A subscription to JoVE is required to view this content. Sign in or start your free trial.
Construction of Out-of-Equilibrium Metabolic Networks in Nano- and Micrometer-Sized Vesicles
In This Article
Summary
We present a protocol for reconstituting membrane proteins and encapsulating enzymes and other water-soluble components in lipid vesicles of sub-micrometer and micrometer size.
Abstract
We present a method to incorporate into vesicles complex protein networks, involving integral membrane proteins, enzymes, and fluorescence-based sensors, using purified components. This method is relevant for the design and construction of bioreactors and the study of complex out-of-equilibrium metabolic reaction networks. We start by reconstituting (multiple) membrane proteins into large unilamellar vesicles (LUVs) according to a previously developed protocol. We then encapsulate a mixture of purified enzymes, metabolites, and fluorescence-based sensors (fluorescent proteins or dyes) via freeze-thaw-extrusion and remove non-incorporated components by centrifugation and/or size-exclusion chromatography. The performance of the metabolic networks is measured in real time by monitoring the ATP/ADP ratio, metabolite concentration, internal pH, or other parameters by fluorescence readout. Our membrane protein-containing vesicles of 100-400 nm diameter can be converted into giant-unilamellar vesicles (GUVs), using existing but optimized procedures. The approach enables the inclusion of soluble components (enzymes, metabolites, sensors) into micrometer-size vesicles, thus upscaling the volume of the bioreactors by orders of magnitude. The metabolic network containing GUVs are trapped in microfluidic devices for analysis by optical microscopy.
Introduction
The field of bottom-up synthetic biology focuses on constructing (minimal) cells1,2 and metabolic bioreactors for biotechnological3,4 or biomedical purposes5,6,7,8. The construction of synthetic cells provides a unique platform that allows researchers to study (membrane) proteins in well-defined conditions mimicking those of native environments, enabling the discovery of emergent properties and concealed biochemical functions of proteins and reaction networks9. As an intermediate step towards an autonomously functioning synthetic cell, modules are developed that capture essential features of living cells such as metabolic energy conservation, protein and lipid synthesis, and homeostasis. Such modules not only enhance our understanding of life but also have potential applications in the fields of medicine8 and biotechnology10.
Transmembrane proteins are at the heart of virtually any metabolic network as they transport molecules in or out of the cell, signal, and respond to the quality of the environment, and play numerous biosynthetic roles. Thus, the engineering of metabolic modules in synthetic cells requires in most cases the reconstitution of integral and/or peripheral membrane proteins into a membrane bilayer composed of specific lipids and high integrity (low permeability). The handling of these membrane proteins is challenging and requires specific knowledge and experimental skills.
Several methods have been developed to reconstitute membrane proteins within phospholipid vesicles, most often with the purpose of studying the function11,12, regulation13, kinetic properties14,15, lipid dependence15,16, and/or stability17 of a specific protein. These methods involve the rapid dilution of detergent-solubilized protein into aqueous media in the presence of lipids18, the removal of detergents by incubating detergent-solubilized protein with detergent-destabilized lipid vesicles and absorption of the detergent(s) onto polystyrene beads19, or the removal of detergents by dialysis or size-exclusion chromatography20. Organic solvents have been used to form lipid vesicles, for example, via the formation of oil-water interphases21, but the majority of integral membrane proteins are inactivated when exposed to such solvents.
In our laboratory, we mostly reconstitute membrane proteins by the detergent-absorption method to form large-unilamellar vesicles (LUVs)19. This method allows the co-reconstitution of multiple membrane proteins and the encapsulation in the vesicle lumen of enzymes, metabolites, and probes22,23. The membrane protein-containing LUVs can be converted into giant-unilamellar vesicles (GUVs) with/without encapsulation of water-soluble components, using either electroformation24 or gel-assisted swelling25 and specific conditions to preserve the integrity of the membrane proteins26.
This paper presents a protocol for the reconstitution in LUVs of an out-of-equilibrium metabolic network that regenerates ATP through the breakdown of L-arginine into L-ornithine27. The formation of ATP is coupled to the production of glycerol-3-phosphate (G3P), an important building block for phospholipid synthesis22,28. The metabolic pathway consists of two integral membrane proteins, an arginine/ornithine (ArcD) and a G3P/Pi antiporter (GlpT). In addition, three soluble enzymes (ArcA, ArcB, ArcC) are required for the recycling of ATP, and GlpK is used to convert glycerol into glycerol 3-phosphate, using the ATP from the breakdown of L-arginine, see Figure 1 for a schematic overview of the pathway. This protocol represents a good starting point for the future construction of even more complex reaction networks-for the synthesis of lipids or proteins or the division of cells. The lipid composition of the vesicles supports the activity of a wide variety of integral membrane proteins and has been optimized for the transport of diverse molecules into or out of the vesicles27,29,30.

Figure 1: Overview of the pathway for ATP production and glycerol 3-phosphate synthesis and excretion. Please click here to view a larger version of this figure.
{kind=link}
In short, purified membrane proteins (solubilized in dodecyl-β-D-maltoside, DDM) are added to preformed lipid vesicles that have been destabilized with Triton X-100, which allows the insertion of the proteins into the membrane. The detergent molecules are subsequently (slowly) removed by the addition of activated polystyrene beads, resulting in the formation of well-sealed proteoliposomes. Soluble components can then be added to the vesicles and encapsulated via freeze-thaw cycles, which traps the molecules in the process of membrane fusion. The obtained vesicles are highly heterogeneous and many are multilamellar. They are then extruded through a polycarbonate filter with a pore size of 400, 200, or 100 nm, which yields more uniformly sized vesicles; the smaller the pore size, the more homogeneous and unilamellar the vesicles but at the price of a smaller internal volume. Non-incorporated proteins and small molecules are removed from the external solution by size-exclusion chromatography. The proteoLUVs can be converted into micrometer size vesicles by gel-assisted swelling, and these proteoGUVs are then collected and trapped in a microfluidic chip for microscopic characterization and manipulation. Figure 2 shows a schematic overview of the full protocol.

Figure 2: Overview of the protocol for reconstituting membrane proteins and encapsulating enzymes and water-soluble components in lipid vesicles of sub-micrometer (LUVs) and micrometer size (GUVs). Please click here to view a larger version of this figure.
{kind=link}
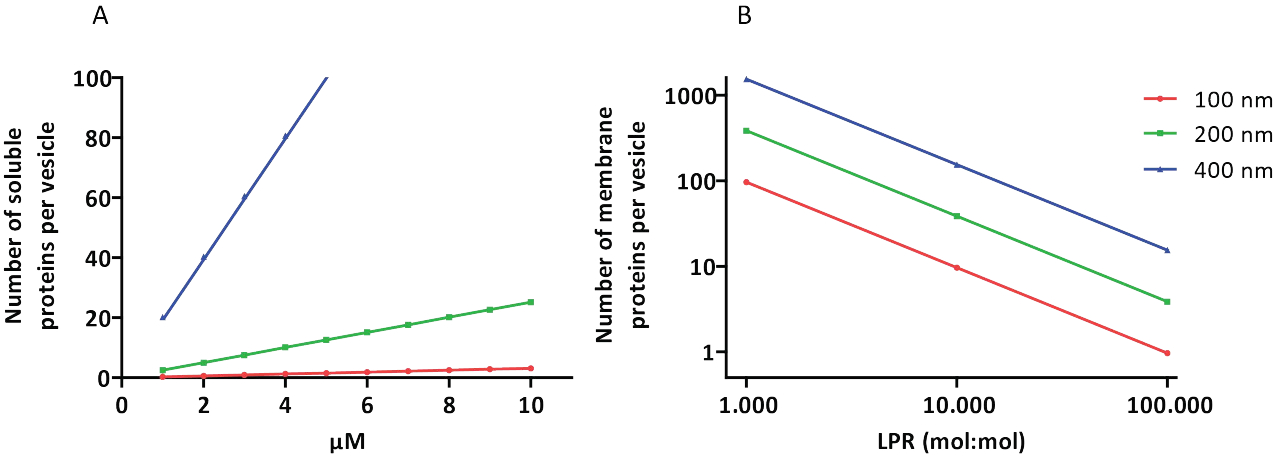
The reconstitution and encapsulation protocols work well and the functionality of the proteins is retained, but the proteoLUVs and proteoGUVs are heterogeneous in size. Microfluidic approaches31,32 allow the formation of micrometer-sized vesicles that are more homogeneous in size, but functional reconstitution of membrane proteins is generally not possible because residual solvent in the bilayer inactivates the proteins. The proteoLUVs range in size from 100 to 400 nm, and at low concentrations of enzymes, the encapsulation may lead to vesicles with incomplete metabolic pathways (stochastic effects; see Figure 3). LUVs are ideal for constructing specific metabolic modules, as shown here for the production of ATP and building blocks like G3P. Such proteoLUVs can potentially be encapsulated in GUVs and serve as organelle-like compartments for the host vesicles.

Figure 3: Number of molecules per vesicle with a diameter of 100, 200, or 400 nm. (A) When the encapsulated proteins (enzymes, probes) are in the range of 1-10 µM. (B) The reconstitution is done at 1 to 1,000, 1 to 10,000, and 1 to 100,000 membrane proteins per lipid (mol/mol). We make the assumption that molecules are encapsulated at the indicated concentrations and incorporated in the membrane at these protein-to-lipid ratios. For some enzymes, we have seen that they bind to membranes, which can increase their apparent concentration in the vesicles. Abbreviation: LPR = Lipid-Protein-Ratio Please click here to view a larger version of this figure.
{kind=link}
Protocol
1. General preparation
- Chemicals
- Dissolve lipids (in powder form) to 25 mg/mL in CHCl3 for making preformed liposomes.
NOTE: It is preferable to prepare fresh lipid stocks, but the stock solutions can also be stored at -20 °C for a few weeks. Working with lipids in powder form is more accurate than using lipids already solubilized in CHCl3. CHCl3 should be handled using glass pipettes and/or syringes and stored in glass containers as CHCl3 dissolves plastics. - Dissolve small molecules (nucleotides, amino acids, fluorescence probes) for the encapsulation procedure in 50 mM KPi (Buffer A, see Table 1) and adjust the pH to 7.00 ± 0.01. Dissolve MgCl2 in deionized water to avoid the formation of magnesium phosphate precipitates.
NOTE: Stock solutions can be stored at -20 °C for a few weeks, with the exception of DTT, which is prepared freshly on the day of the experiment. - Dissolve ionophores (e.g., valinomycin, nigericin) in either DMSO or EtOH at a stock concentration of 100-500 µM. Store at -20 °C for a few weeks; avoid evaporation.
NOTE: DMSO is not volatile and thus preferred over EtOH. Use glass instead of plastic vials to avoid adhesion of the ionophores to the surface of the vials.
- Dissolve lipids (in powder form) to 25 mg/mL in CHCl3 for making preformed liposomes.
- Buffers
- Prepare fresh buffers on the day of the experiment (Table 1). Do not store for longer than 24 h.
- Purification of soluble proteins
- Express ArcA, ArcB, ArcC (we use a specific variant named ArcC1), PercevalHR, and GlpK as described previously27,28,33. Make sure to add 10% v/v glycerol to the cell lysis buffer, which enhances the stability of the proteins. Purify the soluble proteins as described27,28,33 and shortly reported hereafter.
- Thaw 10 mL cell lysate (~5 g wet weight) in an ice-water bath. In the meantime, apply 2 mL (1 CV) Ni2+-Sepharose resin onto a gravity flow column (20 mL capacity) with deionized water (12 CVs) and Buffer B (4 CVs) to wash. Transfer the thawed lysate to ice; work on ice, unless otherwise stated.
- Add imidazole to a final concentration of 10 mM to the thawed lysate, then pour the solution onto the gravity-flow column. Incubate for 1 hour at 4 °C with gentle nutation.
- After 1 h, discard the flow through and wash the resin with Buffer C (20 CVs).
- Elute the protein with Buffer D. Use 60% CV for the first elution step, followed by 4-6 steps of 40% CVs.
- Determine the protein concentration and add Na-EDTA to a final concentration of 5 mM.
- Spin down the purified protein in a refrigerated tabletop centrifuge (max speed, 10 minutes, 4 °C). Purify by size-exclusion chromatography using Buffer E. Pool together the elution fractions and concentrate them to ~10 mg/mL with a concentrating filter with a cut-off of 30 kDa. Prepare aliquots of appropriate size (~ 20 µL), flash-freeze with liquid nitrogen, and store at -80 °C for later use.
NOTE: It is important to concentrate the enzymes to 50-100 µM to minimize the volume needed for the encapsulation.
- Purification of membrane proteins
- Overexpress ArcD and GlpT as described previously22,27,33. Make sure to add 10% v/v glycerol to the cell lysis buffer; for purification of ArcD, include 2 mM reducing agent (e.g., DTT) in the buffer. Purify the affinity-tagged proteins by Ni2+-Sepharose chromatography.
- Thaw an aliquot of crude membrane vesicles (10-20 mg of total membrane protein) in an ice-water bath.
NOTE: Once thawed, always work on ice unless otherwise stated. See Table 1 for the buffers used in this section. - Add the membrane vesicles to Buffer F (ArcD) or Buffer G (GlpT) to a final volume of 6 mL. Incubate the sample for 1 h at 4 °C with gentle nutation.
- Separate the solubilized membrane proteins from the membrane debris by ultracentrifugation (337,000 × g, 30 min, 4 °C). In the meantime, apply 0.25 mL (1 CV) of Ni2+-Sepharose resin onto a gravity flow column (10 mL capacity) with deionized water (40 CVs) and 20 CVs of Buffer H (ArcD) or Buffer I (GlpT).
- Pour the solubilized protein onto the gravity flow column and add imidazole to a final concentration of 10 mM. Incubate for 1 h at 4 °C with gentle nutation.
- After 1 h, discard the flowthrough and wash the resin with 20 CVs of Buffer J (ArcD) or Buffer K (GlpT).
- Elute the membrane protein in 60% CV (1st) and 40% CV (2nd-6th) steps with Buffer L (ArcD) or Buffer M (GlpT).
- Determine the protein concentration and continue to section 2.2. for membrane reconstitution.
NOTE: The size-exclusion purification is not necessarily performed for membrane proteins since the membrane reconstitution yields a similar purification. Steps 1.4 and 2.2 can be performed in 1 day of work. Start with protein purification (step 1.4) in the morning and continue with reconstitution (step 2.2) in the afternoon. Reconstitution ends on the following day (see section 2.2 for details). STOPPING POINT: Purified and DDM-solublized ArcD and GlpT can be stored at -80 oC for later use, but this is not true for all membrane proteins. Prepare aliquots of appropriate size (50-200 µL), flash-freeze with liquid nitrogen, and store at -80 °C for later use. These proteins are active for several months when stored at -80 °C in the presence of 10% v/v glycerol.
- Thaw an aliquot of crude membrane vesicles (10-20 mg of total membrane protein) in an ice-water bath.
- Overexpress ArcD and GlpT as described previously22,27,33. Make sure to add 10% v/v glycerol to the cell lysis buffer; for purification of ArcD, include 2 mM reducing agent (e.g., DTT) in the buffer. Purify the affinity-tagged proteins by Ni2+-Sepharose chromatography.
- Preparation of β-casein for passivation purposes
- Resuspend 100 mg of β-casein in 20 mL of deionized water and titrate with 1 M NaOH until the β-casein is completely dissolved. Then, add 1 M acetic acid to adjust the pH to 7.0 and fill the volume to 50 mL with deionized water. Filter the solution through a 0.2 µm syringe filter and make aliquots of 500 µL.
NOTE The β-casein can be stored at -20 °C for 6 months. It is recommended to filter the β-casein again before use to avoid β-casein aggregates from clogging the microfluidic chip.
- Resuspend 100 mg of β-casein in 20 mL of deionized water and titrate with 1 M NaOH until the β-casein is completely dissolved. Then, add 1 M acetic acid to adjust the pH to 7.0 and fill the volume to 50 mL with deionized water. Filter the solution through a 0.2 µm syringe filter and make aliquots of 500 µL.
| Buffer | Composition | ||
| Buffer A | 50 mM KPi pH 7.0 | ||
| Buffer B | 50 mM KPi, 100 mM KCl, 10% v/v glycerol, 10 mM imidazole, pH 7.5 | ||
| Buffer C | 50 mM KPi, 100 mM KCl, 10% v/v glycerol, 50 mM imidazole, pH 7.5 | ||
| Buffer D | 50 mM KPi, 100 mM KCl, 10% v/v glycerol, 500 mM imidazole, pH 7.5 | ||
| Buffer E | 50 mM KPi, 100 mM KCl, 10% v/v glycerol, pH 7.0 | ||
| Buffer F | 50 mM KPi, 100 mM KCl, 0.5% w/v DDM, 10% v/v glycerol, 2 mM β-mercaptoethanol, pH 7.5 | ||
| Buffer G | 50 mM Tris-HCl, 0.5% w/v DDM, 20% v/v glycerol, pH 8 | ||
| Buffer H | 50 mM KPi, 100 mM KCl, 0.02% w/v DDM, 10% v/v glycerol, 2 mM β-mercaptoethanol, 10 mM imidazole, pH 7.5 | ||
| Buffer I | 50 mM Tris-HCl, 0.04% w/v DDM, 20% v/v glycerol, 10 mM imidazole, pH 8.0 | ||
| Buffer J | 50 mM KPi, 200 mM KCl, 0.02% w/v DDM, 10% v/v glycerol, 2 mM β-mercaptoethanol, 50 mM imidazole, pH 7.5 | ||
| Buffer K | 50 mM Tris-HCl, 0.04% w/v DDM, 20% v/v glycerol, 50 mM imidazole, pH 8 | ||
| Buffer L | 50 mM KPi, 200 mM KCl, 0.02% w/v DDM, 10% v/v glycerol, 2 mM β-mercaptoethanol, 500 mM imidazole, pH 7.5 | ||
| Buffer M | 50 mM Tris-HCl, 0.04% w/v DDM, 20% v/v glycerol, 500 mM imidazole, pH 8 | ||
| Buffer N | 50 mM KPi, 58 mM NaCl, 2 mM DTT, pH 7.0 | ||
| Buffer O | 50 mM KPi, 0.5 mM L-ornithine, 10 mM Na-ADP, 10 mM MgCl2, 2 mM DTT, pH 7.0 | ||
| Buffer P | 50 mM KPi pH 7.0, 2 mM DTT, x mM glucose (x is varied to match the osmolarity of the external and internal medium) | ||
| Buffer Q | 50 mM KPi pH 7.0, 0.5 mM Sucrose, 2 mM DTT | ||
| Buffer R | 50 mM KPi pH 7.0, 2 mM DTT, 10 mM L-arginine, x mM glucose | ||
Table 1: Buffers used in this protocol.
2. Proteoliposomes: reconstitution of purified membrane proteins into preformed lipid vesicles
- Day 1
- Preparation of preformed lipid vesicles
- Choose the lipid composition (e.g., synthetic phospholipids, E. coli polar lipids) on the basis of the requirements of the membrane proteins.
NOTE: A mixture of DOPE, DOPG, and DOPC (25:25:50 mol%) is a good starting point, but sterols or cardiolipin may be required for some proteins; for yeast plasma membrane proteins, we include palmitoyl-oleoyl lipids instead of dioleoyl30. A DOPE, DOPG, and DOPC (25:25:50 mol%) mixture suffices for the reconstitution of ArcD and GlpT. - Mix the desired lipids (solubilized in CHCl3) and evaporate the CHCl3 in a rotary evaporator until a lipid film is formed. Wash the lipids by adding diethyl ether in equal volume as the CHCl3. Evaporate the diethyl ether and obtain a dry lipid film.
- Resuspend the lipid film to 20 mg/mL of total lipids in aqueous media (Buffer A). Start with half of the total volume and shake gently; then, carefully transfer the lipids to a clean tube or bottle of adequate size. Add fresh Buffer A to the flask and repeat the procedure to dissolve the remaining lipids and transfer them to a new vessel. Add additional Buffer A to reach a final concentration of 20 mg/mL.
- Sonicate the resuspended lipids using a probe sonicator. Use the following parameters for a sonicator tip with a diameter of 6 mm: 4 µm intensity, 70% amplitude, 5 s on, 45 s off, 16 cycles. Immerse the lipids in an ice-water bath containing EtOH to avoid overheating by sonication.
- Flash-freeze the sonicated sample (volume of 40 mL in a 50 mL centrifuge tube) in liquid nitrogen, and thaw the sample in a water bath at room temperature. Repeat once; then, aliquot the liposomes in desired volumes (e.g., 1 mL or 20 mg total lipids in a 1.5 mL plastic tube).
STOPPING POINT: The procedure can be stopped at this point. Flash-freeze each aliquot once more (third cycle) and store in liquid nitrogen for up to a few months. Take care of piercing the tube lids twice with a needle to avoid the explosion of the tube upon fast boiling of the liquid nitrogen.
- Choose the lipid composition (e.g., synthetic phospholipids, E. coli polar lipids) on the basis of the requirements of the membrane proteins.
- Preparation of preformed lipid vesicles
- Day 2
- Reconstitution of purified membrane proteins into preformed liposomes
- Thaw one aliquot of liposomes (20 mg total lipids) in a water bath at room temperature. In the meantime, prepare an extruder by applying a filter of choice (e.g., polycarbonate, pore diameter of 400 nm); pre-equilibrate the extruder with Buffer A; and load the thawed liposomes ('milky solution") into the extruder and pass them 13x through the filter. Collect the extruded liposomes (now large unilamellar vesicles; "opaque solution") in a glass or plastic vessel of adequate size (e.g., 15 mL). Dilute the liposomes to 4 mg/mL with Buffer A supplemented with 2 mM DTT.
- Transfer 1 mL of 4 mg/mL liposomes into a 1 mL transparent cuvette. In a spectrophotometer, measure the initial optical density at 540 nm. Pour the measured sample back and add 50 µL of 10% v/v Triton X-100 to the liposomes.
NOTE: A titration volume of 50 µL of 10% Triton-X100 is suitable for 20 mg of lipids in a volume of 5 mL; the addition of Triton X-100 will dilute the lipids by ~5%. Adjust the titration volume when working with different amounts of liposomes. For stable optical density signals, titrate the liposomes with Triton X-100 at room temperature. - Repeat step 2.2.1.2 and note when a maximum optical density is reached (Rsat). Proceed further with the titration until an optical density of approximately 60% Rsat is reached (Figure 4). Pour the detergent-destabilized vesicles back into the glass/plastic tube (the final volume is now approximately 5.2 mL), transfer the sample to ice, and allow it to cool down.
- Add the purified membrane protein(s) to the destabilized liposomes to reach a desired lipid-to-protein ratio (w/w). Use a ratio of 400:1 down to 100:1 w/w; since both ArcD and GlpT have molecular weights of ~55 kDa, a lipid-to-protein ratio of 400:1 w/w corresponds to ~30,000 lipids per protein, and ~ 50 molecules of ArcD and GlpT each per vesicles with a diameter of 400 nm.
NOTE: Here we use 400:1 w/w, corresponding to 50 µg of each protein per 20 mg of lipids. - Nutate the samples at 4 °C for 15 min to allow the membrane proteins to insert into the destabilized liposomal membrane.
- To remove the detergent, add 200 mg of dry polystyrene beads, prepared as per the manufacturer's instructions. Nutate at 4 °C for another 15 min.
NOTE: An amount of 200 mg of dry beads is suitable for 20 mg of lipids but should be adjusted when a different sample size is used. - Repeat step 2.2.1.6 2x, for a total of three additions of polystyrene beads. Then, incubate overnight at 4 °C with gentle nutation.
- Reconstitution of purified membrane proteins into preformed liposomes
- Day 3
- Repeat step 2.2.1.6; however, this time, nutate at 4 °C for 1 h.
- Secure an empty gravity flow column (10 mL capacity) above an empty 6.5 mL ultracentrifugation tube on ice. Pour the sample into the column and collect the proteoliposomes in the ultracentrifugation tube (the beads are retained in the column).
- Wash the beads with 0.5 mL of buffer A supplemented with 2 mM DTT and collect the filtrate in the ultracentrifugation tube.
- Concentrate the proteoliposomes by ultracentrifugation (337,000 × g, 30 min, 4 oC). Resuspend the proteoliposomes in a total volume of 200 µL (100 mg of lipid/mL) in Buffer A supplemented with 2 mM DTT; the dry volume of the vesicles after centrifugation is ~40 to 120 µL27. Split into aliquots of desired size (e.g., three aliquots of 6.66 mg of total lipids).
NOTE: The aliquot size is arbitrary but will affect the pellet size in later steps (see step 3.2.3.1). STOPPING POINT: The procedure can be stopped here. Flash-freeze each aliquot and store in liquid nitrogen for up to a few weeks. Take care to pierce the tube lids twice with a needle to avoid explosion of the tube upon fast boiling of the liquid nitrogen.

Figure 4: Titration of preformed liposomes with Triton X-100. Liposomes at 5 mg of lipids/mL are extruded through a polycarbonate filter (400 nm) in 50 mM KPi (pH 7.0) and then titrated with Triton X-100 (protocol step 2.2.1.2). The turbidity of the vesicles is measured at A540. The arrow indicates the Triton X-100 concentration at which the vesicles are sufficiently destabilized for spontaneous insertion of membrane proteins as described in19. Please click here to view a larger version of this figure.
{kind=link}
3. Encapsulation of a metabolic network for ATP recycling and glycerol 3-P synthesis in submicron-size vesicles
- Day 1
- Mixing of components
- For a standard encapsulation, use 66.6 µL of proteoliposomes in a final volume of 200 µL (33.33 mg/mL of total lipids). Calculate the volume of each component (enzyme, cofactor) needed to reach the desired concentration, add these components to the proteoliposomes, and adjust the volume to 200 µL with Buffer A (Table 2).
NOTE: The protein concentration can be adjusted based on the experimental setup. However, care should be taken that several copies (>10) of each enzyme are on average present per vesicle, to avoid undesired stochastic effects. Concentrations > 1 µM are generally safe; 1 µM in a vesicle with a diameter of 400 nm corresponds to approximately 20 copies (Figure 3). - Pipette buffer A into an empty 1.5 mL tube and add the DTT, Na-ADP, MgCl2, L-ornithine (and pyranine, if needed for internal pH measurements). Next, add the enzymes and mix gently. Add the solution on top of the preformed proteoliposomes, and briefly vortex at low speed.
NOTE: It is important to add Na-ADP before MgCl2 to avoid the undesired formation of magnesium phosphate precipitates. Vortexing is required for proper mixing of the viscous solution; however, minimize the duration and speed to avoid mechanical damage to the proteins. PercevalHR and pyranine cannot be co-encapsulated because their spectra overlap.
- For a standard encapsulation, use 66.6 µL of proteoliposomes in a final volume of 200 µL (33.33 mg/mL of total lipids). Calculate the volume of each component (enzyme, cofactor) needed to reach the desired concentration, add these components to the proteoliposomes, and adjust the volume to 200 µL with Buffer A (Table 2).
- Determination of internal osmolality
- Prepare a 50 µL solution as described in step 3.1.1.1 but without proteoliposomes, and measure the osmolality with a freezing-point osmometer.
- Prepare a calibration curve using buffer (50 mM KPi pH 7.0) and varying concentrations of salt (e.g., NaCl or NaCl) or sugar. Determine the osmolyte concentration that matches the internal osmolality (Buffer N).
NOTE: Membrane permeable components (e.g., glycerol) cannot be used to match the internal osmolality. For proteins solubilized in glycerol-containing buffer (e.g., Buffer E), the same buffer should be used without glycerol. The osmolyte chosen for the preparation of an iso-osmotic external buffer should be membrane-impermeable and not interfere with the metabolic network.
- Freeze-thawing
- Flash-freeze (in liquid nitrogen) the proteoliposomes together with the soluble components, and thaw in a water-ice bath at approximately 10 °C.
- Repeat step 3.1.3.1 for a total of 5x.
STOPPING POINT: The procedure can be stopped here. Skip the last thawing step, and store the frozen sample in liquid nitrogen for 1-3 days. Take care to pierce the tube lids 2x with a needle to avoid explosion of the tube upon fast boiling of the liquid nitrogen. Storage in liquid nitrogen is preferred over -80 °C to minimize lipid oxidation.
- Mixing of components
- Day 2
- Extrusion
NOTE: All extrusion steps are performed at room temperature, as the gastight syringes will otherwise leak.- Prepare an extruder by applying a filter of choice (e.g., polycarbonate, pore diameter of 400 nm). Wash the extruder with a solution containing the same buffer and metabolites used for the encapsulation of the vesicles (e.g., Buffer O plus 0.1 mM pyranine).
NOTE: Use a dedicated extruder for the loading of proteoliposomes with pyranine as the dye sticks to the extruder surface and can cause contamination in later samples. - Load the encapsulation mixture in the extruder, and pass it through the filter 13x. Collect the extruded solution in a 1.5 mL tube.
- Prepare an extruder by applying a filter of choice (e.g., polycarbonate, pore diameter of 400 nm). Wash the extruder with a solution containing the same buffer and metabolites used for the encapsulation of the vesicles (e.g., Buffer O plus 0.1 mM pyranine).
- Size-exclusion chromatography (optional)
NOTE: This step is performed to remove external molecules such as dyes like pyranine by size-exclusion chromatography. If dyes are not present in the system, and other components are not interfering at low concentrations (notice that the vesicles are afterward also washed by ultracentrifugation), then step 3.2.2. can be skipped.- Rehydrate Sephadex G-75 resin and pour it into a glass column (22 cm long, 1.5 cm wide). Equilibrate the resin with an excess of external buffer (e.g., Buffer N).
- Load the extruded proteoliposomes from step 3.2.1.2 onto the preequilibrated size-exclusion chromatography column and apply a gravity flow of external buffer. Discard the void volume (approximately 7 mL); then, collect ten 1 mL aliquots. Visualize the proteoliposome-containing aliquots by short exposure to a UV lamp. Pool the fractions containing most of the proteoliposomes (2-4 mL).
- Washing and resuspension
- Wash the extruded proteoliposomes by ultracentrifugation. Fill a 6.5 mL ultracentrifugation tube with 5.8 mL of Buffer N, and apply the extruded sample on top. If size-exclusion chromatography was performed, fill the tube with the pooled elution samples (2-4 mL) and add Buffer N to a final volume of 6 mL.
- Centrifuge at 337,000 × g, 30 min, 4 °C. Discard the supernatant, and thoroughly dry the ultracentrifugation tube with a dust-free tissue whilst paying attention not to touch the pellet. Resuspend the pellet in a small volume of Buffer N (200 µL). When the pellet is fully resuspended, fill the tube up to 6 mL with Buffer N.
NOTE: resuspending the pellet will take time and should be done carefully. - Repeat steps 3.2.3.1-3.2.3.2 2x, for a total of three washes, unless size-exclusion chromatography is performed (in which case one centrifugation step suffices). Ultimately, resuspend the pellet to a desired concentration (e.g., 5.55 mg/mL total lipid) by adding an appropriate volume of Buffer N.
STOPPING POINT: The proteoliposomes can be used immediately or stored at 4 oC for at least 48 h. The size of ultracentrifugation tubes should be chosen based on the sample size. For a pellet of 6.66 mg of total lipids, a 6.5 mL tube is appropriate. Washing 200 µL of proteoliposomes (33.33 mg of lipids/mL in total; dry pellet volume ~40 µL)28 for 3 x 6 mL of buffer dilutes the external components by a factor of 100 for each washing step. If tubes of different sizes are used, it is desirable to adjust the number of washes accordingly.
- Extrusion
- Day 3
- Detection of ATP synthesis by fluorescence
- Mix the reaction components to a final volume of 120 µL (Table 3) in a black quartz cuvette with a window of 3 x 5 mm and a minimal internal volume of 100 µL.
NOTE: Ionophores can be added to dissipate electrochemical ion gradients. A mixture of valinomycin and nigericin (1 µM each) effectively dissipates any proton and potassium gradients. - Prewarm the sample in a fluorometer set to 30 °C and acquire excitation spectra of PercevalHR (excitation 400-520 nm, bandwidth 5 nm; emission 550 nm, bandwidth 5 nm). Once the probe signal is constant, start the metabolic network by the addition of an excess of L-arginine (5-10 mM), and glycerol (400 µM) when the recycling of ATP is coupled to the synthesis of glycerol 3-phosphate. Follow the reaction over time.
- Mix the reaction components to a final volume of 120 µL (Table 3) in a black quartz cuvette with a window of 3 x 5 mm and a minimal internal volume of 100 µL.
- Data analysis
- Plot the ratio F500/F430 as a function of time, which is a qualitative indicator of the ATP/ADP ratio. For a more quantitative assessment of ATP formation, construct a calibration curve in proteoliposomes or use a complementary approach (e.g., ATP quantification by chemiluminescence28).
- Detection of ATP synthesis by fluorescence
| Component | Final concentration |
| Buffer A | 50 mM KPi pH 7.0 |
| DTT | 2 mM |
| Na-ADP | 10 mM |
| MgCl2 | 10 mM |
| L-ornithine | 0.5 mM |
| Fluorescent probe (PercevalHR or pyranine) | 5.8 µM or 0.1 mM, respectively |
| ArcA (Arginine deiminase) | 1 µM |
| ArcB (Ornithine carbamoyltransferase) | 2 µM |
| ArcC1 (Carbamate kinase) | 5 µM |
| GlpK (Glycerol kinase) | 1.6 µM |
| Proteoliposomes | 33.33 mg/mL total lipids |
Table 2: Encapsulation components. The components are listed in order of addition. Soluble proteins are in Buffer E; all other components (except for MgCl2, in deionized water) are in Buffer A.
| Component | Final concentration |
| Buffer K | 50 mM KPi pH 7.0, 58 mM NaCl, 2 mM DTT, pH 7.0 |
| Proteoliposomes (5.55 mg/mL of lipids) | 2.7 mg/mL of lipids |
| Ionophores (valinomycin, nigericin) | 1 µM each |
Table 3: Experimental conditions. The components are listed in order of addition. Proteoliposomes are in Buffer N, ionophores in DMSO or EtOH.
4. Upscaling a metabolic network for micrometer-sized vesicles
- Day 1
- Microfluidic chip preparation
NOTE: This experiment uses a microfluidic device developed by Robinson et al.34. Other designs of microfluidic chips are available35,36,37 and can easily be implemented in this protocol.- Cut a 200 µL pipet tip one third from the bottom and insert the bottom part of the tip into a premade microfluidic trapping device.
- To the inlet reservoir of the chip, add 400 µL of β-casein solution (2 mg/mL; see step 1.5), taking care to prevent the introduction of air at this step. Place a 96-well plate bucket and add a laboratory tissue in a tabletop conical tube centrifuge. Place the chip on top of the tissue and centrifuge for 6 min at 900 × g to allow passivation of the microfluidic chip. After the centrifugation step, the liquid level should be equal on the inlet reservoir and the outlet tip of the microfluidic chip; check for leakage of the chip. Incubate the β-casein solution in the microfluidic chip for at least 30 min.
- Remove most of the passivation solution without introducing air and add 400 µl of washing buffer (buffer P). Place the chip on the 96-well plate bucket and centrifuge at 900 × g for 6 min. Leave the chip in washing solution until use (for a maximum of 4h).
- Microfluidic chip preparation
- Gel preparation for the making of proteo-GUVs
NOTE: The following description is taken and adapted from25,38.- Dissolve 0.5% (w/w) of a low-gelling temperature (LGT) agarose in deionized water by heating the solution in the microwave; make sure the agarose has completely dissolved and avoid boiling the solution. Keep the agarose at 50 °C until further use.
NOTE: Dissolved agarose can be stored at room temperature for several weeks. To re-use the agarose, simply melt the gel using a microwave. - Take two objective slides and draw the outline of the spacer on the slides. Make the slides hydrophilic by plasma cleaning, using high oxygen plasma for 1 min.
- Add LGT agarose on top of the slide until it is fully covered with agarose (~500 µL); then, tilt the slide at an angle of 90 ° and drain excess agarose onto a tissue. Leave the slides for 30 min at 50 °C.
- Take proteoliposomes stored in liquid nitrogen (section 3) and thaw on ice. Dilute the vesicles to 5 mg/mL of lipids using buffer Q.
NOTE: Proteoliposomes prepared in section 3 do not contain soluble proteins and can be prepared well in advance if stored in liquid nitrogen. When working with proteoGUVs, make sure to always filter the working solutions to prevent clogging of the microfluidic device. - Sonicate the proteo-liposomes using a handheld probe sonicator with a 1 mm probe. Sonicate for 10 cycles of 0.5 s on and 0.5 s off at 70% amplitude. Keep the vesicles, hereafter referred to as proteoSUVs, on ice for 30 s, and repeat the sonication process 5x.
- Fill a 100 µL syringe (in a handheld LCP Dispenser) with the proteo-SUV suspension. Deposit 0.5 µL droplets of proteo-SUVs on the previously prepared agarose gel; be careful to not disturb the agarose layer and to keep enough distance to prevent droplets from fusing.
NOTE Using a hand-held LCP Dispenser allows droplet deposition in a reproducible manner, but alternative pipetting systems can also be used. Spotting with a glass capillary is less suitable because it disturbs the dried agarose gel. - Dry the SUV droplets in ~10 min using a nitrogen flow instead of compressed air to reduce the possibility of oxidizing the lipids.
- Prepare 1 mL of a 1.25x concentrated buffer O (Table 4) in buffer A and pass through a 0.2 µm cellulose acetate filter. Take 800 µL of the 1.25x concentrated solution and add the soluble enzymes and probes to a concentration as indicated in Table 4. Use filtered deionized water to make the final volume 1 mL.
- Prepare 100 µL of swelling solution containing all components of Table 4, with the exception of proteins and glycerol. Measure the osmolality of the swelling solution using a 3-point calibrated freezing point osmometer.
NOTE: Prepare 100 µL of swelling solution without protein and glycerol to accurately determine the osmolality of this solution. Glycerol, which is present in the protein solution, affects the osmolality, but, in GUVs, glycerol rapidly diffuses across the membrane and only leads to transient osmotic differences. - Assemble the GUV swelling chamber by making a sandwich of two objective glasses containing the gel and dried SUVs with a 1.5 or 3.0 mm thick Teflon spacer in between. Then, add the swelling solution in the chamber through the small hole in the side using a syringe and needle.
NOTE: The volume of the swelling solution can be adjusted by varying the spacers from 1.5 to 3.0 mm.
- Dissolve 0.5% (w/w) of a low-gelling temperature (LGT) agarose in deionized water by heating the solution in the microwave; make sure the agarose has completely dissolved and avoid boiling the solution. Keep the agarose at 50 °C until further use.
- Swelling of vesicles and harvesting of GUVs
- Allow swelling of vesicles by putting the chamber at 22 °C for at least 30 min.
NOTE: The swelling of the vesicles can be followed by light microscopy, (e.g., a tabletop phase contrast microscope or a widefield fluorescence microscope) if the proteins or lipids are fluorescently labeled. - Harvest the GUVs from the gel by applying gentle physical agitation by tapping the chamber on a solid surface (e.g., lab bench); take out one third of the volume and use the resulting air bubble to gently induce motion to the remaining liquid thereby detaching the GUVs from the gel.
- While the GUVs are swelling, prepare buffer P and match the osmolality with the swelling solution by adjusting the concentration of glucose. Filter the buffer through a 0.2 µm syringe filter.
NOTE: In general, the washing and substrate solution should be kept within ± 5 mosmol/kg. Any solution passing through the chip should be very clean as contaminants may clog the channels. Make fresh buffers every time or store prepared buffers at -20 °C and filter before use.
- Allow swelling of vesicles by putting the chamber at 22 °C for at least 30 min.
- Trapping of the GUVs for microscopy experiments
- Mount the passivated microfluidic chip on the sample stage of the microscope. Check for possible defects (e.g., leaks, trapped air, clogged channels).
NOTE: Checking the chip can be done well before is used so that a new chip can be prepared if necessary. - Connect the tubing using a bent needle to the outlet of the reservoir and connect the other end to a 1 mL syringe. Mount the syringe to a pump and set the flow rate to a maximum of 10 µL/min.
- Remove the washing buffer from the reservoir (step 4.1.1.3) and replace with fresh medium (buffer P). Start the flow of buffer through the chip by infusing via the syringe at 1-10 µL/min. Wash with at least 80 µL of osmotically balanced washing buffer (Buffer P).
- Remove excess wash buffer from the reservoir and add the (proteo)GUVs to the reservoir. Adjust the flow rate to 0.1- 1 µL/min to allow the GUVs to flow through the chip. Monitor the chip over time until enough GUVs have been trapped in the chip.
NOTE: Vesicles with relatively large amounts (>20 mol%) of charged lipids such as phosphatidylglycerol (PG), phosphatidyl serine (PS), or phosphatidic acid (PA) or non-bilayer-forming lipids like phosphatidylethanolamine (PE) are introduced at a lower flow rate through the chip to prevent bursting. Vesicles composed of pure phosphatidylcholine (PC) tend to be more stable. - Remove excess GUV solution and add washing buffer P, using a constant flow of 0.1-1 µL/min, to exchange the external medium and wash the trapped GUVs for at least 1 h to remove non-encapsulated compounds and lower the background fluorescence when a fluorophore is encapsulated. Monitor the background fluorescence over time; if the background fluorescence does not decrease, the chip may be blocked.
- Localize traps with sufficient amounts of vesicles and save their positions. Apply the settings on the microscope (e.g., laser intensity, gain, wavelength) and start a time series experiment.
- Add osmotically balanced substrate solution (buffer R) to the reservoir and start a flow rate of 0.5 µL/min.
- Mount the passivated microfluidic chip on the sample stage of the microscope. Check for possible defects (e.g., leaks, trapped air, clogged channels).
| Buffer L components | ||
| Component | 1.25 x concentration | Working concentration |
| Buffer A | 62.5 mM KPi pH 7.0 | 50 mM KPi pH 7.0 |
| Sucrose | 125 mM | 100 mM |
| DTT | 2.5 mM | 2 mM |
| Na-ADP | 12.5 mM | 10 mM |
| MgCl2 | 12.5 mM | 10 mM |
| L-ornithine | 0.625 mM | 0.5 mM |
| Encapsulation components | ||
| Pyranine or PercevalHR | 1 mM or 20 µM | |
| ArcA (Arginine deiminase) | 1 µM | |
| ArcB (Ornithine carbamoyltransferase) | 2 µM | |
| ArcC1 (Carbamate kinase) | 5 µM | |
Table 4: Buffer O and encapsulation components. The components are listed in order of addition. All components (except for MgCl2, in deionized water) are in Buffer A. Encapsulation components are listed in order of addition. All water-soluble proteins are in Buffer E.
Results
The reconstitution of solubilized membrane proteins in liposomes requires the destabilization of preformed vesicles. The addition of low amounts of Triton X-100 initially results in an increase of absorbance at 540 nm (A540) due to an increase in light scattering by the swelling of the vesicles (Figure 4). The maximum A540 value is the point where the liposomes are saturated with detergent (Rsat), after which any further addition of Triton X-100 will...
Discussion
We present a protocol for the synthesis of (membrane) protein containing sub-micrometer size lipid vesicles (proteoLUVs), and the conversion of proteoLUVs into giant-unilamellar vesicles (proteoGUVs). The protocol should be applicable for the reconstitution of other membrane proteins13,19,30,40 and the encapsulation of metabolic networks other than the L-arginine breakdown and glycerol 3-phosph...
Disclosures
The authors declare no competing financial interest.
Acknowledgements
The authors thank Aditya Iyer for the cloning of the pBAD-PercevalHR gene and Gea Schuurman-Wolters for aiding with protein production and purification. The research was funded by the NWO Gravitation program "Building a Synthetic Cell" (BaSyC).
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Sigma Aldrich | A9414-25g | |
| Amicon cut-off filter | Sigma Aldrich | Milipore centrifugal filter units Amicon Ultra | |
| BioBeads | BioRad | 152-3920 | |
| CHCl3 | Macron Fine Chemicals | MFCD00000826 | |
| D(+)-Glucose | Formedium | - | |
| D(+)-Sucrose | Formedium | - | |
| DDM | Glycon | D97002 -C | |
| Diethyl Ether | Biosolve | 52805 | |
| DMSO | Sigma-Aldrich | 276855-100ml | |
| DOPC | Avanti | 850375P-1g | |
| DOPE | Avanti | 850725P-1g | |
| DOPG | Avanti | 840475P-1g | |
| DTT | Formedium | DTT005 | |
| EtOH | J.T.Baker Avantor | MFCD00003568 | |
| Extruder | Avestin Inc | LF-1 | |
| Fluorimeter | Jasco | Spectrofluorometer FP-8300 | |
| Glycerol | BOOM | 51171608 | |
| Gravity flow column | Bio-Rad | 732-1010 | |
| Hamilton syringe 100 µL | Hamilton | 7656-01 | |
| Hamilton syringe 1000 µL | Hamilton | 81320 | |
| Handheld LCP dispenser | Art Robbins Instruments | 620-411-00 | |
| Handheld Sonicator | Hielscher Ultrasound Technology | UP50H | |
| HCl | BOOM | x76021889.1000 | |
| Imidazole | Roth | X998.4-250g | |
| K2HPO4 | Supelco | 1.05099.1000 | |
| KCl | BOOM | 76028270.1 | |
| KH2PO4 | Supelco | 1.04873.1000 | |
| Kimwipe | Kimtech Science | 7552 | |
| Large Falcon tube centrifuge | Eppendorf | Centrifuge 5810 R | |
| L-Arginine | Sigma-Aldrich | A5006-100G | |
| Light microscope | Leica | DM LS2 | |
| L-Ornithine | Roth | T204.1 | |
| LSM Laser Scanning Confocal Microscope | Zeiss | LSM 710 ConfoCor 3 | |
| MgCl2 | Sigma-Aldrich | M2670-1KG | |
| Microfluidic chip | Homemade | PDMS based | DOI: https://doi.org/10.1039/C8LC01275J |
| Na-ADP | Sigma-Aldrich | A2754-1G | |
| NaCl | Supelco | 1.06404.1000 | |
| Nanodrop Spectrometer | Isogen Life Science | ND-1000 spectrophotometer NanoDrop | |
| NaOH | Supelco | 1.06498.1000 | |
| Needles for GUVs | Henke-Ject | 14-14575 | 27 G x 3/4'' 0.4 x 20 mm |
| Needles for microfluidics | Henke-Ject | 14-15538 | 18 G x 1 1/2'' 1.2 x 40 mm |
| Ni2+ Sepharose | Cytiva | 17526802 | |
| Nigericin | Sigma-Aldrich | N7143-5MG | |
| Nutator | VWR | 83007-210 | |
| Osmolality meter | Gonotec Salmenkipp | Osmomat 3000 basic freezing point osmometer | |
| Plasmacleaner | Plasma Etch | PE-Avenger | |
| Polycarbonate filter | Cytiva Whatman | Nuclepor Track-Etch Membrane Product: 10417104 | 0.4 µm |
| Polycarbonate ultracentrifuge tube | Beckman Coulter | 355647 | |
| Pyranine | Acros Organics | H1529-1G | |
| Quartz cuvette (black) | Hellma Analytics | 108B-10-40 | |
| Sephadex G-75 resin | GE Healthcare | 17-0050-01 | |
| Sonicator | Sonics Sonics & Materials INC | Sonics vibra cell | |
| Syringe filter | Sarstedt | Filtropur S plus 0.2 | 0.2 µm |
| Syringe pump | Harvard Apparatus | A-42467 | |
| Tabletop centrifuge | Eppendorf | centrifuge 5418 | |
| Teflon spacer | Homemade | Teflon based | 45 x 26 x 1.5 or 45 x 26 x 3 or 20 x 20 x 3 mm |
| Tris | PanReac AppliChem | A1086.1000 | |
| Triton X-100 | Sigma Aldrich | T8787-100 ml | |
| Ultracentrifuge | Beckman Coulter | Optima Max-E | |
| UV lamp | Spectroline | ENB-280C/FE | |
| UV/VIS Spectrometer | Jasco | V730 spectrophotometer | |
| Valinomycin | Sigma-Aldrich | V0627-10MG | |
| Widefield fluorescence microscope | Zeiss | AxioObserver | |
| β-Casein | Sigma Aldrich | C5890-500g |
References
- Hirschi, S., Ward, T. R., Meier, W. P., Müller, D. J., Fotiadis, D. Synthetic biology: bottom-up assembly of molecular systems. Chem Rev. 122 (21), 16294-16328 (2022).
- Ivanov, I., et al. Bottom-up synthesis of artificial cells: recent highlights and future challenges. Annu Rev Chem Biomol. Eng. 12 (1), 287-308 (2021).
- Clomburg, J. M., Crumbley, A. M., Gonzalez, R. Industrial biomanufacturing: The future of chemical production. Science. 355 (6320), (2017).
- Shi, T., Han, P., You, C., Zhang, Y. -. H. P. J. An in vitro synthetic biology platform for emerging industrial biomanufacturing: Bottom-up pathway design. Synth Syst Biotechnol. 3 (3), 186-195 (2018).
- Wang, A., et al. Liver-target and glucose-responsive polymersomes toward mimicking endogenous insulin secretion with improved hepatic glucose utilization. Adv Funct Mater. 30 (13), 1910168 (2020).
- Kanter, G., et al. Cell-free production of scFv fusion proteins: an efficient approach for personalized lymphoma vaccines. Blood. 109 (8), 3393-3399 (2007).
- Zeltins, A. Construction and characterization of virus-like particles: a review. Mol Biotechnol. 53 (1), 92-107 (2013).
- Jain, K. K. Synthetic biology and personalized medicine. Med Princ Pract. 22 (3), 209-219 (2013).
- Schwille, P., Frohn, B. P. Hidden protein functions and what they may teach us. Trends Cell Biol. 32 (2), 102-109 (2022).
- Sachsenmeier, P. Industry 5.0-The relevance and implications of bionics and synthetic biology. Engineering. 2 (2), 225-229 (2016).
- Schmidt, D., Jiang, Q. -. X., MacKinnon, R. Phospholipids and the origin of cationic gating charges in voltage sensors. Nature. 444 (7120), 775-779 (2006).
- Godoy-Hernandez, A., et al. Rapid and highly stable membrane reconstitution by LAiR enables the study of physiological integral membrane protein functions. ACS Cent Sci. 9 (3), 494-507 (2023).
- Sikkema, H. R., et al. Gating by ionic strength and safety check by cyclic-di-AMP in the ABC transporter OpuA. Sci Adv. 6 (47), 7697 (2020).
- Foucaud, C., Poolman, B. Lactose transport system of Streptococcus thermophilus. Functional reconstitution of the protein and characterization of the kinetic mechanism of transport. J Biol Chem. 267 (31), 22087-22094 (1992).
- Yoneda, J. S., Sebinelli, H. G., Itri, R., Ciancaglini, P. Overview on solubilization and lipid reconstitution of Na,K-ATPase: enzyme kinetic and biophysical characterization. Biophys Rev. 12 (1), 49-64 (2020).
- Simidjiev, I., et al. Self-assembly of large, ordered lamellae from non-bilayer lipids and integral membrane proteins in vitro. Proc Natl Acad Sci. 97 (4), 1473-1476 (2000).
- Harris, N. J., Booth, P. J. Folding and stability of membrane transport proteins in vitro. Biochim Biophys Acta BBA - Biomembr. 1818 (4), 1055-1066 (2012).
- Jackson, M. L., Litman, B. J. Rhodopsin-egg phosphatidylcholine reconstitution by an octyl glucoside dilution procedure. Biochim Biophys Acta BBA - Biomembr. 812 (2), 369-376 (1985).
- Geertsma, E. R., Nik Mahmood, N. A. B., Schuurman-Wolters, G. K., Poolman, B. Membrane reconstitution of ABC transporters and assays of translocator function. Nat Protoc. 3 (2), 256-266 (2008).
- Rigaud, J. -. L., Pitard, B., Levy, D. Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim Biophys Acta BBA - Bioenerg. 1231 (3), 223-246 (1995).
- Szoka, F., Papahadjopoulos, D. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc Natl Acad Sci. 75 (9), 4194-4198 (1978).
- . Synthetic Organelles for Energy Conservation and Delivery of Building Blocks for Lipid Biosynthesis Available from: https://www.researchsquare.com/article/rs-3385355/v1 (2023)
- Lee, K. Y., et al. Photosynthetic artificial organelles sustain and control ATP-dependent reactions in a protocellular system. Nat Biotechnol. 36 (6), 530-535 (2018).
- Méléard, P., Bagatolli, L. A., Pott, T. Giant unilamellar vesicle electroformation. Methods in Enzymology. , 161-176 (2009).
- Garten, M., Aimon, S., Bassereau, P., Toombes, G. E. S. Reconstitution of a transmembrane protein, the voltage-gated ion channel, KvAP, into giant unilamellar vesicles for microscopy and patch clamp studies. J. Vis. Exp. (95), e52281 (2015).
- Doeven, M. K., et al. lateral mobility and function of membrane proteins incorporated into giant unilamellar vesicles. Biophys J. 88 (2), 1134-1142 (2005).
- Pols, T., et al. A synthetic metabolic network for physicochemical homeostasis. Nat Commun. 10 (1), 4239 (2019).
- Bailoni, E., Poolman, B. ATP recycling fuels sustainable glycerol 3-phosphate formation in synthetic cells fed by dynamic dialysis. ACS Synth Biol. 11 (7), 2348-2360 (2022).
- Van Der Heide, T. On the osmotic signal and osmosensing mechanism of an ABC transport system for glycine betaine. EMBO J. 20 (24), 7022-7032 (2001).
- Van'T Klooster, J. S., et al. Membrane lipid requirements of the lysine transporter Lyp1 from Saccharomyces cerevisiae. J Mol Biol. 432 (14), 4023-4031 (2020).
- Lou, G., Anderluzzi, G., Woods, S., Roberts, C. W., Perrie, Y. A novel microfluidic-based approach to formulate size-tuneable large unilamellar cationic liposomes: Formulation, cellular uptake and biodistribution investigations. Eur J Pharm Biopharm. 143, 51-60 (2019).
- Weiss, M., et al. Sequential bottom-up assembly of mechanically stabilized synthetic cells by microfluidics. Nat Mater. 17 (1), 89-96 (2018).
- Pols, T., Singh, S., Deelman-Driessen, C., Gaastra, B. F., Poolman, B. Enzymology of the pathway for ATP production by arginine breakdown. FEBS J. 288 (1), 293-309 (2021).
- Yandrapalli, N., Robinson, T. Ultra-high capacity microfluidic trapping of giant vesicles for high-throughput membrane studies. Lab Chip. 19 (4), 626-633 (2019).
- Elias, M., et al. Microfluidic characterization of biomimetic membrane mechanics with an on-chip micropipette. Micro Nano Eng. 8, 100064 (2020).
- Robinson, T., Kuhn, P., Eyer, K., Dittrich, P. S. Microfluidic trapping of giant unilamellar vesicles to study transport through a membrane pore. Biomicrofluidics. 7 (4), 044105 (2013).
- Cooper, A., Girish, V., Subramaniam, A. B. Osmotic Pressure Enables High-Yield Assembly of Giant Vesicles in Solutions of Physiological Ionic Strengths. Langmuir. 39 (15), 5579-5590 (2023).
- Tantama, M., Martínez-François, J. R., Mongeon, R., Yellen, G. Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat Commun. 4 (1), 2550 (2013).
- Setyawati, I., et al. In vitro reconstitution of dynamically interacting integral membrane subunits of energy-coupling factor transporters. eLife. 9, e64389 (2020).
- Oropeza-Guzman, E., Ríos-Ramírez, M., Ruiz-Suárez, J. C. Leveraging the coffee ring effect for a defect-free electroformation of giant unilamellar vesicles. Langmuir. 35 (50), 16528-16535 (2019).
- Estes, D. J., Mayer, M. Electroformation of giant liposomes from spin-coated films of lipids. Colloids Surf B Biointerfaces. 42 (2), 115-123 (2005).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved