Perfil de epítopos de proteínas de superfície em partículas virais por estratégia de repórter duplo multiplex
In This Article
Summary
Aqui, descrevemos um imunoensaio fluorescente multiplex recém-desenvolvido que usa um sistema de citometria de fluxo de repórter duplo para detectar simultaneamente dois epítopos de proteína spike exclusivos em partículas virais intactas do coronavírus 2 da síndrome respiratória aguda grave (SARS-CoV-2) que foram capturadas por microesferas magnéticas acopladas à enzima conversora de angiotensina 2.
Abstract
As proteínas de membrana em vírus envelopados desempenham um papel importante em muitas funções biológicas envolvendo a ligação do vírus aos receptores da célula-alvo, fusão de partículas virais com células hospedeiras, interações hospedeiro-vírus e patogênese da doença. Além disso, as proteínas da membrana viral nas partículas virais e apresentadas nas superfícies das células hospedeiras provaram ser excelentes alvos para antivirais e vacinas. Aqui, descrevemos um protocolo para investigar proteínas de superfície em partículas intactas do coronavírus 2 da síndrome respiratória aguda grave (SARS-CoV-2) usando o sistema de citometria de fluxo dual-reporter. O ensaio explora a tecnologia multiplex para obter uma detecção tripla de partículas virais por três reações de afinidade independentes. Grânulos magnéticos conjugados à enzima conversora de angiotensina humana recombinante 2 (ACE2) foram usados para capturar partículas virais do sobrenadante de células infectadas com SARS-CoV-2. Em seguida, dois reagentes de detecção marcados com R-ficoeritrina (PE) ou Violeta Brilhante 421 (BV421) foram aplicados simultaneamente. Como prova de conceito, foram usados fragmentos de anticorpos direcionados a diferentes epítopos da proteína de superfície SARS-CoV-2 Spike (S1). A detecção de partículas virais por três reações de afinidade independentes fornece forte especificidade e confirma a captura de partículas virais intactas. Curvas de dependência de dose do sobrenadante de células infectadas por SARS-CoV-2 foram geradas com variâncias de coeficiente de replicação (média/DP) ˂14%. O bom desempenho do ensaio em ambos os canais confirmou que dois epítopos de proteína-alvo de superfície do vírus são detectáveis em paralelo. O protocolo descrito aqui pode ser aplicado para (i) perfil de alto multiplex e alto rendimento de proteínas de superfície expressas em vírus envelopados; ii) detecção de partículas virais intactas ativas; e (iii) avaliação da especificidade e afinidade de anticorpos e drogas antivirais para epítopos de superfície de antígenos virais. A aplicação pode ser potencialmente estendida a qualquer tipo de vesículas extracelulares e biopartículas, expondo antígenos de superfície em fluidos corporais ou outras matrizes líquidas.
Introduction
Os vírus patogênicos mais comuns, como influenza, HIV, citomegalovírus humano e cepas de SARS-CoV, são vírus envelopados. A infecção celular por vírus envelopados requer a fusão das membranas das células virais e hospedeiras, resultando na liberação do genoma viral no citoplasma. O RNA viral se replicará antes de ser empacotado em uma nova partícula viral 1,2. Durante esses processos, não apenas as proteínas virais, mas também as proteínas da membrana do hospedeiro podem ser incorporadas ao envelope, tornando-se parte integrante da nova partícula viral. As proteínas da membrana da célula hospedeira incorporadas ao envelope do vírus podem facilitar a entrada do vírus em uma nova célula hospedeira, explorando os mecanismos de interações célula-célula, homing e escape do sistema imunológico 3,4.
Apesar da importância de investigar proteínas associadas a vírus, a maioria das técnicas atualmente disponíveis para análise de vírus5 não suporta caracterização de alto rendimento e alto multiplex do antígeno de superfície do vírus. Também não são capazes de detectar partículas virais individuais ou de discriminar entre partículas virais intactas infecciosas, RNA não infeccioso, proteínas virais e subpopulações de vírus que expressam antígenos diferentes. Recentemente, a citometria de fluxo foi modificada e adaptada em um novo método para a análise de partículas virais, a saber, a virometria de fluxo. A virometria de fluxo permite a investigação de partículas virais únicas e seus antígenos de superfície. No entanto, as limitações, incluindo baixo rendimento, baixa capacidade multiplex, configuração experimental complicada e análise de dados e detectabilidade limitada de partículas virais de pequeno porte permanecem 6,7.
A quantificação multiplexada baseada em microesferas de proteínas e ácidos nucléicos é uma tecnologia bem estabelecida com inúmeras aplicações que vão desde a quantificação de proteínas em fluidos corporais, estudos de interação proteína-proteína e diagnóstico de infecções virais 8,9,10,11,12,13 . Um instrumento de análise de fluxo recentemente introduzido apresenta um canal repórter duplo, permitindo a medição de duas moléculas repórter fluorescentes no mesmo poço de reação. Essa nova capacidade tem se mostrado particularmente útil para o perfil paralelo de diferentes isotipos de imunoglobulinas14. Aqui, é descrito como o sistema repórter duplo pode ser usado para detectar partículas virais intactas, visando vários antígenos de superfície em paralelo.
Como prova de conceito, este relatório detalha o desenvolvimento de um sistema de detecção tripla para partículas do vírus SARS-CoV-2. O SARS-CoV-2 consiste em quatro proteínas principais, uma é a proteína spike (S), que consiste em duas subunidades. A primeira subunidade, S1, faz a ligação primária à ACE2 expressa nas membranas celulares humanas. A segunda subunidade, S2, facilita a entrada na célula-alvo por um peptídeo de fusão, criando um poro na membrana da célula-alvo pelo qual o vírion pode entrar através de15. Os três blocos de construção restantes do SARS-CoV-2 são o nucleocapsídeo (N), a proteína de membrana (M) e a proteína do envelope (E). O nucleocapsídeo é responsável pelo empacotamento do genoma viral, formando estruturas ribonucleoproteicas com RNA, enquanto as proteínas de membrana e envelope desempenham papéis centrais na montagem do vírus.
O ensaio descrito aqui tem como alvo três epítopos independentes da subunidade S1 expressos na superfície do envelope do SARS-CoV-2. Diluições seriadas de sobrenadantes de células infectadas e não infectadas por SARS-CoV-2 são usadas. As partículas virais são capturadas por meio de microesferas conjugadas com ACE2 que ligam a subunidade S1 no vírus. A proteína S do vírus de superfície é então detectada em paralelo com um fragmento variável de cadeia única de imunoglobulina marcado (scFv) comercialmente disponível e um anticorpo monoclonal humano anti-S1 (Hu-anti-S1) juntamente com um scFv marcado com FLAG desenvolvido internamente. O Hu-anti-S1 é detectado pelo primeiro canal (RP1) no sistema de repórter duplo com anticorpo secundário anti-IgG-Fc humano conjugado com R-ficoeritrina (PE) laranja, e o scFv é detectado pelo segundo canal (RP2) com um anticorpo anti-FLAG secundário conjugado com Brilliant Violet 421 (BV421) azul. O ensaio de partículas virais está representado na Figura 1.
Protocol
1. Conjugação de neutravidina e anticorpos de controle para microesferas magnéticas
NOTA: Grânulos magnéticos tingidos com fluorescência (microesferas de poliestireno de 6,5 μm de diâmetro com magnetita incorporada) com diferentes rótulos fluorescentes, listados na Tabela de Materiais , são usados para gerar os seguintes conjugados e controles de esferas: (1) ACE2 humano recombinante biotinilado ligado a grânulos acoplado a um ligante de neutravidina; (2) Biotina ligada a grânulos acoplada a um ligante neutravidina; (3) IgG de cabra acoplado diretamente às contas; e (4) Contas não conjugadas. A proteína a ser acoplada aos grânulos deve estar isenta de azida sódica, albumina de soro bovino (BSA), glicina, tris (hidroxi-metil) aminometano (Tris), glicerol ou aditivos contendo amina. O tampão de ativação é 0,1 M de fosfato de sódio monobásico, anidro (NaH2PO4), pH 6. Ácido 2-morfolinoletansulfônico (MES; 50 mM) O tampão de pH 5 é usado para diluir conjugados. O tampão de lavagem é PBS-T (1x PBS [solução salina tamponada com fosfato], pH 7,4 + 0,05% (v / v) Tween-20). O tampão de armazenamento é de 2,7 mg / mL de reagente bloqueador para ELISA (BRE) + 0,1% de antibióticos (aqui, ProClin 300).
- Retire do frigorífico o pó de sulfo-N-hidroxisulfosuccinimida (NHS) e 65 mg de cloridrato de 1-etil-3-[3-dimetilaminopropil]carbodiimida (EDC) pré-aliquotado e deixe atingir a temperatura ambiente (RT; 18-22 °C) durante 30 min. Armazene o NHS e o EDC em um envelope contendo esferas de sílica durante esta etapa para evitar a hidrólise da umidade atmosférica.
- Prepare microesferas para ativação e acoplamento.
NOTA: Os corantes fluorescentes dentro das microesferas são sensíveis à luz e as esferas devem ser mantidas no escuro e em temperaturas de geladeira (4-8 ° C) quando não estiverem em uso ativo.- Ressuspenda 4 estoques diferentes das microesferas codificadas por ID de cor (12,5 x 106 / mL) (Tabela de Materiais) por vórtice breve, sonicação ou rotação (15 min a 15-30 rpm), de acordo com a folha de informações do produto.
- Transfira 40 μL de cada suspensão de grânulo (5 x 105 microesferas) para poços atribuídos de uma placa de microtitulação de meio poço, fundo plano e 96 poços (Tabela de Materiais).
- Lave as contas magnéticas.
NOTA: As etapas de lavagem podem ser executadas manualmente ou usando uma lavadora de placas automatizada.- Adicione 80 μL/tampão de ativação de poço aos grânulos e imobilize os grânulos em um separador de placas magnéticas por 30 s. Aspirar o sobrenadante das microesferas enquanto os grânulos são imobilizados no separador de placas magnéticas.
- Remova a placa de microtitulação do separador de placa magnética e ressuspenda os grânulos em 50 μL de tampão de ativação.
- Ative as esferas com Sulfo-NHS e EDC.
- Prepare a solução de trabalho Sulfo-NHS a 50 mg / mL em tampão de ativação em um tubo de microcentrífuga de 1,5 mL. Devolva o pó NHS à geladeira (4-8 °C), protegido da umidade.
- Prepare a solução de trabalho EDC a 50 mg / mL em tampão de ativação em seu tubo de microcentrífuga de 1,5 mL. Dissolva as alíquotas pré-fabricadas de 65 mg de pó de EDC em 1,3 mL de tampão de ativação.
NOTA: Sulfo-NHS e EDC começam a hidrolisar e perder atividade ao serem dissolvidos. Evite interromper o procedimento de acoplamento até que o NHS e o EDC tenham sido adicionados aos grânulos. Não guarde as soluções NHS ou EDC dissolvidas para uso posterior. - Prepare a solução de ativação para a ativação do grânulo combinando volumetricamente a solução estoque de Sulfo-NHS a 20% (50 mg / mL), solução estoque de EDC a 20% (50 mg / mL) e tampão de ativação a 60%. É necessária uma solução de ativação de 50 μL para cada reação de ativação do grânulo (usando 5 × 105 grânulos/reação), além de volume extra suficiente para acomodar as perdas por pipetagem.
- Adicione 50 μL de solução de ativação completa a cada poço contendo grânulos lavados. Com o volume de suspensão de grânulos pré-existente de 50 μL no Tampão de Ativação, por poço, a concentração final de Sulfo-NHS será de 5 mg/mL, e a concentração final de EDC também será de 5 mg/mL.
- Sele a placa de reação da microesfera com um selador de placa adesiva descartável de plástico ou folha alumínio e incube por 20 min em um agitador orbital (650 rpm) à temperatura ambiente (18-22 °C) no escuro.
- Lave o excesso de solução de ativação das contas.
- Centrifugue a placa de microtitulação a 233 × g por 1 min.
- Imobilize os grânulos ativados em um separador de placa magnética por 30 s. Remova o selador de placas e aspire o sobrenadante de esferas imobilizadas com ímã com a placa de microtitulação ainda posicionada no separador magnético.
- Remova a placa de microtitulação do separador magnético e adicione 100 μL de tampão MES a cada poço.
- Repita as etapas 1.4.2 a 1.4.3 mais uma vez para um total de duas lavagens.
- Junte neutravidina e IgG de cabra (controle) a conjuntos de contas apropriados. Preparar soluções de trabalho suficientes para neutravidina e IgG de cabra, planeando 100 μL/reacção e um extra suficiente para ter em conta as perdas por pipetagem do seguinte modo:
NOTA: A proteína neutravidina em pó é reconstituída com água ultrapura e depois diluída em uma solução estoque de 1 mg / mL com PBS antes de aliquotar para armazenamento / uso (a proteína neutravidina não é diretamente solúvel em PBS, mas é solúvel em água até ~ 10 mg / mL).- Prepare a solução de trabalho de neutravidina a uma concentração de 125 μg/mL em tampão MES em um tubo de microcentrífuga de baixa ligação a proteínas de 1,5 mL.
- Prepare a solução de trabalho do anticorpo de controle IgG de cabra a uma concentração de 17,5 μg / mL no tampão MES.
- Prepare a placa de microtitulação contendo grânulos ativados. Imobilize os grânulos em um separador de placa magnética por 30 s. Com a placa de microtitulação ainda posicionada no separador magnético, aspirar o sobrenadante dos grânulos imobilizados por ímãs.
- Adicionar 100 μL de solução de trabalho de neutravidina (125 μg/ml) aos alvéolos apropriados que contenham grânulos (para o acoplamento neutravidina-biotina e neutravidina-ACE2).
- Adicione 100 μL de solução de trabalho de IgG de cabra (17,5 μg/mL) ao poço contendo grânulos atribuídos como controles somente de IgG de cabra.
- Adicione 100 μL de MES Buffer ao poço atribuído como controle de esferas não conjugadas.
- Selar a placa de microtitulação e incubar durante 2 h num agitador orbital (650 rpm) a RT (18-22 °C) na obscuridade. Vortex a placa brevemente após 1 h de incubação para garantir que as esferas permaneçam em suspensão.
- Lave as contas com PBS-T.
- Centrifugue a placa de microtitulação a 233 × g por 1 min.
- Imobilize grânulos acoplados em um separador de placa magnética por 30 s. Com a placa de microtitulação ainda posicionada no separador magnético, aspirar o sobrenadante dos grânulos imobilizados por ímãs.
- Remova a placa de microtitulação do separador magnético.
- Adicione 100 μL de PBS-T a cada poço contendo grânulos.
- Repita as etapas de lavagem 1.6.2-1.6.4 uma vez para um total de duas lavagens com PBS-T.
- Prepare contas conjugadas para armazenamento.
- Imobilize grânulos acoplados em um separador de placa magnética por 30 s. Com a placa de microtitulação ainda posicionada no separador magnético, aspirar o sobrenadante dos grânulos imobilizados por ímãs. Remova a placa de microtitulação do separador magnético.
- Adicione 50 μL de buffer de armazenamento a cada ID de microesfera para extinguir a atividade restante do cordão.
- Incube a placa de microtitulação em temperaturas de geladeira (4-8 ° C) no escuro durante a noite (16-22 h).
- Transfira suspensões de grânulos não conjugados e conjugados com IgG de cabra (50 μL) para tubos de microcentrífugas de baixa ligação a proteínas de 1,5 mL devidamente marcados, combinados com dois enxágues de tampão de armazenamento de 100 μL dos poços para garantir a máxima recuperação do grânulo.
NOTA: As contas não conjugadas e conjugadas com IgG de cabra serão numeradas de 5 × 105 em um volume final de 250 μL (ou seja, 2 × 103 contas/μL). Armazene os tubos de microcentrífugas em temperaturas de geladeira (4-8 °C) até o uso.
- Ligar ACE2 biotinilado e biotina a esferas conjugadas com neutravidina.
- Preparar a solução de trabalho ACE2 biotinilada humana recombinante a 18 μg/ml de ACE2 em PBS 10 mM. Por reação, serão necessários 100 μL. Prepare a solução de trabalho de biotina a 2,4 mg / mL de biotina em PBS 10 mM. Por reação, serão necessários 100 μL.
- Preparar a placa de microtitulação contendo microesferas conjugadas com neutravidina.
- Imobilize as microesferas em um separador de placas magnéticas por 30 s. Com a placa de microtitulação ainda posicionada no separador magnético, remova o selador de placas e aspire o sobrenadante das microesferas imobilizadas por ímã.
- Remova a placa de microtitulação do separador magnético e adicione 50 μL de PBS 10 mM / poço.
- Repita as etapas 1.8.2.1-1.8.2.2 uma vez.
- Adicionar 100 μL da solução de trabalho ACE2 biotinilada aos alvéolos apropriados contendo microesferas conjugadas com neutravidina. Adicionar 100 μL da solução de trabalho de biotina aos poços apropriados contendo microesferas conjugadas com neutravidina.
- Selar a placa de microtitulação e incubar durante 1 h num agitador orbital (650 rpm) a RT (18-22 °C) na obscuridade.
- Lave as microesferas conforme descrito nas etapas 1.6.1 a 1.6.5.
- Prepare e armazene as microesferas conjugadas com ACE2 e biotina conforme descrito nas etapas 1.7.1-1.7.4.
NOTA: Os grânulos conjugados com ACE2 e biotina biotinilados serão ambos de 5 × 105 em um volume final de 250 μL (ou seja, 2 × 103 grânulos/μL).
2. Teste de conjugação
- Prepare uma mistura de esferas combinando todos os quatro tipos de microesferas criadas na seção 1 (ou seja, ACE2 biotinilada conjugada com neutravidina, biotina conjugada com neutravidina, conjugada com IgG de cabra e não conjugada).
NOTA: As microesferas de estoque foram armazenadas a 2 × 103 contas/μL e combinadas de forma que a concentração final de esferas na mistura de esferas de trabalho seja de 40 contas de cada conjunto/μL.- Calcule o volume da mistura de grânulos de trabalho necessária para o teste (5 μL/reação), permitindo que o volume extra acomode as perdas de pipetagem. Vortex breve cada tubo e combine volumes calculados iguais de cada suspensão de grânulo em um novo tubo de microcentrífuga de baixa ligação a proteínas. A concentração de grânulos agora é de 400 grânulos de cada conjunto/μL.
- Crie a mistura de grânulos de trabalho diluindo a suspensão de grânulos combinada mais 10 vezes com o buffer de armazenamento (40 de cada conjunto / μL de concentração de trabalho).
NOTA: Faça uma pequena quantidade de mistura de contas de trabalho primeiro para estimar o número de microesferas / μL para cada ID.
- Incube as microesferas com anticorpo anti-ACE2 de cabra.
- Pipete 5 μL de mistura de esferas de trabalho em 3 poços de uma placa de microtitulação de fundo plano, meio poço e 96 poços.
- Adicione 50 μL de anti-ACE2 caprino (0,4 μg / mL diluído em PBS-T, Tabela de Materiais) cada um aos 3 poços contendo grânulos na placa de microtitulação. Selar a placa de microtitulação, o vórtice e incubar num agitador orbital (650 rpm) a RT (18-22 °C) durante 1 h na escuridão.
- Pulsar a placa de microtitulação a 233 × g durante 1 min e lavar as microesferas três vezes com PBS-T, conforme descrito em 1.6.2-1.6.4.
- Incubar microesferas com anticorpos de detecção.
- Pipete 5 μL de mistura de esferas de trabalho em 6 novos poços da placa de microtitulação.
- Prepare 1 μg/mL de cada uma das misturas de detecção de trabalho: IgG PE anti-cabra, IgG PE anti-camundongo e IgG PE anti-coelho em 3 tubos separados de 1,5 mL, usando PBS-T como diluente.
- Adicione 50 μL das misturas de detecção a 3 alvéolos cada, e o IgG anti-cabra é adicionado aos mesmos alvéolos que o anti-ACE2 da etapa 2.2.
- Sele, vórtice e incube em um agitador orbital (650 rpm) em RT (18-22 ° C) por 30 min.
- Pulsar a placa a 233 × g durante 1 min e lavar as microesferas três vezes com PBS-T, conforme descrito em 1.6.2-1.6.4.
- Adicione 100 μL de PBS-T e execute no instrumento de análise de fluxo de relatório duplo com as seguintes configurações:
Modo: Repórter duplo; Tempo limite: 45 s; DD-gating: 7500-17500; Contagem mínima de microesferas: 100 microesferas/conjunto (menor limite de CQ: 35 microesferas/conjunto).
3. Produção de sobrenadante de células infectadas por SARS-CoV-2
O vírus SARS-CoV-2 é propagado em células Vero E6 do hospedeiro (linha de células epiteliais de rim de macaco; ATCC; Tabela de Materiais). As células Vero E6 são cultivadas em meio Eagles modificado (MEM) a 37 ° C em uma atmosfera de 5% de CO2 e 95% de umidade relativa. Cada litro de MEM é suplementado com 10 mL de L-glutamina (200 mM), 38 mL de NaHCO3 (7,5%), 5 mL de solução de penicilina/estreptomicina e 50 mL de soro fetal bovino (FCS); Tabela de Materiais.
CUIDADO: Use procedimentos e equipamentos de biossegurança apropriados ao manusear o SARS-CoV-2.
- As células Vero E6 são cultivadas até a confluência em dois frascos de cultura detecidos 2 de 150 cm. Infecte um frasco com o vírus SARS-CoV-2 e use o outro infectado por simulação como controle.
- Misture aproximadamente 100.000 partículas infecciosas SARS-CoV-2 de tipo selvagem (WT) com 5 mL de meio Eagles MEM.
- Aspirar o meio deum balão 2 de 150 cm e adicionar 55 ml de MEM completo para gerar sobrenadantes de controlo não infectados. Aspirar o meio do outro balão de 150 cm2 e adicionar a mistura de vírus às células. Incubar as células durante 1 h a 37 °C. Agite levemente o frasco a cada 15 minutos para distribuir o vírus.
- Adicione 50 mL de meio MEM completo ao frasco com SARS-CoV-2 adicionado e incube as células até que os efeitos citopáticos sejam observados, avaliando visualmente os frascos a cada 24 h.
NOTA: Deve levar aproximadamente 3-4 dias após a infecção para que a citopatia ocorra. Os efeitos citopáticos na estrutura da monocamada celular Vero E6 (por exemplo, retração celular, crenação, arredondamento, desadesão, perda de granularidade intracitoplasmática, lise evidente) são avaliados qualitativamente observando as células usando um microscópio de luz invertida, de acordo com as diretrizes da Organização Internacional de Padronização para testes de citotoxicidade in vitro 16. - Coletar o sobrenadante celular de ambos os frascos e girar por 6 min a 253 × g para sedimentar os detritos celulares.
- Vírus SARS-CoV-2 inativado por UV no sobrenadante celular
- Pipete 0,5 mL de sobrenadante por poço para 12 poços em uma placa de microtitulação de 24 poços. Irradiar por UV a placa de microtitulação, sem tampa, por 30 s sob uma lâmpada ultravioleta adequada (Tabela de Materiais).
NOTA: A inativação viral no sobrenadante celular deve ser verificada tentando a propagação do vírus em culturas de células Vero E6.
- Pipete 0,5 mL de sobrenadante por poço para 12 poços em uma placa de microtitulação de 24 poços. Irradiar por UV a placa de microtitulação, sem tampa, por 30 s sob uma lâmpada ultravioleta adequada (Tabela de Materiais).
- Aliquotar o sobrenadante da célula em tubos de 1,5 ml e conservar a -20 °C até nova utilização.
NOTA: O sobrenadante celular pode ser armazenado a -80 °C.
4. Ensaio: Detecção de partículas virais de SARS-CoV-2 no sobrenadante celular
- Prepare o tampão de ensaios misturando 0,1% de caseína, 0,5% de álcool polivinílico, 0,8% de polivinilpirrolidona e 1% de BSA (todos p / v) (pH 7). Prepare o tampão de diluição da amostra preparando 10% de IgG de coelho no tampão de ensaio.
- Calcule e prepare o volume da mistura de grânulos de trabalho (etapa 2.1) necessário para o teste (5 μL / reação), permitindo que o excesso de volume acomode as perdas por pipetagem.
- Preparar séries de diluição do sobrenadante. Calcule os volumes sobrenadantes necessários. Analisar cada ponto de diluição em alvéolos triplicados para cada um dos cinco scFvs, resultando em 15 alvéolos por ponto de diluição e tipo de amostra. Utilizar 45 μL de sobrenadantes diluídos em cada alvéolo, para um total de 675 μL necessários; um único tubo de microcentrífuga de 1,5 mL para cada um dos sobrenadantes de SARS-CoV-2 e controle é suficiente.
- Descongele os sobrenadantes do SARS-CoV-2 e os sobrenadantes de controle a 4 °C por pelo menos 1 h. Manter os sobrenadantes e as suas diluições frios (2-8 °C) até à utilização. Rotule e organize oito tubos de microcentrífugas de 1,5 mL cada para SARS-CoV-2 e sobrenadantes de controle.
NOTA: A concentração mais alta analisada será uma diluição sobrenadante de 1:1 (2 vezes), a partir da qual uma série de diluições de 1:2 (3 vezes) será feita usando o tampão de diluição da amostra, com a diluição mais alta sendo uma diluição de 1:1458. Os brancos contendo tampão de diluição de amostra servem apenas como controles sem sobrenadante. Assim, as diluições testadas de cada tipo de sobrenadante (SARS-CoV-2 ou controle) serão de 2, 6, 18, 54, 162, 486 e 1458 vezes, com um controle somente tampão. - Adicione o tampão de diluição da amostra aos tubos de microcentrífugas rotulados. Os tubos de diluição 1:1 (2 vezes) requerem 600 μL de tampão e os tubos restantes requerem 800 μL de tampão. Crie a diluição mais alta (1:1; 2 vezes) de cada sobrenadante combinando 600 μL do sobrenadante com 600 μL de tampão de diluição de amostra em tubos devidamente marcados, seguido de um breve vórtice do tubo para misturar.
- Continue a série transferindo sequencialmente 400 μL dos sobrenadantes diluídos 1:1 (2 vezes) para o próximo tubo de diluição (ou seja, diluição de 6 vezes) e continue as diluições de 3 vezes até que a diluição mais baixa (1458 vezes) tenha sido criada. Vortex breve de cada sobrenadante diluído antes de prosseguir com a próxima diluição.
- Descongele os sobrenadantes do SARS-CoV-2 e os sobrenadantes de controle a 4 °C por pelo menos 1 h. Manter os sobrenadantes e as suas diluições frios (2-8 °C) até à utilização. Rotule e organize oito tubos de microcentrífugas de 1,5 mL cada para SARS-CoV-2 e sobrenadantes de controle.
- Incube as microesferas com o sobrenadante.
- Vortex a mistura de grânulos de trabalho pré-preparada por 30 s para ressuspender as microesferas e adicione 5 μL da mistura de grânulos a cada poço atribuído de uma placa de microtitulação de fundo plano de 384 poços.
NOTA: Placas de 96 poços também podem ser usadas. - Adicione 45 μL das diluições do sobrenadante preparadas aos poços designados contendo microesferas na placa de 384 poços. Selar a placa e incubar durante a noite (16-22 h) num agitador orbital (650 rpm) a RT (18-22 °C) no escuro.
- Vortex a mistura de grânulos de trabalho pré-preparada por 30 s para ressuspender as microesferas e adicione 5 μL da mistura de grânulos a cada poço atribuído de uma placa de microtitulação de fundo plano de 384 poços.
- Lave o excesso de sobrenadante das contas.
- Remova a placa de microtitulação do agitador orbital e remova o selador de placas. Centrifugue a placa a 931 x g por 1 min.
- Imobilize as esferas colocando a placa de microtitulação em um separador de placa magnética por 30 s. Com a placa de microtitulação ainda posicionada no separador magnético, remova o selador de placas e aspire o sobrenadante dos grânulos imobilizados por ímã.
- Remova a placa de microtitulação do separador magnético.
- Adicione 60 μL de PBS-T a cada poço contendo grânulos.
- Repita as etapas de lavagem 4.5.2-4.5.4 duas vezes para um total de três lavagens PBS-T.
- Prepare as diferentes misturas de detecção em tubos separados de 1,5 mL. Cada mistura de detecção consiste em um anticorpo monoclonal humano comercial anti-S1 (Hu-anti-S1) (1 μg/mL) e um dos cinco diferentes scFvs marcados com FLAG (1 μg/mL) (Arquivo Suplementar 1, Tabela Suplementar 1 e Tabela Suplementar 2) visando a proteína spike na partícula SARS-CoV-2, diluída em tampão de ensaio (etapa 4.1), resultando em um total de cinco misturas de detecção diferentes.
- Repita as etapas 4.5.2-4.5.3. Ressuspenda as microesferas lavadas em 50 μL/poço da mistura de detecção de espículas específica para scFv apropriada. Selar a placa de microtitulação e incubar durante 1 h num agitador orbital (650 rpm) a RT (18-22 °C) na obscuridade.
- Centrifugue a placa de microtitulação a 931 × g durante 1 min. Lave o excesso de Reagente de Detecção de Espinhos das contas. Execute três etapas de lavagem com 60 μL de PBS-T de acordo com as etapas 4.5.2-4.5.5.
- Incubar microesferas com mistura de anticorpos de detecção fluorescente.
- Prepare uma mistura de solução fluorescente consistindo de IgG anti-humana conjugada com PE comercialmente disponível, juntamente com anticorpo anti-FLAG conjugado com BV421 diluído no tampão de ensaio, com concentrações de trabalho de 0.2 μg/mL e 1 μg/mL, respectivamente. Por reação, são necessários 50 μL de reagente de detecção fluorescente.
- Repita as etapas 4.5.2 a 4.5.3. Ressuspenda as microesferas em 50 μL/poço de mistura de solução fluorescente. Selar a placa de microtitulação e incubar durante 30 minutos num agitador orbital (650 rpm) a RT (18-22 °C) na obscuridade.
- Gire a placa de microtitulação a 931 × g por 1 min. Lave o excesso de Mistura de Solução Fluorescente das microesferas. Execute três etapas de lavagem com 60 μL de PBS-T de acordo com as etapas 4.5.2-4.5.5.
- Suspenda as microesferas em 60 μL de PBS-T a partir da última etapa de lavagem. Analise a placa em um sistema de análise de fluxo de relatório duplo com as configurações descritas na etapa 2.6.
Representative Results
Teste de conjugação
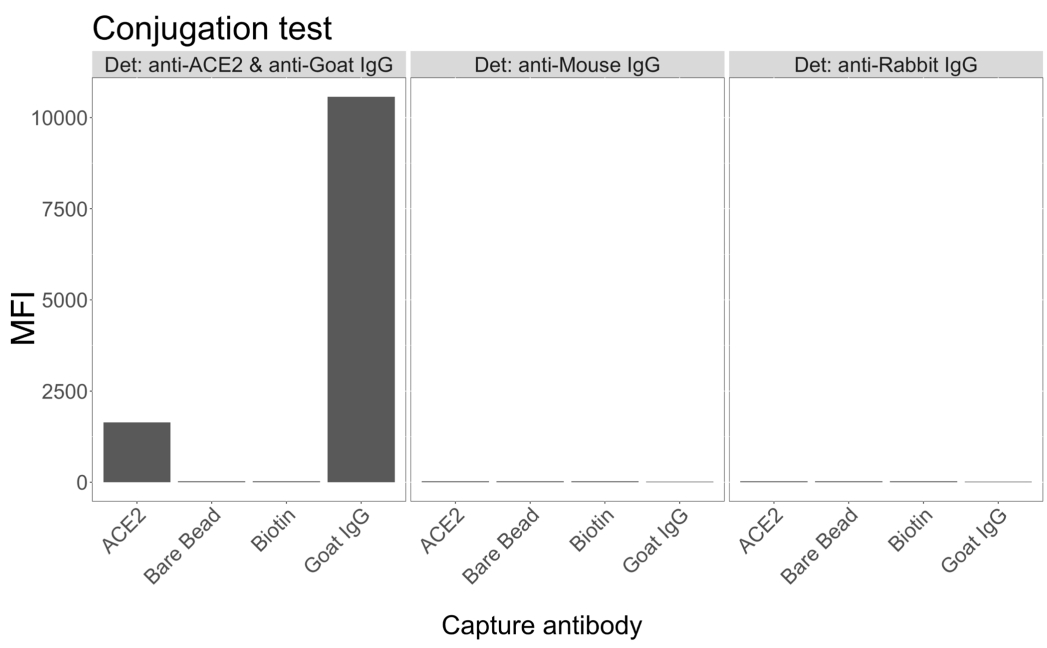
O teste de conjugação mostrou que a ACE2 biotinilada com IgG de cabra e neutravidina-biotinilada foi conjugada com sucesso às microesferas. A especificidade de detecção do ensaio foi confirmada pela sondagem de microesferas conjugadas com ACE2 com anticorpos secundários marcados com PE gerados em diferentes espécies animais (Figura 2). Não foi observada reatividade cruzada entre os diferentes anticorpos de detecção. Quando as misturas de esferas foram sondadas com cabra anti-ACE2 + anti-cabra IgG PE, um valor médio de intensidade de fluorescência (MFI; unidades arbitrárias) acima do fundo foi detectado para as microesferas conjugadas com ACE2 e IgG de cabra, mas não para a microesfera não conjugada (nua) ou para as microesferas revestidas com biotina. IgG PE anti-camundongo e IgG PE anti-coelho foram usados como controles negativos para verificar sinais falso-positivos. Um sinal de fluorescência insignificante foi gerado após a incubação com as microesferas, indicando que os sinais positivos para a ACE2 e a IgG caprina eram específicos.
Detectabilidade de partículas virais em sobrenadantes celulares
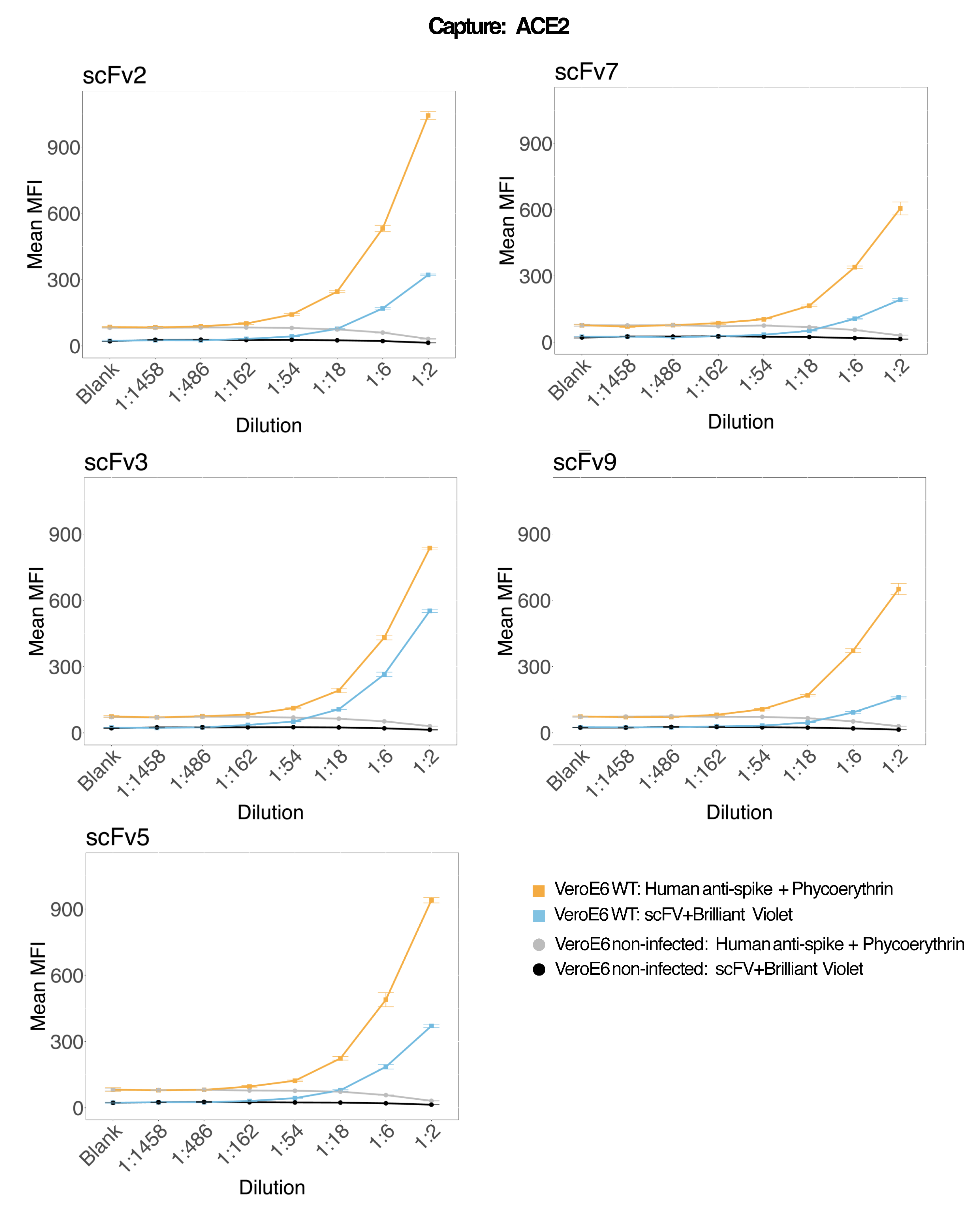
Grânulos magnéticos acoplados a ACE2 humano recombinante foram usados para capturar partículas virais de SARS-CoV-2 de sobrenadantes de cultura de células VeroE6 infectados e de controle (sem vírus) e foram então sondados simultaneamente para duas regiões distintas de pico viral usando um anticorpo monoclonal e um dos cinco scFvs distintos. Um sinal dependente da concentração nas diluições de sobrenadantes de células infectadas por SARS-Cov-2 foi observado em ambos os canais repórteres (RP1 e RP2) (Figura 3), indicando que tanto o anticorpo comercial Hu-anti-S1 quanto os diferentes scFvs detectaram a partícula viral ligada à microesfera conjugada com ACE2. Com três em cada cinco scFvs, o vírus é detectável em diluições de até 1:18 (scFv2, scFv3, scFv5); para os dois scFvs restantes (scFv7 e scFv9), é detectável até diluições de 1:6. Isso pode ser atribuído a uma afinidade diferente com o alvo. Conforme mostrado na Figura 3 e na Tabela 1, scFv3 fornece a maior intensidade de MFI, seguido por scFv5, scFv2, scFv7 e scFv9, respectivamente.
Globalmente, a detecção de scFvs resulta em menor MFI em comparação com o Hu-anti-S1. Isso pode indicar menor afinidade, mas também pode ser um artefato devido à marcação com diferentes corantes fluorescentes (PE e BV421). Outra tendência que pode ser observada para scFv7 e scFv9 é que os valores de MFI são ligeiramente mais baixos para o canal RP1 (anti-spike) também em comparação com as outras três configurações. Isso pode indicar que os scFvs estão reagindo de forma cruzada ou interferindo de outra forma na interação ACE2-Hu-anti-S1, o que também pode explicar o sinal mais baixo no canal RP2. Nenhuma partícula viral foi detectada no sobrenadante das células Vero E6 não infectadas no canal RP1 ou RP2.
A microesfera conjugada neutravidina-biotina, a microesfera de IgG de cabra e as microesferas não conjugadas são usadas como esferas de controle negativo. As partículas virais foram capturadas com microesferas magnéticas acopladas ao ACE2 e testadas com anti-spike humano comercial no canal repórter RP1 e com diferentes scFvs no canal repórter RP2 (scFv é indicado no canto superior esquerdo de cada painel). Nenhuma partícula de vírus foi detectada em nenhuma das amostras infectadas e não infectadas.
Precisão e robustez do ensaio
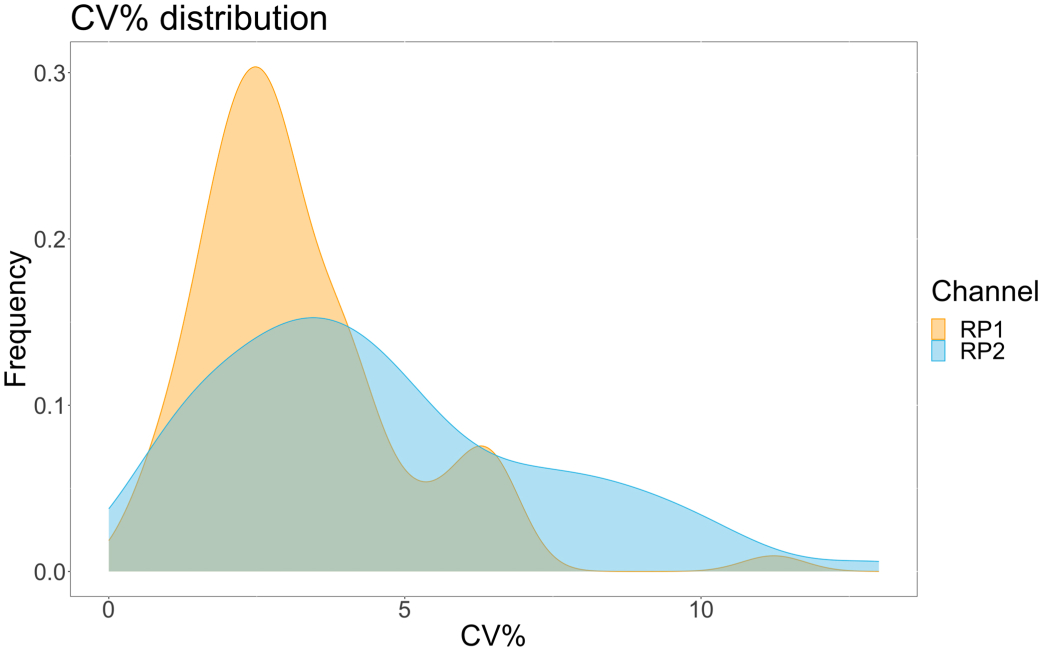
Para avaliar a precisão do ensaio, todas as condições foram executadas em triplicata. Um coeficiente de variância (CV) para a microesfera ACE2 foi calculado para cada ponto de diluição. Todos os CVs calculados para o ensaio estavam abaixo de 15%, onde o CV medido mais alto foi de 13% e o CV mais baixo foi de 1% (Tabela 2). Como pode ser visto no gráfico de densidade (Figura 4) do canal RP1, a detecção de PE do Hu-anti-S1 comercial mostra maior precisão, concentrada principalmente em torno de um CV de 3%. O canal RP2, detecção BV de scFVs, mostra CVs mais altos. No entanto, como pode ser visto na Tabela 2, a maior variação de CVs é impulsionada pelas amostras com baixas concentrações de partículas virais, como o branco. Para testar a robustez do protocolo, o ensaio foi repetido duas vezes por diferentes operadores, utilizando misturas de esferas geradas em dias diferentes e um menor volume de amostra (72% menor). Observou-se uma correlação de Pearson muito boa, variando entre 0,98 e 1, tanto para os canais RP1 quanto para os canais RP2 (p-valor < 0,01), confirmando a robustez do ensaio e a possibilidade de aplicação do ensaio quando houver menos amostra disponível (Figura 5). Essa tecnologia de análise de fluxo segue a "teoria do analito ambiental"17, tornando o ensaio sensível à concentração, mas não ao volume.

Figura 1: O ensaio de partículas de vírus. (A) O sobrenadante celular de células Vero E6 infectadas e não infectadas é adicionado em uma diluição em série a uma placa de 96 ou 384 poços, juntamente com microesferas magnéticas conjugadas com neutravidina e, em seguida, acopladas a ACE2 humano biotinilado ou biotina. Microesferas não conjugadas acopladas com IgG de cabra e microesferas nuas são usadas como controles negativos junto com a microesfera conjugada neutravidina-biotina. (B) Os complexos de partículas de vírus de microesferas que se formaram são detectados com um coquetel de detecção que consiste em Hu-anti-S1 e um dos diferentes scFvs com FLAG-tag. Uma mistura fluorescente com IgG PE anti-humano visando o Hu-anti-S1 e anti-FLAG Brilliant Violet 421 visando o scFvs é então adicionada. (C) O sistema de detecção dupla de três lasers emite um laser vermelho, verde e violeta para detectar o complexo de micropartículas. O laser vermelho detecta o rótulo do corante da microesfera, enquanto os lasers verde e violeta detectam o anti-S1 e o scFvs, respectivamente. Os dados gerados são então analisados. Clique aqui para ver uma versão maior desta figura.
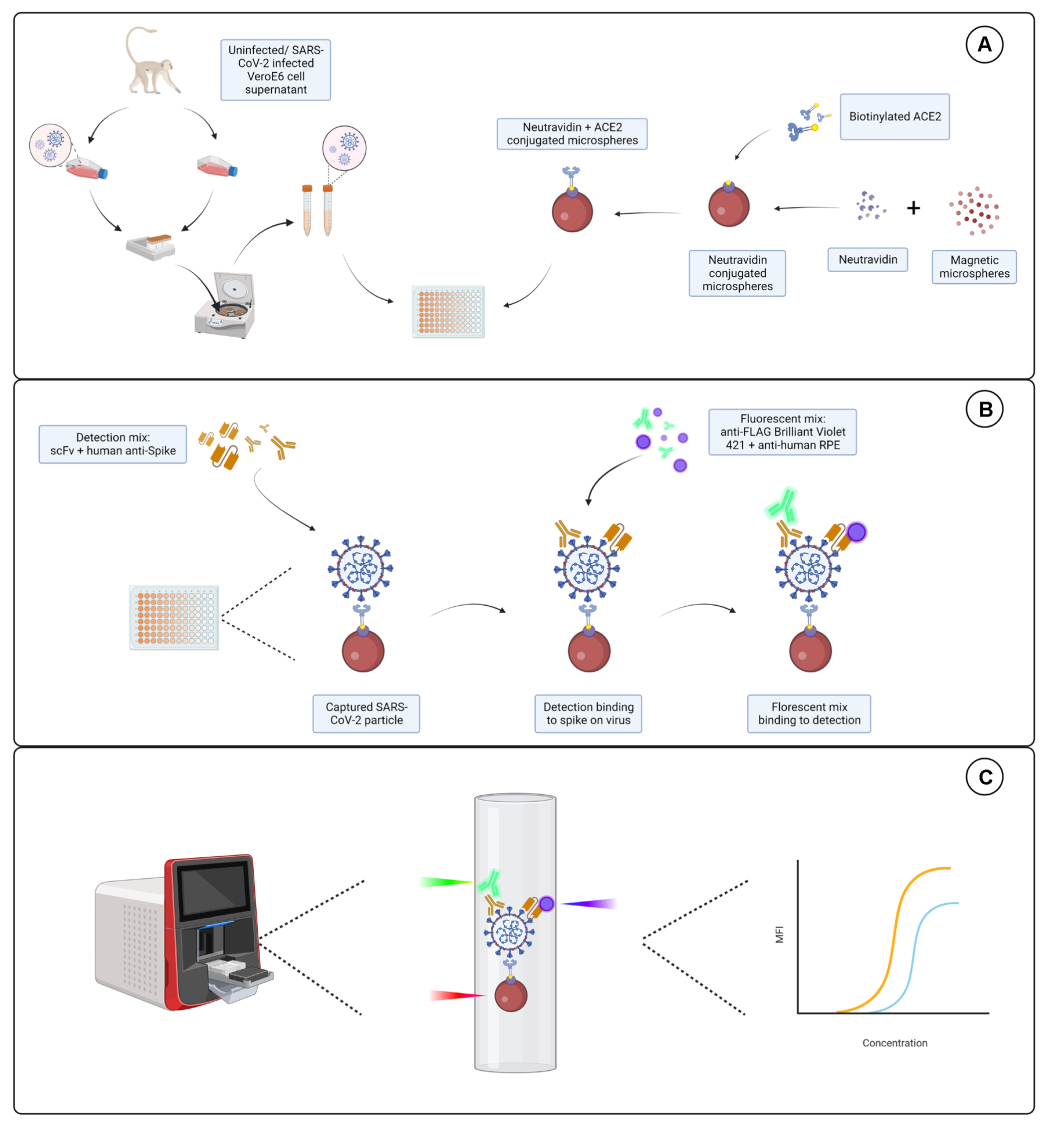{kind=link}

Figura 2: Gráfico de confirmação de conjugação. As misturas de esferas consistiam em quatro IDs de microesferas diferentes, cada uma conjugada com uma proteína diferente: neutravidina-biotina-ACE2 (ACE2), microesfera não conjugada (Bare Bead), neutravidina-biotina (Biotina) e cabra-IgG (IgG de cabra). No teste de conjugação foram utilizadas três configurações diferentes de fluoróforos de detecção. Ou seja, cabra anti-ACE2 + anti-cabra IgG PE, anti-camundongo IgG PE e anti-coelho IgG PE. O eixo Y mostra o sinal médio medido MFI (intensidade média de fluorescência; unidades arbitrárias) de cada microesfera com as três condições diferentes. O eixo X mostra os diferentes anticorpos de captura aplicados. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Detecção multiplexada de proteínas de superfície. Eixo Y: MFI médio (intensidade de fluorescência mediana; unidades arbitrárias ± desvio padrão) para cada amostra, analisado em poços triplicados por condição. Eixo X: Pontos de diluição em série do sobrenadante celular. Laranja: Partículas de vírus no sobrenadante do Vero E6 infectado com SARS-CoV-2 WT detectado com anti-spike humano + anti-PE humano (ficoeritrina). Azul: Sobrenadante de Vero E6 infectado com SARS-CoV-2 WT detectado com os diferentes scFvs + anti-FLAG Brilliant Violet 421. Cinza: sobrenadante de célula não infectada detectado com anti-spike humano + anti-PE humano. Preto: Sobrenadante de célula não infectada detectado com os cinco diferentes scFvs + anti-FLAG Brilliant Violet 421. As partículas virais foram capturadas com microesferas magnéticas acopladas a ACE2 e testadas com anticorpos anti-spike humanos comerciais no canal repórter RP1 e com diferentes scFvs no canal repórter RP2 (scFv é indicado no canto superior esquerdo de cada painel). Nenhuma partícula de vírus foi detectada em nenhuma das amostras não infectadas. O epítopo visado por scFv3 teve a maior afinidade. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 4: Gráfico de dispersão de variação. O eixo Y é a frequência dos eventos e o eixo X mostra o coeficiente de variância (CV) em porcentagem para cada réplica das diferentes amostras. RP1 e RP2 são o primeiro e o segundo canais repórteres que detectam fluorescência associada à ficoeritrina e ao Violeta Brilhante 421, respectivamente. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Matriz de correlação de execução. (A,B) Eixo Y: Matriz de correlação de Pearson em escala log10 entre três ensaios separados, executados por três operadores diferentes e com diferentes misturas de contas. Um menor volume de amostra foi aplicado na terceira execução. Os histogramas mostram a distribuição dos diferentes agrupamentos de variáveis com base no IFM medido. (A) Correlação para o canal de repórter RP1 entre as diferentes execuções. (B) Correlação para o canal de repórter RP2 entre as diferentes execuções. MFI=intensidade média de fluorescência em unidades arbitrárias. p < 0,001. Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Detecção | Reactividade |
| scFv2 | ++ |
| scFv3 | +++ |
| scFv5 | ++ |
| scFv7 | + |
| scFv9 | + |
| IgG anti-Spike humana | ++++ |
Tabela 1: Classificação de scFvs na detecção com base na intensidade do MFI obtida nas curvas padrão.
| RP1 (PE) | RP2 (BV421) | |
| Diluição da amostra | Faixa de CV [%] | Faixa de CV [%] |
| Em branco | 3–11 | 2–13 |
| 1:1458 | 1–7 | 2–7 |
| 1:456 | 4–6 | 3–8 |
| 1:162 | 3–6 | 3–7 |
| 1:54 | 2–4 | 2–4 |
| 1:18 | 2–4 | 1–4 |
| 1:6 | 2–6 | 1–6 |
| 1:2 | 1–5 | 1–3 |
Tabela 2: Intervalo de % CV (média/desvio padrão × 100) de cada ponto de diluição do sobrenadante infectado com SARS-CoV-2 para os canais repórter RP1 e RP2.
Arquivo Suplementar 1: Geração de fragmento variável de cadeia única de imunoglobulina (scFv). Clique aqui para baixar este arquivo.
Tabela Suplementar 1: Triagem de scFvs em pares com Fabs contra diluição serial de Spike recombinante (RBD). Para avaliar o desempenho de diferentes peptídeos de detecção, 12 combinações de proteína spike, Fab, foram utilizadas como captura em tampão enriquecido com RBD recombinante. Dez (10) scFvs direcionados a diferentes epítopos da proteína spike foram aplicados como detecção. Dependendo do desempenho do par de captura-detecção, eles foram marcados como falha (-) ou bem-sucedidos (+). Clique aqui para baixar este arquivo.
Tabela suplementar 2: Triagem de scFvs em pares com Fabs contra diluição serial de sobrenadante de células Calu-3 infectadas com SARS-Cov-2. Para avaliação do desempenho de diferentes peptídeos de detecção, 12 combinações de proteína spike, Fab, foram utilizadas como captura em sobrenadante de células Calu-3 infectadas por SARS-Cov-2. Dez (10) scFvs direcionados a diferentes epítopos da proteína spike foram aplicados como detecção. Dependendo do desempenho do par de captura-detecção, eles foram marcados como falha (-) ou bem-sucedidos (+). Clique aqui para baixar este arquivo.
Discussion
A tecnologia multiplex baseada em grânulos demonstrou ser uma plataforma valiosa para a detecção de patógenos de alto rendimento em várias aplicações clínicas. A alta flexibilidade da plataforma, baseada nos princípios da citometria de fluxo, permite direcionar anticorpos, proteínas e ácidos nucléicos 18,19,20,21,22, multiplexando centenas de analitos simultaneamente. No entanto, até onde sabemos, essa tecnologia não foi aplicada anteriormente para detectar partículas virais intactas. Neste relatório, a tecnologia foi aplicada para a detecção de partículas virais intactas, visando três epítopos de superfície independentes do SARS-CoV-2.
Os vírus de RNA envelopado mostram alta semelhança estrutural com vesículas extracelulares (EVs), pequenas membranas fosfolipídicas que transportam RNA e proteínas virais junto com proteínas hospedeiras23. Imunoensaios sanduíche foram aplicados anteriormente para a detecção de EVs, usando um par de anticorpos direcionado a duas proteínas de superfície distintas24,25. A limitação dos ensaios sanduíche para detectar simultaneamente apenas duas proteínas é removida com abordagens multiplex que permitem a detecção simultânea de mais de duas proteínas por reação.
O sistema de detecção de três lasers de relatório duplo descrito aqui é o instrumento de análise de fluxo baseado em esferas mais avançado até o momento. Com relação aos sistemas de leitura de repórter único, o repórter duplo (canais RP1 e RP2) permite a detecção de três proteínas/epítopos de superfície em paralelo. O direcionamento de várias proteínas e epítopos de superfície viral fornece uma representação mais precisa da carga proteica viral, o que, além de confirmar que o vírus está de fato intacto, também abre a oportunidade de investigar mais detalhadamente os antígenos de superfície viral e os mecanismos das interações virais e proteicas do hospedeiro.
Durante a pandemia de COVID-19, a importância de identificar prontamente os indivíduos portadores de partículas virais ativas foi importante nos esforços para conter a propagação do vírus. O RNA genômico é detectado por RT-PCR quantitativo, independentemente de sua origem (partículas virais intactas ou livres). No entanto, apenas um envelope intacto com proteína S acessível pode mediar a entrada celular e a subsequente replicação do vírus. Estudos anteriores com chips microfluídicos em amostras de pacientes mostraram como a detecção de partículas virais intactas combinada com testes no local de atendimento permitiria testes frequentes e vigilância aprimorada da disseminação da doença, incluindo uma escolha mais informada de indivíduos a serem colocados em quarentena26. A aplicação de um ensaio multiplexado baseado em microesferas permitiria o desenho de ensaios destinados à triagem de múltiplos vírus e suas variantes de antígenos de superfície, obtendo uma imagem mais precisa da disseminação do vírus na população.
A virometria de fluxo é um desenvolvimento recente da citometria de fluxo visando a análise de partículas virais. Apesar de ser capaz de detectar partículas virais discretas, a análise de pequenos vírus representa um problema atual para a virometria de fluxo27,28. Da mesma forma que o método descrito aqui, a virometria de fluxo envolve a captura de vírions intactos por nanopartículas de ouro acopladas a anticorpos. As limitações para ambos os métodos incluem (i) a dependência de reagentes de captura e detecção de alta afinidade para antígenos expressos na superfície direcionados pelas microesferas ou nanopartículas, (ii) capacidade limitada de discriminar entre partículas de vírus e vesículas extracelulares e (iii) falta de padrões para quantificação adequada de partículas.
As células secretam EVs em seus arredores e, quando infectadas por um vírus, também podem secretar vírions de tamanho semelhante aos EVs e podem eventualmente expressar os mesmos antígenos29. Como os EVs terão composições de membrana semelhantes às do vírus, pode ser difícil distingui-los uns dos outros usando apenas métodos baseados em afinidade, como a abordagem de repórter único a laser duplo. No entanto, as estratégias aqui descritas apresentam uma maior capacidade multiplex, permitindo uma investigação mais ampla e profunda da composição proteica das partículas. Os métodos baseados em fluxo permitem o rastreamento de partículas discretas, oferecendo oportunidades para quantificação digital. Uma estratégia para resolver a questão da quantificação em nosso método seria usar vesículas sintéticas bem caracterizadas expressando antígenos de interesse como partículas semelhantes a vírus (VLPs) para preparar curvas padrão.
Um caminho comum de entrada e saída do SARS-CoV-2 das células hospedeiras é através da interação do vírus e da membrana da célula hospedeira 2,15. Nesse processo, a probabilidade de as proteínas da membrana do hospedeiro serem incorporadas à superfície do vírus é alta. Ao rastrear proteínas do hospedeiro incorporadas, pode-se rastrear o caminho da infecção e potencialmente prever o curso da doença para diferentes pacientes de risco, permitindo decisões de tratamento mais precoces. Também permite a caracterização dos vírus em diferentes lotes de amostras em laboratórios de pesquisa. Isso pode ser explorado mais detalhadamente testando se diferentes características estão relacionadas a diferentes níveis de infectividade viral e para triagem de anticorpos e moléculas de medicamentos que têm como alvo as proteínas de superfície viral.
Um aspecto importante sobre o método descrito é que ele depende da afinidade dos reagentes de captura e detecção contra suas proteínas-alvo no vírus. A escolha dos reagentes de afinidade é, portanto, um fator determinante no desempenho do ensaio. Possivelmente, reagentes de afinidade múltipla devem ser rastreados e testados para captura e detecção para selecionar aqueles com a maior afinidade. Aqui, o desempenho de dez scFvs e doze fragmentos de Fab foi avaliado preliminarmente usando RBD recombinante e em partículas virais do sobrenadante de células epiteliais pulmonares Calu-3 infectadas com SARS-Cov-3 (células VeroE6 foram usadas para cultura/avaliação de citotoxicidade em todos os estudos subsequentes). O Anti-FLAG PE foi usado para detectar os scFvs marcados com FLAG (Tabela Suplementar 1 e Tabela Suplementar 2). Os cinco scFvs de melhor desempenho foram então selecionados para serem aplicados no ensaio de repórter duplo, juntamente com Hu-anti-S1 comercial (Tabela 1), em sobrenadantes de células epiteliais renais de macaco verde africano VeroE6 infectadas.
Outro fator crítico para o sucesso do protocolo é o procedimento selecionado para o acoplamento de microesferas. O método de acoplamento deve ser eficiente e, ao mesmo tempo, manter intactos e não modificados os epítopos conformacionais ou resíduos de aminoácidos envolvidos na ligação às proteínas. Aqui, a reação EDC-NHS foi aplicada para acoplar neutravidina diretamente às microesferas, adaptando um protocolo descrito anteriormente30 e um sistema neutravidina + biotina para ligar ACE2 recombinante às microesferas acopladas. Métodos alternativos de acoplamento e sua eficiência podem ser testados e comparados. Finalmente, observou-se que diferentes reagentes de detecção marcados com fluorescência (por exemplo, anti-FLAG PE (ficoeritrina) e anti-FLAG Brilliant Violet 421) podem resultar em diferentes níveis de MFI que podem afetar a sensibilidade do ensaio.
Em conclusão, o método descrito permite a detecção de partículas virais intactas em solução, aplicando uma estratégia de duplo repórter. A análise de três determinantes de superfície em paralelo fornece uma ferramenta mais específica para caracterizar partículas virais e, eventualmente, discriminá-las de outros EVs (por exemplo, não contendo antígenos virais). Essa estratégia é uma alternativa à virometria de fluxo. Embora a abordagem atual não discrimine os tamanhos das partículas, as estratégias de esferas magnéticas usando microesferas codificadas por cores oferecem uma capacidade mais ampla no perfil de antígenos de superfície e no projeto experimental por análise de alto multiplex e alto rendimento. O ensaio apresenta alta precisão e robustez e pode ser estendido para a análise de qualquer tipo de vesícula extracelular e qualquer outro tipo de biopartícula expondo antígenos de superfície em fluidos corporais ou outras matrizes líquidas. Este foi um estudo de prova de conceito que demonstrou a utilidade do uso de scFvs como um reagente de detecção em uma análise multiplex de vários epítopos de proteínas em partículas virais. Estudos futuros são necessários para determinar as características específicas dos scFvs (por exemplo, afinidades de ligação, reatividade cruzada com outros reagentes e alvos) se forem usados para fins quantitativos ou clínicos.
Disclosures
Os autores declaram não haver conflito de interesses.
Acknowledgements
Agradecemos no SciLifeLab, Suécia, a equipe da Unidade Affinity Proteomics-Stockholm Scilifelab por desenvolver e aplicar o método descrito aqui, a Unidade de Terapêutica de Anticorpos Humanos por fornecer reagentes scFvs e Fab e Jonas Klingström pelas células VeroE6 infectadas com isolados de SARS-CoV-2 provenientes de amostras clínicas. Os autores agradecem a Sherry Dunbar, PhD, MBA da Luminex Corporation (Austin, TX), pelo apoio à pesquisa, e Matt Silverman MSci, PhD da Biomedical Publishing Solutions (Cidade do Panamá, FL; mattsilver@yahoo.com) pela assistência científica e de redação. Este trabalho foi apoiado por fundos da Fundação Knut e Alice Wallenberg e do Laboratório Science for Life (SciLifeLab) (VC2020-0015 para Claudia Fredolini e Francesca Chiodi e VC-2022-0028 para Claudia Fredolini).
Materials
| Name | Company | Catalog Number | Comments |
| ACE2-Biotin | Acro Biosystems (Newark, DE) | AC2-H82E6-25 ug | Conc: 340 µg/mL, LOT#BV35376-203HFI-2128 |
| Anti-Goat IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 705-116-147 | Host species: Donkey |
| Anti-Human IgG R-PE | Life Technologies/Thermo Fisher (Waltham, MA) | H10104 | Conc: 0.15 mg/mL, LOT#2079224, Host species: Goat |
| Anti-Mouse IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 115-116-146 | Host species: Goat |
| Anti-Rabbit IgG, PE-conjugated | Jackson ImmunoResearch (West Grove, PA) | 111-116-144 | Host species: Goat |
| Biotin | Thermo-Fisher Scientific (Waltham, MA) | 20RUO | 100 mM, pH 10 Conc. 1 mg/mL |
| Blocker Casein in PBS | Thermo-Fisher Scientific (Waltham, MA) | 37528 | LOT#VD301372 |
| Blocker reagent for ELISA (BRE) | Roche (Basel, Switzerland) | 11112589001 | |
| Brilliant Violet 421 anti-DYKDDDDK Tag Antibody (Anti-FLAG) 0.2 mg/ml, rat IgG2a, λ | BioLegend (Amsterdam, The Netherlands) | 637321 | |
| Bovine serum albumin (BSA) | Saveen & Werner (Limhamn, Sweden) | B2000-500 | LOT#04D5865 |
| EDC (1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) | Proteochem (Hurricane, UT) | C1100-custom (65 mg) | LOT# MK3857 |
| Fetal calf serum (FCS) | Gibco/Thermo Fisher (Waltham, MA) | 10270-106 | |
| Goat anti-ACE2 polyclonal antibody | R&D Systems/Bio-Techne (Minneapolis, MN) | AF933 | Host species: Goat |
| Goat IgG | Bethyl Labs (Montgomery, TX) | P50-200 | LOT#P50-200-6 |
| L-glutamine | Thermo-Fisher Scientific (Waltham, MA) | 25030024 | |
| Low-bind 1.5 mL microfuge tubes | VWR (Radnor, PA) | 525-0133 | |
| MagPlex-C Microspheres | Luminex Corporation (Austin, TX) | MC10XXX-01 | |
| MEM tissue cuture media | Gibco/Thermo Fisher (Waltham, MA) | 21430-020 | |
| Microplate, 96-Well, Polystyrene, Half-area, Clear | Greiner Bio-One (Kremsmünster, Austria) | 675101 | |
| NaHCO3 | Gibco/Thermo Fisher (Waltham, MA) | 25080-060 | |
| Neutravidin | Thermo-Fisher Scientific (Waltham, MA) | 31000 | LOT#UK292857 |
| PBS tablets | Medicago AB (Uppsala, Sweden) | 09-9400-100 | LOT#272320-01 |
| Penicillin/Streptomycin | Gibco/Thermo Fisher (Waltham, MA) | 15140122 | |
| Poly(vinyl alcohol) | Sigma-Aldrich (St. Louis, MO) | 360627 | |
| Polyvinylpyrrolidone | Sigma-Aldrich (St. Louis, MO) | 437190 | |
| ProClin 300 | Sigma-Aldrich (St. Louis, MO) | 48915-U | |
| Rabbit IgG | Bethyl Labs (Montgomery, TX) | P120-301 | LOT#12 |
| scFv-FAb1 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.12 mg/mL. | |
| scFv-FAb2 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc batch1: 0.38 mg/mL. Conc batch2: 0.45 mg/mL | |
| scFv-FAb3 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 0.34 mg/mL. | |
| scFv-FAb4 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc: 2.85 mg/mL. | |
| scFv-FAb5 | In-house production | Human Antibody Therapeutics Unit, Scilifelab, Sweden. Monoclonal scFv. Conc:2.7mg/mL. | |
| SARS-CoV-2 infectious particles, Swedish isolate | In-house production | The Public Health Agency of Sweden | |
| SARS-CoV-2 Spike Antibody (Hu-anti-S1) | Novus Biologicals (Centennial, CO) | NBP2-90980 | Monoclonal antibody. Conc: 1 mg/mL. Host: Human. Clone: CR3022. Isotype: IgG1 Kappa. LOT#T201B06 |
| Sodium phosphate monobasic, anhydrous | Sigma-Aldrich (St. Louis, MO) | S3139 | |
| Sulfo-NHS (N-hydroxysulfosuccinimide) | Thermo-Fisher Scientific (Waltham, MA) | 24510 | LOT# XH321563 |
| Tween | Thermo-Fisher Scientific (Waltham, MA) | BP337-50 | LOT#194435 |
| Ultraviolet lamp | Vilber Lourmat GmbH (Eberhardzell, Germany) | VL-215.G | Wavelength = 254 nm; 2 × 15-watt bulbs |
| Vero E6 cells | ATCC (Manassus, VA) | CRL-1586 | |
| xMAP INTELLIFLEX DR-SE (dual-reporter flow instrument) | Luminex Corporation (Austin, TX) | INTELLIFLEX-DRSE-RUO |
References
- Rey, F. A., Lok, S. M. Common features of enveloped viruses and implications for immunogen design for next-generation vaccines. Cell. 172 (6), 1319-1334 (2018).
- V'kovski, P., Kratzel, A., Steiner, S., Stalder, H., Thiel, V. Coronavirus biology and replication: implications for SARS-CoV-2. Nature Reviews Microbiology. 19 (3), 155-170 (2021).
- Burnie, J., et al. Flow virometry quantification of host proteins on the surface of HIV-1 pseudovirus particles. Viruses. 12 (11), 1296 (2020).
- Gentili, M., et al. Transmission of innate immune signaling by packaging of cGAMP in viral particles. Science. 349 (6253), 1232-1236 (2015).
- Modrow, S., Falke, D., Truyen, U., Schätzl, H. . Viruses: Definition, Structure, Classification. In Molecular Virology. , 163-181 (2013).
- Trinh, K. T. L., Do, H. D. K., Lee, N. Y. Recent advances in molecular and immunological diagnostic platform for virus detection: A review. Biosensors. 13 (4), 490 (2023).
- Zamora, J. L. R., Aguilar, H. C. Flow virometry as a tool to study viruses. Methods. 134-135, 87-97 (2018).
- Graham, H., Chandler, D. J., Dunbar, S. A. The genesis and evolution of bead-based multiplexing. Methods. 158, 2-11 (2019).
- Byström, S., et al. Affinity proteomic profiling of plasma for proteins associated to area-based mammographic breast density. Breast Cancer Research. 20 (1), 14 (2018).
- Rudberg, A. -. S., et al. SARS-CoV-2 exposure, symptoms and seroprevalence in healthcare workers in Sweden. Nature Communications. 11 (1), 5064 (2020).
- Liu, J., et al. Multiplex reverse transcription PCR Luminex assay for detection and quantitation of viral agents of gastroenteritis. Journal of Clinical Virology. 50 (4), 308-313 (2011).
- Gadsby, N. J., Hardie, A., Claas, E. C. J., Templeton, K. E. Comparison of the Luminex respiratory virus panel fast assay with in-house real-time PCR for respiratory viral infection diagnosis. Journal of Clinical Microbiology. 48 (6), 2213-2216 (2010).
- Lorenzen, E., et al. Multiplexed analysis of the secretin-like GPCR-RAMP interactome. Science Advances. 5 (9), (2019).
- Angeloni, S., Cameron, A., Pecora, N. D., Dunbar, S. A rapid, multiplex dual reporter IgG and IgM SARS-CoV-2 neutralization assay for a multiplexed bead-based flow analysis system. Journal of Visualized Experiments: JoVE. (170), e62487 (2021).
- Jackson, C. B., Farzan, M., Chen, B., Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 23 (1), 3-20 (2022).
- ISO10993-5 Biological evaluation of medical devices - Part 5: Tests for in vitro cytotoxicity. International Standardization Organization Available from: https://nhiso.com/wp-content/uploads/2018/05/ISO-10993-5-2009.pdf (2009)
- Poetz, O., et al. Sequential multiplex analyte capturing for phosphoprotein profiling. Molecular & Cellular Proteomics. 9 (11), 2474-2481 (2010).
- Dunbar, S. A., Vander Zee, C. A., Oliver, K. G., Karem, K. L., Jacobson, J. W. Quantitative, multiplexed detection of bacterial pathogens: DNA and protein applications of the Luminex LabMAP system. Journal of Microbiological Methods. 53 (2), 245-252 (2003).
- Taniuchi, M., et al. Multiplex polymerase chain reaction method to detect Cyclospora, Cystoisospora, and Microsporidia in stool samples. Diagnostic Microbiology and Infectious Disease. 71 (4), 386-390 (2011).
- Wu, M., et al. High-throughput Luminex xMAP assay for simultaneous detection of antibodies against rabbit hemorrhagic disease virus, Sendai virus, and rabbit rotavirus. Archives of Virology. 164 (6), 1639-1646 (2019).
- Dias, D., et al. Optimization and validation of a multiplexed Luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clinical and Vaccine Immunology. 12 (8), 959-969 (2005).
- Opalka, D., et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexes lumina assay. Clinical and Diagnostic Laboratory Immunology. 10 (1), 108-115 (2003).
- Nolte-'T Hoen, E., Cremer, T., Gallo, R. C., Margolis, L. B. Extracellular vesicles and viruses: Are they close relatives. Proceedings of the National Academy of Sciences. 113 (33), 9155-9161 (2016).
- Ohmichi, T., et al. Quantification of brain-derived extracellular vesicles in plasma as a biomarker to diagnose Parkinson's and related diseases. Parkinsonism & Related Disorders. 61, 82-87 (2019).
- Ter-Ovanesyan, D., et al. Framework for rapid comparison of extracellular vesicle isolation methods. Elife. 10, e70725 (2021).
- Gamage, S. S. T., et al. Microfluidic affinity selection of active SARS-CoV-2 virus particles. Science Advances. 8 (39), (2022).
- Renner, T. M., Tang, V. A., Burger, D., Langlois, M. -. A. Intact viral particle counts measured by flow virometry provide insight into the infectivity and genome packaging efficiency of moloney murine leukemia virus. Journal of Virology. 94 (2), e01600-01619 (2020).
- Niraja, S., et al. A flow virometry process proposed for detection of SARS-CoV-2 and large-scale screening of COVID-19 cases. Future Virology. 15 (8), 525-532 (2020).
- Lippé, R. Flow virometry: A powerful tool to functionally characterize viruses. Journal of Virology. 92 (3), e01765 (2018).
- Drobin, K., Nilsson, P., Schwenk, J. M. Highly multiplexed antibody suspension bead arrays for plasma protein profiling. Methods in Molecular Biology. 1023, 137-145 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2025 MyJoVE Corporation. All rights reserved