Streptavidin-Affinity Grid Fabrication for Cryo-Electron Microscopy Sample Preparation
In This Article
Summary
A step-by-step protocol for fabricating streptavidin affinity grids is provided for use in structural studies of challenging macromolecular samples by cryo-electron microscopy.
Abstract
Streptavidin affinity grids provide strategies to overcome many commonly encountered cryo-electron microscopy (cryo-EM) sample preparation challenges, including sample denaturation and preferential orientations that can occur due to the air-water interface. Streptavidin affinity grids, however, are currently utilized by few cryo-EM labs because they are not commercially available and require a careful fabrication process. Two-dimensional streptavidin crystals are grown onto a biotinylated lipid monolayer that is applied directly to standard holey-carbon cryo-EM grids. The high-affinity interaction between streptavidin and biotin allows for the subsequent binding of biotinylated samples that are protected from the air-water interface during cryo-EM sample preparation. Additionally, these grids provide a strategy for concentrating samples available in limited quantities and purifying protein complexes of interest directly on the grids. Here, a step-by-step, optimized protocol is provided for the robust fabrication of streptavidin affinity grids for use in cryo-EM and negative-stain experiments. Additionally, a trouble-shooting guide is included for commonly experienced challenges to make the use of streptavidin affinity grids more accessible to the larger cryo-EM community.
Introduction
Electron cryo-microscopy (cryo-EM) has revolutionized the field of structural biology by enabling macromolecular structure determination of large, flexible, and heterogeneous samples that were previously inaccessible by X-ray crystallography or nuclear magnetic resonance1. This method works by flash-freezing macromolecules in solution to create a thin layer of vitreous ice that can be subsequently imaged using an electron microscope. In recent years, significant advances in both microscope hardware and image processing software have further expanded the types of samples suitable for high-resolution structure determination by cryo-EM.
Nevertheless, the preparation of thin, vitrified samples remains one of the most critical steps in macromolecular structure determination by cryo-EM. Biological samples are often dynamic, fragile, prone to denaturation, and sometimes are only available in small quantities for cryo-EM studies. During the blotting process, these particles interact with the hydrophobic air-water interface, which can result in particle-preferred orientations, disassembly of fragile complexes, partial or complete sample denaturation, and aggregation2,3,4. The use of detergents or other surfactants, chemical cross-linking, and adsorption of samples to support layers are common strategies to preserve biological samples during the freezing process. Support layers such as graphene oxide5,6,7 or amorphous carbon8 also function to concentrate particles on the grid by adsorption when the sample is available in limited quantities. However, these methods are not general or reliable, and optimization of grid preparation can be extremely time-consuming or fail altogether.
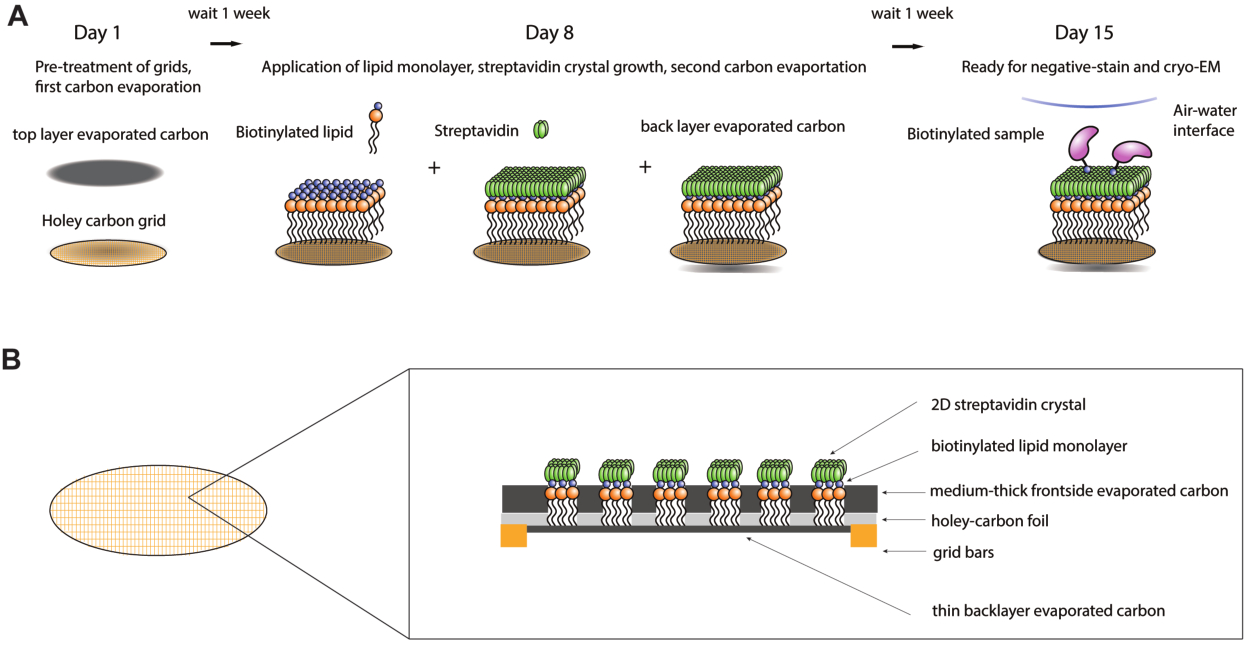
Streptavidin affinity grids9,10 were developed to overcome these shortcomings and to provide a mild and generally applicable method to sequester the complex of interest and protect it from the air-water interface. These grids utilize a two-dimensional (2D) streptavidin crystal lattice grown on a monolayer of biotinylated lipids on the grid. After samples are themselves biotinylated (often sparsely and randomly, meaning one biotin per complex on average), they can be applied to the streptavidin-coated grid. Because sample adsorption relies on the extremely high affinity between streptavidin and biotin, sample concentrations as low as 10 nM can be used with these grids. Commercially available biotinylation kits for proteins and biotinylated primers for DNA-containing complexes make it relatively easy to attach the necessary biotin moieties to most samples of interest. In addition to concentrating the sample and keeping it away from the damaging air-water interface during blotting, the random biotinylation of one or just a few lysine residues can significantly improve the range of orientations of the molecule of interest on the cryo-EM grid, as demonstrated in a number of studies11. While the signal from the underlying streptavidin crystal is present in the raw images, data processing schemes involving Fourier filtration of the sharp Bragg reflections from the crystal can be easily removed during early data processing, ultimately enabling high-resolution reconstructions of the sample of interest11,12,13. Here, an optimized, step-by-step protocol is provided for robust production of streptavidin affinity grids and subsequent use in cryo-EM experiments. The protocol provided is expected to be completed over a 2-week period (Figure 1A). The first parts of the protocol describe the preparation of reagents, the pretreatment of grids, and the first carbon evaporation steps. Next, instructions are described for the preparation of the lipid monolayer and the growth of streptavidin crystals on EM grids. Additionally, instructions are provided for the use of streptavidin affinity grids in negative stain EM and cryo-EM experiments. Finally, procedures are provided for removing streptavidin-signal from micrographs once cryo-EM data has been acquired.
Protocol
1. Preparation of reagents
- Dilute commercially purchased streptavidin to a final concentration of 0.5 mg/mL in crystallization buffer (50 mM HEPES pH 7.5, 150 mM KCl, 5 mM EDTA, 10% trehalose). Make sure that the final trehalose concentration is 10%. Flash-freeze aliquots of 25-50 µL in liquid nitrogen and store at -80 ˚C.
- Dissolve 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) sodium salt into 65:35:8 v/v/v chloroform/methanol/water solvent for a final lipid concentration of 1 mg/mL. Store single use aliquots of 20-30 µL at -80 ˚C in glass vials.
NOTE: Preparation of solvent is more accurate if done by weight. First, combine 1.85 g of ultrapure water with 6.41 g of high-performance liquid chromatography (HPLC) grade methanol and add this to 22.42 g of chloroform to prepare the solvent.
CAUTION: Please read and understand manufacturers' safety data sheets and recommended instructions for the handling and disposal of chloroform and methanol organic solvents before beginning this protocol. Avoid direct contact of methanol with the skin.
2. Pretreatment of grids
- Wash carbon foil on gold mesh grids by dipping twice into 100% chloroform and once into 100% ethanol. Place grids on clean filter paper to dry. Repeat the washing process for a total of three cycles.
NOTE: Reproducible success has been achieved with commercially available carbon foil grids that have a carbon film of 10-12 nm thick carbon (see material list) with hole sizes ranging from 0.6-2 µm. Commercially available gold foil grids and homemade holey-carbon grids prepared following the Rubinstein Laboratory's nanofabrication protocol14 have also been successfully used. Less reproducible results have been obtained with commercially available grids that have thinner carbon films. Do not use copper grids, as copper reacts with any organic amines that may be in the sample. - After the grids dry, evaporate a 2.5-5 nm thick layer of carbon onto the carbon side of the holey-carbon grids. Carbon thickness is controlled with quartz crystal film thickness measurement that is built into the carbon evaporator provided in the Table of Materials file.
- Allow the newly evaporated carbon to age (i.e., become more hydrophobic) for 1 week.
3. Preparation of streptavidin lattice
NOTE: To grow 2D streptavidin crystals on the holey-carbon EM grids, first, a biotinylated lipid monolayer is applied to the holes of the carbon film (Figure 1B) by Langmuir-Schaefer transfer. The lipid monolayer is formed following the subsequent steps (Figure 2). Talcum powder creates a boundary between the castor oil and the Petri dish, and a thin layer of castor oil helps to maintain constant surface pressure when the lipid monolayer is formed. The side containing the evaporated carbon from step 2.2 is used to pick up the monolayer, excess lipids are washed away with crystallization buffer, and streptavidin solution is incubated on the grid for crystallization.
- Clean the bench area with 70% ethanol.
- Rinse a 5 µL glass syringe several times with chloroform to clean. Allow the syringe to completely dry before use.
- Wash carbon-evaporated grids again, i.e., just before use, by dipping them into 100% ethanol. Let the grids dry on clean filter paper.
- While grids are drying, wash each anti-capillary tweezer with 100% chloroform and 100% ethanol. Allow the tweezers to completely dry.
- On the clean side of parafilm, prepare three 50 µL drops of crystallization buffer (50 mM HEPES pH 7.5, 150 mM KCl, 5 mM EDTA, 10% trehalose) for each grid that will be made.
- Fill the lid of a 35 mm uncoated Petri dish with crystallization buffer (50 mM HEPES pH 7.5, 150 mM KCl, 5 mM EDTA, 10% trehalose). Clean the surface with lens paper.
- Sprinkle scientific-grade talcum powder around the perimeter of the Petri dish. This subsequently serves as an indicator, in the next step, of how far the castor oil has spread. Add enough talcum powder to create a barrier that protects the castor oil from touching the edge of the Petri dish. Adding too much talcum powder limits the available space to make the lipid monolayer.
- Dip a 200 µL pipette tip into the castor oil to obtain a medium-sized drop (approximately 20 µL) that hangs from the pipette tip. Touch this drop to the surface of the buffer in the Petri dish, where it will spread to form a uniform film. The resulting thin film of oil serves as a piston15, which maintains a constant surface pressure when a lipid monolayer is formed (step 3.10).
NOTE: It is important to add enough castor oil rather than too little, and it is better to add as one single drop. Allow the castor oil to spread fully before making the lipid monolayer. - Rinse the 5 µL glass syringe one or two times with the dissolved lipid before taking up an aliquot for use. Avoid creating air bubbles in the lipid that fills the syringe.
NOTE: The glass syringe is rinsed with dissolved lipid in case any residual chloroform remains from step 3.2. Residual chloroform can affect the quality of the resulting monolayer. - Dispense the smallest possible volume (~0.5 µL) of lipid from the syringe and gently touch the hanging droplet to the surface of the castor oil film. Note that the lipid solution breaks through the castor oil film at the point of contact and that the resulting lipid monolayer forms a circle in the center of the thin film of castor oil.
- Add several drops of lipid sequentially or add more lipid after making some grids, but if too much is added, the lipid will spread past the boundary of the castor oil and into the perimeter where the talcum powder and any contaminating surfactant have been sequestered. If this occurs, the process will need to be restarted.
- Pick up a grid with an anti-capillary tweezer so that the straight arm of the tweezer and the carbon-evaporated side of the grid both face the monolayer.
- Transfer part of the monolayer to the holey carbon by touching the carbon-evaporated side of the grid to the monolayer for 1-2 s.
NOTE: Successful transfer is indicated by the surface of the grid becoming hydrophilic, causing a thin, spherical cap of buffer to cover the entire grid after it has been lifted away from the Petri dish. - Touch the spherical cap of the buffer that now adheres to the grid, sequentially for 1 s each, to the three 50 µL drops of crystallization buffer that were prepared on parafilm in step 3.5.
NOTE: This step tries to remove as much as possible of the excess lipid, which covers the surface of the spherical cap (rather than the formerly hydrophobic carbon film), in order to avoid formation of lipid vesicles when samples are viewed in the electron microscope. - Carefully add 4 µL of 0.5 mg/mL streptavidin into the remaining spherical cap of crystallization buffer on the grid.
- Place the grid in a humidity chamber. Repeat steps 3.11-3.14 for the entire batch of grids.
- Incubate the grids at room temperature (RT) for 2 h inside a humidity chamber to avoid evaporation.
NOTE: It is important that the gold side of the grid does not become wet at any point after the monolayer has been applied. - After the 2 h incubation, prepare one 300 µL drop of rinse buffer (10 mM HEPES, pH 7.5, 50 mM KCl, 5 mM EDTA, 10% trehalose) for each grid on the clean side of parafilm.
NOTE: The rinse buffer contains 50 mM KCl rather than 150 mM KCl, which is in the crystallization buffer used for steps 3.5-3.6. - Wash the excess (unbound) streptavidin by placing the grid onto the 300 µL drop of rinse buffer. Perform steps 3.18-3.20 for one grid at a time. Avoid leaving the grids on the 300 µL rinse buffer drop for extended periods of time.
NOTE: Only one wash is performed because the streptavidin crystal/monolayer is fragile at this point. Each time that a grid is lifted away from the surface of a drop of wash buffer, the liquid bridge between the grid and the wash drop is ruptured. At the time of rupture, a transient pressure gradient (suction pressure) is applied to the self-supported monolayer crystals that span the open holes of the carbon film. This suction pressure is expected to cause transient doming of the monolayer crystal, accompanied by expansion of the area covered by the crystals. If the increase in the area exceeds the elastic limit of the crystal, the crystal may fracture or become disordered. - Immediately dry the anti-capillary tweezers with a lint-free wipe to facilitate the release of the grid onto filter paper in the following step. Pick up the grid floating on the drop of rinse buffer by sticking the kinked arm of the anti-capillary tweezer into the drop.
- Blot away the excess buffer gently from the side with filter paper. Place the grid on filter paper with the gold side down, repeat steps 3.18-3.20 for each grid, and allow the grids to dry for 15-20 min.
- After the grids are dry, flip them over so that the gold side is now facing up. Evaporate a thin layer of carbon (approximately 0.5-2 nm thick) onto the gold side of the grids.
NOTE: Store grids at a constant humidity, the value of which does not seem to matter, noting that multiple changes in relative humidity are expected to result in cycles of expansion and contraction. Allow grids to age for 1 week before cryo-EM use for best results because the hydrophobicity of the back side of the grid may be important for monolayer stability. Grids are stable for long periods of time, but after 3 months, the backside carbon may become less hydrophobic.
4. Checking streptavidin affinity grid batch quality by negative stain
- Prepare two drops of 50 µL and one drop of 100 µL of sample buffer (50 mM HEPES pH 7.5, 50 mM KCl, 0.5 mM TCEP) on the clean side of parafilm.
- Remove the trehalose and rehydrate streptavidin grids by touching the two 50 µL drops and let the grid float on the 100 µL drop of sample buffer for 10 min.
NOTE: It is best to avoid using detergent during the rehydration and subsequent sample-binding incubations. The buffer will disperse to the gold side of the grids and limit the efficiency of washes to remove excess samples. - After rehydrating, pick the grid up with an anti-capillary tweezer so that the kinked arm of the anti-capillary tweezer is in the drop.
- Gently blot the excess buffer away from the side and quickly reapply 4 µL of 75-100 nM sample (optional).
NOTE: Avoid letting the grid dry after rehydration. - Incubate the sample in a humidity chamber for 1-5 min.
NOTE: Concentration and incubation times will be sample-dependent and should be optimized. The provided sample concentration and incubation time are suggested starting points. - On the clean side of parafilm, set up four drops of 30 µL each of both sample buffer and 1% uranyl formate (UF) stain.
NOTE: Please read and understand manufacturers' safety data sheets and recommended instructions for the handling and disposal of uranyl formate before performing this step. Uranyl formate is a toxic and radioactive compound. Make sure to obtain prior institutional approval for the use of radioactive material. - Wash away the unbound sample by touching each of the four drops of sample buffer.
- Stain the sample using standard negative stain procedures with UF. Gently blot away UF stain from the side, leaving a very thick layer of stain on the grid. An additional 0.5 µL of stain can be applied to the grid after blotting to make the stain thicker if needed. Allow the grid to air dry.
NOTE: Since the streptavidin grids are very hydrophilic, negative stain tends to form a very uniform film everywhere, contrary to what is commonly seen when using glow-discharge treated, continuous carbon grids. As a result, it is recommended to leave a thicker film of stain solution.
5. Freezing streptavidin affinity grids with biotinylated samples
NOTE: The following procedure is intended for a one-sided automated plunger that contains an internal blotting sensor (one-sided manual blotting in other automated plunging apparatus is also possible)
- Prepare two drops of 50 µL and one drop of 100 µL of sample buffer (e.g., 50 mM HEPES pH 7.5, 50 mM KCl, 0.5 mM TCEP) on the clean side of parafilm.
- Rehydrate streptavidin grids by touching the two 50 µL drops and let the grid float on the 100 µL drop of sample buffer for 10 min.
- After rehydrating, pick the grid up with an anti-capillary tweezer so that the kinked arm of the anti-capillary tweezer is in the drop.
- Gently blot the excess buffer away from the side and quickly apply 4 µL of 75-100 nM sample.
- Incubate the sample in a humidity chamber for 1- 5 min.
NOTE: Again, concentration and incubation times will be sample-dependent and should be optimized. The provided sample concentration and incubation time are suggested starting points. For samples at low concentrations, one can incubate for longer time periods and/or bind the sample to the grids several times. - Prepare two 10 µL drops of freezing buffer (e.g., 50 mM HEPES pH 7.5, 50 mM KCl, 0.5 mM TCEP, 3% trehalose, 0.01% NP40) on the clean side of parafilm. After incubation, wash the unbound sample by touching the grid to the first drop of the freezing buffer. Drop the grid onto the second drop of the freezing buffer.
- Quickly grab the edge of the grid with the tweezers attached to the one-sided automated plunger. Gently blot away excess buffer and quickly apply 4 µL of freezing buffer (50 mM HEPES pH 7.5, 50 mM KCl, 0.5 mM TCEP, 3% trehalose, 0.01% NP40) to the grid.
- Attach tweezers to the one-sided automated plunger. The best results have been obtained using a blot sensor that detects the liquid drop on the grid with blot times between 4-6 s. Conditions will vary depending on the sample and buffer used.
6. Data processing of cryo-EM movies collected from streptavidin affinity grids
Data collection on streptavidin affinity grids can be performed as with standard grids, and no special microscope adjustments are necessary. However, the procedure detailed here removes the streptavidin signal only after motion correction of the micrograph. Therefore, data processing steps such as particle polishing that rely on original movie frames cannot be reliably performed. The streptavidin signal subtraction (required for all standard downstream processing except for CTF estimation) here requires Matlab version R2014b or newer running on a Linux environment. Signal subtraction relies on the crystalline nature of the streptavidin layer, which allows for peak masking and, hence, signal removal from the Fourier transform of each micrograph.
- Setting up the processing scripts
- Copy all files from Supplemental Files into a dedicated folder within the project directory
- Prepare a directory called processing_scripts that includes the following files:
lsub.m (Supplemental Coding File 1; the subtraction script)
process_subtration.sh (Supplemental Coding File 2; wrapper script that loops over all available micrographs, spawns parallel jobs, and keeps track of the input and output)
process_subtraction.cfg (Supplemental Coding File 3; configuration file that contains the parameters for the subtraction job, see 6.2) - Prepare a directory called support_scripts that includes the following files:
bg_drill_hole.m (Supplemental Coding File 4)
bg_FastSubtract_standard.m (Supplemental Coding File 5)
bg_Pick_Amp_masked_standard.m (Supplemental Coding File 6)
bg_push_by_rot.m (Supplemental Coding File 7)
ReadMRC.m (Supplemental Coding File 8)
WriteMRC.m (Supplemental Coding File 9)
WriteMRCHeader.m (Supplemental Coding File 10)
- Adjust the configuration file as needed (process_subtraction.cfg, (refer to Figure 3) as needed:
- Input: Path to the directory that contains the motion-corrected micrographs in mrc format. Use either a directory or a pattern including wild cards (?, *).
Example:
input="/path/to/project_directory/micrographs/"
or
input="/path/to/project_directory/micrographs/*.mrc" - output_dir: Path to the directory to which the lattice subtracted micrographs should be written.
- only_do_unfinished: Specify (true|false) if pre-existing subtracted micrographs should be overwritten or skipped. This parameter is useful for starting the lattice subtraction script in the middle of a microscope session (default: true).
- num_parallel_jobs: Specify the number of parallel jobs. This number depends on the hardware specifications used for processing and should not be higher than the number of cores available. On systems with 32 cores and a network filesystem, optimal performance was achieved with 14 parallel jobs. For best performance, optimize this value with a subset of micrographs.
- Pad_Origin_X: 200 for a K3 detector in portrait orientation (image width < image height) or when using a square detector (Falcon III, Falcon IV, K2), 1000 for a K3 detector in landscape orientation (image width > image height). FFT is performed in square boxes and padding is required if the detector has non-square dimensions.
- Pad_Origin_Y: 1000 for a K3 detector in portrait orientation (image width < image height), 200 when using a square detector (Falcon III, Falcon IV, K2) or if using a K3 detector in landscape orientation (image width > image height).
- Do not change the Inside_Radius_Ang (90) and Outside_Radius_Ang (3.0). The lattice subtraction is performed between two resolutions (unit is in angstrom).
- Pixel_Ang: Provide the pixel size as a floating point value.
- Threshold: Subtraction threshold cutoff value to replace the lattice with the background in Fourier space. Successfully used values are between 1.4-1.6. Use 1.42 as a starting point.
- Do not change expand_pixel (10) and pad_out_opt (0). expand_pixel is a diameter value used for masking around pixels with values higher than the threshold cutoff. pad_out_opt is an option that decides whether a padded area is included in the output.
- addpath_m: Path to the support scripts.
- path_to_matlab_bin: Path to the system's Matlab installation bin directory.
- path_matlab_script: Path to the matlab substraction script (lsub.m ) that has been copied above under step 6.1.2.
- Input: Path to the directory that contains the motion-corrected micrographs in mrc format. Use either a directory or a pattern including wild cards (?, *).
- After saving the modified script, run the script (bash ./process_subtraction.sh ) to subtract the streptavidin signal from the input micrographs. The script can be interrupted and/or restarted if the parameter only_do_unfinished is set to true (see in step 6.2.3). If an error occurs, review the parameters specified in the bash script. Ensure that there are no unintended spaces between parameter variables and their assigned values, as this is a common source of mistakes.
- Use the lattice subtracted images for standard cryo-EM image processing.
Representative Results
After the carbon evaporation in step 3.21 (usually after one week), streptavidin affinity grids can be used for cryo-EM or negative-stain sample preparation following the procedures described in steps 4 and 5. A representative micrograph captured using a K3 detector on a titan Krios at 81,000X magnification with a pixel size of 1.05 Å of a biotinylated sample frozen with streptavidin affinity grids is shown in Figure 4A. Successful lattice formation is observed by the continuous grid pattern that appears in the background of the image. This may be more easily observed by the diffraction pattern that appears in the fast Fourier transform (FFT) shown in Figure 4A (right). After data collection, following the procedure outlined in step 6 of this protocol, the signal that is contributed to the image by the streptavidin lattice can be computationally masked to produce a subtracted micrograph (Figure 4B) that can be used for subsequent data processing steps. The FFT in Figure 4B (right) shows that the diffraction pattern observed in Figure 4A (right) has been successfully removed from the original image.
Figure 4C shows an example micrograph taken on a Tecnai 12 microscope at a magnification of 49,000x with a pixel size of 1.6 Å of a biotinylated sample bound to streptavidin affinity grids and negatively stained using uranyl formate. Grids were prepared following the procedure outlined in step 4 of this protocol, leaving a thicker film of stain.
There are several common observations when the streptavidin grid fabrication procedure is unsuccessful that are described further in the discussion section and Table 1. Figure 5 shows several examples of these observations. Figure 5A shows a micrograph of a negatively stained streptavidin affinity grid used from a batch made over six months old. The monolayer has mobilized from the holes in the carbon foil of the quantifoil grids and is observed with similar dimensions to the holes in the grid. Figure 5B shows an example of streptavidin crystals that have high mosaicity that become easily perturbed during sample preparation procedures. Figure 5C shows a representative micrograph from a streptavidin affinity grid in which the carbon evaporation in step 3.21 is too thin or omitted. The streptavidin lattice has become fragmented during the cryo-EM sample preparation process. Figure 5D shows a negatively stained streptavidin affinity grid that has contamination from lipid vesicles, likely due to insufficient washes during step 3.13 or the gold side of the grid becoming wet during steps 3.13-3.20. Please refer to the discussion section and Table 1 for strategies to overcome these common issues.

Figure 1: Schematic of streptavidin affinity grid fabrication procedure. (A) Overview timeline of the streptavidin affinity grid fabrication procedure. The entire procedure can be completed over two weeks requiring two carbon evaporation steps and one week after each to allow the carbon to sufficiently age. (B) Zoomed in view of a grid square of a streptavidin affinity grid showing the layers that make up the grid after completion of the fabrication process, including the standard cryo-EM grid with gold bars and holey-carbon film, the first evaporated carbon layer, lipid monolayer, 2D-streptavidin crystal, and the backside evaporated carbon support layer. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Lipid monolayer and streptavidin crystallization (A) Images demonstrating how to successfully form the lipid monolayer within the small Petri dish utilizing talcum powder to create a boundary for the castor oil that creates constant surface pressure while forming the lipid monolayer. (B) Images demonstrating how to grow streptavidin crystals onto cryo-EM grids. First, the carbon side of the grids is touched to the lipid monolayer and followed by three consecutive washes in the crystallization buffer. Streptavidin is added, and grids are incubated in a humidity chamber for 2 h. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Example parameters for the process_subtraction.cfg file for performing streptavidin lattice subtraction from micrographs (step 6). Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative results of successful streptavidin affinity grid fabrication. (A) Cryo-EM micrograph of biotinylated protein/nucleosome complexes prepared with homemade streptavidin affinity grids. The FFT is shown on the right and shows the streptavidin crystal diffraction pattern. (B) Same micrograph as in panel A is shown following the lattice subtraction procedure to remove the signal from the 2D streptavidin crystal from the original image. (C) An example of a micrograph obtained from negative-staining of a biotinylated sample bound to streptavidin-affinity grids. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative results of unsuccessful streptavidin affinity grid fabrication. (A) Micrograph showing the mobilization of the streptavidin monolayer from the grid holes when grids are older than six months. (B) Micrograph showing streptavidin lattices with high mosaicity that are easily damaged during both cryo-EM and negative-stain sample preparation procedures. (C) Micrograph showing fragmentation of the streptavidin lattice during cryo-EM sample preparation when the carbon evaporation layer in step 3.21 is too thin or omitted. (D) Representative micrograph that is contaminated with lipid vesicles likely due to insufficient washing during step 3.13 or the gold side of the grid becoming wet during steps 3.13-3.20. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Troubleshooting guide to overcome commonly experienced challenges that are encountered during unsuccessful streptavidin affinity grid fabrication. Please click here to download this Table.
Supplemental Coding File 1: lsub.m Please click here to download this File.
Supplemental Coding File 2: process_subtration.sh Please click here to download this File.
Supplemental Coding File 3: process_subtraction.cfg Please click here to download this File.
Supplemental Coding File 4: bg_drill_hole.m Please click here to download this File.
Supplemental Coding File 5: bg_FastSubtract_standard.m Please click here to download this File.
Supplemental Coding File 6: bg_Pick_Amp_masked_standard.m Please click here to download this File.
Supplemental Coding File 7: bg_push_by_rot.m Please click here to download this File.
Supplemental Coding File 8: ReadMRC.m Please click here to download this File.
Supplemental Coding File 9: WriteMRC.m Please click here to download this File.
Supplemental Coding File 10: WriteMRCHeader.m Please click here to download this File.
Discussion
Our protocol describes how to make and use streptavidin affinity grids and how to process the data containing the streptavidin diffraction signal. There are several critical steps in the protocol that require special attention.
Unsuccessful grid batches can be traced back to several common errors. The most common source of error comes from using outdated or poor-quality reagents. It is especially important to prepare the biotinylated lipid solution exactly as described in the protocol. Further, any impurities, such as detergent residue on the labware or naturally present oils on skin, can affect the quality of the streptavidin crystals and, hence, the quality of the resulting images. It is therefore recommended to perform three washing cycles for the grids prior to the first carbon evaporation to remove possible surfactant contamination from the grid manufacturer (step 2.1). Additionally, contamination or impurity of the castor oil has been observed to hinder crystal formation on the grid.
Making and picking up the lipid monolayer is a task that should be practiced a few times to get a feeling for the small lipid volumes. It is not unusual for this step to fail the first two or three times before an adequate lipid monolayer can be produced, as is ultimately reflected in the quality of streptavidin crystals that are seen in negatively stained grids (Figure 4C).
It is important to never let the backside (gold side, lipid side) of the grid get wet during the grid fabrication process (steps 3.12-3.20). In the case the backside does get wet, it is recommended to discard the grid. This can result in the appearance of large lipid vesicles that disrupt the image quality (Figure 5D) (Row 3, Table 1). Lipid vesicles can also be observed due to insufficient washing after the lipid monolayer is applied (step 3.13). Three subsequent washes using crystallization buffer are recommended after touching the lipid monolayer prior to adding streptavidin.
After a thin layer of carbon has been evaporated onto trehalose-embedded grids, the backside of the grid can be wet without damaging the lattice quality. For example, wetting of the backside is frequently observed when the sample buffer contains even minimal amounts of detergent. Wetting of the backside by the sample can result in nonspecific binding of samples to the thin carbon film on the backside, which decreases the effectiveness of preventing particles from diffusing to the air-water interface or the use of affinity binding for on-grid purification strategies. If possible, add the sample to the grid in the absence of detergent. Detergent and other additives can be subsequently added during the grid washing steps or added in a final step before vitrification. This is one limitation to the use of streptavidin affinity grids; however, if the goal is to improve preferential orientation, sample preparation with buffers, including detergent, has been performed successfully11.
A common source of error can be traced to the age of the grids relative to both carbon evaporation steps (step 2.3 and step 3.21). In this protocol, the recommended waiting period is 5-7 days after the backside carbon evaporation before using streptavidin grids for cryo-EM. When grids are used too soon after fabrication, the lattice and lipid monolayer have been observed to mobilize out of the grid holes. A similar observation can be made when the grids are too old and used after six months (Figure 5A) (Row 1, Table 1). We hypothesize that this observation is explained by changes in the hydrophobicity of the carbon backing that is applied to stabilize the lipid monolayer and streptavidin crystals. Additionally, streptavidin lattices may appear mosaic (Figure 5B) (Row 3, Table 1) and broken in both negative-stain and cryo-EM if the monolayer is applied to grids where the first carbon evaporation layer (step 2.3)has not sufficiently aged.
An additional common source of error can be traced to the fragility of the streptavidin crystalline monolayer (the lipid monolayer is itself fluid and can reversibly expand or compress). The backside carbon evaporation (step 3.21) provides critical stability to both the monolayer and streptavidin lattice during the sample adsorption, washing, and cryo-EM blotting process. In the absence of sufficient carbon evaporation to the backside of the grid, streptavidin lattices will often appear fragmented after blotting/freezing (Figure 5C) (Row 2, Table 1). In this protocol, we suggest the use of a one-sided automated plunge freezer for single-sided blotting. This method yields highly reproducible blotting conditions that preserve the streptavidin lattice during the freezing process and allow for streamlined optimization of the sample application and blotting parameters. Alternatively, other automated plunge-freezing apparatuses have been used in combination with manual blotting. To do this, the actual blotting function is disabled in the device settings, and the grid is blotted instead by reaching with a pair of tweezers holding blotting paper through the side entry. This method can achieve high-quality results for laboratories without a single-sided blotting and plunge freezing device; however, grid-to-grid reproducibility is challenging.
While using streptavidin affinity grids has many advantages, some limitations of this method need to be taken into consideration. Owing to the nature of the procedure, it is usually not possible to assess the quality of the streptavidin lattice until the entire process has been completed. It is suggested to quickly screen the quality of each batch by negative stain before freezing samples. Due to the signal contributed by the streptavidin lattice to the raw images, it can be challenging in some cases to assess, from the images alone, whether there is a sufficient number of intact, disperse particles. For the same reason, on-the-fly processing of cryo-EM data during data acquisition is not possible unless the streptavidin signal subtraction procedure has been included in the data pre-processing pipeline. Because the streptavidin signal is usually subtracted from the motion-corrected images and not the frames themselves, Bayesian polishing, as implemented in popular software suites, may fail when using the subtraction procedures as described. It is therefore recommended to perform motion correction in multiple patches to minimize particle movement from the onset of data processing16.
Despite these limitations, streptavidin affinity grids offer many advantages. Two major advantages that streptavidin affinity grids provide over standard open-hole cryo-EM grids are a means to protect samples from the air-water interface and concentrate samples of low abundance (10-100 nM) on the grid. Other support layers, such as carbon and graphene oxide, can also be used as strategies to overcome these sample preparation bottlenecks. In one example, streptavidin affinity grids were the only solution to obtaining an intact reconstruction of a protein-nucleic acid interaction that was incompatible with cross-linking approaches.12
Another major advantage that streptavidin affinity grids provide is a solution to samples that adsorb to other support layers, such as carbon or graphene oxide, with preferred orientations that hinder the ability to obtain a 3D reconstruction of the sample of interest. Random biotinylation of samples using commercially available kits allows the random attachment of the sample of interest to the streptavidin monolayer to obtain more potential views to overcome this challenge.
Additionally, streptavidin affinity grids offer advantages that are unique to affinity-based grids. The high affinity and specificity of the streptavidin-biotin interaction allow the coupling of streptavidin affinity grids with other workflows that involve biotin to purify complexes of interest. In one published example, the authors immobilized a complex on streptavidin affinity grids and incubated a binding partner of unknown stoichiometry in large excess. After washing off any unbound protein, the correctly assembled super-complex was obtained and could be immediately analyzed with cryo-EM11. One possible future application may be to combine streptavidin-binding protein tags, the Avi-tag system, or proximity-labeling approaches with streptavidin affinity grids to extract single proteins and/or protein complexes directly from recombinant or endogenous sources without standard elaborate purification schemes.
By providing this protocol, labs should be able to easily reproduce the fabrication of streptavidin affinity grids and establish them as a more commonly used tool for structural analysis of protein complexes by cryo-EM.
Acknowledgements
T.C. was supported by the National Institute of General Medical Sciences molecular biophysics training grant GM-08295 and the National Science Foundation graduate research fellowship under grant number DGE 2146752. R.G. and B.H. were supported by the National Institute of Health grant R21-GM135666 awarded to R.G. and B.H. This work was partially funded through the National Institute of General Medical Sciences grant R35-GM127018 awarded to E.N. E.N. is a Howard Hughes Medical Institute Investigator.
Materials
| Name | Company | Catalog Number | Comments |
| 1,2-dipalmitoyl-sn-glycero-3- phosphoethanolamine-N-(biotinyl) sodium salt | Avanti Polar Lipids | 870285P | Comes as a powder. Dissolve at concentration of 1–10 mg/mL in chloroform/methanol/water solvent, 65:35:8 v/v and store in single use aliquots at -80 °C |
| 200 proof pure ethanol | Fisher Scientific | 07-678-005 | |
| 5 μL Hamilton Syringe | Hamilton | 87930 | 5 µL Microliter Syringe Model 75 RN, Small Removable Needle, 26 G, 2 in, point style 2 |
| Castor Oil | Sigma Adrich | 259853 | |
| Cell culture dishes untreated (35 mm) | Bio Basic | SP22146 | |
| Chloroform | Sigma Aldrich | 650471 | |
| D-(+)-Trehalose dihydrate,from starch, ≥99% | Sigma Aldrich | T9449 | |
| DUMONT Anti-Capillary Reverse (self-closing) Tweezers Biology Grade | Ted Pella | 510-5NM | |
| HPLC Grade Methanol | Fisher Scientific | A452-4 | |
| Leica EM ACE600 High Vacuum Sputter Coater | Leica | ||
| Leica EM GP2 Automatic Plunge Freezer | Leica | ||
| Quantifoil R 2/1 Au300 | Quantifoil | Q84994 | |
| Steptavidin | New England Bioscience | N7021S | Comes as 1 mg/mL solution. Dilute to final concentration of 0.5 mg /mL in crystallization bufferwith a final trehalose concentration of 10%. Can be flash frozen in 25–50 μL aliquots |
| Talcum powder | Carolina | 896060 | |
| Whatman Grade 1 filter paper | Cytiva | 1001-085 |
References
- Glaeser, R. M., Nogales, E., Chiu, W. . Single-particle Cryo-EM of Biological Macromolecules. IOP Publishing. , (2021).
- Carragher, B., et al. Current outcomes when optimizing 'standard' sample preparation for single-particle cryo-EM. Journal of Microscopy. 276 (1), 39-45 (2019).
- Drulyte, I., et al. Approaches to altering particle distributions in cryo-electron microscopy sample preparation. Acta Crystallographica Section D-Structural Biology. 74, 560-571 (2018).
- Noble, A. J., et al. Routine single particle cryoEM sample and grid characterization by tomography. eLife. 7, e34257 (2018).
- Patel, A., Toso, D., Litvak, A., Nogales, E. Efficient graphene oxide coating improves cryo-EM sample preparation and data collection from tilted grids. bioRxiv. , (2021).
- Wang, F., et al. General and robust covalently linked graphene oxide affinity grids for high-resolution cryo-EM. Proceedings of the National Academy of Sciences. 117 (39), 24269-24273 (2020).
- Pantelic, R. S., Meyer, J. C., Kaiser, U., Baumeister, W., Plitzko, J. M. Graphene oxide: A substrate for optimizing preparations of frozen-hydrated samples. Journal of Structural Biology. 170 (1), 152-156 (2010).
- Williams, R. C., Glaeser, R. M. Ultrathin carbon support films for electron microscopy. Science. 175 (4025), 1000-1001 (1972).
- Han, B. -. G., et al. Long shelf-life streptavidin support-films suitable for electron microscopy of biological macromolecules. Journal of Structural Biology. 195 (2), 238-244 (2016).
- Han, B. -. G., et al. Electron microscopy of biotinylated protein complexes bound to streptavidin monolayer crystals. Journal of Structural Biology. 180 (1), 249-253 (2012).
- Domínguez-Martín, M. A., et al. Structures of a phycobilisome in light-harvesting and photoprotected states. Nature. 609 (7928), 835-845 (2022).
- Kasinath, V., et al. JARID2 and AEBP2 regulate PRC2 in the presence of H2AK119ub1 and other histone modifications. Science. 371 (6527), eabc3393 (2021).
- Lahiri, I., et al. 3.1 structure of yeast RNA polymerase II elongation complex stalled at a cyclobutane pyrimidine dimer lesion solved using streptavidin affinity grids. Journal of Structural Biology. 207 (3), 270-278 (2019).
- Marr, C. R., Benlekbir, S., Rubinstein, J. L. Fabrication of carbon films with ~ 500nm holes for cryo-EM with a direct detector device. Journal of Structural Biology. 185 (1), 42-47 (2014).
- Levine, M. J., Schwarz, J. A. Experimental guidelines for producing molecular assemblies by Langmuir-Blodgett techniques. Journal of Chemical Education. 65 (7), 638 (1988).
- Zheng, S. Q., et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods. 14 (4), 331-332 (2017).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved