Long-Term Mouse Spinal Cord Organotypic Slice Culture as a Platform for Validating Cell Transplantation in Spinal Cord Injury
In This Article
Summary
In this paper, we provide a reproducible method to generate and maintain long-term spinal cord organotypic slices transplanted with neural stem cells as an ex vivo model for testing cellular replacement therapies.
Abstract
Resolutive cures for spinal cord injuries (SCIs) are still lacking, due to the complex pathophysiology. One of the most promising regenerative approaches is based on stem cell transplantation to replace lost tissue and promote functional recovery. This approach should be further explored better in vitro and ex vivo for safety and efficacy before proceeding with more expensive and time-consuming animal testing. In this work, we show the establishment of a long-term platform based on mouse spinal cord (SC) organotypic slices transplanted with human neural stem cells to test cellular replacement therapies for SCIs.
Standard SC organotypic cultures are maintained for around 2 or 3 weeks in vitro. Here, we describe an optimized protocol for long-term maintenance (≥30 days) for up to 90 days. The medium used for long-term culturing of SC slices was also optimized for transplanting neural stem cells into the organotypic model. Human SC-derived neuroepithelial stem (h-SC-NES) cells carrying a green fluorescent protein (GFP) reporter were transplanted into mouse SC slices. Thirty days after the transplant, cells still show GFP expression and a low apoptotic rate, suggesting that the optimized environment sustained their survival and integration inside the tissue. This protocol represents a robust reference for efficiently testing cell replacement therapies in the SC tissue. This platform will allow researchers to perform an ex vivo pre-screening of different cell transplantation therapies, helping them to choose the most appropriate strategy before proceeding with in vivo experiments.
Introduction
Traumatic spinal cord injury (SCI) has devastating physical, psychological, and economic consequences for patients and caregivers1. Many attempts have been made to promote axonal regeneration in SCI with different approaches2,3,4 and some beneficial effects were demonstrated by the formation of neuronal relays between proximal and distal neurons in the injury site through cell replacement therapies. The interest in cell therapies is still growing5 since transplanted cells can play many roles, including providing trophic support, immune modulation, regenerating lost neural circuits through induction of plasticity, cell replacement, and axon remyelination6.
Recently, the main effort in the field focused on human neural stem/progenitor cells (NSCs/NPCs)7. Several studies suggest that NSCs/NPCs modulate astrocyte response8, promote the secretion of proregenerative factors9,10, and replace missing neuronal cells in SCI11,12. However, studies that support the differentiation of transplanted cells into functional neurons are still poor. Moreover, transplanted cell survival and differentiation in the injured spinal cord (SC) are low13, possibly because transplanted cells take several weeks, even months, to differentiate in vivo. Additionally, current studies have not completely elucidated many biochemical, molecular, cellular, and functional aspects of cell replacement therapies. In this context, simple, fast, and cost-effective models are required to study the mechanisms of cell engraftment, the ability of engrafted cells to proliferate, differentiate into specific types or subpopulations of cells, and form synapses with resident neurons.
Integrating histological studies into electrophysiological recording and transcriptome and proteome profiling is necessary for a full comprehension of the molecular cascade occurring after cell transplantation. This certainly will speed up the design and validation of new cell replacement therapies in preclinical models and clinical studies. Indeed, to date, the use of rodents, large animals, and non-human primates has been worthwhile for elucidating many cellular processes after transplantation14. However, due to the high cost, the high ethical impact, as well as the complexity of the organism, their use is often not straightforward or not adequate to unravel biochemical and molecular processes. In addition, they may present many disadvantages correlated with biological differences, both interspecies (metabolism) and intraspecies variability (sex, age). These factors, together with external factors such as stressful situations, could alter the outcome of an experiment and their predictability in terms of therapeutic translation to humans15,16,17.
Thus, many groups employ 2D in vitro cell culture and ex vivo organotypic slices (ex vivo cultures) in addition to animal models. 2D cell culture is the most commonly used system for studying specific biological processes at a single-cell and/or cell population level. Nevertheless, monolayer cell cultures do not reflect the complexity found in a whole organism. The lack of tissue structures and physiological environment does not allow the 2D culture systems to completely emulate key structural, morphological, and functional aspects of the investigated tissue18,19,20. Organotypic cultures can overcome some of these issues. Organotypic models are based on explanting a fragment of a tissue or organ and maintaining it ex vivo for a limited period21,22. In particular, slices of the explanted tissue are generated with a precise thickness that allows nutrients to easily reach almost all the cells in the slices. They can be generated from various regions of the central nervous system, such as the hippocampus, hypothalamus, cerebellum, thalamus, cerebral cortex, substantia nigra and striatum, and spinal cord23. Organotypic cultures retain the tissue architecture, the spatial distribution of cells, the cellular diversity, and the environment (i.e., extracellular matrix composition) of the organ of origin. Moreover, they preserve the original neural activity, connections between cells, and in particular, short-distance circuits after the explant.
These aspects provide some advantages for ex vivo cultures with respect to both monolayer cultures and animal models. They retain key tissue features found in vivo but with the reduction of the costs and the possibility to perform different types of molecular, cellular, and functional experiments with an accurate regulation of the culture environmental parameters24,25,26,27,28,29. Organotypic slices can also be exploited to develop models for different neurological disorders by resembling key histopathological features of specific conditions30. Moreover, the retention of the original multicellular tissue environment renders them appropriate platforms for drug screening and for testing neuroprotective and neuro-regenerative molecules and materials.
In this work, we propose the use of SC organotypic cultures as a model to optimize NSC transplants. This is not trivial since optimal culturing conditions are required to guarantee the survival of both the host (SC tissue) and the transplant (NSCs) for weeks. Different research groups engrafted in organotypic cultures, brain-derived and SC-derived, various types of cells. Most of the works showed the transplantation of mesenchymal stem cells31,32,33, olfactory ensheathing cells34, or NSCs35,36,37,38,39,40 and evaluated the interactions of engrafted cells with the host cells, the survival of the whole system, and whether the transplanted cells differentiated into neurons or neuron-like cells inside the ex vivo tissue environment32,33,41. Some of them evaluated the regenerative potential of cells after transplant, observing their axonal growth inside the tissue37,40,41, the myelinating ability of engrafted precursors of oligodendrocytes42, the migration of engrafted cells into the host tissue43, and whether the transplanted cells released factors pushing towards a proregenerative environment31. One limitation of the current studies is that they do not explore the engraftment for a long-term period.
Considering that NSCs seem to require several weeks to differentiate in vivo44,45, this study focuses on how to generate and maintain long-term (≥30 days) mouse SC slices for up to 90 days. Slices were found to retain their original anatomical structure and to maintain a low and stable apoptotic rate over time and high cell viability. We observed diffuse expression of neuronal markers RNA binding fox-1 homolog 3 (RBFOX3) and neurofilament light chain (NFL), with the latter showing an increasing trend of axonal sprouting around the slices over time, attesting to their healthy condition. Moreover, we successfully transplanted into the SC-slices GFP-expressing human SC-derived neuroepithelial stem (h-SC-NES) cells at the first stages of neuronal differentiation. The NSC graft was maintained for 30 days after transplant and cells showed GFP expression for all the period in culture. The apoptotic rate of cells at day post-transplant (DPT) 30 was also found to be in line with respect to the apoptotic rate value observed at DPT 7 in the same cells40. Cells seemed to engraft into the tissue environment and survived up to several weeks.
In summary, our data demonstrate that it is possible to maintain in culture SC organotypic slices for 3 months without compromising their original cytoarchitecture and the tissue environment. Most importantly, they can be exploited to test cell therapies before proceeding with an in vivo experiment, thus reducing the costs and the experimental time. Here, we illustrate in detail all the passages to generate mouse SC organotypic slices and how to maintain them for long-term periods (≥30 days). Moreover, we deeply explain how to perform NPC transplantation into the slices and how to maintain them for downstream analysis.
Protocol
Animal procedures were performed in strict compliance with protocols approved by the Italian Ministry of Public Health and the local Ethical Committee of the University of Pisa, in conformity with Directive 2010/63/EU (project license no. 39E1C.N.5Q7 released on 30/10/2021). C57BL/6J mice were kept in a regulated environment (23 ± 1 °C, 50 ± 5% humidity) with a 12 h light-dark cycle with food and water ad libitum.
All work related to h-SC-NES cells was performed according to NIH guidelines for the acquisition and distribution of human tissue for biomedical research purposes and with approval by the Human Investigation Committees and Institutional Ethics Committees of each institute from which samples were obtained. Final approval was obtained from the Committee on Bioethics of the University of Pisa (Review No. 29/2020). De-identified human specimens were provided by the Joint MRC/Wellcome Trust grant (099175/Z/12/Z), Human Developmental Biology Resource (www.hdbr.org). Appropriate informed consent was obtained, and all available non-identifying information was recorded for each specimen. Tissue was handled in accordance with ethical guidelines and regulations for the research use of human brain tissue set forth by the NIH (http://bioethics.od.nih.gov/humantissue. html) and the WMA Declaration of Helsinki (http://www.wma.net/en/30publications/10policies/b3/index.html).
1. Preparation of solutions and equipment for spinal cord (SC) isolation and culturing
- Coating solution for membrane inserts
- Prepare the coating solution (Table 1): an aqueous solution with 0.1 mg mL-1 collagen, 0.01 mg mL-1 poly-L-lysine, and 0.01 mg mL-1 laminin.
- Place each membrane insert into a 35 mm dish or a 6-well plate.
- Add to the top of the membrane 1 mL of coating solution: incubate the solution for 4 h at room temperature (RT); then, remove it and let the membrane insert dry overnight (ON). Store the membrane inserts at 4 °C until their use.
NOTE: All the passages should be performed in sterile conditions. Membrane coating should be conserved for a maximum of 1 week at 4 °C before use to avoid protein degradation and suboptimal slice adhesion to the membrane.
- Media preparation: organotypic medium, dissection medium, graft medium
- Prepare organotypic medium (OM, Table 1).
- Prepare the dissection medium as described in Table 1.
- Prepare the graft medium (GM, optimized from Onorati et al.46, Table 1).
NOTE: Solutions should be prepared in sterile conditions and just before their use (1 day before or the same day of the experiment).
- Material preparation for surgery
- In the biosafety cabinet, keep the following ready for use: the dissection stereomicroscope and the surgical instruments: two pairs of micro-scissors, two pairs of straight tweezers, and two pairs of curved tweezers.
- In the biosafety cabinet, prepare the chopper instrument by equipping it with a blade to cut the SC into slices. Rotate a screw of the chopper to lift the metallic arm where the blade should be placed. Place the blade in the designated site, lower the metallic arm with the blade until it contacts the cutting deck, and fix it by tightening the secure screw with a hex key until the blade is firmly secured. Rotate the micrometric screw to the desired slice thickness (generally 350 μm). Check if the blade is placed precisely perpendicular to the cutting deck.
NOTE: Precisely perpendicular placement of the blade with respect to the cutting deck is necessary to perform the cutting correctly. - Prepare in the biosafety cabinet: two plastic Pasteur pipettes (necessary to move isolated SC and the slices), at least four 35 mm and two 60 mm dishes, and a box of fresh ice.
- Sterilize all the instruments with 70% ethanol and UV (one cycle of 20 min) just before their use to preserve culture sterility.
2. Isolation of mouse SC and slice generation
- Isolation of mouse SC
- Sacrifice postnatal day 3 (P3) mouse pups according to the project license.
- Through a midline laparotomy with the micro-scissors, isolate the lumbar region of the backbone from the rest of the mouse body and put it in a cold dissection medium in a 35 mm dish.
- Using a dissection stereomicroscope and the micro-scissors, cut the backbone along the sagittal axis, and use straight tweezers to gently remove the SC from the backbone cavity.
- Carefully peel off the meninges from the isolated lumbar region of the SC by using straight tweezers.
- Transfer and incubate the isolated SC lumbar region in cold and fresh dissection medium for 10-15 min before proceeding with the following step.
- Generation of slices
- By using one plastic Pasteur pipette, take the isolated SC lumbar region from the dissection medium and place it on the cutting deck of the chopper instrument, perpendicularly to the blade.
NOTE: Correct positioning of the SC with respect to the blade (perpendicular) is essential to properly generate SC slices. - Remove the residual dissection medium on the deck around the SC with the help of the Pasteur pipette and sterile absorbent paper. Proceed with SC automated sectioning.
- Once the slices are generated, put some fresh dissection medium with a Pasteur pipette on the cutting deck with the slices. Then, collect the slices into a 35 mm dish with fresh dissection medium and incubate them for 15 min.
- During slice incubation, wash the surface of the membrane inserts 3x with OM using a plastic Pasteur pipette. Then, leave 1 mL of OM at the bottom of each membrane insert.
- Check the slices under the dissection stereomicroscope. Seed the desired number of slices on the conditioned membrane inserts by transferring them with a plastic Pasteur pipette.
- Move the slices in the preferred orientation and desired position onto the membrane inserts with the help of the straight tweezers. Remove the excess of medium with a Pasteur pipette to allow the slices to better adhere to the membrane surface.
NOTE: Moving and orienting the slices with the straight tweezers should be performed gently to avoid tissue or membrane insert damage. - After 30 min incubation at 37 °C, transfer the insert in a new Petri dish.
NOTE: Touch the plastic ring but not the membranes during medium change. - Add 1 mL of fresh OM supplemented with glial cell line-derived neurotrophic factor (GDNF) 100 ug mL-1 at the bottom of the membrane insert.
- Incubate the slices at 37 °C. Refer to the first day in culture as day ex vivo (DEV) 0.
- By using one plastic Pasteur pipette, take the isolated SC lumbar region from the dissection medium and place it on the cutting deck of the chopper instrument, perpendicularly to the blade.
3. Long-term culturing of organotypic slices
- Maintain the slices in culture at 37 °C until the desired time points.
- Replace the medium with fresh OM at DEV 1 as described in steps 2.2.7-2.2.8.
- At DEV 3, switch the medium to the GM to create the appropriate environment for transplanting stem cells the day after. Replace the medium with fresh GM every 48 h (e.g., DEV 5, DEV 7…).
- Add fresh GDNF (final concentration 100 µg mL-1) to the medium every day until DEV 7. After DEV 7, add it only when the medium is changed (step 3.3).
4. h-SC-NES cell culturing
NOTE: h-SC-NES cells are maintained in culture in the presence of growth factors (NES medium, step 4.1.1). Before the transplantation, cells are plated in predifferentiation condition for 7 days by removing the growth factors from the medium (Predifferentiation medium: NES medium without fibroblast growth factor 2 (FGF-2) and epidermal growth factor (EGF), step 4.1.2). Then, cells are plated in differentiation condition (Differentiation medium, step 4.1.3) for 2 days before transplantation. The differentiation is supported by adding neurotrophic supplements (brain-derived neurotrophic factor, BDNF) to the Differentiation medium. The maintenance, the split, the pre-differentiation, and the differentiation of h-SC-NES cells12,46are described in detail below.
- Media preparation: NES, predifferentiation and differentiation media
- Prepare h-SC-NES cell maintenance medium (NES medium, Table 1).
- Prepare h-SC-NES cell predifferentiation medium (Pre-differentiation medium, Table 1).
- Prepare h-SC-NES cell differentiation medium (Differentiation medium, Table 1). Add BDNF freshly when the medium is changed or when the cells are plated for the first time in the differentiation condition.
NOTE: All media should be prepared in sterile conditions and should be filtered with 0.22 µm filters.
- Coating solution for h-SC-NES cells
NOTE: h-SC-NES cells are maintained in POLFN-coated culture supports (POLFN = Poly-L-Ornithine, Laminin, Fibronectin).- Prepare the coating solution in a tube: a Poly-L-Ornithine solution with Laminin (5 µg mL−1) and Fibronectin (1 µg mL−1).
- Transfer the prepared coating solution into the cell culture support, taking care to add enough to cover the entire surface of the cell culture support. Incubate the coating for 1 h at 37 °C or overnight at 4 °C.
- Remove the coating solution from the cell culture supports.
NOTE: POLFN solution could be recycled two more times, but Laminin and Fibronectin should be added freshly each time. - Wash the coating 3x with cell culture grade sterile water. Store the coatings at 4 °C or use it.
NOTE: Coatings should be used within 1 week. After that, the coatings are considered expired because of degradation processes of the added proteins.
- h-SC-NES cell maintenance
- Maintain h-SC-NES cells in culture in NES medium. Check the cells every day under the microscope to monitor when they reach the confluence.
- Change half medium every 2 days: remove half of the conditioned medium and add the fresh one (consider 20% evaporation rate).
- If the cells reach the confluence, proceed with the split, as described in step 4.4.
- h-SC-NES cell passage
NOTE: Cells are split as follows12:- Remove the conditioned medium and wash cells once with Dulbecco's phosphate-buffered saline (DPBS) without Ca2+/Mg2+.
- Remove the DPBS and add trypsin/EDTA solution to the cells to perform enzymatic detachment. Incubate cells at 37 °C for 30 s to 1 min.
- After the incubation, check the cells under the microscope: if they are not detached, slightly tap the cell culture support to perform mechanical detachment and incubate them at 37 °C for 30 s more.
- After the incubation, inactivate the trypsin/EDTA by adding 4 volumes of DPBS/fetal bovine serum (FBS) (10% vol/vol) solution to the cell culture support with cells and trypsin/EDTA. Gently pipette the solution on the cell culture support surface up and down to help all the cells detach. Collect the cell suspension in a tube.
- Centrifuge the cell suspension at 200 × g for 3 min. Discard the supernatant and resuspend the pellet in fresh NES medium.
- Count the cells and plate them on each new POLFN-coated culture support at a density of ̴0.5-1 × 105 cells/cm2.
- Add Y-27632 (10 µM) and place the cells at 37 °C. Check them every day until confluence and then split them again for maintenance/expansion or cell banking.
- h-SC-NES cell predifferentiation
- Split the cells as described in step 4.4.
- Plate the cells on the POLFN-coated cell culture supports at a density of ̴0.5-1 × 105 cells/ cm2 in Predifferentiation medium. Add Y-27632 (10 µM) after the split. Call the first day in predifferentiation day in vitro (DIV) 0.
- Change half of the medium every 2-3 days (see step 4.3.2).
- Maintain the cells in predifferentiation condition until DIV 7 and then proceed with step 4.6.
- h-SC-NES cell differentiation
- At DIV 7 of predifferentiation, split the cells as described in step 4.4.
- Plate the cells on POLFN-coated cell culture supports at a density of ̴1-1.5 × 105cells/cm2in Differentiation medium. Add Y-27632 (10 µM) and BDNF (30 ng mL−1) after the split.
- After 2 days of differentiation (DIV 10), split the cells for the transplantation into slices.
5. h-SC-NES cell transduction with GFP-carrying lentiviral vectors
NOTE: Cell transduction is performed during the maintenance phase of h-SC-NES cells. When cells are correctly transduced, these can be expanded and predifferentiation and differentiation protocols previously described are applied (steps 4.5 and 4.6).
- Preparation of the cell transduction medium
NOTE: Cell transduction medium is prepared by mixing a specific volume of NES medium and a precise volume of lentiviral vector stock (LVS) preparation according to different parameters: the desired MOI (multiplicity of infection = ratio of the numbers of viral particles to the numbers of the host cells in a given infection medium); the number of plated cells; the initial concentration of LVS preparation (=LVS PFU, plaque-forming unit); the surface area of the used culture vessel.- Calculate the correct volume of LVS preparation to be added to NES medium, according to the chosen MOI using equations (1) and (2).
(n cells to plate/cm2) × cm2 of cell culture support × MOI = LVS PFU for the chosen MOI (1)
LVS PFU : Tot Initial LVS Vol (µL) = LVS PFU for MOI : LVS Vol to add to medium (µL)(2)
NOTE: The LVS PFU (initial PFU of LVS) and total initial LVS volume are given by the manufacturer. The LVS PFU for the chosen MOI is calculated as described in equation (1). Thus, we can obtain the volume of LVS preparation that has to be added to the total volume of NES medium (based on cell culture support) for the chosen MOI, as described in equation (2).
Example: We used MOI 3, based on previous lab experience (the MOI could vary depending on the used cell line and the viral preparation). If the desired MOI is 3, the number of cells to be plated is 0.5 × 105/cm2, and the culture support is a 1 well-MW24 (2 cm2), assuming that the initial LVS PFU/TU (plaque forming unit/transducing unit) is 25 × 106 PFU in 1 mL (1,000 μL = initial LVS vol), the calculations are as follows:
Cells plated in 1 well-MW24 (2 cm2) = 0.5 × 105 cells × 2 cm2 = 1 × 105 cells
1 × 105 cells × 3 (MOI) = 3 × 105 PFU = LVS PFU for MOI 3
25 × 106 PFU:1,000 µL = 3 × 105 PFU:x µL
x µL = 12 µL = LVS volume to add to the medium
Thus, to transduce the cells with cell transduction medium with MOI 3 in 1 well-MW24, add 12 µL of initial LVS preparation to the NES medium (238 µL) prepared for 1 well of MW24. The final total volume is 250 µL.
NOTE: The medium is usually prepared freshly on the day of transduction under sterile conditions.
- Calculate the correct volume of LVS preparation to be added to NES medium, according to the chosen MOI using equations (1) and (2).
- h-SC-NES transduction protocol
- Plate h-SC-NES cells at a low passage on POLFN-coated 24-multi well plate (or in any other culturing support) at a density of 0.5 × 105/cm2 in NES medium.
- The next day, collect the conditioned NES medium from the wells where the cells were plated. Depending on the chosen culturing support, add to the cells the lowest volume of fresh cell transduction medium necessary to uniformly cover the seeding surface (e.g., 250 µL/well of a 24-multiwell plate).
- Then, incubate the h-SC-NES cells for 6 h at 37 °C. After that, add the previously collected conditioned medium to the cells (200 µL/well of a 24-multiwell plate) and incubate the cells ON at 37 °C.
- The next day, wash the h-SC-NES cells once with DPBS and perform a total medium change (NES medium).
- The following days, check the cells under a fluorescence microscope to observe GFP expression.
- Expand the h-SC-NES cells for cell banking and transplantation.
6. Cell transplantation into SC slices and co-culturing
- Preparation of glass microneedles
- Use a puller to obtain fine needles from borosilicate glass capillaries. Set the puller as follows: HEAT 990, PULL 350.
NOTE: From one capillary, it is possible to obtain two fine needles.
- Use a puller to obtain fine needles from borosilicate glass capillaries. Set the puller as follows: HEAT 990, PULL 350.
- Cell preparation for transplantation
- Split the cells as described in step 4.4.
NOTE: If the cells do not express a fluorescent reporter, label them with a cell tracking dye to monitor them using a fluorescence microscope after transplantation and during long-term culture. Follow the chosen manufacturer's protocol for the labeling step. - Count the cells after the split and centrifuge at 200 × g for 3 min. Suspend the obtained pellet with fresh medium + Y-27632 (10 µM) to have the desired concentration of cells (usually a range between 30,000-50,000 cells µL-1).
- Transfer the cell suspension into a 500 µL or 1.5 mL tube and place it on ice. Cells are ready for transplantation.
- Split the cells as described in step 4.4.
- Cell transplantation into organotypic slices
NOTE: Perform h-SC-NES cell transplants into the mouse SC organotypic slices using an air microinjector and glass microneedles.- Load a glass microneedle with 4 µL of cell suspension using a micropipette and micro-loader tips.
NOTE: Avoid air bubble formation in the needle since it could hamper the microinjection process. If bubbles are formed, remove them with the micropipette. - Place the needle in the assigned support of the microinjector and break the needle tip using the straight tweezers.
NOTE: Break the glass needle closer to the tip to avoid the formation of big holes. - Before transplanting into the slices, set the microinjection parameters. Set the pressure at 10 psi.
NOTE: The pressure value could be changed based on the microinjector and the operator observations: the pressure should be enough to microinject the cell suspension, avoiding tissue damage. - On a calibrated glass slide, put a drop of mineral oil with a Pasteur pipet and microinject the cell suspension into the drop. The diameter of the obtained sphere of cell suspension in the oil drop correlates with a specific microinjection volume. Change the microinjection parameters as needed to reach a diameter of the cell suspension sphere of 0.2 mm for injecting 4nL.
- After setting the correct volume, microinject the cell suspension rapidly into the slices. Check under the fluorescence stereomicroscope for the presence of the cells in the slices to verify that the microinjection/transplant was successful.
NOTE: The cell suspension could sometimes obstruct the needle: in this case, try to remove the clog of cell suspension by modifying the injection parameters or load a new needle with fresh cell suspension. - After the transplantation, place the slices at 37 °C and 5% CO2until the desired time point and perform medium change every other day as described in steps 2.2.7-2.2.8.
- Load a glass microneedle with 4 µL of cell suspension using a micropipette and micro-loader tips.
7. Immunofluorescence staining
- Day 1
- Remove the medium from the bottom of the insert membrane and wash the slices 3x with prewarmed DPBS.
- Fix the slices with prewarmed 4% formaldehyde (FA): remove the DPBS and add 1.5 mL of 4% FA at the bottom of the membrane insert with the slices. After 15 min incubation at RT, add 1 mL more of 4% FA on the upper surface of the membrane insert and incubate for 15 min at RT. Total fixation time: 30 min at RT
- Remove the 4% FA and wash the slices for 3 x 10 min with DPBS.
- Cut the membrane of the insert circumferentially with a surgical knife, separate the membrane with the slices from the plastic component of the insert, and proceed with the immunofluorescence steps.
NOTE: After this step, the membrane with the slices is floating in DPBS in the dish. - Permeabilize with 1 mL/membrane of a solution with 0.7% Triton in DPBS for 10 min at RT.
- Remove the permeabilization solution and incubate the samples for 4 h at 4 °C with 1 mL/membrane of blocking solution composed of 0.5% Triton, 10% FBS in DPBS.
- Remove the blocking solution and add to the slices the primary antibodies at their working dilution e.g., mouse anti-Neurofilament (NFL) antibody, 1:500; rabbit anti-NFL, 1:500; rabbit anti-RBFOX3 (NeuN) antibody, 1:400; rabbit anti-active caspase-3 (aCASP3), 1:400; mouse anti-human Nuclei, (Hu-Nu), 1:400; rabbit anti-Hu-Nu, 1:400; mouse anti-GFP, 1:400 (as reported Table 1) in 1 mL of antibody solution composed of 0.5% Triton, 1% FBS in DPBS. Incubate ON at 4 °C.
- Day 2
- Wash the membranes for 3 x 10 min with 1-2 mL of DPBS.
- Incubate the membrane with secondary antibodies (e.g., Goat anti-Mouse IgG (H+L) secondary antibody, Alexa Fluor 488, 1:500; Goat anti-Rabbit IgG (H+L) secondary antibody, Alexa Fluor 568, 1:500; Goat anti-Mouse IgG (H+L) secondary antibody, Alexa Fluor 647, 1:500; Goat anti-Rabbit IgG (H+L) secondary antibody, Alexa Fluor 647, 1:500 as reported Table 1) and Hoechst/DAPI for nuclei diluted in 1 mL/membrane of antibody solution (Triton 0.5% + FBS 1% in DPBS) for 3 h at RT.
NOTE: Carefully protect the samples from the light to avoid secondary antibody bleaching during incubation and in the following steps. - Remove the antibody solution and wash for 3 x 10 min with DPBS (1-2 mL).
- Replace the DPBS with fresh DPBS and store at 4 °C in light-protected conditions.
- At the end of the immunofluorescence protocol, mount the membranes on glass slides. Put a drop of 200 µL of mounting solution on a glass slide. With the help of straight tweezers, transfer the floating membrane from the 35 mm dish onto a cover slip and then, transfer the membrane onto the glass slide with the mounting solution.
- Put a drop of 100 µL of mounting solution on a new coverslip and cover the membrane with it, fixing it on the glass slide. Let it dry overnight under the chemical hood in light-protected conditions.
- Store the samples at 4 °C in the dark or perform imaging analysis.
8. Live/Dead assay
- Prepare the working solution by aliquoting 700 µL per dish of fresh medium and add at the correct working dilution the Sytox (e.g., Component B, 1:2,000) and the Calcein AM (e.g., Component A, 1:2,000).
NOTE: As the reagents are light-sensitive, protect the working solution from light. - Evaluate the medium volume at the bottom of the membrane and add the Sytox and the Calcein AM at the same working dilution described in step 8.1.
- Add 2 drops of 30 μL each on the top of each slice of the working solution prepared in step 8.1.
NOTE: Protect the dish from light by placing it in dark conditions. - Incubate the slices for 30 min at RT.
- After the incubation, cut out the membrane circumferentially from the insert with a surgical knife: after that, the membrane with the slices floats in the working solution.
- Place the membrane without the mounting solution upside down on a coverslip with the help of straight tweezers and add 100 µL of DPBS on top of the membranes to keep them hydrated.
- Acquire live images using the confocal microscope as fast as possible.
NOTE: Add 2 drops of 40 µL each of DPBS on the top of the membrane every 30 min during image acquisition to prevent the membrane from drying out.
9. Imaging
- Confocal imaging of fixed samples
- For qualitative analysis, acquire images using a confocal microscope with the following acquisition parameters: set the large image option (choose: 4 x 4), use 10 x objective, no stacks, and a resolution of 3,634 x 3,634 pixels.
- For quantitative analysis (aCASP3, Calcein, and Sytox for slices and aCASP3 for cells), acquire images using the confocal microscope with the following acquisition parameters: 20 x objective, resolution of 1,024 x 1,024 pixels with a Z-step of 3 μm.
- Live imaging of transplanted slices using the stereomicroscope
NOTE: Capture images using the stereomicroscope in brightfield and epifluorescence modes.- Using the brightfield setting, acquire images of the slices (1 x objective with 3x zoom used here).
NOTE: Modify the light depending on the used microscope and use the optical fibers if it is necessary. - Using the fluorescence setting, acquire images of the transplanted cells with the same objective and zoom used for the slices (see step 9.2.1). Use the following parameters for the acquisition: gain 1, exposure 200-500 ms, offset -10.
- Using the brightfield setting, acquire images of the slices (1 x objective with 3x zoom used here).
- Live imaging after Live/Dead assay using the confocal microscope
- Acquire images using a confocal microscope with the following acquisition parameters: 20x objective, resolution 1,024 x 1,024 pixels with a Z-step of 3 μm.
10. Image analysis by ImageJ
- NFL, RBFOX3, and DAPI area analysis
- Open ImageJ software (https://imagej.net/software/imagej/).
- Open the file image by clicking File | open | select file | open.
- In the pop-up window, select Stack viewing | Hyperstack and Color mode | Default (with autoscale).
- In the toolbar, select Image | Color | Split channels.
- Choose the desired channels to analyze: green channel for NFL (axonal marker), red channel for RBFOX 3 (neuronal marker), and blue channel for DAPI (nuclear staining).
- For NFL analysis, proceed with the following steps: in the toolbar select Image | Adjust | Threshold | select the parameters (dark background, algorithm, e.g., default) and move the cursor on the value bar (under/over) to cover and circumscribe all the neurite area (neurites are highlighted in white in a dark background) | Set | Apply.
- Select from the toolbar the tracing tool Wand and use it to automatically define the white area covered by NFL. Press Analyze | Measure | Area value in μm2.
- For DAPI and RBFOX3 analysis, proceed with the following steps: in the toolbar, select Image | Adjust | Threshold | Select the parameters (white background, algorithm, e.g., default) and move the cursor on the value bar (under/over) to cover and circumscribe all the RBFOX3 or DAPI area | Set | Apply.
- In the toolbar, select Process | FFT | Bandpass Filter. Use the threshold value bar to adjust the white area covered by RBFOX3 or DAPI, corresponding to their fluorescence signal.
- From the toolbar, select the tracing tool Wand and use it to automatically define the area covered by RBFOX3 or DAPI. Press Analyze | Measure | Area value in μm2.
- Analysis of apoptosis by ImageJ
- Open ImageJ software (https://imagej.net/software/imagej/).
- Open the Z-stack file image by clicking File | open | select file | open.
- In the pop-up window, select Stack viewing | Hyperstack and Color mode | Default (with autoscale).
- In the toolbar, select Image | Color | Split channels.
- Choose the desired channels: red channel for aCASP3 (apoptosis marker to analyze) and blue or cyan for DAPI or Hu-Nu for nuclei. Then, overlay the channels by selecting in the toolbar Image | Color | Merge channels | create composite.
- Drag the Z-bar at the bottom of the image to browse through the Z-stack of the image and identify stacks in the central region of the slices with aCASP3 positivity.
- In the toolbar, select Plugins | Analyze | Cell counter.
- In the opened pop-up window, select Initialize to prepare the image for the count; then, select a counter type (e.g., Type 1) and rename it as the object to count (e.g., aCASP3+ cells). Rename other counter types as described above to count other objects (e.g., DAPI+ or Hu-Nu+ cells for the total number of cells).
- In the pop-up window, select the counter type corresponding to the object to count (e.g., aCASP3+ cells), then select the Point tool in the toolbar and begin to manually count the number of apoptotic cells, positive for aCASP3, clicking on each positive one in the opened image.
- Select another counter type in the cell counter window and begin to count the total number of cells (DAPI+ cells, for slices; Hu-Nu+ cells, for transplanted cells).
- Analysis of Live/Dead assay by ImageJ
- Open ImageJ software (https://imagej.net/software/imagej/).
- Open the Z-stack file image by clicking on File | open | select file | open.
- In the pop-up window, select Stack viewing | Hyperstack and Color mode | Default (with autoscale).
- In the toolbars, select Image | Color | Split channels.
- Choose the desired channels: green channel for Calcein (vitality marker to analyze) and cyan channel for Sytox (dead marker). Then, overlay the channels by selecting in the toolbar Image | Color | Merge channels | select create composite.
- Drag the Z-bar at the bottom of the image to browse through the Z-stack of the image and identify stacks in the central region of the slices with Calcein and Sytox positivity.
- In the toolbar, select Plugins | Analyze | Cell counter.
- In the opened pop-up window, select Initialize to prepare the image for the count; then select a counter type (e.g., Type 1) and rename it as the object to count (e.g., Calcein+ cells). Rename other counter types as described above if it is necessary to count another object (e.g., Sytox+ cells).
- In the pop-up window, select the counter type corresponding to the object to count; then select the Point tool in the toolbar and begin to manually count the number of Calcein+ cells, clicking on each positive one in the opened image.
- Select another counter type in the cell counter window and count the Sytox+ cells as described for Calcein.
11. Graphs and statistical analysis
- Perform all statistical analysis and plot graphs using the software of choice.
Representative Results
The described methods allow the establishment of SC organotypic slices from mice at stage P3 and their maintenance in culture for a prolonged time in healthy conditions. Moreover, we show a protocol for transplanting cells into the slices and for co-culturing them for up to 30 days (Figure 1). First, we show the optimization of the culture conditions and a protocol suitable for prolonged culturing of the SC slices with transplanted cells (Figure 2A). Slices are generated and maintained from DEV 0 until DEV 2 in the OM, which was originally proposed as an optimal medium for the maintenance of SC slices47. However, due to the presence of serum proteins, this medium could be suboptimal to sustain the neuronal differentiation and maturation of the transplanted neural precursor cells. Indeed, at DEV 3, we tested the switch from OM to the GM, a formulation containing Neurobasal plus B27, which supports neural survival, and without serum, which inhibits the correct neuronal differentiation, promoting instead a glial fate48,49.
Figure 2B shows the results achieved by switching the medium at DEV 3 from OM to GM, compared to the SC slices not receiving the switch (control slices were cultured in OM). We used the distribution of the NFL signal inside the slices as a marker for neuronal integrity (Figure 2B,C). Slices at DEV 7 were healthy in both culturing conditions, showing the diffuse distribution of neurofilament (NFL, in green) inside them. At DEV 10, slices cultured in GM seemed to be healthier with respect to the control slices cultured in OM, as documented by NFL staining distribution. We also estimated the NFL+ area (% NFL+ Area/DAPI+ Area) of the slices shown in the representative images of Figure 2B. The estimated NFL+ area is represented in the histograms in Figure 2C, confirming that the NFL signal is diffusely distributed in the slices at DEV 7 under both conditions. However, at DEV 10, the estimated area covered by NFL staining decreases for the OM culturing condition.
These data suggest that switching to the GM at DEV 3 is well-tolerated for prolonged culturing of SC slices (DEV 10). As a next step, we tested GM at more prolonged time points: DEV 30, DEV 60, and DEV 90. As shown in Figure 3A,B, slices were maintained healthy in culture until DEV 90. NFL staining was found widely present in the slices at each time point, with diffuse sprouting around the slices of neurites departing from the central region. Indeed, we estimated the NFL+ area of the slices shown in Figure 3A and it increased over time as shown in the histograms of Figure 3B. We also observed positivity to the neuronal marker RBFOX3, providing another line of evidence of the neuronal differentiation of the slices. At each time point, we also checked the apoptosis rate by evaluating in different slices the number of cells positive to aCASP3 (Figure 4A,B). The analysis was performed as described in protocol section 10.2. Apoptotic rate (% aCASP3+ cells/total number of DAPI+ cells) was found to be very low at each time point (0.85 ± 0.52%, 0.71 ± 0.27%, 0.66 ± 0.45% for DEV 30, 60, and 90, respectively) with no significant differences between the three considered time points (p-value > 0.05, Figure 4B). These data suggest that the apoptotic rate associated with aCASP3 remains stable during time and, together with the wide distribution of NFL in the slices (Figure 4A), confirm the survival of the slices at each time point.
In support of previous data, we also performed a live/dead assay to evaluate the viability of the slices at the three different time points. We used Calcein (green staining) to label the viable and metabolically active cells and Sytox (cyan staining) to assess cell death. As shown in the histograms in Figure 4C, the percentage of metabolically active cells increases slightly from DEV 30 to DEV 90 (93.17 ± 5.21%, 96.43 ± 3.02%, 96.33 ± 3.10% for DEV 30, 60, and 90, respectively), stabilizing between the last two time points (DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value = 0.99). We found low levels of cell death that decreased over time (6.83 ± 5.21%, 3.57 ± 3.02%, 3.66 ± 3.10% for DEV 30, 60, and 90, respectively) and a significant difference was found between DEV 30 and later time points, DEV 60 and DEV 90 (DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value = 0.99). These data, in association with the apoptosis rate, confirm slice survival over time and support the effectiveness of the long-term culturing protocol performed.
Once the feasibility of prolonged culturing of the SC slices was established, we challenged the system by transplantation of h-SC-NES cells at the first stages of neuronal differentiation. We tested the h-SC-NES cells because they have shown promising results for SCI treatment12. The transplantation procedure of h-SC-NES cells into the mouse SC slices is described in protocol section 6. The SC slices and transplanted h-SC-NES cells were maintained until DPT 30. Cells were grafted at DIV 10 of differentiation (neural precursor stage) into DEV 4 organotypic slices, as shown in the protocol scheme of Figure 5A. Transplanted cells were monitored for the expression of GFP in culture for up to 30 days. Figure 5B shows representative live images, at different DPT, of an SC slice with transplanted GFP+ cells. The stable expression of GFP over time (Figure 5B and Figure 6A) suggests that cells survived into the SC tissue in the previously optimized culture conditions. We also checked the apoptotic rate of transplanted cells as described in protocol section 10.2. The apoptotic rate (% aCASP3+ cells/total number of Hu-Nu+ cells) was found to be very low (0.44 ± 0.34%) after 30 DPT (Figure 6B). Moreover, the apoptotic rate at DPT 30 was found to be in line with that found for the same type of cells at DPT 7, as previously reported40, documenting that the cultures stabilize over time.

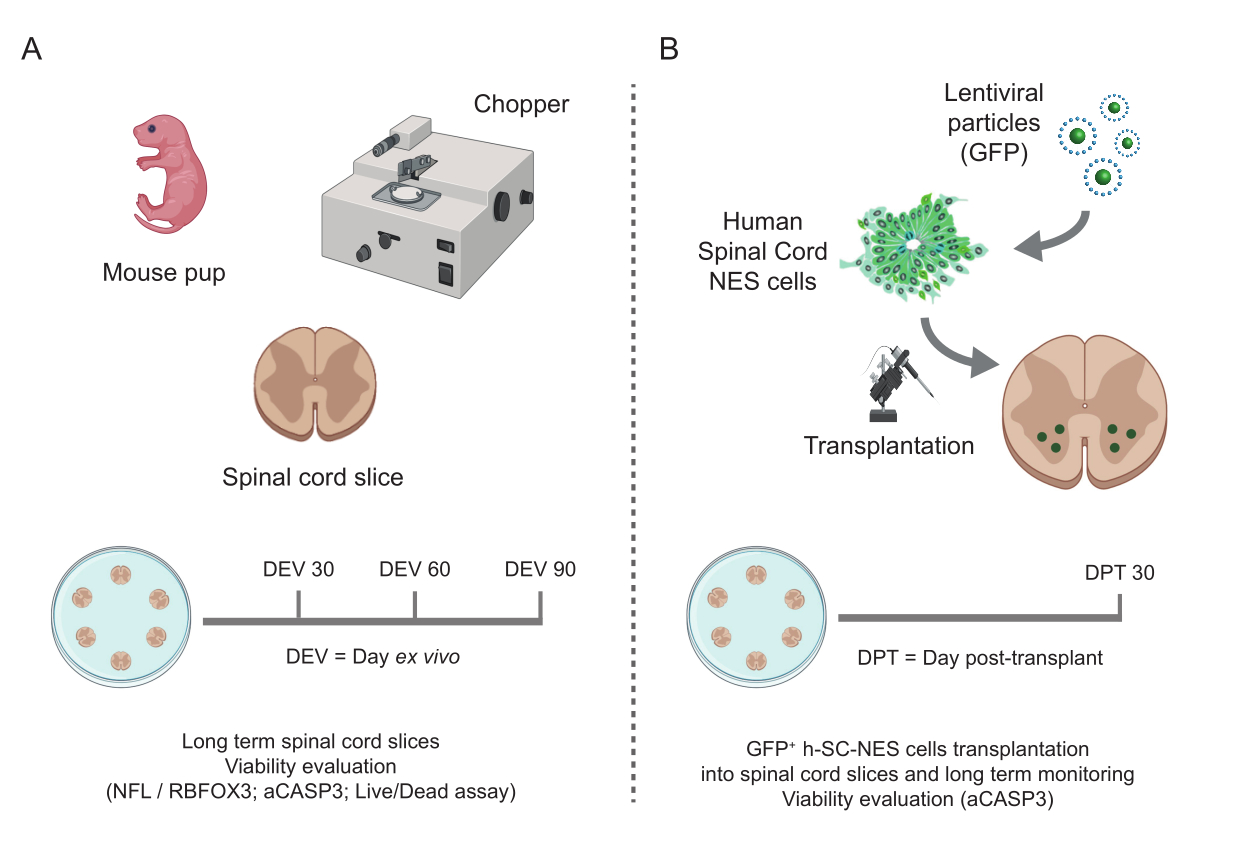
Figure 1: Workflow of the protocol. Representative scheme showing the general workflow of the protocol performed. (A) On the left, a scheme summarizing mouse SC-slice generation from isolated SC of mouse pups at P3 and long-term culturing of SC organotypic slices. (B) On the right, a scheme summarizing the transplantation of h-SC-NES cells expressing GFP into mouse SC-organotypic slices. Grafted cells are maintained for 30 days post-transplant. Abbreviations: h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DEV = day ex vivo; DPT = day post-transplant; NFL = neurofilament light chain; RBFOX3= RNA binding fox-1 homolog 3; aCASP3 = active Caspase-3; SC= spinal cord. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Optimization of long-term culturing conditions. (A) Representative scheme of the protocol for testing OM and GM. OM is maintained until DEV 7-10 for the control group. The medium is switched to the GM at DEV 3 for the treated slices; then, they are fixed at DEV 7-10 for comparison to controls. (B) Representative images comparing mouse SC organotypic slices at DEV 7 and 10 cultured in different conditions. Slices are stained for the cytoskeletal marker neurofilament (NFL, green). The wide distribution of NFL staining in slices cultured with GM suggests an overall survival and differentiation. Nuclei are counterstained with DAPI. Scale bar = 500 µm. (C) Representative histograms of the estimate of the area covered by NFL in the slices shown in Figure 1B. At DEV 10, NFL surface area decreases in the OM culturing condition. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain.; OM = organotypic medium; GM = graft medium; SC = spinal cord. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Long-term cultured mouse SC organotypic slices. (A) Slices are maintained in culture until DEV 90. Immunofluorescence assay reveals a wide distribution of the cytoskeletal marker neurofilament (NFL, green) and the nuclear neuronal marker RBFOX3 (red), attesting to their healthy condition and neuronal identity after long-term culturing. Of note, NFL+ axons sprout out diffusely around the slices over time. Nuclei are counterstained with DAPI. Scale bar = 500 µm. (B) Representative histograms of the estimate of NFL+ area and time and, RBFOX3+ area of the slices shown in panel A. NFL+ neurite area increases over time. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; SC = spinal cord; RBFOX3= RNA binding fox-1 homolog 3. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Evaluation of cell viability in the SC slices over time. (A) Representative images of organotypic slices at DEV 60 stained for aCASP3 (red) and NFL (green). Scale Bar = 100 µm. NFL shows a diffuse pattern. Rare cells are positive for the apoptotic marker aCASP3 (Insets: 1-2-3). (B) Analysis of the apoptosis rate in slices at different time points. Mean ± SD, N (replicates) = 6 slices, n (total cells) > 1,000 for each slice, Kruskal-Wallis test, multiple comparison, p-value > 0.05. The apoptotic rate is stable over time. In the insets 1-2-3 of panel A, it is possible to observe details of cells positive for aCASP3 (red staining, white arrows). Small red dots label cell debris and pyknotic nuclei. Scale bar = 50 µm. (C) Representative images of live/dead assay performed on SC slices at DEV 90: metabolically active cells are labeled in green with Calcein, while dead and damaged cells are labeled in light blue (cyan) with Sytox. The two histograms show the % of cells positive for Calcein (on the left) and Sytox (on the right) on the total number of cells. For both mean ± SD, N (replicates) = 6 slices, n (total cells) > 1,000 for each slice, Kruskal-Wallis test, multiple comparison, DEV 30 vs DEV 60 p-value = 0.018; DEV 30 vs DEV 90 p-value = 0.027; DEV 60 vs DEV 90 p-value > 0.99. Abbreviations: DEV = day ex vivo; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; SC = spinal cord; aCASP3 = active caspase-3. Please click here to view a larger version of this figure.
{kind=link}

Figure 5: h-SC-NES cell transplantation into mouse organotypic slices. (A) Representative scheme of the transplantation protocol. Cells are transplanted as neural precursors at DIV 10 of differentiation into DEV 4 organotypic slices. (B) Representative images of mouse organotypic slices transplanted with GFP-expressing h-SC-NES cells over time until DPT 30. Cells are transduced with a lentiviral vector carrying the GFP gene. GFP expression over time confirms their viability and adaptation to the slice environment. Scale bar = 500 µm. Abbreviations: DIV = first day in pre-differentiation; h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DEV = day ex vivo; OM = organotypic medium; GM = graft medium; DPT = days post-transplant. Please click here to view a larger version of this figure.
{kind=link}

Figure 6: Apoptosis rate evaluation of transplanted h-SC-NES cells after 30 days from transplant. (A) Representative image of a mouse organotypic slice transplanted with GFP-expressing h-SC-NES cells. Cells are transduced with a lentiviral vector carrying the GFP gene for monitoring them into the slices after transplantation. GFP expression over time confirms their viability and adaptation to the slice environment. The time point shown is DPT 30; cells are stained for human nuclei (cyan) and aCASP3 (red). Scale Bar = 150 µm. (B) On the left, representative pie chart of the apoptosis analysis of cells transplanted in slices at DPT 30 (N (replicates) = 5 slices, n (cells) = 5,000), and on the right, an inset of Hu-Nu+ cells and a detail of a cell positive to aCASP3 (white arrow). Scale bar = 75 µm. Small red dots label cell debris and pyknotic nuclei. Abbreviations: h-SC-NES = human spinal cord-derived neuroepithelial stem; GFP = green fluorescent protein; DPT = day post-transplant; DAPI = 4',6-diamidino-2-phenylindole; NFL = neurofilament light chain; aCASP3 = active caspase-3; Hu-Nu = human nuclei. Please click here to view a larger version of this figure.
{kind=link}
Table 1: Composition of solutions used in this protocol. Please click here to download this Table.
Discussion
There is still no effective treatment for patients with SCI. Different approaches have been tested and one of the most promising is based on a regenerative strategy-cell replacement. Currently, the advancements in the regenerative medicine field ask for novel platforms to test the efficacy and safety of cell transplants, alone or in combination with other approaches. Their preclinical validation is essential to pursue further clinical studies. SC organotypic cultures are a useful platform for studying different aspects of neurodegeneration, neural regeneration, and neurodevelopment, and for investigating the effectiveness of novel therapeutic approaches23. In particular, specific features of the organotypic cultures such as the maintenance of original histoarchitecture and cell and microenvironment composition are advantageous to unravel transplantation dynamics, such as cell engraftment, integration, differentiation, and maturation.
Consistently with published protocols, SC organotypic slices can be maintained in culture for approximately 2-3 weeks in healthy conditions, which limits their use for the long-term investigations and functional screening required for testing schemes of cell therapy. Exploring important processes such as differentiation and maturation towards the correct fate of transplanted cells inside SC tissue requires long-term monitoring. These cellular processes are critical during common transplants in animal models. The availability of an ex vivo system that mimics many features present in vivo would be helpful in the preclinical screening phase.
For this reason, in this work, we propose an optimal long-term (≥30 days) SC organotypic culture method that allows to maintain viable SC slices for up to 90 days, tripling their usual culture time frame. Moreover, we show stable h-SC-NES cell engraftment inside SC slices and the maintenance of the transplant culture for up to 30 days. We monitored cell engraftment over time by observing GFP expression to verify cell survival up to DPT 30. After 30 DPT, we evaluated the cell apoptosis rate. In literature, the evaluation of apoptosis of transplanted h-SC-NES cells in SC-slices at 7 DPT has been reported40. Here, we extended cell apoptosis analysis at DPT 30 to compare the apoptotic rate with respect to the earlier time-point (DPT 7). We found out that our data are in line with the literature, suggesting that transplanted h-SC-NES cells survive also at a later time point if they are maintained in the culture condition optimized in our work. This improved long-term ex vivo platform alone and in the transplant configuration will help researchers in preclinical screening for stem cell-based transplants for SCI. This will allow them to identify the best cell candidate for further in vivo studies promoting the success of the transplants. Moreover, after initial screening, SC organotypic slices could also be used in parallel to the in vivo studies to confirm and corroborate long-term cellular dynamics and behaviors observed in animal models or to support mechanistic studies.
Our protocol describes in detail how to generate this long-term organotypic model, but some critical steps should also be discussed. Concerning the generation of the SC organotypic cultures, there are some challenges during the surgery and the first stages of culture. A well-performed surgery procedure is essential to generate slices that maintain the original histoarchitecture. If the SC is ruined during the isolation, slices can lose their typical anatomic structure and tissue damage can induce an excessive pro-inflammatory insult leading to unhealthy conditions and cell death. The most challenging phase during the surgery is the extraction of the SC from the backbone and the removal of meninges from the isolated SC. The success of these steps depends on the experience of the operator; therefore, a training period before starting with the experiments is recommended.
Coronal sectioning of the SC through a chopper is also a challenging phase. The isolated SC should be placed on the cutting deck exactly perpendicular to the blade. The operator should also place the blade perpendicularly to the cutting deck. These precautions are necessary to ensure the generation of reproducible slices among the same and different experiments. Another important issue is that the time for surgery is limited: the entire slice generation procedure must take ~30 min. If the operator spends more time on surgery and cutting, the SC tissue will suffer and this can impair the success of the culture and the next steps of the experiment.
Once slices are placed on the culture membrane, it is important to feed them correctly. GDNF is necessary to sustain tissue recovery and survival. Cutting with a chopper is traumatic for the tissue and, for this reason, slices are placed soon after the cut into an ice-cold dissection medium to clean away the excess of pro-inflammatory and death-promoting molecules. Then, slices are placed on the culture membranes (cell culture inserts) with fresh medium modified with GDNF to promote a faster recovery and slice adhesion to the membrane. GDNF should be added to the medium every day for the first week in culture because of its short half-life50,51. We observed that slices need the continous presence of GDNF during the first days in culture to promote tissue recovery and viability. In any case, as GDNF presence is important for the entire culturing period, it is strongly discouraged to interrupt GDNF administration at further time points.
During the first week in culture, it is also important to check the slices macroscopically by eye and at the microscope. Translucent tissue and transparency of the borders are signs of proper adhesion of the slices to the membrane and of viable tissue. The necrotic tissue will appear extremely white at first macroscopic sight and the necrotic areas will appear dark grey at the microscope. After some weeks in culture, the morphology of tissue may change: cell movements and tissue adhesion to the membrane can influence this process. We observed, for example, the loss of the central lumen in some slices filled with cells and the loss of dorsal and ventral horn morphology. This happens mainly with smaller slices, while most of them will maintain an anatomic structure close to the original one. Slices are usually generated from the lumbar or thoracic regions because in this way they can have the appropriate size to maintain their original histoarchitecture over time: if they are too small, they lose their architecture while, if too big, the central region can undergo into necrosis. Thus, we used the lumbar region of mouse pups to generate slices with the appropriate size for optimal long-term culturing but, in principle, other segments can be considered. Moreover, we opted to use the lumbar region, because ventral and dorsal regions are more distinguishable from each other. In addition, this region presents tissue areas with a higher percentage of motor neurons and grey matter, which are sites of interest for cell replacement therapies in SCI. Concerning transplanting cells into the slices, the main issue is related to the break of the glass microneedle tip. If the hole for the passage of cells is too big, it can cause damage to SC tissue during the microinjection. If it is too small, cell stacking can obstruct the needle, hampering the transplantation process. The transplantation procedure should be completed within 1 h to minimize cell suffering and death.
The proposed protocol provides an optimal and versatile tool for different types of investigations. Here, we apply our long-term platform to validate the transplantation of h-SC-NES cells at the first stages of differentiation inside mouse SC tissue for 30 days. The main novelty of the proposed approach is the optimization of the co-culture protocol. The components of GM sustain long-term neuronal survival of the SC-slices and the transplanted h-SC-NES cells. Indeed, GM, being a serum-free medium, sustains the differentiation of the transplanted cells towards the neuronal fate with respect to the medium previously used for organotypic slice culture47.
Regarding the proposed models for SCI, experiments are usually performed on adult mice. So far, the most important differences between neonatal and adult SC are related to the higher regenerative potential found in neonatal with respect to the adult mice52. However, such differences have no impact on the type of protocol that we are proposing, since here we focus on the response of grafted cells to the hosting tissue environment rather than to the regeneration capabilities of the resident neurons. Another difference between neonatal and adult mice after a SCI is related to the formation of the glial scar that occurs in adults. This aspect is not taken into account in the proposed model, which does not consider the complex physiopathological processes resulting from primary and secondary injuries.
Regarding the applications, the platform could also be used to investigate the integration between the transplanted cells with resident circuits present in the SC organotypic model. Genetic engineering tools were already used in the CNS to evaluate synaptic connectivity and could be exploited in this regard53,54,55. In particular, the integration could be investigated and validated by assessing the formation of synapses between the engrafted cells and the SC ex vivo tissue. These long-term organotypic cultures could also be exploited for testing neuroprotective and neuroregenerative agents or novel molecules/materials or to study neurodegenerative disorders that involve the SC. To study specific neurodegenerative disorders, the protocol needs to be adapted for culturing SC slices generated from relevant models, such as transgenic mice carrying specific pathology-associated mutations, at the relevant stage for the pathology (i.e., neonatal, juvenile, adult). In conclusion, our protocol and organotypic cultures in general, being explants of a specific organ, present features that bridge the gap between 2D cell cultures and in vivo models, confirming them as an invaluable tool for both basic research and preclinical testing.
Acknowledgements
The study was supported by the Wings for Life Foundation (WFL-IT- 20/21), the European Union Next-Generation EU-National Recovery and Resilience Plan (NRRP)-mission 4 component 2, investment n. 1.4-CUP N. B83C22003930001 (Tuscany Health Ecosystem-THE, Spoke 8), and the Marina Romoli Onlus. This manuscript reflects only the authors' views and opinions, neither the European Union nor the European Commission can be considered responsible for them. Data and metadata are available on Zenodo 10.5281/zenodo.10433147. Images were generated with Biorender https://www.biorender.com/.
Materials
| Name | Company | Catalog Number | Comments |
| anti-cleaved Caspase-3, (Asp175) (5A1E) (Rabbit) | Cell Signaling Technology | 9661S | 1:400 |
| anti-GFP (Mouse) - monoclonal | Sigma/Merck | G6539 | 1:400 |
| anti-Human Nuclei (Mouse) - monoclonal, clone 235-1 | Sigma/Merck | MAB1281 | 1:400 |
| anti-Human Nuclei (Rabbit) | NeoBiotechnologies | RBM5-346-P1 | 1:400 |
| anti-NeuN (RBFOX3) (Rabbit) - polyclonal | Sigma/Merck | ABN78 | 1:400 |
| anti-NFL (Mouse) | Sigma/Merck | MAB1615 | 1:400 |
| anti-NFL H-Phospho (Rabbit) -polyclonal | Biologend | 840801 | 1:500 |
| Aqua Polymount | Poly-sciences | 18606-20 | |
| B-27 | Gibco | 17504-044 | |
| BDNF | Gibco | PHC7074 | |
| Blades | Leica | 118364227 | |
| Cell culture graded water | Sigma/Merck | W3500-500ML | |
| Collagen from rat tail | Sigma/Merck | C7661 | |
| Confocal microscope - A1 Confocal Microscope (Eclipse Ti) | Nikon | ||
| D(+)-Glucose | Sigma/Merck | G7021 | |
| Dissecting Forceps | World Precision Instruments | 15915 | |
| DMEM/F12 | Gibco | 31330 | |
| DPBS | Sigma/Merck | D8537 | |
| EGF | Sigma/Merck | gf144 | |
| FBS | Gibco | 10270-106 | |
| FGF-2 | Stemgent | 03-0002 | |
| GDNF | Sigma/Merck | SRP3200 | |
| Glass capillaries, 3.5" | Drummond Scientific Company | 3-000-203-G/X | |
| Glutamax | Gibco | 35050-038 | |
| Goat-anti Mouse IgG Alexa Fluor 488 | Thermo Fisher Scientific | A11029 | |
| Goat-anti Mouse IgG Alexa Fluor 647 | Thermo Fisher Scientific | A21236 | 1:500 |
| Goat-anti Rabbit IgG Alexa Fluor 568 | Thermo Fisher Scientific | A11011 | 1:500 |
| Goat-anti Rabbit IgG Alexa Fluor 647 | Thermo Fisher Scientific | A21244 | 1:500 |
| Graph Pad-Prism | Dotmatics | Software for Statistical Analysis | |
| HBSS | Gibco | 14025-050 | 1:500 |
| HEPES | Gibco | 15630-056 | |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | |
| Horse Serum | Gibco | 16050-122 | |
| Insulin | Sigma/Merck | I9278 | |
| Laminin | Sigma/Merck | L2020 | |
| Lentiviral prep | Addgene | 17446-LV | |
| L-Glutamine | Thermo Fisher Scientific | 25030024 | |
| LIVE/DEAD Viability/Cytotoxicity assay kit | Thermo Fisher Scientific | L32250 | |
| McIlwain Tissue Chopper | World Precision Instruments | ||
| MEM | Gibco | 11090-081 | |
| Microloader tips | Eppendorf | 5242956003 | to load cells in the needle for transplantation |
| Microscope slides | VWR | 631-0909 | |
| Millicell cell culture membrane | Sigma/Merck | PICM0RG50 | |
| Miscroscope cover glasses | VWR | ECN 631-1572 | |
| N-2 | Gibco | 17502-048 | |
| Neurobasal | Gibco | 21103-049 | |
| Penicillin/Streptomycin | Thermo Fisher Scientific | 15140122 | |
| Petri dish (35mm) | VWR | 734-2317 | |
| PFA | Sigma/Merck | P6148-500G | |
| Plastic pasteur pipette | Sarstedt | 86.1171.010 | |
| Pneumatic PicoPump | World Precision Instruments | PV830 | Microinjector for transplantation |
| Poly-L-lysine | Sigma/Merck | P4707 | |
| Scalpel blade No 10 Sterile Stainless Steel | VWR International | SWAN3001 | |
| Scalpel handle #3 | World Precision Instruments | 500236 | |
| Spring Scissors | World Precision Instruments | 501235 | |
| Stereomicroscope for imaging and acquisition | Nikon | SMZ18 | |
| Stereomicroscope for surgery | VWR | ||
| Triton X-100 | Merck | T8787 | |
| Tweezers-Dumont #5-inox | World Precision Instruments | 501985 | |
| Vannas Scissors, 8.5 cm | World Precision Instruments | 500086 | |
| Vertical micropipette puller | Shutter Instrument | P-30 | |
| Y-27632 | R&D Systems | 1254/50 |
References
- Ding, W., et al. Spinal cord injury: The global incidence, prevalence, and disability from the Global Burden of Disease Study 2019. Spine. 47 (21), 1532-1540 (2022).
- Yang, B., et al. Strategies and prospects of effective neural circuits reconstruction after spinal cord injury. Cell Death Dis. 11 (6), 439 (2020).
- Liu, K., et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 13 (9), 1075-1081 (2010).
- Anderson, M. A., et al. Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature. 561 (7723), 396-400 (2018).
- de Freria, C. M., Van Niekerk, E., Blesch, A., Lu, P. Neural stem cells: promoting axonal regeneration and spinal cord connectivity. Cells. 10 (12), 3296 (2021).
- Badner, A., Siddiqui, A. M., Fehlings, M. G. Spinal cord injuries: how could cell therapy help. Expert Opin Biol Ther. 17 (5), 529-541 (2017).
- Assinck, P., Duncan, G. J., Hilton, B. J., Plemel, J. R., Tetzlaff, W. Cell transplantation therapy for spinal cord injury. Nat Neurosci. 20 (5), 637-647 (2017).
- Ishii, K., et al. Neutralization of ciliary neurotrophic factor reduces astrocyte production from transplanted neural stem cells and promotes regeneration of corticospinal tract fibers in spinal cord injury. J Neurosci Res. 84 (8), 1669-1681 (2006).
- Zhang, Y. W., Denham, J., Thies, R. S. Oligodendrocyte progenitor cells derived from human embryonic stem cells express neurotrophic factors. Stem Cells Dev. 15 (6), 943-952 (2006).
- Faulkner, J., Keirstead, H. S. Human embryonic stem cell-derived oligodendrocyte progenitors for the treatment of spinal cord injury. Transpl Immunol. 15 (2), 131-142 (2005).
- Kadoya, K., et al. Spinal cord reconstitution with homologous neural grafts enables robust corticospinal regeneration. Nat Med. 22 (5), 479-487 (2016).
- Dell' Anno, M. T., et al. Human neuroepithelial stem cell regional specificity enables spinal cord repair through a relay circuit. Nat Commun. 9 (1), 3419 (2018).
- Wu, S., FitzGerald, K. T., Giordano, J. On the viability and potential value of stem cells for repair and treatment of central neurotrauma: overview and speculations. Front Neurol. 9, 602 (2018).
- Nardone, R., et al. Rodent, large animal and non-human primate models of spinal cord injury. Zoology. 123, 101-114 (2017).
- Hartung, T. Thoughts on limitations of animal models. Parkinsonism Relat Disord. 14, (2008).
- Shanks, N., Greek, R., Greek, J. Are animal models predictive for humans. Philosophy, Ethics, and Humanities in Medicine. 4 (1), 2 (2009).
- Dawson, T. M., Golde, T. E., Lagier-Tourenne, C. Animal models of neurodegenerative diseases. Nat Neurosci. 21 (10), 1370-1379 (2018).
- Hayden, P. J., Harbell, J. W. Special review series on 3D organotypic culture models: Introduction and historical perspective. In Vitro Cell Dev Biol Anim. 57 (2), 95 (2021).
- Jensen, C., Teng, Y. Is it time to start transitioning from 2D to 3D cell culture. Front Mol Biosci. 7, 33 (2020).
- Mirbagheri, M., et al. Advanced cell culture platforms: a growing quest for emulating natural tissues. Materials Horizons. 6 (1), 45-71 (2019).
- Gähwiler, B. H. Organotypic monolayer cultures of nervous tissue. J Neurosci Methods. 4 (4), 329-342 (1981).
- Stoppini, L., Buchs, P. -. A., Muller, D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 37 (2), 173-182 (1991).
- Pandamooz, S., Nabiuni, M., Miyan, J., Ahmadiani, A., Dargahi, L. Organotypic spinal cord culture: a proper platform for the functional screening. Mol Neurobiol. 53 (7), 4659-4674 (2016).
- Fuller, L., Dailey, M. E. Preparation of rodent hippocampal slice cultures. CSH Protoc. 2007, (2007).
- Gertz, C. C., Lui, J. H., LaMonica, B. E., Wang, X., Kriegstein, A. R. Diverse behaviors of outer radial glia in developing ferret and human cortex. J Neurosci. 34 (7), 2559-2570 (2014).
- Ballerini, L., Galante, M. Network bursting by organotypic spinal slice cultures in the presence of bicuculline and/or strychnine is developmentally regulated. Eur J Neurosci. 10 (9), 2871-2879 (1998).
- Avossa, D., Rosato-Siri, M. D., Mazzarol, F., Ballerini, L. Spinal circuits formation: a study of developmentally regulated markers in organotypic cultures of embryonic mouse spinal cord. Neuroscience. 122 (2), 391-405 (2003).
- Lossi, L., Merighi, A. The use of ex vivo rodent platforms in neuroscience translational research with attention to the 3RS philosophy. Front Vet Sci. 5, 164 (2018).
- Nogueira, G. O., Garcez, P. P., Bardy, C., Cunningham, M. O., Sebollela, A. Modeling the human brain with ex vivo slices and in vitro organoids for translational neuroscience. Front Neurosci. 16, 838594 (2022).
- Qi, X. R., et al. Human brain slice culture: a useful tool to study brain disorders and potential therapeutic compounds. Neurosci Bull. 35 (2), 244 (2019).
- Park, H. W., et al. Human mesenchymal stem cell-derived Schwann cell-like cells exhibit neurotrophic effects, via distinct growth factor production, in a model of spinal cord injury. Glia. 58 (9), 1118-1132 (2010).
- Charrière, K., Risold, P. Y., Fellmann, D. In vitro interactions between bone marrow stromal cells and hippocampal slice cultures. C R Biol. 333 (8), 582-590 (2010).
- Jeong, D. K., Taghavi, C. E., Song, K. J., Lee, K. B., Kang, H. W. Organotypic human spinal cord slice culture as an alternative to direct transplantation of human bone marrow precursor cells for treating spinal cord injury. World Neurosurg. 75 (3-4), 533-539 (2011).
- Riggio, C., et al. Generation of magnetized olfactory ensheathing cells for regenerative studies in the central and peripheral nervous tissue. Int J Mol Sci. 14 (6), 10852-10868 (2013).
- Kamei, N., et al. Neural progenitor cells promote corticospinal axon growth in organotypic co-cultures. Neuroreport. 15 (17), 2579-2583 (2004).
- Kamei, N., et al. NGF released from transplanted neural progenitor cells promote corticospinal axon growth in organotypic cocultures. Spine. 32 (12), 1272-1278 (2007).
- Hamasaki, T., et al. Magnetically labeled neural progenitor cells, which are localized by magnetic force, promote axon growth in organotypic cocultures. Spine. 32 (21), 2300-2305 (2007).
- Kim, H. M., Lee, H. J., Lee, M. Y., Kim, S. U., Kim, B. G. Organotypic spinal cord slice culture to study neural stem/progenitor cell microenvironment in the injured spinal cord. Exp Neurobiol. 19 (2), 106-113 (2010).
- Liu, X., Chu, T. H., Su, H., Guo, A., Wu, W. Neural progenitor cell apoptosis and differentiation were affected by activated microglia in spinal cord slice culture. Neurol Sci. 35 (3), 415-419 (2014).
- De Vincentiis, S., et al. Low forces push the maturation of neural precursors into neurons. Small. 19 (30), 2205871 (2023).
- Abouelfetouh, A., Kondoh, T., Ehara, K., Kohmura, E. Morphological differentiation of bone marrow stromal cells into neuron-like cells after co-culture with hippocampal slice. Brain Res. 1029 (1), 114-119 (2004).
- Sypecka, J., Koniusz, S., Kawalec, M., Sarnowska, A. The organotypic longitudinal spinal cord slice culture for stem cell study. Stem Cells Int. 2015, 471216 (2015).
- Tanvig, M., et al. A brain slice culture model for studies of endogenous and exogenous precursor cell migration in the rostral migratory stream. Brain Res. 1295, 1-12 (2009).
- Tennstaedt, A., et al. Human neural stem cell intracerebral grafts show spontaneous early neuronal differentiation after several weeks. Biomaterials. 44, 143-154 (2015).
- Vogel, S., et al. The in vivo timeline of differentiation of engrafted human neural progenitor cells. Stem Cell Res. 37, 101429 (2019).
- Onorati, M., et al. Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 16 (10), 2576-2592 (2016).
- Vyas, A., et al. An in vitro model of adult mammalian nerve repair. Exp Neurol. 223 (1), 112-118 (2010).
- Brewer, G. J., Torricelli, J. R., Evege, E. K., Price, P. J. Optimized survival of hippocampal neurons in B27-supplemented neurobasal, a new serum-free medium combination. J Neurosci Res. 35 (5), 567-576 (1993).
- De Vries, G. H., Boullerne, A. I. Glial cell lines: an overview. Neurochem Res. 35 (12), 1978-2000 (2010).
- Ziv-Polat, O., et al. The role of neurotrophic factors conjugated to iron oxide nanoparticles in peripheral nerve regeneration: in vitro studies. Biomed Res Int. 2014, 267808 (2014).
- Mesa-Infante, V., Afonso-Oramas, D., Salas-Hernández, J., Rodríguez-Núñez, J., Barroso-Chinea, P. Long-term exposure to GDNF induces dephosphorylation of Ret, AKT, and ERK1/2, and is ineffective at protecting midbrain dopaminergic neurons in cellular models of Parkinson's disease. Mol Cell Neurosci. 118, 103684 (2022).
- Montero, A. M., Huang, A. H. The regenerative capacity of neonatal tissues. Development. 149 (12), (2022).
- Feng, L., Kwon, O., Lee, B., Oh, W. C., Kim, J. Using mammalian GFP reconstitution across synaptic partners (mGRASP) to map synaptic connectivity in the mouse brain. Nat Protoc. 9 (10), 2425-2437 (2014).
- Il Choi, D., Kaang, B. -. K. Interrogating structural plasticity among synaptic engrams. Curr Opin Neurobiol. 75, 102552 (2022).
- Choi, J. -. H., et al. Interregional synaptic maps among engram cells underlie memory formation. Science. 360 (6387), 430-435 (2018).
This article has been published
Video Coming Soon
ABOUT JoVE
Copyright © 2024 MyJoVE Corporation. All rights reserved