Kryokonservierung von Primordialen Keimzellen und Wiederbelebung von Drosophila-Stämmen
* These authors contributed equally
In This Article
Summary
Eine Langzeitkonservierungsmethode für Drosophila-Stämme als Alternative zum häufigen Umtopfen von adulten Fliegen in frische Fläschchen ist sehr wünschenswert. Dieses Protokoll beschreibt die Kryokonservierung von Drosophila-Urkeimzellen und die Wiederbelebung von Stämmen durch ihre Transplantation in agametische Wirtsembryonen.
Abstract
Drosophila-Stämme müssen durch häufiges Umfüllen von erwachsenen Fliegen in neue Fläschchen erhalten werden. Dies birgt die Gefahr einer Mutationsverschlechterung und phänotypischer Veränderungen. Die Entwicklung einer alternativen Methode zur Langzeitkonservierung ohne solche Änderungen ist daher zwingend erforderlich. Trotz früherer erfolgreicher Versuche ist die Kryokonservierung von Drosophila-Embryonen aufgrund der geringen Reproduzierbarkeit immer noch nicht von praktischem Nutzen. In dieser Arbeit beschreiben wir ein Protokoll für die Kryokonservierung von primordialen Keimzellen (PGC) und die Wiederbelebung von Stämmen durch Transplantation von kryokonservierten PGCs in agametische Wirtsembryonen von Drosophila melanogaster (D. melanogaster). PGCs sind sehr durchlässig für Kryoprotektiva (CPAs), und die entwicklungsbedingte und morphologische Variation zwischen den Stämmen ist weniger problematisch als bei der Kryokonservierung von Embryonen. Bei dieser Methode werden PGCs von etwa 30 Spenderembryonen entnommen, nach der CPA-Behandlung in eine Nadel geladen und dann in flüssigem Stickstoff kryokonserviert. Um Keimzellen aus Spendern herzustellen, werden die kryokonservierten PGCs in einer Nadel aufgetaut und dann in etwa 15 agametische Wirtsembryonen eingebracht. Mit diesem Protokoll wurde eine Häufigkeit von mindestens 15 % fruchtbarer Fliegen erreicht, und die Anzahl der Nachkommen pro fruchtbarem Paar war immer mehr als ausreichend, um den ursprünglichen Stamm wiederzubeleben (die durchschnittliche Nachkommenzahl betrug 77,2 ± 7,1), was auf die Fähigkeit kryokonservierter PGCs hinweist, sich in Keimbahnstammzellen zu verwandeln. Die durchschnittliche Anzahl der fruchtbaren Fliegen pro Nadel betrug 1,1 ± 0,2, und 9 von 26 Nadeln brachten zwei oder mehr fruchtbare Nachkommen hervor. Es wurde festgestellt, dass 11 Nadeln ausreichen, um 6 oder mehr Nachkommen zu produzieren, in denen wahrscheinlich mindestens ein Weibchen und ein Männchen enthalten sind. Der agametische Wirt ermöglicht es, den Stamm schnell wiederzubeleben, indem er einfach neu geschlüpfte weibliche und männliche Fliegen kreuzt. Darüber hinaus haben PGCs das Potenzial, in gentechnischen Anwendungen, wie z. B. der Genom-Editierung, eingesetzt zu werden.
Introduction
Die Aufrechterhaltung von Drosophila-Stämmen durch den Transfer erwachsener Fliegen in neue Futterfläschchen führt unweigerlich zur Anhäufung von Mutationen und epigenetischen Veränderungen im Laufe der Zeit. Die Entwicklung einer alternativen Methode zur langfristigen Erhaltung von Drosophila-Stämmen ohne solche Veränderungen ist unerlässlich, insbesondere für Referenzstämme, bei denen das gesamte Genom erhalten bleiben muss. Es wurden mehrere erfolgreiche Versuche zur Kryokonservierung von Drosophila-Embryonen oder -Eierstöcken beschrieben 1,2,3. Leider sind sie aufgrund der geringen Reproduzierbarkeit immer noch nicht von praktischem Nutzen. In der Tat haben Embryonen im Frühstadium aufgrund ihres hohen Dottergehalts, der die Permeation und Diffusion von Kryoprotektiva (CPA) behindert, eine niedrige Überlebensrate nach der Kryokonservierung 2,3. Die CPA-Permeabilität ist auch durch die wachsartigen Schichten von Embryonen im Spätstadium stark eingeschränkt. Es ist schwierig und zeitaufwändig, einen stammspezifischen Zeitraum zu finden, in dem Embryonen eine hohe Überlebensrate und eine dünnere Wachsschicht aufweisen. Kürzlich haben Zhan et al.4 die Methoden zur Permeabilisierung von Embryonen, zur CPA-Beladung und zur Vitrifikation verbessert und erfolgreich Embryonen mehrerer Stämme kryokonserviert. Die Methoden sind jedoch nicht einfach anzuwenden, da die Lebensfähigkeit der Embryonen nach der Permeabilisierung tendenziell schlecht ist. Daher sind weitere Verbesserungen und die Entwicklung alternativer Ansätze erforderlich. Methoden zur Kryokonservierung von primordialen Keimzellen (PGCs) sind ein alternativer Ansatz für die langfristige Erhaltung von Drosophila-Stämmen.
Die PGC-Transplantation (auch Polzelltransplantation genannt) wurde zur Erzeugung von Keimbahnchimären, insbesondere von Weibchen, verwendet, um Prozesse wie die mütterlichen Auswirkungen von zygotischen letalen Mutationen und die Geschlechtsbestimmung von Keimzellen zu untersuchen 5,6,7,8,9,10,11,12 . PGCs sind viel kleiner als Embryonen und wahrscheinlich sehr durchlässig für die meisten Kryoprotektiva. Darüber hinaus ist die entwicklungsbedingte und morphologische Variation zwischen den Stämmen weniger problematisch, und ein agametischer Wirt ermöglicht eine schnelle Wiederherstellung ganzer Genome. Wir haben kürzlich eine neue Methode der PGC-Kryokonservierung13 entwickelt, die die sonst unvermeidlichen genetischen und epigenetischen Veränderungen bei Drosophila-Stämmen verhindert. Hier stellen wir Ihnen das detaillierte Protokoll vor.
Diese Kryokonservierungsmethode erfordert spezifisches Fachwissen in der Handhabung und Instrumentierung von PGC. Während ein schrittweiser Ansatz für diejenigen, die damit nicht vertraut sind, eine effiziente Lösung sein kann, kann er für kleine Labore aufgrund der Anforderungen an die Instrumentierung ungeeignet sein. Dieses PGC-Kryokonservierungsprotokoll kann aufgrund geringerer Entwicklungs- und morphologischer Unterschiede leichter für die Verwendung mit verschiedenen Drosophila-Arten und verschiedenen Insektenarten angepasst werden als Embryo-Kryokonservierungsprotokolle. PGCs können möglicherweise auch in gentechnischen Anwendungen eingesetzt werden, wie z. B. bei der Genom-Editierung 14,15,16. Zusammenfassend lässt sich sagen, dass diese Methode in Lagerzentren und anderen Laboratorien eingesetzt werden kann, um Fliegen- und andere Insektenstämme über längere Zeiträume ohne Veränderungen zu erhalten.
Protocol
1. Vorbereitung der Ausrüstung
- Mikromanipulatorsystem: Zusammenbau eines Mikromanipulatorsystems zur Entnahme und Transplantation von Zellen (Abbildung 1A).
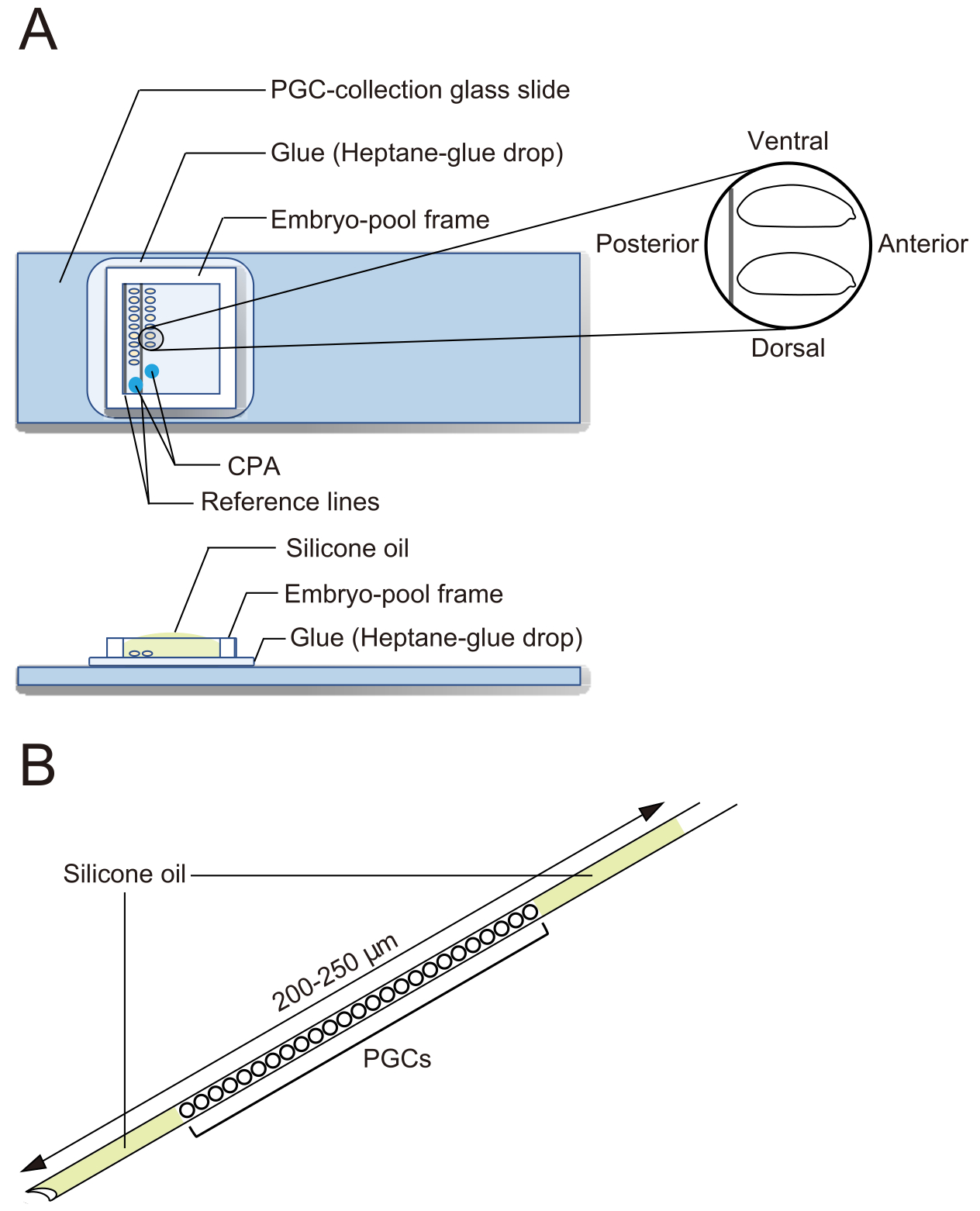
- Objektträger aus PGC-Sammlung (Abbildung 2A)
- Um Heptankleber herzustellen, schneiden Sie ein ca. 30 cm langes doppelseitiges Klebeband ab und weichen Sie es über Nacht in 7 ml technischer (normaler) Heptanlösung ein.
- Zeichnen Sie zwei parallele Referenzlinien für die Ausrichtung des Embryos auf die Rückseite eines Objektträgers.
- Tropfen des obigen Heptanklebers mit einer Pasteurpipette auf dem Objektträger (auf der Seite ohne die Linien) verteilen. Trocknen Sie die Oberfläche des Objektträgers an der Luft, bis sie weiß wird.
- Wiederholen Sie die Zugabe und das Verteilen von Heptan-Leimtropfen und trocknen Sie den Objektträger erneut.
HINWEIS: Der Kleber verhindert, dass sich flüssige Lösungen auf der flachen Oberfläche verteilen, und erleichtert das Einfüllen wässriger Lösungen in eine Nadel. - Um Embryo-Pool-Rahmen herzustellen, kleben Sie drei Lagen 0,2 mm dickes Standard-Vinylband, wie z. B. Isolierband, auf ein Schneidebrett. Schneide das Klebeband in 1,5 cm breite Rechtecke. Schneiden Sie dann alle drei Lagen Klebeband ab und lassen Sie einen Rahmen von 2 bis 3 mm frei.
HINWEIS: Nach dem Ausrichten der Embryonen wird ein Embryo-Pool-Rahmen angebracht, um einen Pool für Embryonen zu bilden.
- Transplantations-Nadeln
HINWEIS: Alle zum Zeitpunkt dieser Studie im Handel erhältlichen Nadeln waren zu schmal oder zu breit für die PGC-Kryokonservierung.- Stellen Sie eine Nadel mit einer Glaskapillare und einem Abzieher her. Wir verwenden einen NARISHIGE PN-31 Abzieher mit einem Heizniveau von 85,0-98,4, einem Magnethauptpegel von 57,8 und einem Magnet-Unterpegel von 45,0.
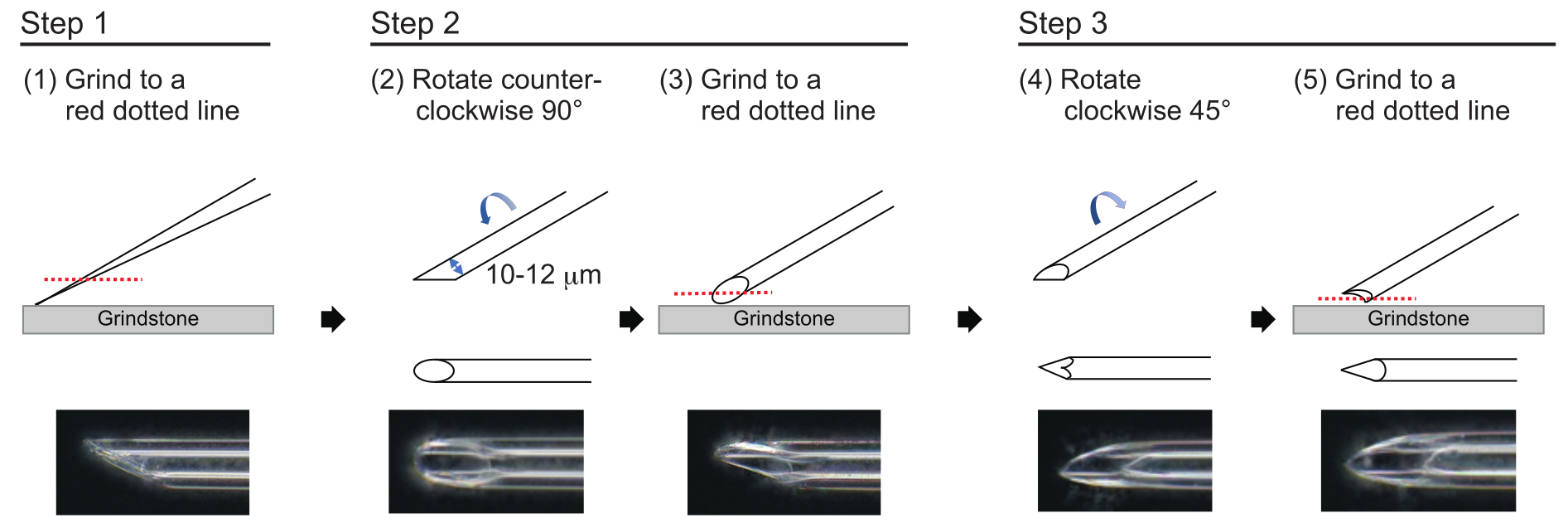
- Um eine Nadel mit einer ungefähren Wandstärke von 1 μm und einer Spitze von ca. 200 μm Länge mit einem Innendurchmesser von 10-12 μm herzustellen, polieren Sie die Nadelspitze im folgenden dreistufigen Prozess (Abbildung 3). Zuerst schleifen Sie die Nadelspitze in einem Winkel von 30° mit einer Drehzahl von 780 U/min nach unten, bis die Spitze einen Innendurchmesser von 10-12 μm hat. Dieser erste Schleifschritt dauert ca. 1 h.
HINWEIS: Um ein Brechen der Nadelspitze zu vermeiden, drehen Sie zuerst den Schleifstein und bewegen Sie dann die Nadel vorsichtig nach unten auf den Schleifstein. - Zeichne eine Linie auf die Oberseite der Nadel, um den gewünschten Winkel zu verfolgen. Drehen Sie die Nadel um 90° gegen den Uhrzeigersinn und polieren Sie sie erneut mit einer Drehzahl von 180 U/min. Dies dauert ca. 5 Minuten.
- Drehen Sie die Nadel im Uhrzeigersinn um 45° und polieren Sie sie eine Sekunde lang bei einer Drehzahl von 180 U/min.
- Legen Sie einen Objektträger mit einem Tropfen Chromsäuremischung (ACHTUNG: giftig) auf den Mikroskoptisch. Befestigen Sie die Nadel in einem Winkel von 10°-13° zur Objektträgeroberfläche am Kapillarhalter (Abbildung 1D), bewegen Sie die Nadel vorsichtig nach unten und tauchen Sie die Spitze in die Chromsäuremischung.
- Durch Ziehen und Drücken des Kolbens (Abbildung 1B) wird die Lösung mehrmals mechanisch von der Nadel geladen und entleert, um Glasreste in der Nadel zu entfernen. Achten Sie darauf, auch die Außenwand zu reinigen.
- Waschen Sie die Innen- und Außenseite der Nadel zweimal mit destilliertem Wasser, um die Chromsäure vollständig zu entfernen.
2. Sammlung und Kryokonservierung von PGCs
- Entnahme von Embryonen
- Eine angemessene Anzahl von Fliegen des betreffenden Spenderstamms (ca. 450 für jedes Geschlecht für den Embryo-Entnahmebecher) wird in einen Embryo-Entnahmebecher mit einer Embryo-Entnahmeplatte (Abbildung 1E) überführt und bei 25 °C inkubiert. Wir verwenden in der Regel 3 bis 5 Tage alte Elternfliegen, die unter weniger überfüllten Bedingungen bei Raumtemperatur (23-25 °C) aufgezogen werden.
- Führen Sie zwei 30-minütige Vorentnahmen durch und werfen Sie alle gelegten Eier weg. Da Weibchen befruchtete Eier, die sich im Eileiter entwickeln, zurückhalten können, ist dieser Schritt erforderlich, um die Eiablage in Schritt 2.1.3 zu synchronisieren (Abbildung 4).
- Nach den beiden Vorentnahmen werden die Embryonen 50 Minuten lang entnommen und dann in einer befeuchteten Kammer bei 25 °C inkubiert, damit sich die Embryonen bis zum Blastodermstadium entwickeln können (Frühstadium 517). Die Inkubationszeit beträgt in der Regel 100 Minuten, kann aber je nach Stamm auf bis zu 120 Minuten verlängert werden (Abbildung 4).
HINWEIS: Eine befeuchtete Kammer wird hergestellt, indem ein feuchtes Papiertuch auf den Boden einer Plastikbox gelegt und vor dem Gebrauch mit einem Wassernebel besprüht wird. Bei Embryonen im frühen Stadium 5 ist die PGC-Bildung abgeschlossen, die somatische Zellularisierung jedoch nicht. Das genaue Stadium eines Embryos wird in Schritt 2.4 unter dem Mikroskop bestimmt.
- Dechorionierung von Embryonen
- Geben Sie einen Tropfen destilliertes Wasser auf ein Sieb aus Edelstahlgewebe (150 mesh, 109 μm Öffnung, 60 μm Drahtdurchmesser; Abbildung 1F). Entnehmen Sie die Embryonen mit einer Pinzette von der Embryo-Entnahmeplatte und legen Sie sie in den Wassertropfen.
- Drücke Seidenpapier von unten gegen das Sieb, um das Wasser aufzusaugen. Die Embryonen werden mit Tröpfchen frischer 5%iger Natriumhypochloritlösung (als Cl) versetzt und 10 s lang ununterbrochen auf das Sieb geklopft.
- Waschen Sie die Embryonen, indem Sie sie direkt mit destilliertem Wasser bespritzen und Seidenpapier von unten gegen das Sieb drücken, um das Wasser aufzusaugen. Wiederholen Sie diesen Schritt 3x.
- Ausrichten dekorierter Embryonen
- Verwenden Sie unter einem Stereomikroskop eine Pinzette, um die Embryonen zu übertragen. Richten Sie die dechorionierten Embryonen in zwei Reihen auf einem PGC-Objektträger entlang der beiden Referenzlinien aus (Abbildung 2A). Die Embryonen sind mit der vorderen Seite nach rechts (die zu manipulierende Seite) und der ventralen Seite nach oben ausgerichtet.
HINWEIS: Dieser Schritt sollte in 20 Minuten abgeschlossen sein, in denen wir normalerweise etwa 40 Embryonen ausrichten. - Befestigen Sie einen Embryo-Pool-Rahmen um die Embryonen auf dem Glasobjektträger der PGC-Sammlung. Tropfen Sie 1 μl CPA-Lösung (1x Ephrussi-Beadle-Ringer-Lösung, EBR, enthält 20 % Ethylenglykol und 1 M Saccharose; 1x EBR: 130 mM NaCl, 5 mM KCl, 2 mM CaCl2 und 10 mM Hepes bei pH 6,9) an zwei getrennten Stellen in dem vom Rahmen umschlossenen Bereich und füllen Sie den Pool mit Silikonöl, um ein Austrocknen der Embryonen zu verhindern (Abbildung 2A).
HINWEIS: Zur Herstellung der CPA-Lösung werden 10,26 g Saccharose vollständig in etwa 20 ml destilliertemH2Ogelöst, das 3 ml 10 x EBR-Lösung enthält. Fügen Sie 6 ml Ethylenglykol hinzu und fügen Sie dann destilliertesH2Obis zu 30 ml hinzu. Nach gründlichem Mischen filtrieren Sie die Lösung durch eine 0,22 mm Einwegmembran.
- Verwenden Sie unter einem Stereomikroskop eine Pinzette, um die Embryonen zu übertragen. Richten Sie die dechorionierten Embryonen in zwei Reihen auf einem PGC-Objektträger entlang der beiden Referenzlinien aus (Abbildung 2A). Die Embryonen sind mit der vorderen Seite nach rechts (die zu manipulierende Seite) und der ventralen Seite nach oben ausgerichtet.
- Sammeln von PGCs
- Den Objektträger aus PGC-Sammelglas aus Schritt 2.3.2 auf den Tisch eines Mikroskops legen, das mit einem Mikromanipulatorsystem ausgestattet ist. Befestigen Sie die Nadel am Kapillarhalter und bringen Sie den ersten Embryo in der linken Reihe und die Nadelspitze in die gleiche Fokusebene. Silikonöl für 2-3 s in die Nadel geben.
- Beginnen Sie mit der PGC-Entnahme von Embryonen in der linken Reihe. Bewegen Sie mit einer 20-fachen Objektivlinse die Nadelspitze vorsichtig an die Oberfläche des vorderen Endes des Embryos und dringen Sie in Richtung des hinteren Endes ein, nicht durch Bewegen der Nadel, sondern durch Bewegen des Mikroskoptisches.
- Wenn die Nadelspitze das hintere Ende erreicht, ziehen Sie die Nadel leicht zurück und lassen Sie das Eigelb in der Nadel direkt in der somatischen Zellschicht vollständig ab.
- Halten Sie den Druck in der Nadel konstant, bewegen Sie die Nadelspitze zu den PGCs direkt im hinteren Pol und belasten Sie die PGCs vorsichtig, aber ohne viel Zeit in Anspruch zu nehmen.
- Ziehen Sie die Nadel schnell aus dem Embryo heraus und leiten Sie Eigelb und andere Verunreinigungen aus der Nadel in den Silikonölpool ab, wobei die PGCs in der Nadel bleiben. Laden Sie dann sauberes Silikonöl aus dem Pool.
- Wiederholen Sie die Schritte 2.4.2 bis 2.4.5 für die anderen Embryonen in der linken Reihe. Bevor Sie PGCs von einem neuen Embryo entnehmen, geben Sie so viel wie möglich von dem in Schritt 2.4.5 geladenen Silikonöl in die somatische Zellschicht, während die geladenen PGCs in der Nadel bleiben. Dadurch wird sichergestellt, dass neu geladene PGCs an die zuvor gesammelten PGCs angrenzen, ohne dass sich dazwischen Material befindet.
- Nachdem Sie die PGC-Entnahme von Embryonen in der linken Reihe abgeschlossen haben, trennen Sie PGCs so weit wie möglich vom Eigelb und anderen Verunreinigungen. Um dies zu erreichen, deponieren Sie alle PGCs in der Nadel auf der Oberfläche eines Embryos und entfernen Sie jegliches Eigelb oder andere Verunreinigungen von einem anderen benachbarten Embryo.
- Als nächstes entnehmen Sie PGCs von Embryonen in der rechten Reihe. Kombinieren Sie die PGCs aus der rechten und linken Reihe.
- Auftragen von Kryoprotektivum (CPA) auf PGCs
- Nachdem Sie die Nadel mit CPA in einem Tropfen gewaschen haben, füllen Sie frisches CPA in einem anderen Tropfen in die Nadel und fügen Sie CPA zu den PGCs hinzu, die sich auf dem Embryo abgelagert haben. Das CPA-Volumen sollte dem der PGCs entsprechen.
- Entfernen Sie 1-2 s nach der Zugabe von CPA so viel CPA wie möglich aus dem PGC-Cluster. PGCs schrumpfen leicht und nehmen unmittelbar nach der CPA-Zugabe eine quadratische Form an.
- Entleeren Sie die Nadel und füllen Sie dann Silikonöl für 5 s oder länger. Laden Sie alle gesammelten PGCs und laden Sie dann Silikonöl erneut für 5 s oder länger. PGCs befinden sich nun zwischen zwei Schichten Silikonöl (Abbildung 2B).
HINWEIS: Es ist wichtig, so viel Eigelb, CPA und andere Verunreinigungen wie möglich zu entfernen.
- Kryokonservierende PGCs
- Öffnen Sie den Dreiwegehahn (Abbildung 1C) und lösen Sie dann die Nadel vom Mikromanipulator. Tupfen Sie das Öl mit weichem Seidenpapier von der Oberfläche der Nadel ab. Berühren Sie die Nadelspitze nicht direkt mit dem Taschentuch.
- Befestigen Sie die Nadel an einem Nadelhalter und arretieren Sie sie mit Vinylklebeband an der Basis (Abbildung 1H). Kleben Sie ein Etikett auf das Halterrohr.
- Frieren Sie den Halter mit der Nadel nach unten ein, indem Sie ihn in flüssigen Stickstoff eintauchen. Lassen Sie den Halter erst los, wenn die Flüssigkeit nicht mehr aus dem Gestell sprudelt.
- Lagern Sie den Halter in einem Flüssigstickstoff-Vorratsbehälter im Flüssigphasenbereich, nicht im Dampfphasenbereich.
3. Auftauen und Umpflanzen von PGCs
- Sammeln, Dehorionieren und Ausrichten von Embryonen aus agametischen Wirtsfliegen
- Sammeln und dekorieren Sie Embryonen von agametischen Wirtsfliegen gemäß Schritt 2.
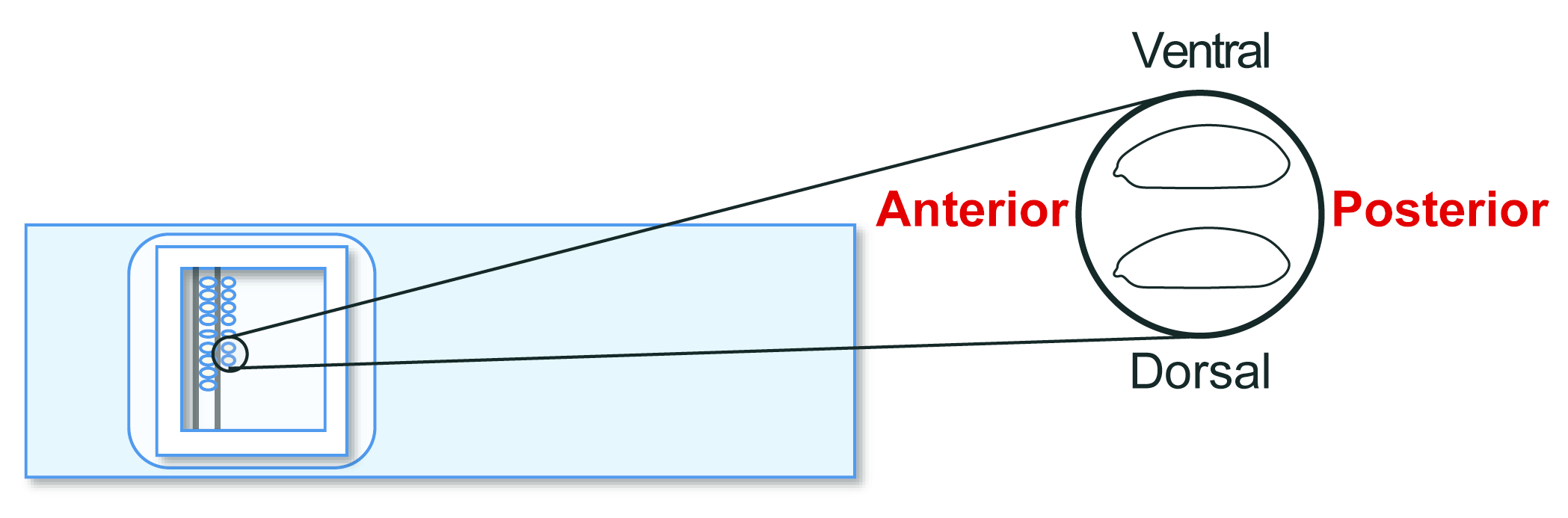
- Richten Sie die agametischen Wirtsembryonen im Stadium 5 auf einem Transplantationsobjektträger aus. Richten Sie dieses Mal jedoch das Seitenzahn nach rechts (die zu manipulierende Seite) und ventral nach oben aus (Abbildung 5). Bringen Sie etwa 30 Embryonen in 20 Minuten in zwei Reihen in eine Reihe.
- Während der Ausrichtung der Embryonen sollte ein Luftbefeuchter für 2-10 Minuten betrieben werden, wenn die Raumfeuchtigkeit dies erfordert (Tabelle 1). Die ideale Luftfeuchtigkeit liegt bei 30 % bis 40 %, kann jedoch je nach thermischen Bedingungen variieren.
- Auftauen und Transplantieren von PGCs in Wirtsembryonen
- Um kryokonservierte PGCs schnell aufzutauen, schieben Sie den Halter mit der Nadel nach unten in eine 1x EBR-Lösung bei Raumtemperatur und halten Sie sie 10 s lang untergetaucht.
- Platzieren Sie den Objektträger des Transplantationsglases auf dem Tisch eines Mikroskops. Befestigen Sie die gefrieraufgetaute Nadel am Kapillarhalter und bringen Sie den ersten Embryo in der linken Reihe und die Nadelspitze in dieselbe Fokusebene.
- Bewegen Sie die Nadelspitze mit einer 20-fachen Objektivlinse vorsichtig an die Oberfläche des hinteren Endes des Embryos.
- Stoßen Sie jeden Embryo vorsichtig an die Außenseite und stellen Sie sicher, dass er langsam in seine ursprüngliche Form zurückkehrt. Das Anstoßen bestätigt, dass der Innendruck des Embryos nicht zu hoch oder zu niedrig ist.
- Bewegen Sie die Nadel vorsichtig und dringen Sie vom hinteren Pol aus in einen Embryo ein.
- Geben Sie vorsichtig etwa 10-20 PGCs direkt im hinteren Pol ab, genau zwischen der Vitellinmembran und der somatischen Zellschicht des Embryos. Vermeiden Sie es, sie in der somatischen Zellschicht abzulagern. Wenn die perivitelline Flüssigkeit aus dem Embryo austritt, saugen Sie die ausgetretene Flüssigkeit in die Nadel und entfernen Sie sie.
- Ziehen Sie die Nadel vom Embryo zurück. Wiederholen Sie die Schritte 3.2.5 und 3.2.6 für nachfolgende Embryonen.
4. Inkubation von Embryonen und Wiederherstellung von Spenderstämmen
- Entfernen Sie alle Embryonen, die keine transplantierten PGCs erhalten, und inkubieren Sie die verbleibenden Embryonen in einer befeuchteten Kammer (Abbildung 1G) bei 25 °C.
- Mindestens 24 Stunden nach der Transplantation und so bald wie möglich nach dem Schlüpfen werden die geschlüpften Larven mit einer Pinzette aufgenommen und in Standard-Drosophila-Futterfläschchen überführt und bei 25 °C inkubiert.
- Um den Stamm wiederzubeleben, kreuzen Sie neu geschlüpfte Weibchen und Männchen (Abbildung 6).
HINWEIS: Agametische Wirte ermöglichen es, das gesamte Genom auf einmal wiederherzustellen, ohne sich mit Balancer-Chromosomen-Stämmen zu kreuzen. Die Koexistenz von agametischen Männchen in einem Fläschchen spielt keine Rolle, da Weibchen, selbst wenn sie mit ihnen gepaart werden, keine langfristigen Reaktionen nach der Paarung zeigen, einschließlich einer verminderten Empfänglichkeit für Remating18,19.
Representative Results
Die Wirksamkeit der kryokonservierten PGC-Transplantation wurde von Asaoka et al.13 berichtet und ist in Tabelle 2 für die Transplantation von PGCs angegeben, die für 1 Tag oder länger in flüssigem Stickstoff kryokonserviert wurden. Die Schlupfrate betrug 168/208 transplantierte Embryonen (80,8%) und die Lebensfähigkeit vom Embryo bis zum Erwachsenen 87/208 (41,8%). Die Häufigkeit der fruchtbaren Fliegen betrug 28/87 (32,2%). Diese Häufigkeit unterschied sich nicht zwischen PGCs, die 8 bis 30 Tage lang kryokonserviert wurden, und solchen, die 31-150 Tage lang kryokonserviert wurden (20/57 vs. 8/30, G' = 0,63, p >0,1, d.f. = 1). Die durchschnittliche Anzahl der Nachkommen pro Paar betrug 77,2 ± 7,1 (n = 18, 28-122), was auf die Fähigkeit kryokonservierter PGCs hinweist, sich in Keimbahnstammzellen zu verwandeln. Von den 26 Nadeln brachten 10 keine fruchtbaren Nachkommen, 7 Nadeln 1 fruchtbare Nachkommenschaft, 7 Nadeln 2 fruchtbare Nachkommen und 2 Nadeln 3 oder 4 fruchtbare Nachkommen. Die durchschnittliche Anzahl der fruchtbaren Fliegen pro Nadel betrug 1,1 ± 0,2. Basierend auf diesen Daten reichen 11 Nadeln mit einer Sicherheit von 95 % aus, um 6 oder mehr Nachkommen zu produzieren, in denen wahrscheinlich mindestens ein Weibchen und ein Männchen enthalten sind.
In den obigen Experimenten haben wir Embryonen, die ovo-A-mRNA in PGCs (Nanos>ovo-A, OvoA_OE Embryonen) exprimieren, als agametischen Wirt verwendet. Von 669 F1-Weibchen und 720 F1-Männchen, die aus transplantierten Nanos>Ovo-A-Paaren hervorgingen, gab es keinen Escaper, der von den Wirts-PGCs abstammte. Mehrere Oskar (osk)-Mutanten sind auch temperaturempfindlich agametisch20,21. Da eine osk-Mutante mit hoher homozygoter Lebensfähigkeit und dem agametischen Phänotyp nicht mehr verfügbar ist, haben wir die osk[8]-Missense-Mutante20 durch CRISPR/Cas9-gestützte Genom-Editierung nachgebaut. Diese Fliegen waren bei 25 °C völlig agametisch (0 Ausbrecher von 230 Weibchen und 192 Männchen), aber einige wenige Ausbrecher schlüpften bei 23 °C (1 von 248 Weibchen und 1 von 290 Männchen). nanos>ovo-A werden daher als agametische Wirtsembryonen empfohlen. Sowohl UASp-ovo-A als auch nanos-Gal4 Stock13 werden in Kürze im KYOTO Drosophila Stock Center erhältlich sein.

Abbildung 1: Erforderliche Ausrüstung. (A) Ein Mikromanipulatorsystem zum Sammeln und Transplantieren von Zellen. i) inverses Mikroskop, ii) mechanischer Mikromanipulator, iii) Spritze, iv) Kapillarhalter, v) Dreiwegehahn, vi) Luftbefeuchter und vii) Stereomikroskop. (B) Eine Spritze. (C) Ein Dreiwegehahn und Silikonschläuche verbinden eine Spritze und einen Kapillarhalter. (D) Eine Nadel und ein Kapillarhalter sind an einem Mikromanipulator befestigt. (E) Ein Embryo-Entnahmebecher mit einer Embryo-Entnahmeplatte (6 cm Durchmesser, 7,7 cm hoch). (F) Ein Sieb aus Edelstahlgewebe. (G) Ein Behälter, der als Feuchtkammer mit einem Objektträger aus Glas verwendet wird. Um die Luftfeuchtigkeit zu erhalten, legen Sie nasses Papier auf den Boden und schließen Sie den Deckel. (H) einen Nadelhalter mit einer Nadel für die Kryokonservierung. (I) Ein Lagergestell für die Kryokonservierung und eine Kiste mit Nadeln. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Ein PGC-Sammelglasobjektträger und eine Kryokonservierungsnadel. (A) Ein mit Klebstoff beschichteter Objektträger zur Sammlung von primordialen Keimzellen (PGC). Die dechorionierten Embryonen werden in zwei Reihen ausgerichtet und mit der vorderen nach rechts (die zu manipulierende Seite) und der ventralen Seite nach oben ausgerichtet. Ein Embryo-Pool-Rahmen wird angebracht, zwei Tropfen Kryoprotektiva (CPA)-Lösung werden abgegeben und der Pool wird mit Silikonöl gefüllt. (B) Eine Nadel sollte eine möglichst geringe Menge an Eigelb und anderen Verunreinigungen enthalten. PGCs werden zwischen zwei Schichten Silikonöl eingebettet, wenn sie in flüssigem Stickstoff kryokonserviert werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Herstellung der Nadel. Dreistufige Methode zum Polieren der Spitze, um eine Nadel mit einer geeigneten Lochgröße und einer scharfen Spitze herzustellen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Schema der Embryonenentnahme. Nach zwei Vorabsammlungen sammeln wir in der Regel drei- bis viermal pro Tag. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Ausrichtung des Wirtsembryos. Ausrichtung von Wirtsembryonen auf einem Objektträger. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Ein Überblick über die PGC-Kryokonservierungsmethode. Ein Überblick über alle Schritte, die zur Durchführung der Kryokonservierung der primordialen Keimzellen (PGC) befolgt wurden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
| Luftfeuchtigkeit im Raum | |||
| < 30% | ~ 30% | > 30% | |
| Wirtsembryonen ausrichten (~20 Minuten) | Verwenden Sie einen Luftbefeuchter für 2 - 10 min | Verwenden Sie einen Luftbefeuchter intermittierend für 1 Minute | Verwenden Sie keinen Luftbefeuchter |
| PGCs von Spendern auftauen | Nicht zutreffend | Nicht zutreffend | Nicht zutreffend |
| Lufttrocknende PGCs | Lassen Sie diesen Schritt aus | Lassen Sie diesen Schritt aus | 5 Minuten |
| Silikonöl auftragen | Nicht zutreffend | Nicht zutreffend | Nicht zutreffend |
| PGCs transplantieren | Nicht zutreffend | Nicht zutreffend | Nicht zutreffend |
| Alle diese Schritte sollten in 50 Minuten abgeschlossen sein. | |||
Tabelle 1: Trocknung der Embryonen während der Embryonenausrichtung und des PGC-Auftauens.
| Stamm des Spenders | Zeitraum der Kryokonservierung | Anzahl der transplantierten Embryonen (A) | Anzahl der geschlüpften Larven (B) (Schlupffähigkeit, B/A) | Anzahl der geschlossenen Erwachsenen (C) (Lebensfähigkeit von der Eizelle bis zum erwachsenen Tier, C/A) | Anzahl der fruchtbaren Erwachsenen (D) (Häufigkeit fruchtbarer Fliegen, D/C) |
| M17-KARTON | 8 - 30 Tage | 134 | 108 (80.6%) | 57 (42.5%) | 20 (35.1%) |
| M17-KARTON | 31 - 150 Tage | 74 | 60 (81.1%) | 30 (40.5%) | 8 (26.7%) |
| M17: igitt; TM6B, P{Dfd-GMR-nvYFP}4, Sb[1] Tb[1] ca[1]/ Pri[1] | |||||
Tabelle 2: Effizienz der kryokonservierten PGC-Transplantation. Diese Tabelle wurde von13 geändert. Alle Daten stammen von agametischen Wirten.
Discussion
Ein entscheidender Faktor für den Erfolg der PGC-Kryokonservierung und -Wiederbelebung ist die Verwendung guter Embryonen. Junge Weibchen (z. B. 3 bis 5 Tage alt) sollten für die Embryonenentnahme verwendet werden. Sowohl Spender- als auch Wirtsembryonen werden durch mikroskopische Inspektion beurteilt, und es werden nur solche im Blastodermstadium (Stadium 5) verwendet12. Für die PGC-Entnahme richten wir in der Regel ca. 40 Spenderembryonen in einem Zeitraum von 20 Minuten aus und entnehmen PGCs von ca. 30 Embryonen im Frühstadium 5; Ältere und defekte Embryonen werden nicht verwendet. Nach der Kryokonservierung und dem Auftauen sollten PGCs ihre Form behalten. PGCs reißen bei erfolgloser Konservierung. Die Wirtsembryonen sollten sich ebenfalls im Stadium 5 befinden und einen moderaten Innendruck haben. Die Embryonen sollten nach sanftem Anstoßen langsam in ihre ursprüngliche Form zurückkehren. Übermäßig und unzureichend getrocknete Embryonen entwickeln sich nach der Transplantation nicht normal. Da die heterosexuelle Transplantation von PGCs in Drosophila 5,10 keine Gameten produziert, ist es wahrscheinlicher, dass die Transplantation von PGCs von mehreren Spenderembryonen in Wirtsembryonen fruchtbare Erwachsene hervorbringt. Zu diesem Zweck sammeln wir in der Regel PGCs von ca. 30 Embryonen pro Nadel.
Als Kryoprotektiva haben wir Ethylenglykol, Dimethylsulfoxid und Glycerin zusammen mit Saccharose in verschiedenen Konzentrationen ausprobiert. Wir stellten fest, dass EBR mit 20 % Ethylenglykol und 1 M Saccharose die besten13 sind. Die Verwendung verschiedener Kryoprotektoren kann jedoch die PGC-Konservierungverbessern 22.
Diese Kryokonservierungsmethode erfordert spezielle Fähigkeiten im Umgang mit PGCs, und es sind etwa 6 Wochen Training erforderlich, um PGCs bequem zu entnehmen und zu transplantieren. Um die Fertigkeiten zu beurteilen und zu verbessern, kann dies in sechs Trainingsschritte unterteilt werden: 1) Ausrichten von Embryonen auf einem Glasobjektträger, 2) Steuern eines Manipulators, 3) Transplantation von PGCs aus einem Embryo in einen anderen Embryo ohne Kryokonservierung, 4) Transplantation von PGCs von 10 oder mehr Embryonen in 5 bis 10 Embryonen, 5) Transplantation von PGCs nach der Anwendung von CPA und 6) Transplantation von PGCs nach dem Einfrierentauen. Jeder Schritt kann 1 Woche dauern. Die kurzfristigen Ziele in Schritt 3 sind eine Schlupfrate von 40 %, eine Lebensfähigkeit vom Embryo bis zum Erwachsenen von 10 % bis 20 % und eine Häufigkeit fruchtbarer Fliegen von 20 %.
Die PGC-Kryokonservierung erfordert teure Instrumente und hochqualifiziertes Personal. Daher wird diese Methode möglicherweise nicht von vielen Laboratorien übernommen. Die derzeitige PGC-Methode hat jedoch mehrere wichtige Aspekte. Erstens sind PGCs viel kleiner als Embryonen und sehr durchlässig für Kryoprotektoren. Im Gegensatz dazu ist die Permeabilität von Kryoprotektiva durch die wachsartigen Schichten von Drosophila-Embryonen stark eingeschränkt, was das schwerwiegendste Problem bei der Kryokonservierung von Embryonen darstellt. In der Tat haben frühere Studien große Anstrengungen unternommen, um ein Zeitfenster zu finden, in dem Embryonen eine hohe Überlebensrate und eine dünnere Wachsschicht aufweisen. Die zweite befasst sich mit der Entwicklung und morphologischen Variation zwischen den Stämmen. PGCs werden von Embryonen im frühen Stadium 5 (2 h 30 min-3 h 20 min nach der Eiablage) entnommen, während die Kryokonservierung von Embryonen im Stadium 16 (14-22 h nach der Eiablage) durchgeführt wird. Die Embryonen sind daher viel älter und zeigen im Vergleich zur PGC-Kryokonservierung eine viel größere Stammvariation im optimalen Zeitfenster für die Kryokonservierung. Tatsächlich variierte die Häufigkeit der Wirte, die von Spendern stammende Nachkommen produzierten, nicht zwischen fünf Stämmen, die von Asaoka et al. untersucht wurden.13, obwohl die Wirte nicht agametisch waren. Darüber hinaus haben PGCs das Potenzial, in gentechnischen Anwendungen wie der Genom-Editierung eingesetzt zu werden 14,15,16.
Acknowledgements
Wir danken dem KYOTO Drosophila Stock Center für die Fliegenstämme. Wir danken auch Frau Wanda Miyata für das englischsprachige Lektorat des Manuskripts und Dr. Jeremy Allen von Edanz (https://jp.edanz.com/ac) für die Bearbeitung eines Entwurfs dieses Manuskripts. Diese Arbeit wurde unterstützt durch Zuschüsse (JP16km0210072, JP17km0210146, JP18km0210146) der Japan Agency for Medical Research and Development (AMED) an T.T.-S.-K., Zuschüsse (JP16km0210073, JP17km0210147, JP18km0210145) von AMED an S.K., einen Zuschuss (JP20km0210172) von AMED an T.T.-S.-K. und S.K., ein Grant-in-Aid for Scientific Research (C) (JP19K06780) von der Japan Society for the Promotion of Science (JSPS) an T.T.-S.-K. und ein Grant-in-Aid for Scientific Research on Innovative Areas (JP18H05552) von JSPS an S.K.
Materials
| Name | Company | Catalog Number | Comments |
| Acetic acid | FUJIFILM Wako Pure Chemical Corporation | 017-00256 | For embryo collection |
| Agar powder | FUJIFILM Wako Pure Chemical Corporation | 010-08725 | For embryo collection |
| Calcium chloride | FUJIFILM Wako Pure Chemical Corporation | 038-24985 | For EBR solution |
| Capillary | Sutter Instrument | B100-75-10-PT | BOROSILICATE GLASS; O.D: 1.0mm, I.D: 0.75mm , length: 10cm, 225Pcs |
| Capillary holder | Eppendorf | 5196 081.005 | Capillary holder 4; for micromanipulation |
| Chromic acid mixture | FUJIFILM Wako Pure Chemical Corporation | 037-05415 | For needle washing |
| CPA solution | 1x EBR containing 20% ethylene glycol and 1M sucrose | ||
| Double-sided tape | 3M | Scotch w-12 | For glue extracting |
| Ephrussi–Beadle Ringer solution (EBR) | 130 mM NaCl, 5 mM KCl, 2 mM CaCl2, and 10 mM Hepes at pH 6.9 | ||
| Ethanol (99.5) | FUJIFILM Wako Pure Chemical Corporation | 057-00451 | For embryo collection |
| Ethylene glycol | FUJIFILM Wako Pure Chemical Corporation | 054-00983 | For CPA solution |
| Falcon 50 mm x 9 mm bacteriological petri dish | Corning Inc. | 351006 | For embryo collection |
| Forceps | Vigor | Type5 Titan | For embryo handling |
| Grape juice | Asahi Soft Drinks Co., LTD. | Welch's Grape 100 | For embryo collection |
| Grape juice agar plate | 50% grape juice, 2% agar, 1% ethanol, 1% acetic acid | ||
| Heptane | FUJIFILM Wako Pure Chemical Corporation | 084-08105 | For glue extracting |
| Humidifier | APIX INTERNATIONAL CO., LTD. | FSWD2201-WH | For embryo preparation |
| Inverted microscope | Leica Microsystems GmbH | Leica DM IL LED | For micromanipulation |
| Luer-lock glass syringe | Tokyo Garasu Kikai Co., Ltd. | 0550 14 71 08 | Coat a plunger with silicon oil (FL-100-450CS);for micromanipulation |
| Mechanical micromanipulator | Leica Microsystems GmbH | For micromanipulation | |
| Micro slide glass | Matsunami Glass Ind., Ltd. | S-2441 | For embryo aligning |
| Microgrinder | NARISHIGE Group | Custom order | EG-401-S combined EG-401 and MF2 (with ocular lens MF2-LE15 ); for needle preparation |
| Microscope camera | Leica Microsystems GmbH | Leica MC170 HD | For micromanipulation |
| Needle holder | Merck KGaA | Eppendorf TransferTip (ES) | For cryopreservation |
| Potassium chloride | Nacalai Tesque, Inc. | 28514-75 | For EBR solution |
| Puller | NARISHIGE Group | PN-31 | For needle preparation; the heater level is set to 85.0-98.4, the magnet main level to 57.8, and the magnet sub level to 45.0. |
| PVC adhesive tape for electric insulation | Nitto Denko Corporation | J2515 | For embryo-pool frame |
| Silicon oil | Shin-Etsu Chemical, Co, Ltd. | FL-100-450CS | For embryo handling |
| Sodium chloride | Nacalai Tesque, Inc. | 31320-05 | For EBR solution |
| Sodium hypochlorite solution | FUJIFILM Wako Pure Chemical Corporation | 197-02206 | Undiluted and freshly prepared; for embryo breaching |
| Sucrose | Nacalai Tesque, Inc. | 30404-45 | For CPA solution |
References
- Brüschweiler, W., Gehring, W. A method for freezing living ovaries of Drosophila melanogaster larvae and its application to the storage of mutant stocks. Experientia. 29, 134-135 (1973).
- Steponkus, P. L., et al. Cryopreservation of Drosophila melanogaster embryos. Nature. 345, 170-172 (1990).
- Mazur, P., Cole, K. W., Hall, J. W., Schreuders, P. D., Mahowald, A. P. Cryobiological preservation of Drosophila embryos. Science. 258 (5090), 1932-1935 (1992).
- Zhan, L., Li, M. G., Hays, T., Bischof, J. Cryopreservation method for Drosophila melanogaster embryos. Nat Comm. 12, 2412 (2021).
- Van Deusen, E. B. Sex determination in germ line chimeras of Drosophila melanogaster. Development. 37 (1), 173-185 (1977).
- Breen, T. R., Duncan, I. M. Maternal expression of genes that regulate the bithorax complex of Drosophila melanogaster. Dev Biol. 118, 442-456 (1986).
- Schupbach, T., Wieschaus, E. Germline autonomy of maternal-effect mutations altering the embryonic body pattern of Drosophila. Dev Biol. 113, 443-448 (1986).
- Irish, V., Lehmann, R., Akam, M. The Drosophila posterior-group gene nanos functions by repressing hunchback activity. Nature. 338, 646-648 (1989).
- Hülskamp, M., Schröder, C., Pfeifle, C., Jäckle, H., Tautz, D. Posterior segmentation of the Drosophila embryo in the absence of a maternal posterior organizer gene. Nature. 338, 629-632 (1989).
- Steinmann-Zwicky, M., Schmid, H., Nöthiger, R. Cell-autonomous and inductive signals can determine the sex of the germ line of Drosophila by regulating the gene Sxl. Cell. 57 (1), 157-166 (1989).
- Stein, D., Roth, S., Vogelsang, E., Nüsslein-Volhard, C. The polarity of the dorsoventral axis in the drosophila embryo is defined by an extracellular signal. Cell. 65 (5), 725-735 (1991).
- Kobayashi, S., Yamada, M., Asaoka, M., Kitamura, T. Essential role of the posterior morphogen nanos for germline development in Drosophila. Nature. 380, 708-711 (1996).
- Asaoka, M., et al. Offspring production from cryopreserved primordial germ cells in Drosophila. Comm Biol. 4 (1), 1159 (2021).
- Blitz, I. L., Fish, M. B., Cho, K. W. Y. Leapfrogging: primordial germ cell transplantation permits recovery of CRISPR/Cas9-induced mutations in essential genes. Development. 143 (15), 2868-2875 (2016).
- Koslová, A., et al. Precise CRISPR/Cas9 editing of the NHE1 gene renders chickens resistant to the J subgroup of avian leukosis virus. Proc Natl Acad Sci U S A. 117 (4), 2108-2112 (2020).
- Zhang, F. Efficient generation of zebrafish maternal-zygotic mutants through transplantation of ectopically induced and Cas9/gRNA targeted primordial germ cells. J Genet Genom. 47 (1), 37-47 (2020).
- Campos-Ortega, J. A., Hartenstein, V. Stages of Drosophila Embryogenesis. The Embryonic Development of Drosophila. , (1997).
- Manning, A. A sperm factor affecting the receptivity of Drosophila melanogaster females. Nature. 194, 252-253 (1962).
- Kubli, E. Sex-peptides: seminal peptides of the Drosophila male. Cell Mol Life Sci. 60, 1689-1704 (2003).
- Lehmann, R., Nüsslein-Volhard, C. Abdominal segmentation, pole cell formation, and embryonic polarity require the localized activity of oskar, a maternal gene in drosophila. Cell. 47 (1), 141-152 (1986).
- Kiger, A. A., Gigliotti, S., Fuller, M. T. Developmental genetics of the essential Drosophila Nucleoporin nup154: allelic differences due to an outward-directed promoter in the P-element 3′ end. Genetics. 153 (2), 799-812 (1999).
- Rienzi, L. F., et al. Perspectives in gamete and embryo cryopreservation. Semin Reprod Med. 36 (5), 253-264 (2018).
Explore More Articles
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved