículas (ddPCR), incluídas apenas quando consideradas relevantes. Recomendações mais recentes têm se concentrado em considerações para ensaios de ddPCR, mas têm se concentrado em grande parte em suas aplicações para quantificação do genoma vetorial em um ambiente de fabricação, em vez de nas matrizes biológicas complexas encontradas em estudos de bioshedding.
Dependendo da aplicação clínica e dos objetivos, a ddPCR pode ser preferida em relação à qPCR em apoio a estudos de biodistribuição e eliminação devido à maior sensibilidade e capacidade da ddPCR de lidar com interferência de matriz em comparação com a qPCR. Além disso, devido ao particionamento das amostras em aproximadamente 20.000 gotículas, a quantificação precisa do número de cópias pode ser obtida sem o uso de uma curva padrão usando a estatística de Poisson, simplificando o desenvolvimento e validação do método. O objetivo deste protocolo é descrever uma abordagem padronizada para o desenvolvimento e validação de um método baseado em ddPCR para a detecção de vetores de AAV em lágrimas coletadas da superfície ocular em apoio a estudos clínicos de bioshedding.
Protocol
1. Preparação de um fragmento de ADN sintético
- Projetar e encomendar um fragmento de DNA sintético contendo a região alvo de amplificação para uso como controle de qualidade.
- Certifique-se de que a sequência contenha toda a sequência amplicon do primer para frente até o primer reverso do gene alvo de interesse, com uma extensão de quatro a seis pares de base de sequência nas extremidades 5' de cada sequência de ligação do primer.
- Evitar homopolímeros de adenina e timina maiores que 12 pares de bases ou guanina e citosina pares de bases maiores que oito pares de bases, pois desde que homopolímeros possam interferir na síntese do fragmento gênico.
NOTA: Se os amplicons contiverem tais sequências, as substituições de base podem ser feitas desde que os locais de recozimento para os primers e sondas sejam mantidos. - Alternativamente, prepare um plasmídeo linearizado contendo o amplicon usando estratégias típicas de clonagem.
- Centrifugar o tubo contendo o fragmento de DNA sintético em uma microcentrífuga por ~10 s para garantir que o material seja coletado no fundo do tubo.
- Ressuspender o fragmento de DNA sintético usando tampão tris-EDTA (TE) para uma concentração de 1,0 × 1010 cópias/μL, ou conforme apropriado com base na faixa de ensaio alvo.
- Vórtice brevemente e, em seguida, incube a 50 °C por 20 ± 5 min. Resfriar no gelo.
- Prepare alíquotas múltiplas, idealmente de uso único, e guarde a -70 a -90 °C até o uso.
NOTA: Os fragmentos de ADN sintéticos preparados desta forma são normalmente estáveis durante, pelo menos, 24 meses a contar da data de ressuspensão. - Se desejado, determinar a concentração exata do estoque de DNA sintético preparado antes de usá-lo como controle de qualidade, ou estimar a concentração nominal com base na ressuspensão utilizada.
2. Preparação de primers e sonda
- Projeta e ordena primers e uma sonda de hidrólise para atingir a região de amplificação desejada usando estratégias típicas de projeto26,27.
- Utilize um corante fluorescente de repórter de 5' (por exemplo, FAM) e um quencher de 3' (por exemplo, quencher escuro Iowa Black) compatível com o sistema ddPCR.
NOTA: Existem vários pacotes de software de design de ensaio de PCR e qualquer um pode ser utilizado. Por exemplo, o Primer-BLAST do National Center for Biotechnology Information28 é amplamente utilizado devido às opções robustas para o desenho do ensaio e à facilidade com que a especificidade pode ser avaliada bioinformaticamente para identificar possíveis efeitos fora do alvo. Deve-se notar que a preparação de primers e sondas pode variar das etapas listadas aqui, dependendo do formato em que são fornecidas.
- Utilize um corante fluorescente de repórter de 5' (por exemplo, FAM) e um quencher de 3' (por exemplo, quencher escuro Iowa Black) compatível com o sistema ddPCR.
- Centrifugar os tubos contendo o primer para frente, primer reverso e sonda em uma microcentrífuga por ~10 s para o material da pelota para o fundo do tubo.
- Ressuspender os primers para 20 μM usando tampão TE. Vórtice brevemente.
- Ressuspenda a sonda para 10 μM usando o buffer TE. Vórtice brevemente.
- Prepare alíquotas múltiplas, idealmente de uso único, e guarde a um mínimo de -20 °C até o uso.
NOTA: Primers e sondas preparados desta forma são normalmente estáveis durante pelo menos 24 meses a partir da data de ressuspensão.
3. Preparação do tampão de diluição da amostra
- Tampão de PCR de descongelamento e DNA de espermatozoides de salmão cortado à temperatura ambiente. Vórtice bem para misturar.
- Preparar um tampão de diluição da amostra, conforme Tabela 1.
- Vórtice completamente. Conservar a 2-8 °C até 1 mês após a preparação.
Quadro 1: Preparação do tampão de diluição da amostra. Clique aqui para baixar esta tabela.
4. Preparação do master mix
- Descongele a mistura mestra ddPCR para sondas, primer para frente, primer reverso e sonda à temperatura ambiente e deixe aquecer por pelo menos 10 minutos após o descongelamento antes do uso. Conservar à temperatura ambiente até à utilização.
NOTA: Estes reagentes devem ser totalmente levados à temperatura ambiente para garantir a formação eficiente de gotículas. Não mantenha os reagentes no gelo durante a preparação.- Vórtice centrífuga completa e brevemente em uma mini centrífuga antes de usar.
NOTA: As enzimas de restrição são normalmente fornecidas em glicerol e devem ser removidas do armazenamento imediatamente antes do uso. Misture delicadamente. Não vórtice.
- Vórtice centrífuga completa e brevemente em uma mini centrífuga antes de usar.
- Prepare uma mistura mestre de PCR para cada alvo de amplificação. Veja a Tabela 2 para uma composição sugerida da mistura mestre de PCR e modifique as concentrações de primers e sondas conforme necessário.
- Vórtice completamente e centrifugar brevemente antes da adição da enzima de restrição. Adicione a enzima de restrição e inverta para misturar.
NOTA: Nesta etapa, 22 μL de reação de PCR são necessários para obter um volume final de 40 μL de reação de PCR após a formação de gotículas (consistindo de 15 μL de mistura mestre de PCR, 5,0 μL de molde e 20 μL de óleo de geração de gotículas).
- Vórtice completamente e centrifugar brevemente antes da adição da enzima de restrição. Adicione a enzima de restrição e inverta para misturar.
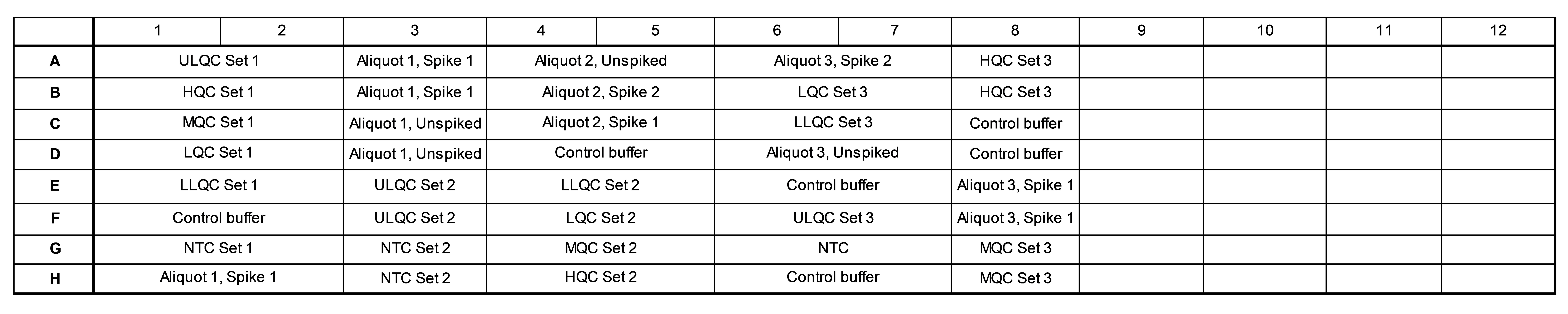
- Adicione 16,5 μL de mistura mestre a cada poço de acordo com o mapa da placa. Consulte a Figura 1 para obter um exemplo de mapa de placa para obter uma precisão de validação e uma execução de precisão.
- Certifique-se de que uma placa contenha três preparações independentes da série de controle de qualidade (QC), três alíquotas endógenas de lágrima testadas independentemente, cravadas em um nível alto e baixo e sem pontas, e três controles independentes sem modelo (NTCs).
- Varie a disposição desses poços ao longo da placa, onde o Conjunto 1 é carregado em ordem decrescente de concentração, o Conjunto 2 é carregado em ordem crescente de concentração e o Conjunto 3 é carregado em uma ordem aleatória para avaliar se há algum efeito específico de localização da placa.
- Organize as amostras para preencher o máximo possível de uma coluna e preencha poços não utilizados dentro de uma coluna com buffer de controle. Inclua vários lotes de controle endógenos (por exemplo, mais poças de lágrimas ou lágrimas coletadas de indivíduos) nos poços restantes, se desejado.
- Sele a placa com filme adesivo transparente. Segure a placa à temperatura ambiente durante a preparação do modelo. Alternativamente, segure a placa por até 4 h a 2-8 °C, mas leve-a de volta à temperatura ambiente por pelo menos 10 minutos antes da adição do modelo.
Tabela 2: Exemplo de preparação do mix mestre de PCR. Clique aqui para baixar esta tabela.

Figura 1: Exemplo de mapa de placa para precisão de validação e precisão de execução. Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para ver uma versão maior desta figura.
{kind=link}
5. Preparação de CQ
- Descongelar fragmentos de DNA sintético ou plasmídeos linearizados à temperatura ambiente e deixar aquecer por pelo menos 10 minutos após o descongelamento antes do uso. Leve os moldes à temperatura ambiente para garantir a formação eficiente de gotículas.
- Conservar à temperatura ambiente até à utilização. Vórtice centrífuga completa e brevemente em uma mini centrífuga antes de usar.
- Preparar diluições de CQ utilizando o tampão de diluição da amostra como diluente. Um exemplo das concentrações recomendadas para se preparar para uma execução de precisão e precisão de validação é apresentado na Tabela 3.
NOTA: Após a conclusão bem-sucedida das execuções de precisão e precisão, somente o controle de alta qualidade (HQC), o controle de qualidade médio (MQC) e o baixo controle de qualidade (LQC) precisam ser executados em cada placa. Para ensaios de exatidão e precisão, pelo menos três diluições independentes dos CQ são incluídas para a avaliação da exatidão e precisão intraensaio. Após execuções de exatidão e precisão, apenas uma série de diluição precisa ser incluída. - Após a preparação, conservar as diluições à temperatura ambiente até serem adicionadas ao prato.
- Conservar as diluições no gelo ou a 2-8 °C, se necessário. Antes da utilização subsequente, deixe as diluições aquecerem à temperatura ambiente durante, pelo menos, 10 minutos antes da utilização. Descarte os QCs no final do dia.
Tabela 3: Exemplo de preparação de controle de qualidade (CQ) usando fragmentos sintéticos de DNA de fita dupla. Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
6. Preparação das amostras
- Descongelar amostras de lágrima coletadas de um ensaio clínico à temperatura ambiente até descongelar e deixar aquecer por pelo menos 10 minutos após o descongelamento antes do uso.
- Conservar à temperatura ambiente até à utilização. Centrífuga completa e breve em uma microcentrífuga antes do uso.
- Diluir amostras lacrimais 1:10 (ou superior) utilizando tampão de diluição da amostra como diluente em tubos de PCR de 0,2 ml ou tiras de PCR de 8 poços. Sele os tubos.
NOTA: Dependendo da concentração esperada do alvo em rasgos, pode ser necessário diluir ainda mais as amostras ou testar diluições múltiplas de cada amostra. - Aquecer as amostras num termociclador a 95 °C durante 10 min, seguido de manter a 4 °C durante, pelo menos, 5 minutos para arrefecer. Use uma taxa de rampa de 3 °C/s.
NOTA: As amostras podem permanecer no termociclador a 4 °C até serem utilizadas no mesmo dia ou podem ser congeladas a -70 °C a -90 °C para armazenamento mais prolongado. Essa etapa serve para desnaturar o capsídeo vetorial, liberando o genoma. Como fragmentos de DNA sintético QC ou plasmídeos linearizados são de fita dupla, eles não devem passar por essa etapa de aquecimento. - Devolver as amostras após o arrefecimento à temperatura ambiente (ou, se congeladas, descongelar à temperatura ambiente) e deixar aquecer durante, pelo menos, 10 min.
NOTA: As amostras devem ser totalmente levadas à temperatura ambiente para garantir a formação eficiente de gotículas.
7. Adição de modelo
- Recupere a placa ddPCR que contém a mistura mestre. Vórtice cada amostra ou tubo de diluição QC completamente e centrifugar brevemente para recolher novamente o material.
- Remova o filme adesivo e adicione 5,5 μL de QCs ou amostras aos poços apropriados da placa de 96 poços, de acordo com o mapa da placa.
Observação : consulte a etapa 4.2.1 para obter explicação dos volumes necessários - Adicionar 5,5 μL de tampão de diluição da amostra aos poços NTC.
- A geração de gotículas requer que todos os poços de uma coluna tenham um controle de reação ou tampão. Se algum poço de uma coluna não contiver reações de amostra, diluir 2x o controle de tampão ddPCR 1:2 usando água livre de nucleases. Adicione 22 μL de controle de buffer ddPCR 1x a quaisquer poços vazios de uma coluna.
Observação : se uma coluna inteira não for usada, não é necessário adicionar controle de buffer para esses poços. - Adicione um selo de folha perfurável ao prato. Coloque a placa no selador de chapas e sele por 5 s a 180 °C.
- Alternativamente, sele a placa de acordo com as recomendações do fabricante do sistema ddPCR.
- Vórtice a placa à velocidade máxima por pelo menos 30 s (usando a configuração de vórtice contínuo; não use vórtice de toque) e centrifugue brevemente em um girador de placa.
NOTA: A mistura completa e completa da placa nesta etapa é fundamental para o particionamento adequado da reação de PCR em gotículas. Certifique-se de que não há bolhas visíveis nos poços. Se necessário, a placa pode ser mantida a 2-8 °C antes da geração de gotículas por no máximo 4 h. Se mantida, deixe a placa chegar à temperatura ambiente por um mínimo de 10 minutos antes da geração de gotículas.
8. Geração automatizada de gotículas, ciclagem térmica e leitura de gotículas
- Gere gotículas no gerador de gotículas automatizado da seguinte maneira.
- Na tela sensível ao toque, selecione as colunas no mapa da placa contendo amostras. O convés do instrumento acenderá para indicar quais consumíveis (cartuchos DG32, pontas, recipiente de resíduos, óleo de geração de gotículas) são necessários. As luzes amarelas indicam que é necessário adicionar um consumível, enquanto as luzes verdes indicam que consumíveis suficientes estão disponíveis.
- Carregue o gerador de gotículas de trás para frente.
- Para sondas de hidrólise, certifique-se de que o óleo de geração de gotículas para sondas esteja instalado e óleo suficiente para o número de poços restantes. Se forem utilizados produtos químicos de PCR alternativos, certifique-se de que um óleo de geração de gotículas compatível esteja instalado.
- Coloque um bloco frio no suporte da placa de gotículas. Certifique-se de que o bloco é totalmente azul e nenhum rosa está visível. Coloque uma nova placa ddPCR de 96 poços no bloco frio.
- Coloque a placa de PCR preparada no suporte da placa de amostra. Feche a tampa da máquina. Pressione start para geração de gotículas.
- Após a formação de gotículas, um total de 40 μL por reação é transferido automaticamente para a nova placa de PCR.
- Dentro de 30 minutos após a conclusão da geração de gotículas, remova a placa que contém as gotículas do bloco frio. Trabalhe suavemente, pois as gotículas são mais frágeis nesta fase.
- Adicione um selo de folha perfurável ao prato. Coloque a placa no selador de chapas e sele por 5 s a 180 °C.
- Alternativamente, sele a placa de acordo com as recomendações do fabricante do sistema ddPCR.
- Coloque a placa em um termociclador compatível. Insira as condições de ciclismo (ver Tabela 4).
- Após o término da ciclagem térmica, segure a placa no termociclador, transferida para 2-8 °C, ou leia-a imediatamente.
NOTA: Segurar a placa por 12 h a 4-12 °C pode melhorar a contagem de gotículas, mas isso não é necessário. Gotículas suficientes devem ser obtidas sem a retenção. - Coloque a placa no leitor de gotículas, garantindo que o óleo do leitor permaneça suficiente e que o recipiente de resíduos tenha espaço suficiente. Leia as gotículas. Realizar a leitura das gotículas dentro de 24 h após o início da ciclagem térmica.
Tabela 4: Condições típicas de ciclagem térmica. Clique aqui para baixar esta tabela.
9. Análise dos dados
NOTA: Um mínimo de 10.000 gotículas por poço é necessário para o cálculo adequado da concentração usando a estatística de Poisson. Não tente analisar em poços com menos de 10.000 gotículas.
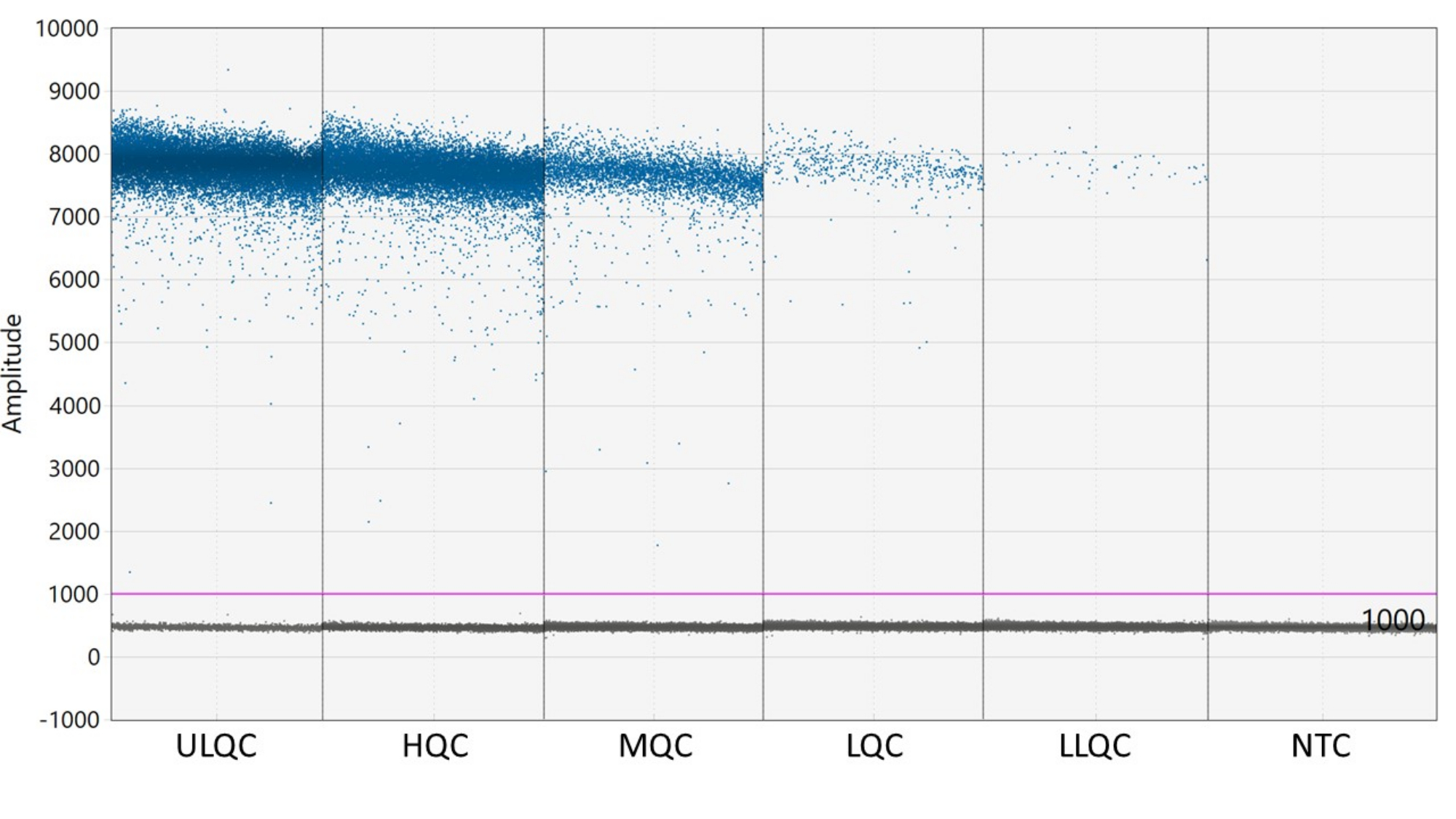
- Um limiar é necessário para definir as gotículas como positivas ou negativas. O software de análise ddPCR aplica automaticamente um limite que pode variar entre poços. No entanto, defina manualmente um limite para todos os poços da placa ligeiramente acima da intensidade fluorescente dos poços NTC para obter resultados mais consistentes, precisos e precisos.
NOTA: A colocação adequada do limiar pode exigir otimização, dependendo da separação das gotículas positivas e negativas e da quantidade de chuva de gotículas existente (ver Figura 2). Neste exemplo, o gráfico de amplitude de gotículas mostra poços de exemplo em cada nível de CQ e o NTC. A linha roxa indica um limiar de 1.000, definido ligeiramente acima da população de gotículas negativas. - A modelagem estatística de Poisson requer pelo menos três gotículas positivas para calcular a concentração com 95% de confiança. Considere todos os poços contendo zero, uma ou duas gotículas positivas como negativos e definidos para uma concentração dezero 27.
- Calcule o número de cópias em cada amostra de lágrima.
- A concentração, em cópias/μL, é fornecida no relatório de dados. Use esse valor para determinar a concentração em cópias/μL da amostra original (ou seja, na amostra de lágrima).
- Para calcular a diluição da reação de ddPCR, divida o volume inicial da reação de PCR antes da formação de gotículas pelo volume de molde adicionado. Quando os volumes apresentados neste método são utilizados, obtém-se um valor de 4.

- Determinar o factor de diluição em série a partir da amostra original (passo 6.2).
- Para determinar as cópias/μL na amostra, multiplique as cópias/μL pela diluição da reação ddPCR e, em seguida, pelo fator de diluição seriado. Por exemplo, a concentração em cópias/μL gerada no relatório de dados foi de 966; 5,5 μL de molde foi adicionado por 22 μL de reação. Utilizou-se uma diluição seriada de 1:50.000 da amostra.


- Se forem testadas várias diluições da mesma amostra, analise todas as diluições válidas no intervalo e calcule a média.
- Para cada CQ, calcular as cópias esperadas/μL da reação de PCR dividindo a concentração da diluição dada de QC (em cópias/μL) pelo volume da reação de ddPCR (20 μL). Isso permite a comparação direta desse valor nominal com o valor de cópias/μL fornecido no relatório de dados sem cálculos adicionais.
NOTA: Esta abordagem também foi utilizada para a análise das amostras de lágrima fortificada utilizadas nos resultados representativos. - Determine o valor médio, o desvio padrão, o coeficiente de variação (%CV) e o erro relativo percentual à concentração nominal (%RE) da amostra ou do valor do CQ usando os poços de replicação (incluir diluições múltiplas, se aplicável).
- Para a avaliação da precisão entre poços, determine-a para cada uma das duplicatas de poço, se incluídas.
- Para a avaliação da exatidão e precisão intraensaio, determine-a para cada série de diluição ou alíquota utilizada dentro de um lote.
- Para a avaliação da exatidão e precisão interensaios, determine-a utilizando as médias intra-ensaio de cada um dos lotes incluídos.

Figura 2: Exemplo de definição de limite. Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para ver uma versão maior desta figura.
{kind=link}
10. Critérios de aceitação do ensaio
- Utilize as seguintes especificações para os dados calculados para cada lote para determinar se o lote é aceitável. Se essas condições não forem atendidas, invalide e repita o lote.
NOTA: Estes critérios foram determinados como consenso a partir de white papers publicados sobre validação de ensaios baseados em PCR 18,19,20,21,22,23,24,25. Pode ser necessário modificar os critérios-alvo conforme apropriado para aplicação clínica. - Nenhum controle de modelo (NTC)
- Certifique-se de que cada poço NTC tenha pelo menos 10.000 gotículas.
- Certifique-se de que cada poço NTC tenha menos de 3 gotículas positivas.
- QCs e faixa de ensaio
- Certifique-se de que cada poço QC tenha pelo menos 10.000 gotículas.
- Certifique-se de que a precisão dos poços replicados de uma concentração de CQ seja de ≤25,0% CV, exceto nos limites superior e inferior de quantificação, onde ≤30,0% é aceitável. Avalie isso independentemente para cada conjunto de CQ e nível de concentração.
- Assegurar que o erro relativo da concentração retrocalculada em cada nível médio de CQ esteja dentro de ±25,0% de ER da concentração nominal (cópias/reação de PCR), exceto nos limites superior e inferior de quantificação, onde ±30,0% de ER é aceitável. Avalie isso independentemente para cada conjunto de CQ e nível de concentração.
- Certifique-se de que pelo menos 2/3 das amostras de CQ (por exemplo, quatro de seis resultados) e 50% das amostras de CQ em cada nível (baixo, médio, alto) atendam a essas diretrizes.
- Amostras
- Certifique-se de que os poços de amostra a serem analisados tenham pelo menos 10.000 gotículas.
- Certifique-se de que a precisão dos poços replicados de uma diluição de amostra a ser analisada seja de ≤25,0% CV.
- Assegurar que pelo menos uma diluição incluída da amostra em causa está dentro do intervalo de quantificação definido do ensaio, tal como acima definido com base nos QC de limite superior e inferior.
- Se todas as diluições incluírem resultados de rendimento superiores ao limite superior de quantificação definido e se restar um volume de amostra suficiente, repetir o ensaio utilizando uma diluição mais elevada da amostra.
- Se todas as diluições incluídas produzirem um resultado inferior ao limite inferior de quantificação, e se restar um volume de amostra suficiente, repetir o ensaio utilizando uma diluição inferior da amostra.
NOTA: Amostras contendo mais de três gotículas positivas, mas que têm uma concentração abaixo do limite inferior de quantificação, podem ser descritas como detectáveis, mas não quantificáveis.
Representative Results
Para fins demonstrativos, um ensaio projetado para detectar um vetor AAV2 que expressa proteína fluorescente verde (eGFP) comercialmente disponível, com um fragmento sintético de DNA de fita dupla contendo eGFP como controle de qualidade, foi desenvolvido. Atualmente, há um debate em andamento sobre se o vetor em si ou um fragmento de DNA sintético ou plasmídeo linearizado é mais apropriado para uso como CQ. Geralmente, um fragmento de DNA sintético ou plasmídeo linearizado pode ser usado se a equivalência ao vetor for demonstrada no desenvolvimento do método (dados não mostrados). Primers e sondas foram projetados e otimizados para detectar o transgene eGFP. Consulte a Tabela Suplementar S1 para ver as sequências utilizadas neste trabalho. A concentração do estoque de fragmentos de CQ foi determinada empiricamente por ddPCR. Todos os ensaios foram realizados utilizando as concentrações e condições de PCR dadas como exemplos na seção de protocolo.
Para ensaios de qPCR, recomenda-se avaliar a linearidade, sensibilidade, faixa dinâmica, exatidão e precisão da curva padrão. Como a ddPCR não depende de uma curva padrão para quantificação do alvo, essas recomendações devem ser modificadas. Em vez disso, QCs consistindo de fragmentos sintéticos de DNA de fita dupla diluídos em várias concentrações para cobrir a faixa quantificável esperada de uma reação de ddPCR baseada na modelagem estatística de Poisson foram utilizados 29,30,31,32 para definir a faixa dinâmica e a sensibilidade e avaliar a acurácia e a precisão. A escolha das concentrações de CQ foi baseada principalmente na proporção esperada de gotículas positivas para totais dentro de um poço em uma dada concentração. Matematicamente, a ddPCR é teoricamente mais precisa quando aproximadamente 80% das partições amplificam positivamente. À medida que a razão gotículas positiva/total aumenta acima de 0,8, a precisão diminui devido à saturação das partições, não sendo possível a quantificação, uma vez que 100% das gotículas são positivas. Na extremidade baixa, teoricamente, apenas uma gotícula positiva pode ser detectada e quantificada, embora a acurácia seja menor e o ensaio esteja sujeito a falsos positivos de baixo nível. Normalmente, pelo menos três gotículas devem ser positivas para que um resultado seja calculado com 95% de confiança, que é o limite para calcular uma concentração que usamos aqui.
Uma série de cinco diferentes concentrações de CQ foi preparada, com o número de cópias alvo/volumes de reação de PCR μL calculados esperando-se que produzam razões de gotículas positivas para totais abrangendo a faixa quantificável de ddPCR, como mostrado na Tabela 5. Estes foram utilizados para avaliar a exatidão e precisão do ensaio. Na avaliação em questão, os limites superior e inferior de quantificação não foram levados ao máximo teórico possível na ddPCR. A quantificação precisa pode ser possível em níveis mais altos e mais baixos do que os demonstrados aqui. A gama deve ser desenvolvida em alinhamento com as aplicações a jusante deste método.
Um total de três séries de diluição desses QCs preparadas independentemente foram preparadas em tampão de diluição de amostra para cada lote para avaliar a exatidão e precisão intra-ensaio. Poços duplicados de cada diluição de CQ foram incluídos. Para simular um protocolo de validação real, um total de seis lotes de precisão e exatidão foram realizados por vários analistas ao longo de vários dias. Os resultados desses seis lotes foram analisados para definir a exatidão e precisão intra-ensaio e interensaio do método e para definir a faixa dinâmica do ensaio.
O desempenho intra-ensaio foi avaliado para cada lote em cada nível de CQ. Esperávamos que todos os poços QC e NTC tivessem pelo menos 10.000 gotículas. Isso foi atendido em 216 dos 216 poços testados em todos os seis lotes, com uma contagem média de gotículas de 19.748 gotículas/poço (Tabela 6). Em seguida, esperava-se que o %CV interpoços de cada conjunto de poços duplicados de cada CQ fosse de ≤25,0%, exceto para o QC dos limites superior e inferior, onde ≤30,0% era esperado. Isso foi atendido em conjuntos de 90 dos 90 poços testados em todos os seis lotes para os CQs, com uma média de %CV entre poços de 3,9% em todos os níveis de CQ (Tabela 7). Todos os CQ produziram razões médias positivas em relação ao total de gotículas dentro dos intervalos esperados acima (Tabela 6).
Dentro de cada lote, a média intra-ensaio e o desvio padrão foram calculados para cada um dos pontos da série de diluição preparados independentemente e estes foram usados para calcular uma média intra-ensaio para cada concentração em cada ensaio. Este foi utilizado para avaliar a acurácia e precisão do ensaio (Tabela 8). A precisão refere-se à variabilidade nos dados de réplicas de uma mesma amostra homogênea em condições normais de ensaio e é avaliada pelo cálculo do %CV das alíquotas múltiplas incluídas. Esperava-se que as três alíquotas testadas dentro de cada lote produzissem um %CV intra-ensaio ≤25,0%, exceto para o limite superior e inferior do QC, onde ≤30,0% era esperado. Isso foi atendido para todos os cinco níveis de CQ em cada um dos 60 lotes (30 de 30 desempenhos totais). De modo geral, foi possível alcançar maior precisão intraensaio do que os critérios-alvo, com uma média intra-ensaio %CV de 7,7% em todos os níveis de CQ. A exatidão refere-se à proximidade de concordância entre o valor determinado experimentalmente e o valor nominal. Isso é avaliado calculando-se o erro relativo percentual (%RE, ou %Viés) entre as concentrações calculadas de cada CQ e suas concentrações nominais teoricamente esperadas. Esperava-se que a média intra-ensaio das três alíquotas fosse de ±25,0% ER da concentração nominal, exceto para o QC dos limites superior e inferior, onde ±30,0% era esperado. Isso foi atendido para todos os cinco níveis de CQ em cada um dos 60 lotes (30 de 30 desempenhos totais). De modo geral, maior acurácia intra-ensaio do que nossa meta poderia ser alcançada, com uma média absoluta intra-ensaio %ER de 4,2% em todos os níveis de CQ. Em todos os desempenhos das NTC (30 no total), não foram detectadas gotículas positivas.
A exatidão e a precisão interensaio também foram calculadas usando a média intraensaio de cada nível de CQ dentro de cada lote. A precisão interensaio foi esperada para ≤25,0% CV, exceto para o limite superior e inferior do QC, onde ≤30,0% era esperado. Da mesma forma, para a acurácia interensaios, esperava-se ±25,0% de ER, exceto para o limite superior e inferior do QC, onde ±30,0% era esperado. Observou-se acurácia e precisão interensaio significativamente maiores do que esses alvos (Tabela 9), com precisão interensaio variando de 4,0% a 8,5% e acurácia absoluta interensaio variando de 1,0% a 3,2%. Coletivamente, esses resultados demonstram que esse método pode alcançar precisão e precisão intra e interensaios suficientes dentro das metas atuais da indústria. Uma faixa dinâmica deste ensaio de 2.500-2,5 cópias por μL de reação de PCR pode ser definida com base nesses resultados, com uma sensibilidade geral do ensaio de 2,5 cópias por μL de reação de PCR. Como mencionado anteriormente, pode ser possível validar faixas dinâmicas mais amplas.
Em seguida, foi necessário avaliar a acurácia e precisão do ensaio dentro da matriz alvo - no caso, as lágrimas. Normalmente, os ensaios são validados antes do início dos estudos clínicos, o que significa que é improvável que as lágrimas coletadas de pacientes tratados com vetor estejam disponíveis para fins de validação. Isso pode ser criado artificialmente ao aumentar o vetor AAV alvo em lágrimas coletadas de doadores voluntários para criar QCs com pico de matriz. Para prova de princípio, um vetor AAV2 que expressa eGFP adquirido de uma fonte comercial foi utilizado. A concentração do estoque do vetor AAV2 foi determinada empiricamente por meio de ddPCR, sem a utilização de uma etapa de isolamento de DNA, como descrito neste protocolo. Em cada corrida, o AAV2 foi independentemente cravado nas três alíquotas lacrimais em um nível alto (esperado 1,41 x 103 cópias/μL de reação de PCR) e baixo (28,2 cópias/μL de reação de PCR). Alíquotas não cravadas foram incluídas como controle para demonstrar a especificidade do método.
O desempenho intra-ensaio foi avaliado para cada lote em cada nível de espiga . Esperava-se que todas as amostras lacrimais tivessem pelo menos 10.000 gotículas. Isso foi encontrado em 108 dos 108 poços testados em todos os seis lotes, com um número total médio de gotículas de 20.208 gotículas/poço (Tabela 10). Em seguida, esperava-se que o %CV interpoços de cada conjunto de poços duplicados de cada CQ fosse de ≤25,0% para os níveis de espiga alto e baixo. Isso foi atendido em 36 dos 36 conjuntos de poços testados em todos os seis lotes para os CQs, com uma média de %CV entre poços de 3,2% (Tabela 11).
Dentro de cada lote, a média intra-ensaio e o desvio padrão foram calculados para cada uma das pontas lacrimais preparadas independentemente, e estes foram usados para calcular uma média intra-ensaio para cada concentração em cada ensaio. Este foi utilizado para avaliar a acurácia e precisão do ensaio em matriz (Tabela 12). Esperávamos que o %CV intra-ensaio fosse de ≤25,0% e níveis de pico altos e baixos. Isso foi cumprido em seis dos seis lotes para cada nível. De modo geral, foi possível atingir maior precisão intra-ensaio na matriz do que a meta, com média intra-ensaio %CV de 3,7% no nível alto e 12,2% no nível baixo (8,0% no nível geral). Também era esperado que o %RE intraensaio fosse de ±25,0% em ambos os níveis de pico. Isso foi cumprido em seis dos seis lotes para cada nível. Da mesma forma, verificou-se, de modo geral, que a acurácia intra-ensaio na matriz maior do que a meta poderia ser alcançada, com %ER absoluto intraensaio médio de 8,1% no nível baixo e 11,3% no nível alto (9,7% no nível geral). Para o controle sem ponta, nenhum sinal de eGFP foi detectável em nenhuma das alíquotas (Tabela 12), demonstrando a especificidade do método na matriz lacrimal humana.
A exatidão interensaio e a precisão na matriz lacrimal também foram calculadas usando a média intra-ensaio de cada nível de espícula dentro de cada lote. Esperava-se que a precisão interensaio fosse de ≤25,0% CV e, para a acurácia interensaio, esperava-se ±25,0% de ER. Observou-se acurácia interensaio e precisão significativamente maiores do que esses alvos (Tabela 13), com precisão interensaio de 5,5% no nível alto e 7,1% no nível baixo, e acurácia absoluta interensaio de 11,3% no nível alto e 8,1% no nível baixo. Coletivamente, esses resultados demonstram a acurácia, precisão e especificidade do método em matriz lacrimal.
Tabela 5: Controles de qualidade utilizados para definir a faixa dinâmica do ensaio. Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
Tabela 6: Contagem total de gotículas e razão entre gotículas positivas e totais do controle de qualidade de DNA sintético de fita dupla e NTC. Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
Tabela 7: Estatísticas entre poços de CQ (alvos de cópia/reação de PCR μL). Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
Tabela 8: Acurácia intra-ensaio e precisão dos CQ (alvos de cópia/reação de PCR μL). Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
Tabela 9: Acurácia interensaio e precisão dos CQ (alvos de cópia/reação de PCR μL). Abreviaturas: ULQC = limite superior de controle de qualidade; HQC = alto controle de qualidade; MQC = controle de qualidade médio; CQL = baixo controle de qualidade; LLQC = controle de qualidade limite inferior; NTC = nenhum controle de modelo. Clique aqui para baixar esta tabela.
Tabela 10: Contagem total de gotículas das amostras lacrimais. Clique aqui para baixar esta tabela.
Tabela 11: Estatísticas entre poços de amostras lacrimais (alvos de cópia/reação de PCR μL). Clique aqui para baixar esta tabela.
Tabela 12: Acurácia intra-ensaio e precisão das amostras lacrimais (alvos de cópia/reação de PCR μL). Clique aqui para baixar esta tabela.
Tabela 13: Exatidão interensaio e precisão das amostras lacrimais (alvos de cópia/reação de PCR μL). Clique aqui para baixar esta tabela.
Tabela Suplementar S1: Sequências de primer, sondas e controle de qualidade sintético de DNA de fita dupla utilizadas neste estudo. Clique aqui para baixar esta tabela.
Discussion
Existem várias etapas do protocolo ddPCR que são críticas para o bom desempenho do ensaio. O primeiro passo crítico é o projeto e otimização dos primers e da sonda. Em geral, o uso de química baseada em sonda de hidrólise sobre química baseada em corante (por exemplo, SYBR Green) em um ambiente pré-clínico ou clínico é recomendado devido à sua especificidade superior. Além disso, a escolha do alvo de amplificação é crítica. Normalmente, o transgene de interesse do vetor é direcionado. No entanto, em estágios pré-clínicos mais precoces ou em vetores onde pode não ser possível distinguir o transgene do vetor versus o DNA genômico, pode ser apropriado usar alvos vetoriais padronizados. Por exemplo, pode-se visar a região de repetição terminal invertida, o promotor, a cauda poli-A ou as junções intersegmentos entre esses componentes vetoriais. A escolha do alvo varia de acordo com o design vetorial. As estratégias e o software tradicionais de projeto de sonda e primer qPCR são tipicamente apropriados para ddPCR. Os parâmetros de projeto que devem produzir uma temperatura de recozimento consistente (por exemplo, 60°C) devem ser selecionados para reduzir a quantidade de otimização necessária. Também foi recomendado projetar, ordenar e avaliar pelo menos três conjuntos diferentes para cada alvo. Deve-se, então, selecionar o conjunto que apresenta maior especificidade (ausência de amplificação no poço de controle negativo ou em uma matriz de DNA-alvo relacionado) e sensibilidade (i.e., limite de detecção)20.
Se for vantajoso poder fazer a transição do ensaio entre qPCR e ddPCR, recomenda-se otimizar primeiro as condições do ensaio usando qPCR e identificar condições para o conjunto selecionado que resultem em eficiências de amplificação de 90%-110% com um R2 ≥ 0,98. No entanto, a ddPCR como um método de endpoint é tipicamente menos sensível do que a qPCR devido a variações nas eficiências de amplificação. No mínimo, recomenda-se executar um gradiente térmico de temperatura na etapa de recozimento/extensão para cobrir temperaturas acima e abaixo das temperaturas de recozimento esperadas e avaliar a separação de amplitude de chuva e fluorescente entre os aglomerados de gotículas negativas e positivas em função da temperatura. Se o espaço de trabalho permitir, é recomendável ter estações de trabalho dedicadas individuais para preparação de mistura mestre, adição de modelo e amplificação. Sempre que possível, estes devem ser fisicamente segregados por um fluxo de trabalho unidirecional com controlos de engenharia incorporados, tais como acesso controlado e pressões de ar diferenciais, para reduzir o risco de contaminação cruzada e falsos positivos. Se isso não for possível, deve-se tomar extrema cautela para evitar a contaminação cruzada.
Existem duas etapas neste protocolo que podem parecer incomuns para aqueles que estão mais acostumados com o desenvolvimento do ensaio de qPCR. A primeira é a inclusão de uma enzima de restrição no master mix de PCR. Durante a amplificação da ddPCR, cada gotícula é termociclada até o ponto final. Em um ensaio devidamente otimizado, isso resulta em duas populações de gotículas, um conjunto exibindo um nível consistentemente alto de sinais fluorescentes - os positivos - e outro exibindo consistentemente um baixo nível de sinal fluorescente - os negativos. Se ocorrer interferência da PCR, ela pode dessincronizar o início da amplificação da PCR, fazendo com que a gota não atinja um platô de amplificação e, portanto, desfechos fluorescentes inconsistentes. Nesse caso, as gotículas serão distribuídas entre os negativos e positivos, resultando em um fenômeno chamado chuva ddPCR. Isso pode resultar em quantificação imprecisa do alvo e limiares aplicados de forma inconsistente e subjetiva. Nossa recomendação é definir o limite ligeiramente acima do sinal do NTC, o que deve minimizar os efeitos da chuva na quantificação final, pois todas as gotículas ainda são consideradas positivas, mesmo que não totalmente cicladas até o ponto final. Os AAVs possuem uma estrutura secundária de alta complexidade que, dependendo do alvo de amplificação, pode reduzir a acessibilidade aos primers e sondas, resultando em interferência da PCR e, consequentemente, chuva. A inclusão da enzima de restrição na mistura mestre cliva essa estrutura secundária para aumentar o acesso dos primers e sondas, reduzindo a chuva, o que pode melhorar a precisão do ensaio. Os efeitos da inclusão de uma enzima de restrição na reação de ddPCR foram descritos anteriormente25,32. Qualquer enzima de restrição pode ser usada, desde que seja confirmado que não corta dentro da região alvo de amplificação. Não são necessárias etapas de pré-digestão ou composições alternativas de tampão.
O segundo passo incomum é a preparação da amostra lacrimal contendo AAV. Nesse protocolo, uma proporção de 1:10 (ou maior) de lágrimas foi utilizada e, posteriormente, a amostra foi aquecida. Tipicamente, quando as lágrimas são coletadas por meio de um tubo capilar, que é um método de coleta amplamente utilizado, em média podem ser coletados aproximadamente 10,0 μL33. A diluição ajuda a resolver o volume limitado da amostra e fornece material suficiente para o teste de poços duplicados. Embora isso reduza o limite teórico de detecção, a sensibilidade robusta da ddPCR ainda deve resultar na detecção de todos, exceto um número extremamente pequeno de partículas vetoriais. Essa abordagem também cria um poço de "backup" se um falhar inesperadamente. Nesse caso, ou em casos de volume de amostra insuficiente para executar dois poços, o erro de Poisson poderia ser usado para avaliar a precisão. Além disso, nos casos em que a concentração está abaixo do limite de detecção, cria-se uma oportunidade de mesclar dados de poços para determinar uma concentração. É necessário liberar os vetores AAV dos capsídeos virais para detecção de ddPCR. Alguns métodos para a quantificação do AAV incluíram uma etapa de digestão da proteinase K para remoção do capsídeo viral34,35,36. Todos os sorotipos de AAV de ocorrência natural têm temperaturas de fusão iguais ou inferiores a aproximadamente 90 °C, com a maioria caindo abaixo de 80 °C; portanto, essa parece ser uma inclusão desnecessária37. O aquecimento por si só parece ser suficiente para liberar DNA vetorial.
Além disso, a ddPCR é geralmente menos suscetível aos inibidores de PCR que podem estar presentes em uma amostra que pode afetar um ensaio de qPCR. Se uma etapa específica de isolamento de DNA for incluída, isso também exigiria validação específica, o que é evitado neste protocolo. As amostras são diluídas antes do aquecimento devido à cinética de difusão dos genomas do vetor em um líquido. Durante o processo de aquecimento e resfriamento subsequente, as cadeias de sentido positivo e negativo do genoma de DNA de fita simples podem se agrupar para produzir um intermediário de fita dupla se as concentrações forem suficientemente altas. A diluição antes do aquecimento reduz as concentrações e torna matematicamente improvável que se formem intermediários de fita dupla suficientes para ter um efeito adverso na precisão da quantificação. Deve-se notar que os fragmentos de DNA sintético ou plasmídeos linearizados usados como controles de qualidade não devem ser submetidos a essa etapa de aquecimento. Como estes são de fita dupla, o aquecimento resultaria na conversão para intermediários de fita simples. Após a partição independente desses QCs de fita simples em gotículas, espera-se que isso resulte em um aumento de duas vezes na concentração de QC em relação à concentração nominal. Alternativamente, se os CQ devem ser aquecidos para padronizar o método, isso deve ser levado em consideração na reconstituição e atribuição de uma concentração nominal.
Finalmente, com relação à preparação da amostra, muitos protocolos também recomendam a inclusão de uma etapa de tratamento com DNase para remover qualquer DNA vetorial não capturado. Esta etapa é crítica nos casos em que não se deseja quantificar o DNA livre associado à preparação do vetor (como durante a quantificação para fins de dosagem). No entanto, no contexto de estudos de biodistribuição e bioshedding, normalmente deseja-se saber para onde qualquer DNA vetorial viajou, independentemente de estar encapsidado ou não. Portanto, sugere-se que normalmente não se realize uma etapa de tratamento com DNase durante tais estudos. Se for determinado que é necessário incluir uma etapa de DNase, esta etapa deve ser anterior às diluições e ao aquecimento.
Neste artigo, são apresentados dados representativos da abordagem para avaliação da faixa dinâmica, sensibilidade, exatidão e precisão do método no contexto de uma validação adequada às boas práticas de laboratório em conformidade com o propósito. A atual falta de orientação sobre esse tema faz com que os laboratórios validadores determinem os critérios de ensaio alvo para si mesmos, em linha com o pensamento atual da indústria. Diferentes grupos apresentaram critérios-alvo mais altos e inferiores aos utilizados neste estudo 19,20,21,22,23,24,25. Os critérios de ensaio alvo, até que sejam mais rigidamente definidos, devem ser selecionados antes da validação com base nas aplicações clínicas pretendidas do método. Dependendo das decisões a jusante a tomar com base nos dados, podem ser necessários níveis mais elevados de exatidão e precisão. Por outro lado, um simples resultado positivo versus negativo pode ser suficiente.
A abordagem também abordou recomendações para a avaliação da especificidade e de um efeito de matriz. Um pool de lágrimas coletadas de indivíduos não tratados não produziu um resultado positivo neste ensaio, enquanto o alvo pôde ser detectado quando o vetor foi cravado nas lágrimas em uma concentração alta e baixa dentro das taxas de recuperação recomendadas. Idealmente, matriz contendo vetor endógeno (por exemplo, coletado após tratamento com vetor viral) também seria incluída nessas avaliações. No entanto, é improvável que tais amostras estejam disponíveis para uso em uma validação. Para aumentar a robustez da validação, vários pools de lágrimas, ou lágrimas coletadas de uma variedade de indivíduos, poderiam ser avaliados para determinar se um efeito de matriz específico do paciente ocorre. Por fim, recomenda-se avaliar a estabilidade. Em fluxos de trabalho onde a extração de DNA ocorre fora da matriz biológica, pode ser necessário avaliar a estabilidade tanto da amostra quanto do DNA extraído. Nesse fluxo de trabalho, a amostra é testada diretamente no ensaio, sem a necessidade de extração de DNA. Portanto, considerando a avaliação da estabilidade desse método, deve-se avaliar a estabilidade das amostras lacrimais. Normalmente, recomendam-se avaliações de bancada, geladeira, congelamento/descongelamento e estabilidade de longo prazo. Estes não foram realizados como parte deste estudo, mas os métodos aqui desenvolvidos podem ser utilizados nesta avaliação, após manipulações das amostras de entrada.
Em geral, este método demonstrou ser um ensaio robusto, repetível e validável para detectar vetores baseados em AAV em amostras lacrimais. Pode servir como uma plataforma a ser adaptada a vetores específicos para apoiar ensaios clínicos e fornece uma base para a validação de um ensaio consistente com boas práticas de laboratório.
Disclosures
Todos os autores são funcionários da KCAS Bioanalytical and Biomarker Services, uma organização de pesquisa contratada que fornece serviços abrangentes de desenvolvimento em conformidade com Boas Práticas de Laboratório/Boas Práticas Clínicas, desde o suporte à descoberta precoce até o registro do produto, incluindo o suporte a ensaios baseados em AAV, conforme descrito neste protocolo. O KCAS financiou este projeto e a publicação deste protocolo. Embora não haja diretrizes atuais da FDA para o desenho e validação dos experimentos apresentados neste artigo, os especialistas e líderes de pensamento que desenvolvem tais estudos no KCAS adotaram essa abordagem como a abordagem de base. Critérios e parâmetros adicionais são discutidos projeto a projeto e podem ser incluídos com base no uso pretendido.
Acknowledgements
Gostaríamos de agradecer a Nick Russell e Brandon McKethan da Bio-Rad por suas discussões úteis durante o desenvolvimento deste método.
Materials
| Name | Company | Catalog Number | Comments |
| AAV-eGFP Vector | Charles River Laboratories | RS-AAV2-FL | Lot AAV2-0720-FL, used as a proof of principle vector |
| AutoDG Droplet Digital PCR system | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| AutoDG Oil for Probes | Bio-Rad | 1864110 | Or use material compatible with ddPCR system. |
| ddPCR Buffer Control for Probes | Bio-Rad | 1863052 | Or use material compatible with ddPCR system and PCR chemistry. |
| ddPCR Droplet Reader Oil | Bio-Rad | 1863004 | Or use material compatible with ddPCR system. |
| ddPCR Piercable Foil Seals | Bio-Rad | 1814040 | Or use material compatible with ddPCR system. |
| ddPCR Plates 96-Well, Semi-Skirted | Bio-Rad | 12001925 | Or use material compatible with ddPCR system. |
| ddPCR Supermix for Probes (no dUTP) | Bio-Rad | 1863023, 1863024, or 1863025 | Use master mix compatible with primers/probes and ddPCR system. |
| DG32 AutoDG Cartidges | Bio-Rad | 1864108 | Or use material compatible with ddPCR system. |
| Droplet Reader | Bio-Rad | QX200 | Alternative ddPCR system may be used following manufacturer’s protocol. |
| GeneAmp PCR Buffer | Applied Biosystems | N8080129 | N/A |
| Nuclease-Free Water | Ambion | AM9906 | N/A |
| PCR Plate Sealer | Bio-Rad | PX1 | Or use material compatible with ddPCR system. |
| Pipet Tips for AutoDG | Bio-Rad | 1864120 | Or use material compatible with ddPCR system. |
| Pluronic F-68 Non-ionic Surfactant | Gibco | 24040 | N/A |
| Primer and Hydrolysis Probes | Various | Various | Design based on target sequence using general approaches for primer/probe design. Select fluorphores and quenchers compatible with ddPCR system. |
| Restriction Enzyme | Various | Various | Varies with target amplification sequence. Use restriction enzyme that does not cut in the amplified sequence |
| Sheared salmon sperm DNA | ThermoFisher | AM9680 | N/A |
| Synthetic DNA gene fragment or linearized plasmid | Various | Various | Design a synthetic DNA fragment containing the target amplification region for use as a quality control |
| TE Buffer | Teknova | T0224 | Ensure prepared or purchases nuclease free. 10 mM Tris-HCl, 1.0 mM EDTA, pH=8.0 |
| Touch Thermal Cycler | Bio-Rad | C1000 | Or use material compatible with ddPCR system. |
References
- Sherkow, J. S., Zettler, P. J., Greeley, H. T. Is it 'gene therapy. Journal of Law and the Biosciences. 5 (3), 786-793 (2018).
- Ginn, S. L., Amaya, A. K., Alexander, I. E., Edelstein, M., Abedi, M. R. Gene therapy clinical trials worldwide to 2017: an update. The Journal of Gene Medicine. 20 (5), 3015 (2018).
- Ghosh, S., Brown, A. M., Jenkins, C., Campbell, K. Viral vector systems for gene therapy: a comprehensive literature review of progress and biosafety challenges. Applied Biosafety. 25 (1), 7-18 (2020).
- Gene, Cell, & RNA therapy landscape, Q4 2022 quarterly data report. American Society for Gene & Cell Therapy Available from: https://asgct.org/global/documents/asgct_citeline-q4-2022-report_final.aspx (2022)
- Approved Cellular and Gene Therapy Products. United States Food and Drug Administration Available from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products (2022)
- Au, H. K. E., Isalan, M., Meilcarek, M. Gene therapy advances: a meta-analysis of AAV usage in clinical settings. Frontiers in Medicine. 8, 809118 (2022).
- Lundstrom, K. Viral vectors in gene therapy. Diseases. 6 (2), 42 (2018).
- Naso, M. F., Tomkowicz, B., Perry, W. L., Strohl, W. R. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 31, 317-334 (2017).
- Russell, S., et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomized, controlled, open-label, phase 3 trial. Lancet. 390 (10097), 849-860 (2017).
- Chemistry, manufacturing, and control (CMC) information for human gene therapy investigational new drugs (INDs). United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/chemistry-manufacturing-and-control-cmc-information-human-gene-therapy-investigationsal-new-drug (2020)
- Long term follow-up after administration of human gene therapy products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-adminstration-human-gene-therapy-products (2020)
- Testing of retroviral vector-based human gene therapy products for replication competent retrovirus during product manufacture and patient follow-up. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/testing-retroviral-vector-based-human-gene-therapy-products-replication-competent-retrovirus-during (2020)
- Recommendations for microbial vectors used in gene therapy. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/recommendations-microbial-vectors-used-gene-therapy (2016)
- Multidisciplinary: gene therapy. European Medicines Agency Available from: https://www.europa.eu/en/human-regulatory-development/scientific-guidelines/multidisciplinary/multidisciplinary-gene-therapy (2023)
- Human gene therapy for retinal disorders. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/human-gene-therapy-retinal-disorders (2020)
- Design and analysis of shedding studies for virus or bacterial-based gene therapy and oncolytic products. United States Food and Drug Administration Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-and-analysis-shedding-studies-virus-or-bacteria-based-gene-therapy-and-oncolytic-products (2015)
- Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. European Medicines Agency Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-quality-non-clinical-clinical-aspects-gene-therapy-medicinal-products_en.pdf (2018)
- Kaur, S. white paper on recent issues in bioanalysis: mass spec of proteins, extracellular vesicles, CRISPR, chiral assays, oligos; nanomedicines bioanalysis; ICH M10 section 7.1; non-liquid & rare matrices; regulatory inputs (part 1A - recommendations on endogenous compounds, small molecules, complex methods, regulated mass spec of large molecules, small molecule, PoC & part 1B - regulatory agencies' inputs on bioanalysis, biomarkers, immunogenicity, gene & cell therapy and vaccine). Bioanalysis. 14 (9), 505-580 (2022).
- Hays, A., Islam, R., Matys, K., Williams, D. Best practices in qPCR and qPCR validation in regulated bio analytical laboratories. The AAPS Journal. 24 (2), 36 (2022).
- Ma, H., Bell, K. N., Loker, R. N. qPCR and qRT-PCR analysis: Regulatory points to consider when conducting biodistribution and vector shedding studies. Molecular Therapy. Methods & Clinical Development. 17 (20), 152-168 (2020).
- Wissel, M. Recommendations on qPCR/ddPCR assay validation by GCC. Bioanalysis. 14 (12), 853-863 (2022).
- Expectations for biodistribution (BD) assessments for gene therapy (GT) products. International Pharmaceutical Regulators Programme Available from: https://admin.iprp.global/sites/default/files/2018-09/IPRP_GTWG_ReflectionPaper_BD_Final_2018_0713.pdf (2018)
- Pinheiro, L., Emslie, K. R. Basic concepts and validation of digital PCR measurements. Methods in Molecular Biology. 1768, 11-24 (2018).
- Tzonev, S. Fundamentals of counting statistics in digital PCR: I measured two target copies-what does it mean. Methods in Molecular Biology. 1768, 25-43 (2018).
- Prantner, A., Marr, D. Genome concentration, characterization, and integrity analysis of recombinant adeno-associated viral vectors using droplet digital PCR. PLoS One. 18, 0280242 (2023).
- Primer-BLAST. National Library of Medicine, National Center for Biotechnology Information Available from: https://www.ncbi.nim.nih.gov/tools/primer-blast/ (2023)
- Koressaar, T., Remm, M. Enhancements and modifications of primer design program Primer 3. Bioinformatics. 23 (10), 1289-1291 (2007).
- Ye, J. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 13, 134 (2012).
- Droplet Digital PCR Application Guide. Bio-Rad Available from: https://www.bio-rad.com/webroot/we/pdf/lsr/literature/Bulletin_6407.pdf (2023)
- Qian, P. L., Sauzade, M., Brouzes, E. dPCR: A technology review. Sensors. 18 (4), 1271 (2018).
- Basu, A. Digital assays part I: portioning statistics and digital PCR. SLAS Technology. 22 (4), 369-386 (2017).
- Sanmiguel, J., Gao, G., Vandeberghe, L. H. Quantitative and digital droplet-based AAV genome titration. Methods in Molecular Biology. 1950, 51-83 (2019).
- Bachhuber, F., Huss, A., Senel, M., Tumani, H. Diagnostic biomarkers in tear fluid: from sampling to preanalytical processing. Scientific Reports. 11, 10064 (2021).
- Martinez-Fernandez de la Camara, C., McClements, M. E., MacLaren, R. E. Accurate quantification of AAV vector genomes by quantitative PCR. Genes. 12 (4), 601 (2021).
- Ai, J., Ibraheim, R., Tai, P. W. L., Gao, G. A scalable and accurate method for quantifying vector genomes of recombinant adeno-associated viruses in crude lysate. Human Gene TherapyMethods. 28 (3), 139-147 (2017).
- Dobnik, D., et al. Accurate quantification and characterization of adeno-associated viral vectors. Frontiers in Microbiology. 10, 1570 (2019).
- Bennett, A., et al. Thermal stability as a determinant of AAV serotype identity. Molecular Therapy Methods and Clinical Development. 6, 171-182 (2017).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved